

Abstract
Traumatic brain injury, a silent epidemic of modern societies, is a largely neglected area in drug development and no drug is currently available for the treatment of patients suffering from brain trauma. Despite this grim situation, much progress has been made over the last two decades in closely related medical indications, such as spinal cord injury, giving rise to a more optimistic approach to drug development in brain trauma. Fundamental insights have been gained with animal models of central nervous system (CNS) trauma and spinal cord injury. Neuroregenerative drug candidates have been identified and two of these have progressed to clinical development for spinal cord injury patients. If successful, these drug candidates may be used to treat brain trauma patients. Significant progress has also been made in understanding the fundamental molecular mechanism underlying irreversible axonal growth arrest in the injured CNS of higher mammals. From these studies, we have learned that the axonal retraction bulb, previously regarded as a marker for failure of regenerative growth, is not static but dynamic and, therefore, amenable to pharmacotherapeutic approaches. With the development of modified magnetic resonance imaging methods, fibre tracts can be visualised in the living human brain and such imaging methods will soon be used to evaluate the neuroregenerative potential of drug candidates. These significant advances are expected to fundamentally change the often hopeless situation of brain trauma patients and will be the first step towards overcoming the silent epidemic of brain injury.
Keywords: traumatic brain injury, spinal cord injury, neuroregeneration, neuroplasticity, diffusion tensor imaging, retraction bulb, neuronal growth cone
Neuroregenerative approaches for central nervous system injury
For thousands of years, damage to the adult central nervous system (CNS) in humans has been regarded as an ‘ailment which cannot be treated’, a phrase used to characterize the hopeless situation of two spinal cord injury victims in the 4500-year-old Edwin Smith Papyrus (Donovan, 2007). Only recently has this dogma and therapeutic nihilism been overcome and fundamental insight gained, making neuroregenerative drug approaches feasible. The amazing progress seen over the last 30 years stems from the remarkable discovery of Albert Aguayo and his co-workers that adult CNS neurons are able to grow extensively after injury if offered a permissive peripheral nerve transplant (Richardson et al., 1980; David and Aguayo, 1981; Aguayo et al., 1991; Xie and Zheng, 2008). These experiments not only helped to overcome the dogma that injured adult CNS axons have lost their axonal growth capability but also induced a shift in focus towards the micro-environment of injured axons, giving rise to the question, ‘What makes the CNS micro-environment so hostile for regrowth of injured fibers?’ One of the first to answer this question was Martin Berry who postulated that breakdown products of the CNS myelin are responsible for the failure of injured CNS fibres to regrow. Martin Schwab and colleagues soon discovered the differences between CNS myelin and that of the peripheral nervous system where regeneration of injured nerve fibres is known to occur (Berry, 1982; Schwab and Thoenen, 1985; Schwab and Bartholdi, 1996; Xie and Zheng, 2008). The inhibitory activity of CNS myelin was overcome in a seminal rat spinal cord injury experiment conducted by Schnell and Schwab. They showed that intraventricular injection of hybridoma cells, producing a monoclonal antibody directed against inhibitory proteins of the CNS myelin, later called NOGO A, enhanced regenerative growth of injured corticospinal tract fibres across the spinal lesion site and improved functional recovery (Schnell and Schwab, 1990). Since these pioneering studies, we have learned that, besides CNS myelin, glial scar tissue forming at the lesion site in the spinal cord or brain also impedes fibre growth. Some of its molecular constituents will be discussed later in more detail (Silver and Miller, 2004; Mueller et al., 2005). Based on experiments with antagonists of myelin-associated