

Abstract
Untreated type 1 diabetes increases hepatic drug metabolism in both human patients and rodent models. We used mouse knockouts to test the role of the nuclear xenobiotic receptors CAR and PXR in this process. Streptozotocin induced diabetes resulted in increased expression of drug metabolizing cytochrome P450's and also increased the clearance of the cytochrome P450 substrate zoxazolamine. This induction was completely absent in Car-/- mice, but was not affected by the loss of PXR. Among the many effects of diabetes on the liver, we identified elevations in bile acids and activated AMP kinase as potential CAR activating stimuli. Expression of the CAR coactivator PGC-1α was also increased in mouse models of type 1 diabetes. The CAR-dependent induction of drug metabolism in newly diagnosed or poorly managed type 1 diabetes has the potential for significant impact on the efficacy or toxicity of therapeutic agents.
Keywords: Bile acids, CAR, cytochrome P450, streptozotocin, drug metabolism
The primary determinant of the half-life of many therapeutic agents is hepatic metabolism and clearance. It has been known for decades that metabolic capacity can be increased by a wide variety of chemical inducers (1). Much more recently, the two nuclear hormone receptors CAR and PXR have emerged as primary mediators of such inductive effects, and their species-specific responses to particular xenobiotic and endobiotic inducers have been well characterized (2-5). An increase in drug metabolism in rodent models of type 1 diabetes was first described nearly 50 years ago (6). Although there are some species differences in responses of specific cytochrome CYPs, as expected, a number of subsequent studies indicate that the predominant effect of type 1 diabetes is induction of a range of different enzymes of drug metabolism, particularly CYPs that are known CAR or PXR targets (7-10).
The impact of diabetes on drug metabolism in humans was examined by comparing xenobiotic clearance in nearly 300 patients with type 1 or type 2 diabetes and more than 200 controls (11). These studies demonstrated a 2-fold increase in the rate of clearance of a single oral dose of antipyrine in poorly controlled, hypoinsulinemic male type 1 diabetics. Drug metabolism was also increased by this criterion in a smaller group of newly diagnosed adult males with untreated type l diabetes (11), and this effect was eliminated by therapeutically effective insulin treatment. Male type 2 diabetics showed the opposite effect, with a moderate decrease in drug metabolism. Although there are conflicting findings potentially due to a variety of factors, such as degree of diabetic control (12), the increased drug metabolism in poorly managed type 1 diabetes is quite consistent with smaller prior human studies (13, 14).
In this study we used knockout mice to test the hypothesis that CAR or PXR activation could account for the induction of drug metabolism in Type I diabetes. Streptozotocin (STZ) induced diabetes resulted in increased expression of Cyp2B10, Cyp3A11, and Cyp2C29, and this response was reversed by insulin treatment. The induction was completely absent in Car-/- mice, indicating a central role of CAR in the enhanced drug metabolism. In addition, CAR targets are upregulated in both a humanized CAR mouse model treated with STZ and in the non-obese diabetic (NOD) model, a widely used type 1 diabetes model that shares major disease characteristics with the human disease (15). CAR can be activated indirectly by bile acids (16) via a nuclear translocation pathway dependent on adenosine monophosphate activated protein kinase (AMPK) (17, 18). We found that streptozotocin-induced diabetes results in elevation of hepatic bile acids, elevated AMPK phosphorylation and increased expression of PGC-1α, a CAR coactivator (19). We conclude that CAR activation, potentially due to elevated bile acids and AMPK activation and supported by induced expression of PGC-1α, mediates the induction of drug metabolism in response to type 1 diabetes.
Results
Enhanced drug clearance in mouse models of type 1 diabetes is CAR dependent
To critically test the potential role of CAR in the induction of drug metabolism observed in human type 1 diabetes (11, 13, 14) and rodent models of the disease (6, 9, 10), wild type and Car-/- mice were injected with 2 doses of STZ on consecutive days. One week after STZ treatment, when serum glucose was dramatically elevated (Fig. 1A), subgroups of wild type and Car-/- mice received an insulin pellet or sham operation. Loss of CAR had no effect on glucose levels in STZ treated or control mice, but serum glucose was restored to normal in insulin treated mice, as expected (Fig. 1A). In accord with previous results (9, 10), expression of Cyp2B10, Cyp3A11 and Cyp2C29 was elevated in the diabetic wild- type mice (Fig. 1B). As also expected, CAR activity was reverted to the basal, inactive state following reversal of diabetes by insulin treatment. The induction of these CAR target genes was completely absent in the diabetic Car-/- mice (Fig. 1B), indicating that the induced Cyp gene expression in this mouse model of type 1 diabetes is CAR dependent.
Figure 1. CAR is activated in STZ-induced type 1 diabetic mice and drug clearance is enhanced.
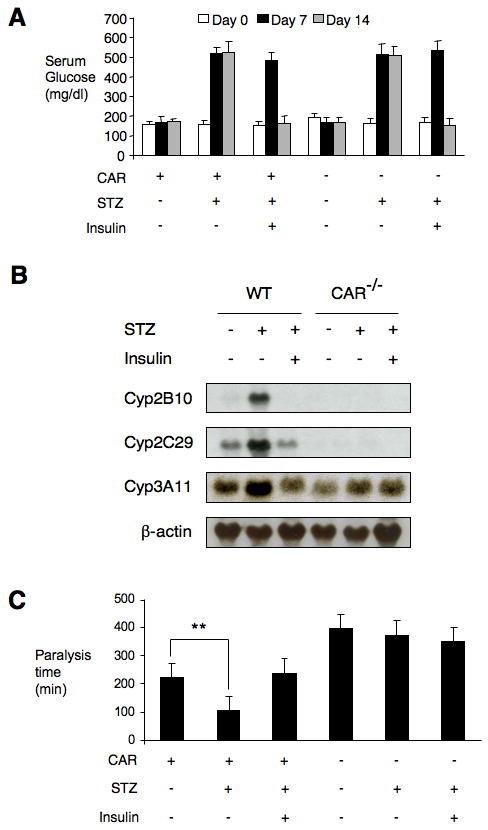
(A) Wild type and CAR-/- mice (n=5) were treated with or without STZ for 7 days and subgroups of diabetic mice were treated with insulin for 7 days. Blood glucose levels were measured on indicated days. (B) Liver total RNA was isolated from mice of different treatments and equal amounts of RNA were pooled from individual mice. Northern blot was performed with indicated probes. (C) Zoxazolamine was dosed to non-diabetic mice, diabetic mice and diabetic mice with insulin treatment. Paralysis time was compared in each group. (**p < 0.01).
