

Abstract
The MUC1 oncoprotein is aberrantly overexpressed by approximately 90% of human breast cancers. However, there are no effective agents that directly inhibit MUC1 and induce death of breast cancer cells. We have synthesized a MUC1 inhibitor, designated GO-201, that binds to the MUC1 cytoplasmic domain and blocks the formation of MUC1 oligomers in cells. GO-201, and not an altered version, attenuates targeting of MUC1 to the nucleus of human breast cancer cells, disrupts redox balance and activates the DNA damage response. GO-201 also arrests growth and induces necrotic death. By contrast the MUC1 inhibitor has no effect on cells null for MUC1 expression or non-malignant mammary epithelial cells. Administration of GO-201 to nude mice bearing human breast tumor xenografts was associated with loss of tumorigenicity and extensive necrosis that results in prolonged regression of tumor growth. These findings demonstrate that targeting the MUC1 oncoprotein is effective in inducing death of human breast cancer cells in vitro and in tumor models.
Introduction
Mucins are extensively O-glycosylated proteins that are predominantly expressed by epithelial cells. The secreted and membrane-bound mucins form a physical barrier that protects the apical borders of epithelial cells from damage induced by toxins, microorganisms and other forms of stress that occur at the interface with the external environment. The transmembrane mucin 1 (MUC1) has no sequence similarity with other membrane-bound mucins, except for the presence of a sea urchin sperm protein-enterokinase-agrin (SEA) domain (1). MUC1 is translated as a single polypeptide and then undergoes autocleavage at the SEA domain with the generation of two subunits that form a stable heterodimer (2, 3). The MUC1 N-terminal subunit (MUC1-N) contains variable numbers of tandem repeats that are modified by O-glycosylation (4). MUC1-N extends beyond the glycocalyx of the cell and is tethered to the cell surface through noncovalent binding to the transmembrane MUC1 C-terminal subunit (MUC1-C) (5). MUC1-C consists of a 58 amino acid extracellular domain, a 28 amino acid transmembrane domain and a 72 amino acid cytoplasmic domain that interacts with diverse signaling molecules (6). Shedding of MUC1-N into the protective physical barrier leaves MUC1-C at the cell surface as a putative receptor to transduce intracellular signals that confer growth and survival (7, 8).
With transformation and loss of polarity, MUC1 is expressed at high levels on the entire cell surface in a wide range of carcinomas of the breast, lung, prostate, gastrointestinal tract and other epithelia (9). Loss of restriction to the apical membrane allows for the formation of complexes with the epidermal growth factor receptor (EGFR) and coactivation of EGFR-mediated signaling (7, 10). Overexpression of MUC1 by carcinoma cells is associated with accumulation of MUC1-C in the cytosol and targeting of this subunit to the nucleus (11-13) and mitochondria (14, 15). Importantly, the MUC1-C cytoplasmic domain activates expression of gene signatures that are predictive of both response to tamoxifen and overall survival in breast cancer patients (16, 17). In this context, oligomerization of MUC1-C is necessary for its nuclear targeting and interaction with diverse effectors (18). For example, the MUC1-C cytoplasmic domain (MUC1-CD) functions as a substrate for c-Src (19), c-Abl (20), protein kinase Cδ (21) and glycogen synthase kinase 3β (22) and interacts directly with the Wnt pathway effector, β-catenin (23, 24), and the p53 tumor suppressor (25). Other work has shown that overexpression of the MUC1 heterodimer confers anchorage-independent growth and tumorigenicity (12, 14, 25, 26), at least in part through stabilization of β-catenin (24). Moreover, consistent with a survival function for normal epithelial cells, overexpression of the MUC1 heterodimer confers resistance of carcinoma cells to stress-induced apoptosis by a mechanism mediated in part through suppression of intracellular reactive oxygen species (ROS) (14, 27-29).
The present results demonstrate that targeting MUC1-C oligomerization blocks nuclear localization of MUC1-C and induces growth arrest and death of human breast cancer cells. The findings also demonstrate that inhibiting MUC1-C is highly effective in the treatment of human breast tumor xenografts in nude mice.
Materials and Methods
Cell culture
Human ZR-75-1 and MDA-MB-231 cell lines were grown in RPMI1640 medium supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. Human MCF-7 breast cancer cells and 293 cells were grown in Dulbecco’s modified Eagle’s medium with 10% HI-FBS, antibiotics and 2 mM L-glutamine. Primary human breast cancer cells isolated from a malignant pleural effusion were cultured in RPMI1640 medium containing 10% serum. Human MCF-10A breast epithelial cells were grown in mammary epithelial cell growth medium (MEGM; Lonza). Cells were treated with the GO-201, GO-202 or CP-1 peptides synthesized by the MIT Biopolymer Laboratory (Cambridge, MA) and by AnaSpec, Inc. (San Jose, CA). Viability was determined by trypan blue exclusion.
Analysis of cell cycle distribution, apoptosis and necrosis
Cells were harvested, washed with PBS, fixed with 80% ethanol, and incubated in PBS containing 40 μg/ml RNAse and 40 μg/ml propidium iodide for 30 min at 37°C. Cell cycle distribution and sub-G1 DNA content was determined by flow cytometry. For assessment of cell membrane integrity, cells were incubated with 1 μg/ml propidium iodide/PBS for 5 min at room temperature and then monitored by flow cytometry as described (29, 30). In the absence of other markers of apoptosis, uptake of propidium iodide reflects loss of membrane integrity indicative of necrosis (30).