inhibitors or glial scar-associated inhibitors or pharmacological modifiers of the neuronal growth response, performed primarily in animals with spinal injury, several mechanisms have been described to account for drug-induced axon growth or rearrangement-induced functional improvement. A recent classification distinguishes regeneration from sprouting and plasticity based on the inciting event, the timing of axonal growth and rearrangement and distance of axonal regrowth (Cafferty et al., 2008). The term regeneration is used to describe the growth of injured or damaged axons over longer distances and over longer periods of time, whereas sprouting refers to growth from injured or damaged fibres or from intact fibres over moderate distances. Plasticity, in a narrow sense, describes changes in the underlying network induced by damaged fibres and correlated functional loss (Cafferty et al., 2008). The most time-consuming of these is the process of regeneration which takes weeks to months to bring about functional improvements, whereas sprouting and plasticity occur at a faster rate, that is, within days or even hours of the damaging event (Cafferty et al., 2008). Most of these different mechanisms have been identified in animals with spinal cord injury and another important insight gained from these experiments was that, despite their often meandering course within the spinal cord tissue, regrowing fibres are able to synapse with their proper target neurons. Just how they are guided to their synaptic partners is not actually known but re-expression of many classes of developmentally active attractive and repulsive axon guidance molecules at the site of lesion or damage might be an explanation for the proper pathfinding and paucity of aberrant connections formed. This constitutes a very encouraging sign for the development of axonal growth therapeutics and several neuroregenerative drug candidates are currently being evaluated in clinical trials with spinal cord injury patients (Baptiste and Fehlings, 2008; Gonzenbach and Schwab, 2008). Table 1 provides a summary of potential axonal growth therapeutics including their proposed mode of action and investigational status in spinal cord injury. If successful, it is envisioned that these regenerative drug approaches will be extended to other CNS injuries such as traumatic brain injury (TBI).
Table 1.
Regenerative strategies under evaluation for spinal cord injury treatment
| Therapeutic agent | Proposed mechanism of action | Status |
|---|---|---|
| Rho inhibitor (Cethrin®) | Blockade of growth inhibitory pathway | phase I/IIa |
| Anti-Nogo A antibodies | Neutralization of growth inhibitory myelin protein | phase I/IIa |
| Chondroitinase ABC | Degradation of glycosaminoglycan chains of growth inhibitory proteoglycans | preclinical |
| Anti-NgR antibodies or antagonists | Blockade of growth inhibition mediating receptor | preclinical |
| LINGO-1 antagonist | Blockade of growth inhibition mediating receptor complex | preclinical |
| Anti-RGM A antibodies | Neutralization of growth inhibitory protein | preclinical |
| Rho kinase (ROCK) inhibitors (Fasudil) | Blockade of growth inhibitory pathway | preclinical |
| Sema3A inhibitor (SM-216289) | Neutralization of growth inhibitory protein | preclinical |
| Neurotrophins (BDNF, CNTF, GDNF) | Growth promotion by neurotrophin receptor stimulation | preclinical |
| PDE4 inhibitor (Rolipram®) | Growth promotion by increasing cAMP levels | preclinical |
| Anti-RYK antibodies | Neutralization of growth inhibitory proteins | preclinical |
| Noggin (BMP-antagonist) | Neutralization of inhibitory proteins | preclinical |
| EGFR inhibitor (PD168393) | Blockade of growth inhibitory pathway | preclinical |
| GSK-3β inhibitor (Lithium, SB415286) | Blockade of growth inhibitory pathway | preclinical |
| PKC inhibitor (Gö6976) | Blockade of growth inhibitory pathway | preclinical |
| L1 cell adhesion molecule | Neurite growth promotion | preclinical |
| Iron-chelator (Cordaneurin®) | Inhibition of collagen scar formation | preclinical |
Summary of regenerative drug approaches in spinal cord injury with proposed working mechanism and status of development.