The level of induction of the Cyp genes in the STZ treated mice is less than that observed with the potent CAR agonist TCPOBOP, for example. Thus, the zoxazolamine paralysis test was used to determine whether this induction is associated with a physiological increase in drug clearance. Zoxazolamine is a muscle relaxant, as well as a substrate of several CYP enzymes, and the duration of zoxazolamine-induced paralysis provides a simple indicator of drug clearance (16, 20, 21). Control groups, diabetic groups and insulin treated diabetic groups of wild type and Car-/- mice were treated with zoxazolamine (200 mg/kg) and paralysis times were recorded. Consistent with our previous results (16, 21), untreated Car-/- mice showed a 2-fold increase in paralysis time compared with untreated wild type mice (Fig. 1C). In contrast, induction of diabetes significantly decreased paralysis time in the wild type mice (2-fold) and insulin treatment completely reversed this effect, however, diabetic condition had no effect in the absence of CAR (Fig. 1C). We conclude that the induction of drug clearance in STZ induced diabetes is CAR-dependent.
Based on the species specific differences in xenobiotic responses (20, 22), we studied the response of human CAR (hCAR) to STZ induced diabetes in previously described “humanized” transgenic mice that express hCAR in the liver in a Car-/- background (22, 23). The induction of Cyp2B10 and Cyp3A11 expression in these mice confirms that hCAR can also be activated in STZ induced diabetes (Fig. 2A). We also studied CAR activation in NOD mice, which spontaneously develop autoimmune diabetes with a 20%-30% incidence in males (15). Again, we observed an upregulation of CAR targets in diabetic NOD mice, which was absent in nondiabetic NOD controls (Fig. 2B). Due to the multigenic etiology of the diabetes phenotype in the NOD mice (15), it was not practical to introduce the Car-/- allele into this strain.
Figure 2. CAR activation maintains in diabetic hCAR mice, NOD mice and PXR-/- mice.
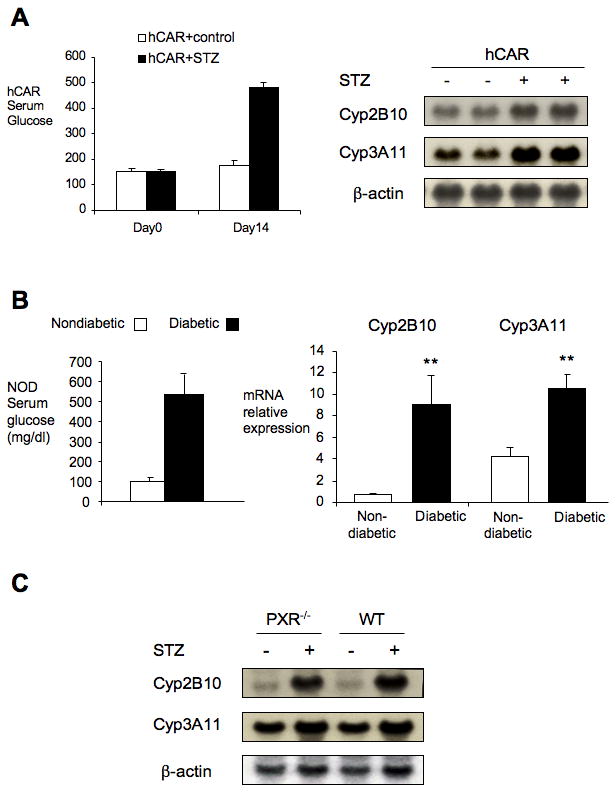
Total liver RNA of each different group was isolated. Northern blot or qPCR was performed with indicated probes. (A) hCAR mice (n=4) were treated with or without STZ for 2 weeks. Glucose and gene expression were studied. (B) 22-week male NOD non diabetic and diabetic mice (n=3-4) were checked for glucose and CAR target gene expression. (C) Wild type and PXR-/- mice (n=3-4) were treated with or without STZ for 2 weeks. Gene expression of CAR targets were studied for CAR activation. (**p<0.01)
To test whether PXR is also involved in the activation of CYP enzymes in STZ induced diabetes, Pxr-/- mice were similarly treated with STZ. In contrast to the results with the Car-/- mice, loss of PXR function did not prevent induction of Cyp2B10 or Cyp3A11 (Fig. 2C). In addition, the increased drug clearance demonstrated by the zoxazolamine paralysis test was not lost in diabetic Pxr-/- mice (Supp. 1). Thus, PXR is not required for the induction of Cyp enzymes in STZ induced diabetes.
Mechanism of CAR activation
In addition to the conventional agonist dependent response, CAR can be activated indirectly by a pathway dependent on translocation from the hepatocyte cytoplasm to the nucleus (5, 24-26). Although the molecular basis for this indirect pathway remains to be defined, it can be activated by a variety of stimuli including elevated levels of xenobiotics that do not function as agonists, notably phenobarbital (24). Elevated levels of potentially toxic endobiotics, such as bile acids (16) or bilirubin (27) can also indirectly activate CAR. It has recently been suggested that activation of AMP kinase is both necessary and sufficient for this indirect activation (17, 18).
Type 1 diabetes (28) or loss of insulin receptor signaling (29) results in myriad changes in the liver, and consequently many potential stimuli for CAR activation. In the disease state, it is not technically possible to systematically study the impact of individual stimuli on CAR, due to the impracticality of altering one such stimulus, such as hyperglycemia, without affecting others. Thus, our strategy was not to identify which of a number of potential hepatic stresses are necessary for CAR activation, but instead to identify one or more potential stimuli in the diabetic liver that are known to be sufficient to activate CAR in a normal liver. This strategy identified 2 of the 5 stimuli tested as potential contributors to CAR activation.