Human breast tumor xenograft models
Balb-c nu/nu female mice (Charles River Laboratories, Wilmington, MA) were implanted subcutaneously with 17-β-estradiol plugs (0.72 mg; Innovative Research, Sarasota, FL) using a trocar gun. After 24 h, 1 × 107 ZR-75-1 or MCF-7 cells were injected subcutaneously in the flank. MDA-MB-231 cells (1 × 107) were similarly injected into mice without estradiol implants. When tumors were detectable, the mice were pair matched into treatment and control groups. Mice bearing tumors that were not within 10% of the mean volume were not included in the treatment and control groups. Each group contained 5-10 mice, each of which was ear tagged and followed throughout the study. Initial dosing was administered at the time of pair matching (day 1). Phosphate-buffered saline (vehicle), GO-201 and CP-1 were administered daily by intraperitoneal injection. Mice were weighed twice weekly. Tumor volume (V) was calculated using the formula V=L2 × W/2, where L and W are the larger and smaller diameters, respectively.
Results
Effects of GO-201 on MUC1 oligomer formation
The MUC1 cytoplasmic domain (MUC1-CD) contains a CQC motif that is necessary for the formation of oligomers (18). To determine whether MUC1 oligomerization is druggable, we synthesized a peptide derived from the N-terminal region of MUC1-CD that contains the CQC motif (GO-201; Fig. 1A). A poly D-arginine transduction domain was included in the synthesis to facilitate entry of the peptide into cells (31) (Fig. 1A). As a control, a similar peptide was synthesized in which the CQC motif was altered to AQA (CP-1; Fig. 1A). To assess binding of the peptides to MUC1-CD, we immobilized His-tagged MUC1-CD to a BIAcore sensor chip. GO-201 bound to His-MUC1-CD with a dissociation constant (Kd) of 30 nM (Fig. 1B), which is similar to that obtained for binding of full-length MUC1-CD dimers (18). By contrast, there was no apparent binding of CP-1 (data not shown). Purified His-tagged MUC1-CD forms oligomers as detected by electrophoresis in polyacrylamide gels (Fig. 1C). Incubation of His-MUC1-CD with GO-201 substantially decreased oligomer formation with an increase in monomers (Fig. 1C). Moreover, incubation with CP-1 had little effect (Fig. 1C). To assess effects on MUC1 oligomerization in vivo, 293 cells were transfected with vectors expressing GFP-MUC1-CD and Flag-MUC1-CD (Fig. 1D, left). Complexes of GFP-MUC1-CD and Flag-MUC1-CD were detectable by coprecipitation of lysates from cells not exposed to GO-201 (Fig. 1D, right). In concert with the in vitro results, incubation of the transfected 293 cells with GO-201 was associated with disruption of the interaction between Flag-MUC1-CD and GFP-MUC1-CD (Fig. 1D, right). In addition, CP-1 had no apparent effect (Fig. 1D, right). These results indicate that GO-201 binds to MUC1-CD and blocks formation of MUC1-CD oligomers in vitro and in cells.
Figure 1. GO-201 blocks MUC1 oligomerization.
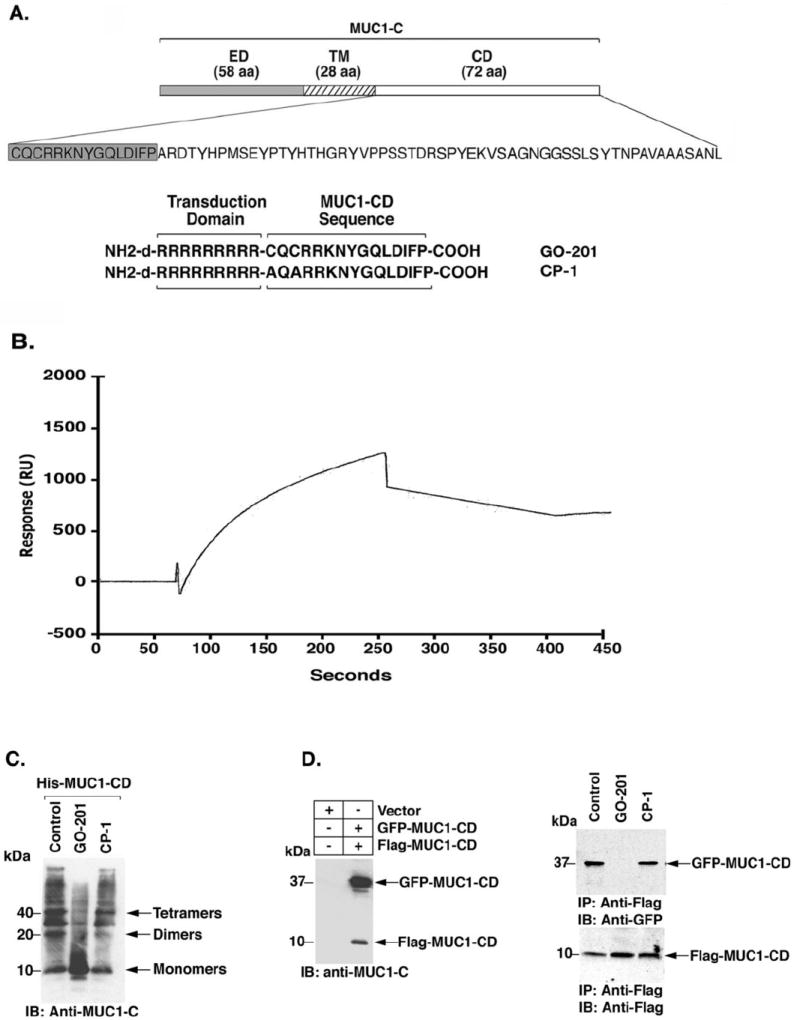
A. Schematic representation of the MUC1-C subunit and the 72 amino acid sequence of MUC1-CD are shown. The N-terminal 15 amino acid (shaded sequence) GO-201 and mutated CP-1 peptides were synthesized with the poly-dArg transduction domain. B. His-MUC1-CD (1.4 mg/ml) was immobilized on a sensor chip in a BIAcore. GO-201 was injected over the chip at 10 μM. Raw binding data were analyzed by BIAevaluation software version 3.0 and fit to a 1:1 Languir binding model. C. Purified His-MUC1-CD was incubated with PBS (Control), GO-201 or CP-1 for 1 h at room temperature. The proteins were separated in a non-reducing SDS-polyacrylamide gel and analyzed by immunoblotting with anti-MUC1-C. D. 293 cells were transiently transfected to express an empty vector or GFP-MUC1-CD and Flag-MUC1-CD. At 48 h after transfection, the cells were left untreated (Control), and treated with 5 μM GO-201 or CP-1 each day for 3 d. The cells were then harvested for immunoblotting with anti-MUC1-C (left panel). Whole cell lysates were also precipitated with anti-Flag and the precipitates were immunoblotted with the indicated antibodies (right panels).