Traumatic brain injury
Traumatic brain injury constitutes a major public health problem with a reported annual incidence of 150–500 cases per 100 000 (Bruns and Hauser, 2003; Kraus and Chu, 2005; Bazarian et al., 2007) and is regarded as a silent epidemic of modern societies with death from TBI accounting for 1–2% of all deaths from all causes (Graham et al., 2002 in Greenfield's Neuropathology; Goldstein, 1990). In the United States, 1.4 million people suffer from TBI each year. A total of 50 000 TBI victims die, 235 000 are hospitalized and 1.1 million are treated and released from emergency units [Centers for Disease Control and Prevention (CDC), 2008]. Several high-risk groups have been described: children aged 0–4 years, young people aged 15–19 years and people older than 64 years (Kraus and Chu, 2005; CDC, 2008). Males are more affected than females with ratios between 1.6 and 2.8 (Kraus and Chu, 2005) or 1.5 (CDC, 2008). Due to high incidence and the tender age of the TBI victims, the burden on the healthcare systems is heavy with medical and related costs amounting to $60 billion per year in the United States in 2000 (Corso et al., 2006; CDC, 2008). TBI is clinically classified based on the Glasgow Coma Score, loss of consciousness, and post-traumatic amnesia into three categories – mild, moderate and severe – the approximate percentage distribution being 50%, 30% and 20% respectively (Kraus and Chu, 2005; Kraus et al., 2007). The most common TBI causes in the US are falls (28%), motor vehicle accidents (20%), struck-by events (19%) and assault (11%) (CDC, 2008). To exemplify the hidden or silent epidemic of TBI, estimates of ‘The Centers of Disease Control and Prevention’ suggest that approximately 5.3 million TBI victims in the US require long-term or even lifelong help to perform their daily living activities (CDC, 2008).
Despite significant medical need, no pharmacotherapy is currently available to fundamentally relieve or improve the situation of TBI victims. This may result from the heterogeneity of brain injuries, the precise type, location and extent of the primary injury, the contribution of different pathological mechanisms, delayed secondary injury mechanisms and compounding conditions such as brain swelling, increases in intracranial pressure, and hypoxia. Depending on the severity of the injury, acute TBI may induce coma or a vegetative or minimally conscious state (MCS), whereas mild to moderate TBI may lead to neurobehavioural deficits, especially in cognition. Cognitive symptoms may manifest as deficits in verbal fluency, mental flexibility or working memory as well as impaired attention, planning inability and increased impulsivity (Kraus et al., 2007; CDC, 2008). Long-term sequelae of TBI may include the induction of epileptic seizures and increased risk for Alzheimer's and Parkinson's diseases.
The large variety of symptoms caused by TBI impedes identification of the clinically relevant neuropathology which is responsible for the symptoms occurring in the brain injury patients. An important concept developed over recent decades focuses on the degeneration of cerebral white matter and is known as diffuse axonal injury (DAI) or traumatic axonal injury (Graham et al., 2002; Gennarelli and Graham, 2005). Implicit to this concept is its ability to explain the progressive severity of TBI symptoms with an increasing amount of axonal damage, ranging from concussive syndromes to the most severe brain impairment characterized by immediate or prolonged coma (Graham et al., 2002). In addition, the concept is supported by human neuropathology and experimental brain injury models and has enabled the development of a grading system of increasing severity: DAI I is characterized by widespread axon damage in the corpus callosum, cerebral hemispheres and brainstem. DAI II comprises, in addition, focal abnormalities that are often associated with haemorrhaging in the corpus callosum. The most severe form, DAI III, encompasses DAI II characteristics plus axonal abnormalities resulting from haemorrhaging in the rostral brainstem (Graham et al., 2002). Focal lesions in the corpus callosum and brainstem can be identified macroscopically in DAI III, whereas DAI may be inferred from imaging studies but might require light microscopic analysis of post-mortem brain tissue for conclusive confirmation (Graham et al., 2002; Kraus et al., 2007).