The simplest potential stimulus is STZ acting as a xenobiotic in the liver, rather than via β-cell elimination. Since STZ is specifically taken up into pancreatic islets and is rapidly metabolized and eliminated (30), it is unlikely that this could account for the CAR activation observed 2 weeks after the treatment. Nonetheless, wild type and Car-/- mice were treated with 2 doses of STZ by intraperitoneal injection on consecutive days, and livers were harvested on the third day. This treatment did not alter serum glucose levels or induce acute liver damage, as indicated by the absence of any effect on serum alanine aminotransferase (data not shown). It also failed to induce either Cyp2B10 or Cyp3A11 expression (Fig. 3A). Thus, STZ does not act directly as a xenobiotic to activate CAR.
Figure 3. CAR activation is not observed in mice with either acute exposure to STZ or β-hydroxybutyrate.
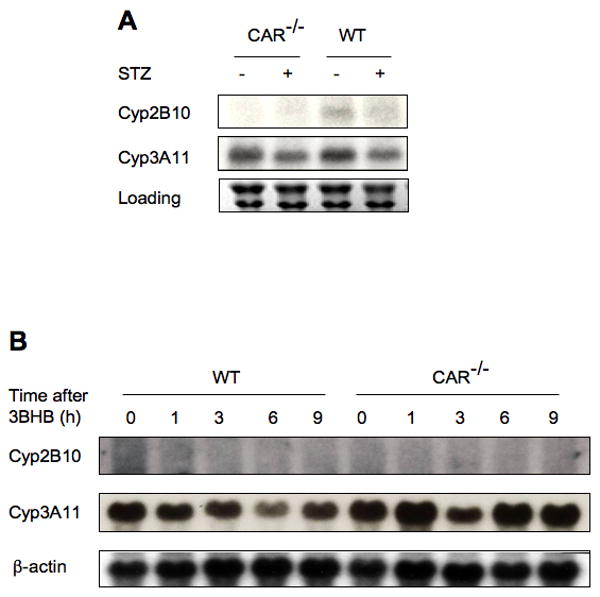
(A) Wild type and CAR-/- mice (n=5) were treated with or without STZ for 2 days and on the third day, mice were sacrificed and their liver samples were harvested. Total RNA were isolated from each group and northern blots were performed with indicated probes. (B) Wild type and CAR-/- mice (n=5) were treated with β-hydroxybutyrate and mice were sacrificed at indicated hours. Liver samples were harvested and total RNA were isolated and prepared. Northern analysis was performed with indicated probes.
Hyperglycemia is the most obvious consequence of the loss of β-cells and insulin and glucose challenge can turn on numerous genes. To test whether CAR can be activated by hyperglycemia, we challenged the mice with 6 doses of glucose in a day, a standard method for inducing hyperglycemia in nondiabetic mice (31). We did not observe any induction of CAR target genes in response to hyperglycemia with this treatment (Supp. 2), or a longer 7 day induction of hyperglycemia in mice (data not shown).
Previous studies in rats have suggested that the induction of drug metabolism in type 1 diabetes is dependent on elevation of ketone bodies (32), a well known consequence of the increased fatty acid metabolic flux in the diabetic liver. Therefore, we challenged wild type and Car-/- mice with β-hydroxybutyrate by intraperitoneal injection. In contrast to reports of increased Cyp2B levels in similarly treated rats, we observed a moderate reduction of Cyp gene expression in ketone body challenged wild type mice; this reduction was absent in similarly treated Car-/- mice (Fig. 3B). The lack of an inductive effect was not due to ineffective delivery or more rapid clearance in mice, since increasing the β-hydroxybutyrate dose led to lethality. In addition, no CAR activation was observed in primary hepatocytes treated with increasing doses of ketone bodies (Supp. 3). We conclude that elevated levels of ketone bodies are not sufficient to activate CAR in mice, and are unlikely to account for the diabetes-induced CAR activation.
Bile acids have been identified as CAR activators (16) and diabetes has been associated with elevated levels of bile acids or bile flow in a number of studies in rats and mice (33-35). Thus, we compared serum and hepatic levels of bile acids in diabetic and non-diabetic mice. Consistent with the prior studies, we observed a significant elevation of both serum and hepatic bile acids in both wild type and Car-/- diabetic mice (Fig. 4). This elevation of hepatic bile acids may contribute to CAR activation in type 1 diabetes.
Figure 4. Elevation of bile acid levels in STZ-induced diabetic mice.
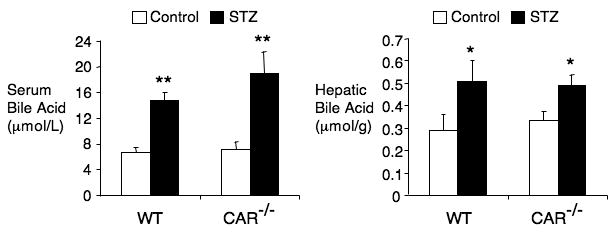
Wild type and CAR-/- mice (n=7) were treated with or without STZ for 2 weeks and serum and hepatic bile acid levels were measured. (*p<0.05; **p<0.01)
The last stimulus examined was AMP kinase (AMPK). AMPK is activated when cellular ATP levels are depleted and has been identified as a target of the type 2 diabetes treatment metformin. The impact of type 1 diabetes on AMPK activation has not been explored, but recent reports described ATP decreases and AMP/ATP ratio increases in STZ induced diabetic rodent livers (36, 37), strongly suggesting that AMPK may be activated in type 1 diabetes. Using an anti-phospho threonine 172 antibody, we found that AMPK is activated in STZ induced diabetic liver of both wild type and Car-/- mice (Fig. 5). This indicates that type 1 diabetes leads to AMPK activation, which is CAR independent.
Figure 5. AMPK is activated in STZ-induced diabetic mice.