GO-201 disrupts MUC1-C function
To assess peptide uptake, ZR-75-1 cells were incubated with 5 μM FITC-labeled GO-201 (Fig. 2A). At 2 h, analysis of the cells by flow cytometry showed a substantial increase in fluorescence intensity with a mean (MFI) of 145 (Fig. 2A). Further increases in MFI were identified at 6 and 24 h (Fig. 2A). Treatment of ZR-75-1 cells with 5 μM GO-201 or CP-1 for 3 d had no effect on cellular MUC1-C levels (Fig. 2B). However, in concert with effects on oligomerization, treatment with GO-201, and not CP-1, was associated with decreases in nuclear MUC1-C (Fig. 2B). Downregulation of nuclear MUC1-C levels was also observed in the response of MCF-7 cells to GO-201 treatment (Supplemental Fig. S1A). Previous work has demonstrated that MUC1-C decreases intracellular ROS levels (27-29). To determine whether targeting of MUC1-C with GO-201 disrupts redox balance, we incubated GO-201-treated ZR-75-1 cells with c-H2DCFDA, and ROS-mediated oxidation of the fluorochrome was assayed by flow cytometry. The results demonstrate that treatment with GO-201, and not CP-1, increases intracellular ROS levels (Fig. 2C). Similar results were obtained with MCF-7 cells (Supplemental Fig. S1B). Increases in ROS above the reducing capacity of the cell can result in the formation of DNA double-strand breaks. The recognition of such DNA damage is associated with activation of the ataxia telangiectasia-mutated (ATM) and Rad-3 related kinases, which in turn phosphorylate the histone variant H2AX and the checkpoint-1 kinase (Chk1) (32). In concert with this model and the observed increases in ROS, treatment of ZR-75-1 breast cancer cells with GO-201 was associated with activation of ATM and phosphorylation of H2AX and Chk1 (Fig. 2D). Similar results were obtained when MCF-7 cells were treated with GO-201 (Supplemental Fig. S1C). These findings indicate that GO-201 disrupts MUC1 function and redox balance and, in turn, activates the DNA damage response.
Figure 2. GO-201 blocks MUC1-C function.
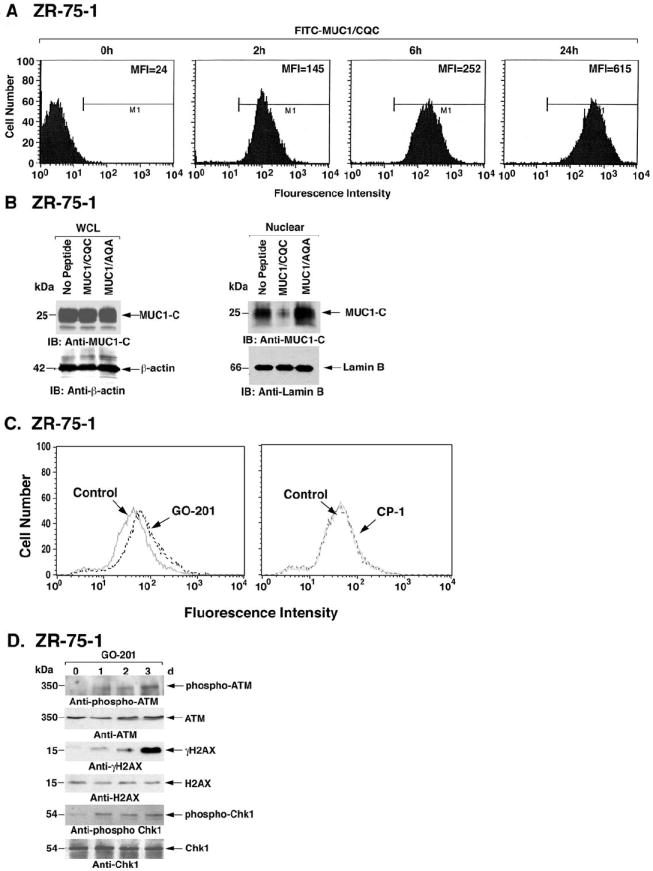
A. ZR-75-1 cells were incubated with 5 μM FITC-labeled GO-201 for the indicated times and then analyzed by flow cytometry. The mean fluorescence index (MFI) is included in each of the panels. B. ZR-75-1 cells were left untreated, and treated with 5 μM GO-201 or CP-1 each day for 3 d. Whole cell lysates (WCL) (left panels) and nuclear lysates (right panels) were immunoblotted with the indicated antibodies. C. ZR-75-1 cells were left untreated (Control), and treated with 5 μM GO-201 or CP-1 for 36 h. The cells were then incubated with c-H2DCFDA for 30 min and fluorescence of oxidized c-H2DCF was measured by flow cytometry. D. ZR-75-1 cells were treated with 5 μM GO-201 or CP-1 each day for 3 d. Lysates were subjected to immunoblotting with the indicated antibodies against ATM, H2AX and Chk1.