Diffuse axonal injury may be caused directly by a traumatic incident or indirectly by ischemia and could be the only significant pathology in certain, mild TBI cases (Kraus et al., 2007). Continuous efforts over the last 20 years have revealed many mechanistic aspects of DAI and, as a result, the original opinion – that axons are disrupted by shear forces at the moment of injury – was discarded. This was replaced by a refined picture of a progressive process wherein the initial acceleration-deceleration insult first triggers local axolemmal permeability. This was evidenced by the influx of tracer molecules into the axon, for example, horseradish peroxidase or fluorescent dextranes, which are normally excluded due to their high molecular weight (Pettus et al., 1994; Stone et al., 2004). At the same time, local axolemmal permeability would permit a massive influx of calcium which, in turn, activates Ca-dependent proteases, for example, calpain. In support of this hypothesis, an alteration of the neurofilament and microtubular axonal cytoskeleton including the occurrence of calpain-specific breakdown products of spectrin has been reported (Saatman et al., 1996; Posmantur et al., 1997; Büki et al., 1999). Calcium influx is buffered by uptake into the mitochondria, eventually building up to levels inside their matrix high enough to trigger opening of the mitochondrial permeability transition pore (MPTP). Indeed, swollen mitochondria with ruptured membranes, hallmarks of MPTP opening, are observed at sites of axonal injury (Colicos and Dash, 1996; Pettus and Povlishock, 1996; Okonkwo and Povlishock, 1998). Mitochondrial membrane rupture not only impairs ATP production but leads to the release of cytochrome c and the activation of caspases, further contributing to the cytoskeletal breakdown observed in experimental TBI (Büki et al., 2000). As a consequence of the proteolytic attack on the microtubule and neurofilament network, anterograde axonal transport is stopped at these sites, leading to the accumulation of organelles. Experimentally, this accumulation is easily visualized using antibodies to the amyloid precursor protein (APP) which serves as a marker for anterograde axonal transport. Accumulation of transport vesicles and organelles results in concomitant swelling of the axon, ultimately leading to disconnection of the distal axonal segment which is removed by Wallerian degeneration over time. The remaining neuron is characterized by a structure known as an axonal end bulb, described in more detail below. Interestingly, dying back of the neuronal soma does not appear to occur even in cases of perisomal axonal disconnection (Kelley et al., 2006) despite the activation of caspases involved in the induction of apopotosis. The elucidation of such a hierarchical order of events ranging from the initial insult to axonal disconnection represents considerable progress in understanding the mechanism of DAI (Büki and Povlishock, 2006). However, this is probably an oversimplification of the process as, for instance, recent studies have shown apparent disconnections, that is, early breakdown in axolemmal integrity can occur without producing axonal swelling for several hours and swelling can be evidenced in axons in which there are no signs of axolemmal permeability (Stone et al., 2004; Kelley et al., 2006). Moreover, several questions still remain to be answered. For instance, why is the injury diffuse, that is, why do we see damaged axons next to intact ones which seem to have experienced the same initial insult? Does confinement of the bulk of injury to myelinated fibre tracts reflect higher susceptibility of these or does it just mirror the ease of observation? How long after the insult does TBI result in ongoing axonal damage – just for the number of hours the experimental data cover or for several days? The last question, in particular, might be of considerable significance in the light of therapeutic intervention. The success of a neuroprotective or axoprotective strategy, for example, by inhibiting calpain or acting at the mitochondrial level (Posmantur et al., 1997; Okonkwo and Povlishock, 1998; Büki et al., 1999), will most likely be limited to the time of axonal disconnection. In contrast, neuroregenerative approaches might range far beyond this point, increasing the attractiveness of such therapeutic interventions. Documentation of axonoprotective or neuroregenerative effects of new drug candidates now seems feasible thanks to new imaging techniques.
A recently developed magnetic resonance imaging technique, called diffusion tensor imaging (DTI), has become an extremely valuable tool for evaluating patients with brain injuries. DTI enables measurement of the integrity of white matter fibre tracts making the axonal organization of the brain visible. It measures the diffusion of water molecules that prefer to diffuse along white matter fibre tracts (anisotropically) and not perpendicularly to the course of the fibre tracts (Mori and Zhang, 2006). Values for fractional anisotropy (FA) range between 0 and 1, with 0 standing for isotropic, lack of directional diffusion, and 1 representing anisotropic (directional) diffusion. Factors influencing FA are myelination of fibre tracts and axonal integrity and density (Mori and Zhang, 2006; Kraus et al., 2007). In a recent study, DTI was validated in a controlled cortical mouse TBI model using histology and electronmicroscopic analysis. The technique was able to detect perilesional white matter damage in a range of acute to subacute time points, thereby predicting the approximate time since trauma (Mac Donald et al., 2007). DTI was also able to correlate recovery of the damaged corticospinal tract with functional improvement in a patient suffering from paraparesis after TBI (Han et al., 2007). These remarkable abilities of DTI make it an extremely valuable tool in the evaluation of restorative and axon regenerative therapies in animal models of TBI. DTI has recently been used to document myelin-protective activities of a therapeutic antibody candidate in an animal multiple sclerosis model (Mi et al., 2007) and is extensively used in rodent models characterized by axonal or myelin damage.