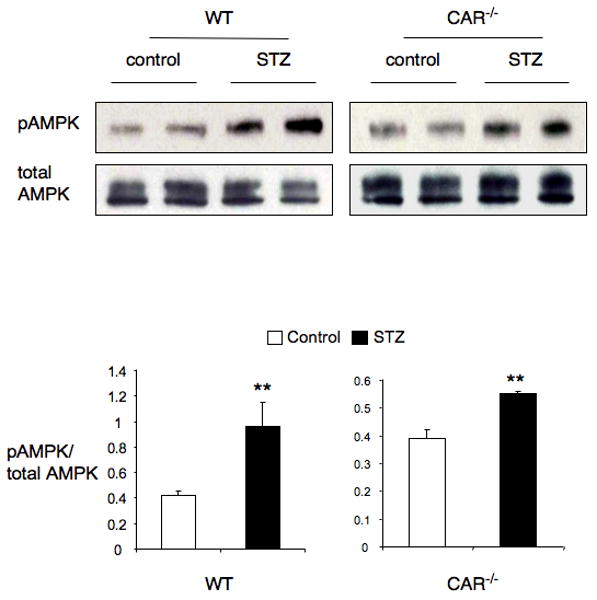
Wild type and CAR-/- mice (n=4) were treated with or without STZ for 2 weeks and liver samples were harvested. Liver tissue from different groups was homogenized and every two samples from same groups were pooled together and 30 μg of protein was separated on SDS gel. Phospho-AMPK and total AMPK level were determined by Western blot using antibodies as indicated. The graphs demonstrate the quantification of phosphorylation of AMPK. (**p<0.01)
In addition to these activating stimuli, upregulated expression of CAR or its coactivators, such as PGC-1α (19), could contribute to the response of CAR target genes. Although a statistically significant increase in CAR mRNA expression was observed in several experiments, this response was not completely consistent. In agreement with a previous study in STZ treated mice (38), however, we consistently observed a substantial induction of PGC-1α in both the STZ treated and NOD diabetic mouse models (Fig 6).
Figure 6. PGC-1α expression is induced in mouse models of type 1 diabetes.
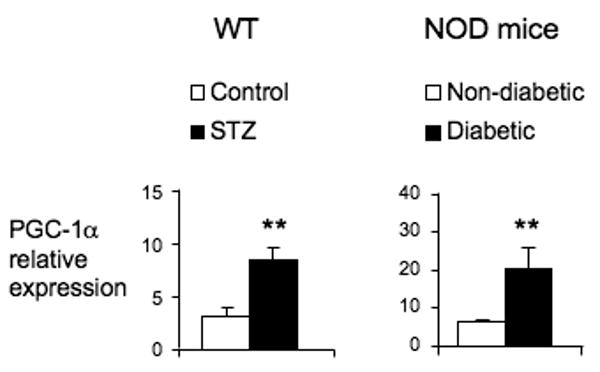
Liver RNA from WT mice (n=4) treated with or without STZ for 2 weeks and NOD nondiabetic and diabetic mice (n=3-4) was extracted and analyzed for PGC-1α by quantitative RT-PCR. (**p<0.01)
Discussion
CAR and its close relative PXR are major regulators of drug metabolism (2-5). They control expression of a common set of genes including those catalyzing phase I and phase II drug clearance, suggesting overlapping roles. Consistent with this, activation of either receptor can protect against potentially toxic endobiotics, such as bile acids and bilirubin. However, functional studies using knockout mice have often revealed more specific functions (e.g. (16)). In general, PXR appears to act primarily in xenobiotic induction of drug metabolism, while CAR may be more important in response to endogenous stimuli. This is consistent with the response of the indirect pathway of CAR activation to a range of endobiotic stresses (5, 16, 27).
Type 1 diabetes is accompanied by many liver abnormalities, including alterations in carbohydrate, lipid and protein metabolism (28). Although it has been known for many years (6), it is less well recognized that drug metabolism is induced in the type 1 diabetic liver. More recently, CAR targets were among the numerous genes whose expression were increased in mice lacking both Irs1 and Irs2 in the liver (39), but the molecular mechanism that underlies this induction has not been characterized. Here we show that the activation of cytochrome P450 gene expression by STZ induced diabetes is absent in Car-/- mice, but is unaffected by the loss of PXR. The physiologic relevance of this induction is strongly supported by the observation that the STZ treated mice, despite their numerous liver problems, show increased zoxazolamine clearance. We conclude that CAR is an essential mediator of the enhanced drug metabolism in mouse models of type 1 diabetes.
There are a number of potential CAR activating stimuli in the type 1 diabetic liver. Theoretically, it might be simplest to address the question of what activates CAR by systematically eliminating each candidate individually. However, this is not possible due to the complexity of the impact of type 1 diabetes on the liver and the tight interconnection of metabolic pathways. Moreover, the strategy of identifying a particular stimulus necessary for the induction would fail if each of two or more stimuli were sufficient for CAR activation. We therefore focused on identifying stimuli present in the diabetic liver that, individually, would be sufficient to activate CAR in the normal liver.
Among the five quite different candidates tested, the potential contribution of STZ acting in the liver as a xenobiotic was clearly ruled out by its lack of an acute effect on CAR target genes in the STZ induced type 1 diabetes model. Even if STZ were able to acutely activate CAR, it could not account for the CAR response observed two weeks after the treatment since it is very rapidly eliminated (30). Moreover, CAR is also activated robustly in the NOD model.