GO-201 induces S phase arrest and death
To determine whether exposure to GO-201 affects growth, ZR-75-1 cells were treated with 5 μM GO-201 for 3 d and monitored for cell cycle distribution. Significantly, and consistent with activation of the DNA damage response, there was a substantial arrest in S phase as compared to that in cells left untreated or treated with CP-1 (Fig. 3A). By day 4, the S phase population was decreased, potentially through attrition by cell death (Fig. 3A). There was little if any accumulation of cells with a distinct sub-G1 DNA peak to support the induction of apoptosis (Fig. 3A). Moreover, there was no detectable activation of caspase-3 (data not shown). However, oxidative stress is also associated with non-apoptotic forms of death that include necrosis (33). In this regard, treatment of ZR-75-1 cells with GO-201, and not CP-1, was associated with the appearance of cells with DNA that had undergone extensive degradation consistent with necrosis (Fig. 3B). In addition, treatment with GO-201 was associated with uptake of propidium iodide as a measure of loss of membrane integrity, which was detectable by day 3 and more prominent by day 4 (Fig. 3B). The MCF-7 cells responded similarly to GO-201 with arrest of growth in S phase and the absence of cells with a distinct sub-G1 peak (Fig. 3C). Treatment of MCF-7 cells with GO-201 was also associated with loss of membrane integrity (Fig. 3D). These findings indicate that GO-201 inhibits growth and induces necrosis of human breast cancer cells. However, given potential difficulties in distinguishing late apoptosis from necrosis (30), the results do not exclude the possibility that some of the cells may have died by a late apoptotic response.
Figure 3. GO-201 induces S phase arrest and necrosis.
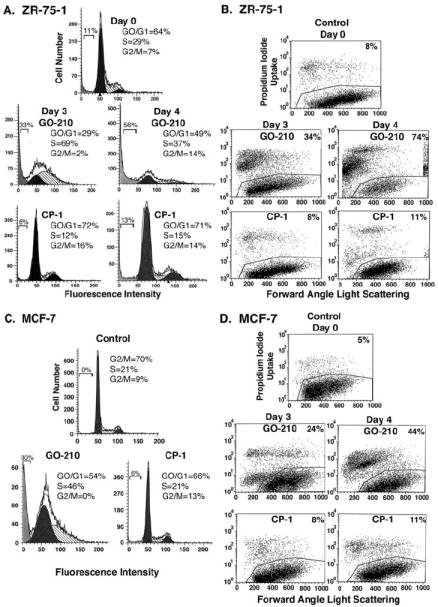
A-D. ZR-75-1 (A and B) and MCF-7 (C and D) cells were treated with 5 μM GO-201 or CP-1 each day for 3 and 4 d. Cells were fixed and analyzed for cell cycle distribution by flow cytometry (A and C). The percentage of diploid cells in G0/G1, S and G2/M phases is included in the panels. There were no distinct sub-G1 peaks supporting induction of apoptosis; whereas, debris in this region reflects cells with DNA degradation and is included as a percentage of diploid cells. Cells were also stained with propidium iodide and analyzed by flow cytometry for necrosis (B and D). The percentage of necrotic cells is included in the panels.
GO-201 targets MUC1 expressing breast carcinoma cells
Human MDA-MB-231 breast cancer cells express endogenous MUC1 (7), but unlike ZR-75-1 and MCF-7 cells, these cells are negative for ER, PR and ErbB2 (triple-negative) (34). Notably, as found for ZR-75-1 and MCF-7 cells, GO-201 and not CP-1 treatment was associated with arrest of MDA-MB-231 cell growth (Fig. 4A), accumulation of cells in S phase with the absence of a distinct sub-G1 peak (data not shown) and the loss of cell membrane integrity (Supplemental Fig. S2). Moreover, GO-201 treatment of MUC1-expressing primary breast cancer cells in short-term culture resulted in growth arrest (Fig. 4B) and death (Fig. 4C). In contrast to these results, GO-201 had no effect on growth of MUC1-negative 293 kidney epithelial cells (Supplemental Fig. S3). Studies were also performed on the MCF-10A non-transformed mammary epithelial cell line (35, 36), which expresses MUC1, but at levels lower than that found in ZR-75-1 and MCF-7 cells (8). Unlike the breast cancer cells, GO-201 had no effect on MCF-10A cell growth (Fig. 4D), cell cycle distribution (Supplemental Fig. S4) or death (data not shown). These findings indicate that GO-201 targets breast carcinoma cells that are addicted to endogenous MUC1.
Figure 4. Selectivity of GO-201 for MUC1 expressing breast cancer cells.

A. MDA-MB-231 cells were left untreated (diamonds), and treated with 5 μM GO-201 (squares) or CP-1 (triangles) each day for the indicated times. Viable cell number was determined by trypan blue exclusion. B. Primary human breast cancer cells growing in short term culture were stained with an IgG1 control antibody or anti-MUC1 (9) (upper panels). The cells were left untreated (diamonds), and treated with 5 μM GO-201 (squares) or CP-1 (triangles) each day for the indicated times. Viable cell number was determined by trypan blue exclusion. C. The primary breast cancer cells were treated with 5 μM GO-201 or CP-1 each day for 3 d. Cells were stained with propidium iodide and analyzed by flow cytometry for necrosis. The percentage of necrotic cells is included in the panels. D. MCF-10A cells were left untreated (diamonds), and treated with 5 μM GO-201 (squares) or CP-1 (triangles) each day for the indicated times. Viable cell number was determined by trypan blue exclusion.