The axonal retraction bulb of the injured central nervous system
Immunohistochemical analysis of post-mortem brain sections of TBI victims very often reveal DAI characterized by axonal swelling, varicosities, transected axons with terminal end bulbs, also called retraction balls, retraction bulbs or dystrophic axons (a generic term for misshapen axons carrying larger spheroids and smaller varicosities) in the corpus callosum, thalamus, brainstem, cerebellum or cerebral hemispheres (Graham et al., 2002; Coleman, 2005; Gennarelli and Graham, 2005). These axonal bulbs or varicosities as indicators of failed or reduced axonal transport (Coleman, 2005) are not visible in DTI but become visible when immunostained with markers like β-APP (APP), or with certain neurofilament markers (SMI-32). β-APP positive axonal pathology has been observed not only in TBI but in multiple sclerosis, spinal cord injury, stroke, Alzheimer's disease and several other CNS disorders, suggesting the existence of convergence points (reduced axonal transport, mitochondrial dysfunction, intra-axonal calcium accumulation) of axonal degeneration, despite very different primary insults (Coleman, 2005).
Historically, silver staining techniques have enabled visualization of axonal retraction bulbs in injured CNS tissue where Wallerian degeneration, a proactive axon death mechanism, has resulted in removal of the distal axon, leaving the proximal axon equipped with a large terminal retraction bulb (Coleman, 2005). Despite their characteristic appearance, our knowledge of these structures is still fragmentary. Questions arising in this regard address the reversibility of the retraction bulbs, sensitivity of different axons to bulb formation, relationship of Wallerian degeneration mechanisms and acute axonal damage programmes to retraction bulb formation and their static nature. More recent results from various groups suggest that terminal retraction bulbs may not be as static as originally considered, pointing to the possibility of therapeutic intervention by enhancing their structural plasticity. During development of the central and peripheral nervous system, the tips of neurites are not equipped with end bulb-like terminals, but form highly mobile structures, known as neuronal growth cones. These structures regulate and direct neurite growth, are equipped with remarkable abilities to respond to a large variety of guidance cues and are responsible for proper wiring of the embryonic nervous system (Mueller, 1999). In the adult CNS, damage to axons does not result in the generation of a new growth cone but results in the formation of axonal retraction bulbs. These structures seem to be remarkably stable as they can be found in brain injury sites often years after damage has occurred and are regarded as the visible static marker for failed regenerative growth responses of damaged axons (Li and Raisman, 1995; Tom et al., 2004). Results from recent in vitro, in situ slice culture, and in vivo experiments suggest that dystrophic endings or retraction bulbs are, however, dynamic, that they contain both F-actin and disorganized microtubules and are able to extend fine, potentially regenerative sprouts, pointing to regenerative, albeit truncated, behaviour (Tom et al., 2004; Dickson et al., 2007; Ertürk et al., 2007).