There was also no evidence to support an important role for either hyperglycemia or hyperketonemia. Inducing sustained hyperglycemia in normal mice is complicated by the effect of insulin, which necessitates multiple treatments and results in fluctuating serum levels. In addition, insulin has been reported to modestly suppress the phenobarbital induction of CAR target genes in primary hepatocytes (40). The lack of an effect of hyperglycemia is consistent with the observation that CAR shows little or no activation in Ob/Ob mice (BD and DDM, unpublished), which have serum glucose levels comparable to those in the STZ treated mice. Despite reported stimulatory effects of elevated ketone bodies on expression of drug metabolizing enzymes in rats (41), we were unable to reproduce similar responses to elevated β-hydroxybutyrate in normal mice or in primary hepatocytes contrasts. We were also unable to reproduce the reported inductive effects of another prominent ketone body, acetone, due to problems of lethality with both normal mice and primary hepatocytes. While these negative results cannot rule out a contributory effect in the context of the type 1 diabetic liver, particularly for hyperglycemia and hyperketonemia, we conclude that none of these three stimuli is sufficient to activate CAR.
In contrast to these negative results, we observed significant elevations of both bile acids and activated AMPK in the diabetic livers. The increased bile acid levels are consistent with previous studies in diabetic rodents (33-35), as well as the reported repressive effect of insulin on Cyp7A1 expression and bile acid production (42, 43). Since the hepatic bile acid levels in the STZ treated mice are comparable to those observed in either Fxr-/- mice or Little mice (Ghrhrlit/lit), which are thought to account for CAR activation and induction of drug metabolism in both cases (44, 45) we conclude that they are sufficient to activate CAR.
Since AMPK is the target for metformin and a well known metabolic regulator (46), it is somewhat surprising that we were unable to identify prior studies on the effect of type 1 diabetes on AMPK activity. Consistent with reports that the AMP/ATP ratio increases in STZ induced diabetes (36, 37), however, our studies revealed a reproducible increase in the levels of activated threonine 172 phosphorylated AMPK. This approximately 2-3 fold phosphorylation response is less than that described for the CAR activator phenobarbital in cell lines (47), but comparable to or greater than the increase in AMPK enzyme activity in phenobarbital treated liver (17). Thus, the observed elevations of both bile acids and AMPK activity are sufficient to activate CAR in normal liver, and we conclude that both are likely to contribute to the activation of CAR in the type 1 diabetic liver. Increased expression of PGC-1α presumably also contributes to CAR transcriptional activity in the mouse models of type 1 diabetes.
It is also surprising that the clear impact of poorly managed type 1 diabetes on drug metabolism in human patients (11, 13, 14) has not been well understood or appreciated. As recently reviewed (48), there has been only a limited linkage of the type 1 disease to altered drug metabolism or hepatotoxicity in human patients. One possible example is the association of type 1 diabetes with an increase in methotrexate-induced hepatotoxicity (49-52). In rats, however, this has been ascribed to decreased drug clearance (53), which seems more likely to be a reflection of other complications of the disease than to an induction of drug metabolism. Nonetheless, the complications of diabetes result in increased exposure to a variety of drugs, some of which have a narrow therapeutic range (54). Particularly since many patients fail to achieve complete glucose control, the CAR-dependent induction of drug metabolism in newly diagnosed or poorly managed type 1 diabetes has the potential for substantial clinical impact.
Experimental Procedures
Animal treatment
Mice were fed with a standard diet and maintained in a pathogen-free animal facility. All studies were approved by the BCM Institutional Animal Care and Use Committee. 10-week old wild type (C57BL/6) mice, Car-/- mice (21) (>10 backcrosses to C57BL/6), humanized CAR mice (22) and Pxr-/- mice (20) were used in the experiments. Type 1 diabetes was induced by 2 daily intraperitoneal doses of streptozotocin (STZ) 125 mg/kg body weight. STZ was dissolved in 0.01 M sodium citrate buffer (pH 4.3) immediately before administration. All of the mice developed diabetes at one week (blood glucose >350 mg/dl). Subsets of the diabetic mice received insulin treatment or sham operation. In the insulin treatment group, mice received a subcutaneous implant of a sustained release insulin pellet (Linplant, LinShin Canada, Inc.) for 1 week. A control group of diabetic mice was maintained on normal diet after sham operation. NOD mice were provided by Drs. Yechoor and Chan at BCM. For drug clearance, mice were treated intraperitoneally with zoxazolamine (200 mg/kg) or 3-β-hydroxybutyrate (20 mmol/kg) for indicated time periods. All drugs were purchased from Sigma.
Serum Glucose, ALT and Bile Acid Measurement
Blood glucose was measured by tail bleeds and values were determined by One-Touch Ultra glucometer (Lifescan, Milpitas, CA). For serum bile acid and ALT, blood was collected after 6 hours fasting. Serum was separated by centrifugation at 6000 × g for 10 min. Alanine aminotransferase (ALT) levels and total bile acid were measured as previously reported (16, 23). At least four mice were used for each treatment group.
RNA Preparation and Gene Expresssion Analysis
Total liver RNA was extracted using Trizol (Invitrogen, Carlsbad, CA). Equivalent amounts of RNA from each treatment group were pooled, and 20 μg was used for Northern blot analysis. Primers used for generation of cDNA probes were described (16, 23). All blots were stripped and rehybridized with β-actin as internal control. For qPCR, RNA was reverse-transcribed using SuperScript™ III RT (Invitrogen). Samples were analyzed using an ABI prism 7500 with SYBR green and TaqMan reagents (Applied Biosystems) and compared to GAPDH control. Primers and probe used:
GAPDH
F: 5′-CCTACCCCCAATGTGTCCG-3′
R: 5′-CCTTCTTGATGTCATCATACTTGGC-3′
CYP2B10
F: 5′-GACTTTGGGATGGGAAAGAG-3′
R: 5′-CCAAACACAATGGAGCAGAT-3′
Fluorogenic probe (FAM-TAGTGGAGGAACTGCGGAAATCCC-BHQ1)
CYP3A11
F: 5′-AGATTGGTTTTGATGCCTGGTT-3′
R: 5′-GCAAATTTCCTGTGCTGTCACT-3′
Primary hepatocyte culture
Mouse primary hepatocytes were prepared and maintained in William's E medium (Invitrogen, Carlsbad, CA), supplemented with 10 μg/ml insulin (Sigma) and 10-7 M triamcinolone actinide. Cells were treated with different concentrations of 3-β-hydroxybutyrate for indicated time.