GO-201 inhibits tumorigenicity of estrogen-dependent and - independent breast cancers
To assess anti-tumor activity, ZR-75-1 cells were implanted subcutaneously into the flanks of nude mice. Mice bearing tumors of approximately 150 mm3 were treated with GO-201 at doses of 10 and 50 mg/kg/d. Administration of GO-201 at 10 mg/kg/d × 21 d slowed growth as compared to that obtained with vehicle (PBS) alone or with CP-1 given at 50 mg/kg/d × 21 d (Fig. 5A). In addition, administration of GO-201 at 50 mg/kg/d had no effect on body weight (Supplemental Fig. S5) and blocked tumor growth over the initial 6 d of treatment (Fig. 5A). Consequently, treatment was stopped and there was no detectable growth of the tumors over the next 17 d (Fig. 5A). To assess in part the basis for the activity, tumors harvested on day 24 from control and GO-201-treated (50 mg/kg/d) mice were examined by histopathology. Tumors from the treated mice were markedly necrotic compared to that from mice treated with the vehicle or CP-1 (Fig. 5B). In other studies with somewhat larger tumors (~275 mm3), administration of GO-201 at an intermediate dose of 30 mg/kg/d × 21 d was also associated with arrest of tumor growth (Fig. 5C). Moreover, these tumors exhibited areas with necrosis, cellular debris and swollen cells with cytoplasmic vacuoles (Fig. 5D).
Figure 5. GO-201 blocks growth of ZR-75-1 breast tumor xenografts.

A. Four to six week old female Balb-c nu/nu mice were implanted with 17-β-estradiol plugs. After 24 h, ZR-75-1 breast cancer cells (imbedded in matrigel) were injected subcutaneously in the flank. When tumors were ~150 mm3 (range 140-170 mm3) the mice were pair matched into groups of 5 mice and injected intraperitoneally with PBS (vehicle control; closed squares), 50 mg/kg CP-1 each day for 21 d (open squares), 10 mg/kg GO-201 each day for 21 d (closed triangles) or 50 mg/kg GO-201 each day for 6 d (open triangles). Mice were weighed twice weekly and tumor measurements were performed every 4 d. Tumor volumes are expressed as the mean±SE. B. Tumors harvested on day 24 from the control group and the group treated with 50 mg/kg/d GO-201 × 6 d were stained with H&E. C. Female Balb-c nu/nu mice were implanted with 17-β-estradiol plugs. After 24 h, ZR-75-1 breast cancer cells (imbedded in matrigel) were injected subcutaneously in the flank. When tumors were ~275 mm3 (range: 240-310 mm3) the mice were pair matched into groups of 5 mice and injected intraperitoneally with PBS (closed squares) or 30 mg/kg GO-201 (open squares) each day for 21 d. There was no evidence for weight loss in the treated group. D. Tumors harvested on day 28 from the group treated with GO-210 were stained with H&E.
To extend these findings, additional groups of 10 mice bearing ZR-75-1 tumors were treated with GO-201 at 30 mg/kg/d × 21 d and followed for longer periods (Fig. 6A). The GO-201 dose of 30 mg/kg/d was selected based on the findings that 10 mg/kg/d partially inhibits tumor growth compared to complete growth inhibition at both 30 and 50 mg/kg/d. In addition and to determine whether this activity is dependent on all 15 MUC1 amino acids in GO-201, we synthesized GO-202, a shorter CQCRRKN peptide with the poly D-arginine transduction domain (Supplemental Fig. S6). As found with GO-201, treatment of ZR-75-1 cells with GO-202 in vitro was associated with growth arrest and induction of necrosis (Supplemental Fig. S6 and data not shown). Moreover, like GO-201, treatment of ZR-75-1 tumors with GO-202 at 30 mg/kg/d × 21 d resulted in arrest of growth (Fig. 6A). Significantly, tumors treated with GO-201 or GO-202 were no longer palpable by days 49-56 (Fig. 6A). On day 63, one mouse from each treated group was sacrificed to assess the subcutaneous region implanted with ZR-75-1 tumor cells. There was no visual evidence for remaining tumor at the implantation site or spread to other organs. Histopathologic examination of the implantation site further supported the absence of tumor cells (Fig. 6B). The remaining 9 mice in each treatment group are being followed for the reemergence of tumors. As of day 152, none of the 9 remaining mice in each treated group had evidence for tumor regrowth. To further assess anti-tumor activity, mice bearing MCF-7 xenografts were similarly treated with GO-201 and GO-202. As found for ZR-75-1 tumors, growth of the MCF-7 xenografts was arrested by both GO-201 and GO-202 treatment (Fig. 6C). By day 49, none of the treated mice had palpable tumors (Fig. 6C). One mouse from each treatment group sacrificed on day 49 had no visual evidence of remaining tumor at the implantation site or spread to other organs. There was also no evidence for remaining tumor cells at the implantation site by histopathologic analysis (Supplemental Fig. S7A). Moreover, none of the remaining mice have had recurrence of tumors as of day 136. To determine whether targeting MUC1 also inhibits tumorigenicity of estrogen-independent breast cancer cells, we treated MDA-MB-231 tumor xenografts with GO-201 or GO-202. As found for ZR-75-1 and MCF-7 tumors, growth of the MDA-MB-231 xenografts was arrested by GO-201 and GO-202 treatment (Fig. 6D). Tumors in the treated mice were no longer palpable by day 42 and, on day 49, animals were sacrificed to assess remaining tumor. Again, there was no visual evidence for tumor at the implantation site or in other organs. Moreover, histopathology confirmed the absence of tumor cells at the implantation site (Supplemental Fig. S7B). None of the 9 remaining mice in each treated group had evidence for tumor progression on day 138 and are being followed for recurrence. These findings with MDA-MB-231 and the estrogen-dependent ZR-75-1 and MCF-7 tumors indicate that targeting MUC1 with GO-201 or GO-202 is associated with regression and prolonged arrest of tumor regrowth.
Figure 6. GO-201 and GO-202 induce regression of ZR-75-1, MCF-7 and MDA-MB-231 tumors.

A-C. Female Balb-c nu/nu mice were implanted with 17-β-estradiol plugs. After 24 h, ZR-75-1 (A) or MCF-7 (C) breast cancer cells (1 × 107) derived from xenografts were injected subcutaneously in the flank. When ZR-75-1 and MCF-7 tumors were 140-170 mm3 and 175-225 mm3, respectively, the mice were pair matched into groups of 10 mice and injected intraperitoneally with PBS (closed squares), 30 mg/kg GO-201 each day for 21 d (closed triangles) or 30 mg/kg GO-202 each day for 21 d (open circles). Mice were weighed twice weekly and tumor measurements were performed every 2 d. After 5 weeks, the tumors were measured once each week. There was no evidence of weight loss in the treated groups. Tumor volumes are expressed as the mean and had a SE of less than 10%. For H&E staining: (i) control ZR-75-1 and MCF-7 tumors were harvested on day 14; (ii) the ZR-75-1 cell implantation sites from treated mice were obtained on day 63 (B); and (iii) the MCF-7 cell implantation sites from treated mice were obtained on day 49 (Supplemental Fig. S7A). D. Four to six week old female Balb-c nu/nu mice were injected subcutaneously in the flank with MDA-MB-231 cells (1 × 107) derived from xenografts. When tumors were 240-280 mm3, the mice were pair matched into groups of 10 mice and injected intraperitoneally with PBS (closed squares), 30 mg/kg GO-201 each day for 21 d (closed triangles) or 30 mg/kg GO-202 each day for 21 d (open circles). Control tumors harvested on day 14 and the MDA-MB-231 cell implantation sites from the treated mice obtained on day 49 were stained with H&E (Supplemental Fig. S7B).
Discussion
GO-201 blocks MUC1 oligomerization
MUC1-induced transformation is abrogated by mutation of the CQC sequence in the cytoplasmic domain to AQA, indicating that this motif is of importance to the transforming function (18). MUC1 forms oligomers and the CQC motif is necessary for this oligomerization (18). In addition, oligomer formation is necessary for targeting of the MUC1-C subunit to the nucleus (18) and function of MUC1-C in maintaining redox balance in carcinoma cells (27-29). Based on these findings, we reasoned that disruption of MUC1 oligomerization would have the potential to block the MUC1 transforming function. The present studies demonstrate that GO-201 inhibits oligomerization of MUC1-CD in vitro. MUC1-CD forms dimers with a dissociation constant (Kd) of 33 nM (18). GO-201 similarly bound to MUC1-CD with a Kd of 30 nM. In addition, the demonstration that the mutated control CP-1 peptide has little if any effect on MUC1 oligomerization provided support for dependence on the CQC motif. GO-201, and not CP-1, was also effective in blocking MUC1-C oligomerization and suppression of ROS in cells. While completing these studies, a peptide (PMIP) corresponding to another region of MUC1-CD was shown to inhibit binding of MUC1 to β-catenin and EGFR (37). In contrast to GO-201, which interacts directly with MUC1, PMIP acts as a decoy to interact with MUC1 binding partners. Thus, GO-201 and PMIP represent different strategies to inhibit MUC1 function.
Selectivity of GO-201 for MUC1 overexpressing carcinoma cells
Consistent with nuclear targeting of MUC1 being dependent on oligomerization (18), uptake of GO-201 in ZR-75-1 cells was associated with down-regulation of MUC1-C levels in the nucleus. Similar results were obtained with MCF-7 breast cancer cells, indicating that this response to GO-201 is not cell specific. Moreover, exposure of these cells to GO-201, and not CP-1, was associated with ROS-induced activation of the DNA damage response, S phase growth arrest and induction of late apoptosis/necrosis. These effects of directly inhibiting MUC1 with GO-201 are in contrast to that obtained with the PIMP decoy peptide that reduces proliferation without evidence for inducing cell death (37). Of importance is whether GO-201 induces death by a mechanism dependent on expression of its intended target or if it functions as a non-specific cytotoxin. In that context, GO-201 had no effect on MUC1-negative 293 cells. In addition, exposure of non-malignant MCF-10A mammary epithelial cells to GO-201 had no apparent effect. These findings indicate that sensitivity to GO-201 is dependent on a function of MUC1, such as suppression of ROS, associated with the malignant phenotype. GO-201 thus appears to have activity that is selective for carcinoma cells that overexpress MUC1.
Anti-tumor activity associated with disrupting MUC1 function
An overriding question was whether GO-201 could be delivered in vivo with an effective therapeutic index, that is anti-tumor activity and an acceptable toxicity profile. In addressing this issue, we found that administration of GO-201 was well-tolerated without apparent acute toxicities. We also found that GO-201 treatment is effective in inducing tumor regression and prolonged delays in regrowth. These results were in contrast to administration of the control CP-1, which had no anti-tumor activity. These results are explained, at least in part, by the finding that treatment with GO-201 is associated with the induction of tumor necrosis. Similar effects were observed with the ZR-75-1, MCF-7 and MDA-MB-231 tumor models, indicating that GO-201 is active against estrogen-dependent and triple-negative breast cancer cells, the latter being insensitive to currently available targeted therapies (38). The effects of directly inhibiting MUC1 with GO-201 are in contrast to that obtained with the PIMP decoy peptide that inhibits growth of MDA-MB-231 tumors without inducing complete regressions (37). Importantly in this regard, the ZR-75-1, MCF-7 and MDA-MB-231 tumors treated with GO-201 have had no evidence for recurrence as of days 136-152.