Protein Analysis and Western blot
Frozen liver samples were excised and homogenized in ice-cold lysis buffer (25 mM Tris-HCl (pH 7.4), 10 mM sodium orthovanadate, 10 mM sodium pyrophosphate, 100 mM sodium fluoride, 10 mM EDTA, 10 mM EGTA, and 1 mM phenylmethylsulfonyl fluoride). Samples were resolved by 10% PAGE and transferred to a Hybond-P polyvinylidene difluoride transfer membrane (Amersham Biosciences, Piscataway, NJ). Membranes were immunoblotted with antibodies specific for phospho-AMPK and anti-AMPK (Cell Signaling Technology, Inc., Beverly, MA) and bands were detected using ECL kit (Amersham Biosciences).
Statistics
Data are presented in all figures as mean ± S.E. The number of subjects is indicated by n. Figures shown are representative of consistent results; differences between different genotypes were calculated by the 2-tailed Student's t test. All statistical tests with P<0.05 were considered significant.
Supplementary Material
Figure 1. Enhanced drug clearance is maintained in STZ treated PXR-/- mice. Wild type and PXR-/- mice (n=6) were treated with or without STZ for 2 weeks and zoxazolamine was dosed to each groups. Paralysis time of wild type and PXR-/- diabetic mice and their controls was monitored. (**p<0.01).
Figure 2. Hyperglycemia does not activate CAR. Wild type mice (n=4) were IP injected with 2.0 g/kg glucose 6 times in 1 day experiment. Mice were sacrificed and liver samples were harvested. Total RNA was isolated and equal amounts of RNA from individual mice were pooled. Northern blot was performed with indicated probes.
Figure 3. Ketone bodies do not activate CAR. Primary hepatocytes from wild type and CAR-/- mice were cultured in William's E medium and incubated with different concentrations of 3-beta-hydroxybutyrate for indicated time. Gene expression was analyzed by RT-PCR using Super-Script One Step RT-PCR system (Invitrogen) with the GAPDH as the internal control.
Acknowledgments
Financial Support: NIH R01 DK46546
Abbreviations
- CAR
constitutive androstane receptor
- PXR
pregnane and xenobiotic receptor
- AMPK
AMP activated protein kinase
- STZ
streptozotocin
- NOD
non-obese diabetes
- CYP
cytochrome P40
References
- 1.Remmer H, Merker HJ. Drug-Induced Changes In The Liver Endoplasmic Reticulum: Association With Drug-Metabolizing Enzymes. Science. 1963;142:1657–1658. doi: 10.1126/science.142.3600.1657. [DOI] [PubMed] [Google Scholar]
- 2.Willson TM, Kliewer SA. PXR, CAR and drug metabolism. Nat Rev Drug Discov. 2002;1:259–266. doi: 10.1038/nrd753. [DOI] [PubMed] [Google Scholar]
- 3.Sonoda J, Rosenfeld JM, Xu L, Evans RM, Xie W. A nuclear receptor-mediated xenobiotic response and its implication in drug metabolism and host protection. Curr Drug Metab. 2003;4:59–72. doi: 10.2174/1389200033336739. [DOI] [PubMed] [Google Scholar]
- 4.Honkakoski P, Sueyoshi T, Negishi M. Drug-activated nuclear receptors CAR and PXR. Ann Med. 2003;35:172–182. doi: 10.1080/07853890310008224. [DOI] [PubMed] [Google Scholar]
- 5.Qatanani M, Moore DD. CAR, the continuously advancing receptor, in drug metabolism and disease. Curr Drug Metab. 2005;6:329–339. doi: 10.2174/1389200054633899. [DOI] [PubMed] [Google Scholar]
- 6.Dixon RL, Hart LG, Fouts JR. The metabolism of drugs by liver microsomes from alloxan-diabetic rats. J Pharmacol Exp Ther. 1961;133:7–11. [PubMed] [Google Scholar]
- 7.Schenkman JB, Thummel KE, Favreau LV. Physiological and pathophysiological alterations in rat hepatic cytochrome P-450. Drug Metab Rev. 1989;20:557–584. doi: 10.3109/03602538909103562. [DOI] [PubMed] [Google Scholar]
- 8.Shimojo N. Cytochrome P450 changes in rats with streptozocin-induced diabetes. Int J Biochem. 1994;26:1261–1268. doi: 10.1016/0020-711x(94)90095-7. [DOI] [PubMed] [Google Scholar]
- 9.Sakuma T, Honma R, Maguchi S, Tamaki H, Nemoto N. Different expression of hepatic and renal cytochrome P450s between the streptozotocin-induced diabetic mouse and rat. Xenobiotica. 2001;31:223–237. doi: 10.1080/00498250110046451. [DOI] [PubMed] [Google Scholar]
- 10.Sindhu RK, Koo JR, Sindhu KK, Ehdaie A, Farmand F, Roberts CK. Differential regulation of hepatic cytochrome P450 monooxygenases in streptozotocin-induced diabetic rats. Free Radic Res. 2006;40:921–928. doi: 10.1080/10715760600801272. [DOI] [PubMed] [Google Scholar]
- 11.Sotaniemi EA, Pelkonen O, Arranto AJ, Tapanainen P, Rautio A, Pasanen M. Diabetes and elimination of antipyrine in man: an analysis of 298 patients classified by type of diabetes, age, sex, duration of disease and liver involvement. Pharmacol Toxicol. 2002;90:155–160. doi: 10.1034/j.1600-0773.2002.900308.x. [DOI] [PubMed] [Google Scholar]
- 12.Cheng PY, Morgan ET. Hepatic cytochrome P450 regulation in disease states. Curr Drug Metab. 2001;2:165–183. doi: 10.2174/1389200013338676. [DOI] [PubMed] [Google Scholar]
- 13.Zysset T, Wietholtz H. Differential effect of type I and type II diabetes on antipyrine disposition in man. Eur J Clin Pharmacol. 1988;34:369–375. doi: 10.1007/BF00542438. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein S, Simpson A, Saenger P. Hepatic drug metabolism is increased in poorly controlled insulin-dependent diabetes mellitus. Acta Endocrinol (Copenh) 1990;123:550–556. doi: 10.1530/acta.0.1230550. [DOI] [PubMed] [Google Scholar]