Targeting of MUC1 function
Overexpression of MUC1 as found in human breast tumors blocks death in the response to oxidative stress, DNA damage and hypoxia (14, 20, 26-29). Overexpression of MUC1 also induces anchorage-independent growth and tumorigenicity (12, 24). However, there has been no evidence that targeting MUC1 oligomerization can affect survival or tumorigenicity of human breast cancer cells. The present results with GO-201 emphasize the importance of MUC1 in contributing to breast cancer cell survival and tumorigenicity. To provide further support for the observed activity with GO-201, we synthesized GO-202, which also targets the CQC motif. We found that GO-202 behaves like GO-201 in vitro and in the xenograft models, indicating that the CQC motif is indeed an Achilles heel of the MUC1 oncoprotein. The present results also suggest that the breast cancer cells studied here are addicted to MUC1 function. In this context, oncogene addiction has been defined as dependency of a cancer cell on a single gene product for maintenance of the malignant phenotype (39). Notably, therapies that target addiction to specific oncoproteins have been associated with the emergence of (i) circumventing mutations and/or (ii) dependence on other transforming genes. Thus, it is conceivable that targeting the MUC1 CQC motif could result in resistant mutants. Alternatively, targeting MUC1 could select for the emergence of breast cancer cells that are dependent on other oncoproteins. Whereas these issues will need to be addressed in subsequent studies, the loss of tumorigenicity in the xenograft models suggests that targeting MUC1 by blocking oligomerization can have prolonged effects.
Supplementary Material
Acknowledgments
This work was supported in part by NCI Grants CA97098 and CA42802. D. Kufe is a founder of Genus Oncology and a consultant to the company.
References
- 1.Duraisamy S, Ramasamy S, Kharbanda S, Kufe D. Distinct evolution of the human carcinoma-associated transmembrane mucins, MUC1, MUC4 AND MUC16. Gene. 2006;373:28–34. doi: 10.1016/j.gene.2005.12.021. [DOI] [PubMed] [Google Scholar]
- 2.Ligtenberg MJ, Kruijshaar L, Buijs F, van Meijer M, Litvinov SV, Hilkens J. Cell-associated episialin is a complex containing two proteins derived from a common precursor. J Biol Chem. 1992;267:6171–7. [PubMed] [Google Scholar]
- 3.Macao B, Johansson DG, Hansson GC, Hard T. Autoproteolysis coupled to protein folding in the SEA domain of the membrane-bound MUC1 mucin. Nat Struct Mol Biol. 2006;13:71–6. doi: 10.1038/nsmb1035. [DOI] [PubMed] [Google Scholar]
- 4.Siddiqui J, Abe M, Hayes D, Shani E, Yunis E, Kufe D. Isolation and sequencing of a cDNA coding for the human DF3 breast carcinoma-associated antigen. Proc Natl Acad Sci USA. 1988;85:2320–3. doi: 10.1073/pnas.85.7.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merlo G, Siddiqui J, Cropp C, et al. DF3 tumor-associated antigen gene is located in a region on chromosome 1q frequently altered in primary human breast cancer. Cancer Res. 1989;49:6966–71. [PubMed] [Google Scholar]
- 6.Kufe D. Targeting the MUC1 oncoprotein: a tale of two proteins. Cancer Biol Ther. 2008;7:81–4. doi: 10.4161/cbt.7.1.5631. [DOI] [PubMed] [Google Scholar]
- 7.Ramasamy S, Duraisamy S, Barbashov S, Kawano T, Kharbanda S, Kufe D. The MUC1 and galectin-3 oncoproteins function in a microRNA-dependent regulatory loop. Mol Cell. 2007;27:992–1004. doi: 10.1016/j.molcel.2007.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmad R, Raina D, Trivedi V, et al. MUC1 oncoprotein activates the I| B kinase β complex and constitutive NF-| B signaling. Nat Cell Biol. 2007;9:1419–27. doi: 10.1038/ncb1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kufe D, Inghirami G, Abe M, Hayes D, Justi-Wheeler H, Schlom J. Differential reactivity of a novel monoclonal antibody (DF3) with human malignant versus benign breast tumors. Hybridoma. 1984;3:223–32. doi: 10.1089/hyb.1984.3.223. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Ren J, Yu W-H, et al. The EGF receptor regulates interaction of the human DF3/MUC1 carcinoma antigen with c-Src and β-catenin. J Biol Chem. 2001;276:35239–42. doi: 10.1074/jbc.C100359200. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Chen W, Ren J, et al. DF3/MUC1 signaling in multiple myeloma cells is regulated by interleukin-7. Cancer Biol Ther. 2003;2:187–93. doi: 10.4161/cbt.2.2.282. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Liu D, Chen D, Kharbanda S, Kufe D. Human DF3/MUC1 carcinoma-associated protein functions as an oncogene. Oncogene. 2003;22:6107–10. doi: 10.1038/sj.onc.1206732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Yu W-H, Ren J, et al. Heregulin targets γ-catenin to the nucleolus by a mechanism dependent on the DF3/MUC1 protein. Mol Cancer Res. 2003;1:765–75. [PubMed] [Google Scholar]
- 14.Ren J, Agata N, Chen D, et al. Human MUC1 carcinoma-associated protein confers resistance to genotoxic anti-cancer agents. Cancer Cell. 2004;5:163–75. doi: 10.1016/s1535-6108(04)00020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren J, Bharti A, Raina D, Chen W, Ahmad R, Kufe D. MUC1 oncoprotein is targeted to mitochondria by heregulin-induced activation of c-Src and the molecular chaperone HSP90. Oncogene. 2006;25:20–31. doi: 10.1038/sj.onc.1209012. [DOI] [PubMed] [Google Scholar]
- 16.Pitroda S, Khodarev N, Beckett M, Kufe D, Weichselbaum R. MUC1-induced alterations in a lipid metabolic gene network predict response of human breast cancers to tamoxifen treatment. Proc Natl Acad Sci USA. 2009;106:5837–41. doi: 10.1073/pnas.0812029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khodarev N, Pitroda S, Beckett M, et al. MUC1-induced transcriptional programs associated with tumorigenesis predict outcome in breast and lung cancer. Cancer Res. 2009 doi: 10.1158/0008-5472.CAN-08-4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leng Y, Cao C, Ren J, et al. Nuclear import of the MUC1-C oncoprotein is mediated by nucleoporin Nup62. J Biol Chem. 2007;282:19321–30. doi: 10.1074/jbc.M703222200. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Kuwahara H, Ren J, Wen G, Kufe D. The c-Src tyrosine kinase regulates signaling of the human DF3/MUC1 carcinoma-associated antigen with GSK3β and β-catenin. J Biol Chem. 2001;276:6061–4. doi: 10.1074/jbc.C000754200. [DOI] [PubMed] [Google Scholar]
- 20.Raina D, Ahmad R, Kumar S, et al. MUC1 oncoprotein blocks nuclear targeting of c-Abl in the apoptotic response to DNA damage. EMBO J. 2006;25:3774–83. doi: 10.1038/sj.emboj.7601263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ren J, Li Y, Kufe D. Protein kinase C δ regulates function of the DF3/MUC1 carcinoma antigen in β-catenin signaling. J Biol Chem. 2002;277:17616–22. doi: 10.1074/jbc.M200436200. [DOI] [PubMed] [Google Scholar]
- 22.Li Y, Bharti A, Chen D, Gong J, Kufe D. Interaction of glycogen synthase kinase 3β with the DF3/MUC1 carcinoma-associated antigen and β-catenin. Mol Cell Biol. 1998;18:7216–24. doi: 10.1128/mcb.18.12.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamamoto M, Bharti A, Li Y, Kufe D. Interaction of the DF3/MUC1 breast carcinoma-associated antigen and β-catenin in cell adhesion. J Biol Chem. 1997;272:12492–4. doi: 10.1074/jbc.272.19.12492. [DOI] [PubMed] [Google Scholar]
- 24.Huang L, Chen D, Liu D, Yin L, Kharbanda S, Kufe D. MUC1 oncoprotein blocks GSK3β-mediated phosphorylation and degradation of β-catenin. Cancer Res. 2005;65:10413–22. doi: 10.1158/0008-5472.CAN-05-2474. [DOI] [PubMed] [Google Scholar]
- 25.Wei X, Xu H, Kufe D. Human MUC1 oncoprotein regulates p53-responsive gene transcription in the genotoxic stress response. Cancer Cell. 2005;7:167–78. doi: 10.1016/j.ccr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 26.Raina D, Kharbanda S, Kufe D. The MUC1 oncoprotein activates the anti-apoptotic PI3K/Akt and Bcl-xL pathways in rat 3Y1 fibroblasts. J Biol Chem. 2004;279:20607–12. doi: 10.1074/jbc.M310538200. [DOI] [PubMed] [Google Scholar]
- 27.Yin L, Kufe D. Human MUC1 carcinoma antigen regulates intracellular oxidant levels and the apoptotic response to oxidative stress. J Biol Chem. 2003;278:35458–64. doi: 10.1074/jbc.M301987200. [DOI] [PubMed] [Google Scholar]
- 28.Yin L, Huang L, Kufe D. MUC1 oncoprotein activates the FOXO3a transcription factor in a survival response to oxidative stress. J Biol Chem. 2004;279:45721–7. doi: 10.1074/jbc.M408027200. [DOI] [PubMed] [Google Scholar]
- 29.Yin L, Kharbanda S, Kufe D. Mucin 1 oncoprotein blocks hypoxia-inducible factor 1 alpha activation in a survival response to hypoxia. J Biol Chem. 2007;282:257–66. doi: 10.1074/jbc.M610156200. [DOI] [PubMed] [Google Scholar]
- 30.McGahon AJ, Martin SJ, Bissonnette RP, et al. The end of the (cell) line: methods for the study of apoptosis in vitro. Methods Cell Biol. 1995;46:153–85. doi: 10.1016/s0091-679x(08)61929-9. [DOI] [PubMed] [Google Scholar]
- 31.Fischer PM. Cellular uptake mechanisms and potential therapeutic utility of peptidic cell delivery vectors: progress 2001-2006. Med Res Rev. 2007;27:755–95. doi: 10.1002/med.20093. [DOI] [PubMed] [Google Scholar]
- 32.Lavin MF, Delia D, Chessa L. ATM and the DNA damage response. EMBO Reports. 2006;7:154–60. doi: 10.1038/sj.embor.7400629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Amaravadi R, Thompson C. The roles of therapy-induced autophagy and necrosis in cancer treatment. Clin Cancer Res. 2007;13:7271–9. doi: 10.1158/1078-0432.CCR-07-1595. [DOI] [PubMed] [Google Scholar]
- 34.Walsh MD, Luckie SM, Cummings MC, Antalis TM, McGuckin MA. Heterogeneity of MUC1 expression by human breast carcinoma cell lines in vivo and in vitro. Breast Cancer Res and Treat. 2000;58:255–66. doi: 10.1023/a:1006345301364. [DOI] [PubMed] [Google Scholar]
- 35.Soule HD, Maloney TM, Wolman SR, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990;50:6075–86. [PubMed] [Google Scholar]
- 36.Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3:785–92. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bitler B, Menzl I, Huerta C, et al. Intracellular MUC1 peptides inhibit cancer progression. Clin Cancer Res. 2009;15:100–9. doi: 10.1158/1078-0432.CCR-08-1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rakha E, Reis-Filho J, Ellis I. Basal-like breast cancer: a critical review. J Clin Oncol. 2008;26:2568–81. doi: 10.1200/JCO.2007.13.1748. [DOI] [PubMed] [Google Scholar]
- 39.Weinstein I, Joe A. Oncogene addiction. Cancer Res. 2008;68:3077–80. doi: 10.1158/0008-5472.CAN-07-3293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.