- 15.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Huang W, Qatanani M, Evans RM, Moore DD. The constitutive androstane receptor and pregnane X receptor function coordinately to prevent bile acid-induced hepatotoxicity. J Biol Chem. 2004;279:49517–49522. doi: 10.1074/jbc.M409041200. [DOI] [PubMed] [Google Scholar]
- 17.Rencurel F, Foretz M, Kaufmann MR, Stroka D, Looser R, Leclerc I, da Silva Xavier G, et al. Stimulation of AMP-activated protein kinase is essential for the induction of drug metabolizing enzymes by phenobarbital in human and mouse liver. Mol Pharmacol. 2006 doi: 10.1124/mol.106.029421. [DOI] [PubMed] [Google Scholar]
- 18.Blattler SM, Rencurel F, Kaufmann MR, Meyer UA. In the regulation of cytochrome P450 genes, phenobarbital targets LKB1 for necessary activation of AMP-activated protein kinase. Proc Natl Acad Sci U S A. 2007;104:1045–1050. doi: 10.1073/pnas.0610216104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shiraki T, Sakai N, Kanaya E, Jingami H. Activation of orphan nuclear constitutive androstane receptor requires subnuclear targeting by peroxisome proliferator-activated receptor gamma coactivator-1 alpha. A possible link between xenobiotic response and nutritional state. J Biol Chem. 2003;278:11344–11350. doi: 10.1074/jbc.M212859200. [DOI] [PubMed] [Google Scholar]
- 20.Xie W, Barwick JL, Downes M, Blumberg B, Simon CM, Nelson MC, Neuschwander-Tetri BA, et al. Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature. 2000;406:435–439. doi: 10.1038/35019116. [DOI] [PubMed] [Google Scholar]
- 21.Wei P, Zhang J, Egan-Hafley M, Liang S, Moore DD. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature. 2000;407:920–923. doi: 10.1038/35038112. [DOI] [PubMed] [Google Scholar]
- 22.Huang W, Zhang J, Wei P, Schrader WT, Moore DD. Meclizine Is an Agonist Ligand for Mouse Constitutive Androstane Receptor (CAR) and an Inverse Agonist for Human CAR. Mol Endocrinol. 2004;18:2402–2408. doi: 10.1210/me.2004-0046. [DOI] [PubMed] [Google Scholar]
- 23.Zhang J, Huang W, Chua SS, Wei P, Moore DD. Modulation of acetaminophen-induced hepatotoxicity by the xenobiotic receptor CAR. Science. 2002;298:422–424. doi: 10.1126/science.1073502. [DOI] [PubMed] [Google Scholar]
- 24.Kawamoto T, Sueyoshi T, Zelko I, Moore R, Washburn K, Negishi M. Phenobarbital-responsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol Cell Biol. 1999;19:6318–6322. doi: 10.1128/mcb.19.9.6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tzameli I, Pissios P, Schuetz EG, Moore DD. The xenobiotic compound 1,4-Bis[2-(3,5-Dichloropyridyloxy)]Benzene is an agonist ligand for the nuclear receptor CAR. Mol Cell Biol. 2000;20:2951–2958. doi: 10.1128/mcb.20.9.2951-2958.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kodama S, Negishi M. Phenobarbital confers its diverse effects by activating the orphan nuclear receptor car. Drug Metab Rev. 2006;38:75–87. doi: 10.1080/03602530600569851. [DOI] [PubMed] [Google Scholar]
- 27.Huang W, Zhang J, Chua SS, Qatanani M, Han Y, Granata R, Moore DD. Induction of bilirubin clearance by the constitutive androstane receptor (CAR) Proc Natl Acad Sci U S A. 2003;100:4156–4161. doi: 10.1073/pnas.0630614100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev. 2007;87:507–520. doi: 10.1152/physrev.00024.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michael MD, Kulkarni RN, Postic C, Previs SF, Shulman GI, Magnuson MA, Kahn CR. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6:87–97. [PubMed] [Google Scholar]
- 30.Karunanayake EH, Hearse DJ, Mellows G. Streptozotocin: its excretion and metabolism in the rat. Diabetologia. 1976;12:483–488. doi: 10.1007/BF01219512. [DOI] [PubMed] [Google Scholar]
- 31.Kojima H, Fujimiya M, Matsumura K, Nakahara T, Hara M, Chan L. Extrapancreatic insulin-producing cells in multiple organs in diabetes. Proc Natl Acad Sci U S A. 2004;101:2458–2463. doi: 10.1073/pnas.0308690100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barnett CR, Abbott RA, Bailey CJ, Flatt PR, Ioannides C. Cytochrome P-450-dependent mixed-function oxidase and glutathione S-transferase activities in spontaneous obesity-diabetes. Biochem Pharmacol. 1992;43:1868–1871. doi: 10.1016/0006-2952(92)90724-w. [DOI] [PubMed] [Google Scholar]
- 33.Nervi FO, Severin CH, Valdivieso VD. Bile acid pool changes and regulation of cholate synthesis in experimental diabetes. Biochim Biophys Acta. 1978;529:212–223. doi: 10.1016/0005-2760(78)90064-4. [DOI] [PubMed] [Google Scholar]
- 34.Uchida K, Makino S, Akiyoshi T. Altered bile acid metabolism in nonobese, spontaneously diabetic (NOD) mice. Diabetes. 1985;34:79–83. doi: 10.2337/diab.34.1.79. [DOI] [PubMed] [Google Scholar]
- 35.Watkins JB, 3rd, Dykstra TP. Alterations in biliary excretory function by streptozotocin-induced diabetes. Drug Metab Dispos. 1987;15:177–183. [PubMed] [Google Scholar]
- 36.Fagan TE, Cefaratti C, Romani A. Streptozotocin-induced diabetes impairs Mg2+ homeostasis and uptake in rat liver cells. Am J Physiol Endocrinol Metab. 2004;286:E184–193. doi: 10.1152/ajpendo.00200.2003. [DOI] [PubMed] [Google Scholar]
- 37.Miyamoto A, Takeshita M, Pan-Hou H, Fujimori H. Hepatic changes in adenine nucleotide levels and adenosine 3′-monophosphate forming enzyme in streptozotocin-induced diabetic mice. J Toxicol Sci. 2008;33:209–217. doi: 10.2131/jts.33.209. [DOI] [PubMed] [Google Scholar]
- 38.Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 39.Dong X, Park S, Lin X, Copps K, Yi X, White MF. Irs1 and Irs2 signaling is essential for hepatic glucose homeostasis and systemic growth. J Clin Invest. 2006;116:101–114. doi: 10.1172/JCI25735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sidhu JS, Omiecinski CJ. Insulin-mediated modulation of cytochrome P450 gene induction profiles in primary rat hepatocyte cultures. J Biochem Mol Toxicol. 1999;13:1–9. doi: 10.1002/(sici)1099-0461(1999)13:1<1::aid-jbt1>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 41.Barnett CR, Petrides L, Wilson J, Flatt PR, Ioannides C. Induction of rat hepatic mixed-function oxidases by acetone and other physiological ketones: their role in diabetes-induced changes in cytochrome P450 proteins. Xenobiotica. 1992;22:1441–1450. doi: 10.3109/00498259209056694. [DOI] [PubMed] [Google Scholar]
- 42.Twisk J, Hoekman MF, Lehmann EM, Meijer P, Mager WH, Princen HM. Insulin suppresses bile acid synthesis in cultured rat hepatocytes by down-regulation of cholesterol 7 alpha-hydroxylase and sterol 27-hydroxylase gene transcription. Hepatology. 1995;21:501–510. [PubMed] [Google Scholar]
- 43.Crestani M, Stroup D, Chiang JY. Hormonal regulation of the cholesterol 7 alpha-hydroxylase gene (CYP7) J Lipid Res. 1995;36:2419–2432. [PubMed] [Google Scholar]
- 44.Guo GL, Lambert G, Negishi M, Ward JM, Brewer HB, Jr, Kliewer SA, Gonzalez FJ, et al. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J Biol Chem. 2003;278:45062–45071. doi: 10.1074/jbc.M307145200. [DOI] [PubMed] [Google Scholar]
- 45.Amador-Noguez D, Dean A, Huang W, Setchell K, Moore D, Darlington G. Alterations in xenobiotic metabolism in the long-lived Little mice. Aging Cell. 2007;6:453–470. doi: 10.1111/j.1474-9726.2007.00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 47.Rencurel F, Stenhouse A, Hawley SA, Friedberg T, Hardie DG, Sutherland C, Wolf CR. AMP-activated protein kinase mediates phenobarbital induction of CYP2B gene expression in hepatocytes and a newly derived human hepatoma cell line. J Biol Chem. 2005;280:4367–4373. doi: 10.1074/jbc.M412711200. [DOI] [PubMed] [Google Scholar]
- 48.Wang T, Shankar K, Ronis MJ, Mehendale HM. Mechanisms and outcomes of drug- and toxicant-induced liver toxicity in diabetes. Crit Rev Toxicol. 2007;37:413–459. doi: 10.1080/10408440701215100. [DOI] [PubMed] [Google Scholar]
- 49.Roenigk HH, Jr, Bergfeld WF, St Jacques R, Owens FJ, Hawk WA. Hepatotoxicity of methotrexate in the treatment of psoriasis. Arch Dermatol. 1971;103:250–261. [PubMed] [Google Scholar]
- 50.Nyfors A. Benefits and adverse drug experiences during long-term methotrexate treatment of 248 psoriatics. Dan Med Bull. 1978;25:208–211. [PubMed] [Google Scholar]
- 51.Erickson AR, Reddy V, Vogelgesang SA, West SG. Usefulness of the American College of Rheumatology recommendations for liver biopsy in methotrexate-treated rheumatoid arthritis patients. Arthritis Rheum. 1995;38:1115–1119. doi: 10.1002/art.1780380814. [DOI] [PubMed] [Google Scholar]
- 52.Malatjalian DA, Ross JB, Williams CN, Colwell SJ, Eastwood BJ. Methotrexate hepatotoxicity in psoriatics: report of 104 patients from Nova Scotia, with analysis of risks from obesity, diabetes and alcohol consumption during long term follow-up. Can J Gastroenterol. 1996;10:369–375. doi: 10.1155/1996/213596. [DOI] [PubMed] [Google Scholar]
- 53.Park JM, Moon CH, Lee MG. Pharmacokinetic changes of methotrexate after intravenous administration to streptozotocin-induced diabetes mellitus rats. Res Commun Mol Pathol Pharmacol. 1996;93:343–352. [PubMed] [Google Scholar]
- 54.Isacson D, Stalhammar J. Prescription drug use among diabetics--a population study. J Chronic Dis. 1987;40:651–660. doi: 10.1016/0021-9681(87)90101-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1. Enhanced drug clearance is maintained in STZ treated PXR-/- mice. Wild type and PXR-/- mice (n=6) were treated with or without STZ for 2 weeks and zoxazolamine was dosed to each groups. Paralysis time of wild type and PXR-/- diabetic mice and their controls was monitored. (**p<0.01).
Figure 2. Hyperglycemia does not activate CAR. Wild type mice (n=4) were IP injected with 2.0 g/kg glucose 6 times in 1 day experiment. Mice were sacrificed and liver samples were harvested. Total RNA was isolated and equal amounts of RNA from individual mice were pooled. Northern blot was performed with indicated probes.
Figure 3. Ketone bodies do not activate CAR. Primary hepatocytes from wild type and CAR-/- mice were cultured in William's E medium and incubated with different concentrations of 3-beta-hydroxybutyrate for indicated time. Gene expression was analyzed by RT-PCR using Super-Script One Step RT-PCR system (Invitrogen) with the GAPDH as the internal control.