Injury to the adult CNS results in formation of a glial scar at the lesion site and this scar tissue represents a major barrier to successful nerve regeneration. Regenerating neurites usually do not penetrate scar tissue and avoid this inhibitory area, despite the presence of factors promoting outgrowth. Inhibitory factors of the scar tissue are complex chondroitinsulfate proteoglycans (CSPG), proteins with a chondroitin sulphate glycosaminoglycan moiety, and other inhibitory factors such as ephrins, semaphorins, RGMs (repulsive guidance molecule), tenascins, and other molecules. As only little is known about the response of adult neurites to scar tissue, the development of an in vitro model of scar tissue is a step forward in studying the behaviour of neurites encountering the artificial scar and evaluating drug candidates that promote regeneration (Tom et al., 2004). In an artificial gradient consisting of Aggrecan, a CSPG present at high levels in CNS scars, and the potent outgrowth promoter Laminin, present in a counter-gradient, regenerating adult sensory neurons entered the artificial scar but their outgrowth was stalled by the high CSPG concentration, and the neurites developed a bulbous, dystrophic appearance, very similar to that of the retraction bulb structures observed in the injured CNS (Tom et al., 2004). Besides the in vitro generation of these end bulb-like structures, the authors also clearly demonstrated that the dystrophic axons or truncated growth cones were not immobile but showed extensive dynamic behaviour. This was confirmed by in vivo analysis of fluorescently labelled, injured and dystrophic sensory nerves in the adult rat spinal cord, proving that dynamic behaviour of dystrophic nerves was maintained, even 1 week after injury. Using this in vitro model, a first analysis of signalling cascades underlying the behaviour of dystrophic neurites was attempted but no clear signalling candidate emerged from this study (Tom et al., 2004). Similar conclusions were obtained with respect to the dynamic behaviour of axonal bulbs forming after axonal transection. Dickson and colleagues used live-imaging methods to observe the behaviour of transected adult DiI-labelled neocortical neurites in an in situ rat brain slice model (Dickson et al., 2007). Within a period of 30 min to 2 h, transection of the neurites resulted in a stereotypic response of the cut axons characterized by formation of large bulb-like structures which is very similar and probably identical with the retraction bulbs observed in vivo (Dickson et al., 2007). Again, these terminal end bulbs or retraction bulbs were neither immobile nor static but extended very fine regenerative axonal sprouts. These not only increased in length but changed their growth direction, highlighting not only the dynamic behaviour of the terminal bulbs but also suggesting that axons in vitro and in vivo respond to mechanical injury with regenerative growth responses (Kerschensteiner et al., 2005; Dickson et al., 2007). Currently, nothing is known about the molecular machinery of this response but a direct comparison with the behaviour of regenerating axons in the agnathostom lamprey suggested that it might be the propulsive forces of the neurofilaments invading the terminal bulb which drive their dynamic behaviour (Zhang et al., 2005). In another study, it was suggested that it is the microtubular system, and not neurofilaments, that underlies the formation of retraction bulbs (Ertürk et al., 2007). Using transgenic mice expressing green fluorescent protein-M (GFP-M) and yellow fluorescent protein-H (YFP-H) under the control of the neuron-specific Thy-1 promoter, the authors focused on the in vivo behaviour of the GFP-labelled dorsal root ganglia neurons (Ertürk et al., 2007). These neurons possess independent centrally and peripherally projecting axon branches enabling comparative observation of the in vivo response after transecting the peripheral or the central axon branches. Transection of the peripheral axon resulted in formation of a new growth cone and regeneration of the severed axon, whereas transection of the central dorsal root ganglion (DRG) axon induced formation of an immobile, non-growing retraction bulb and resulted in regenerative response failure. Detailed analysis of these retraction bulbs gave new insights into their behaviour and molecular characterization with the retraction bulb of the proximal cut end growing in size over weeks and containing disorganized microtubules, trans-Golgi vesicles and normal-looking mitochondria (Ertürk et al., 2007). Focusing on microtubules, the retraction bulbs contained both stable (detyrosinated) and dynamic (tyrosinated) microtubules in a disorganized structure in contrast to the highly organized appearance of dynamic and stable microtubules in the peripheral growth cone, pointing to an important role of microtubule organization in the formation of retraction bulbs and growth cones. To support this, Ertürk and colleagues treated the injured peripheral axon at the lesion site with the microtubule disruptor Nocodazole (330 µmol·L−1) 1 day after transection. Twenty-four hours later, more than 40% of the peripheral growth cones had transformed their growth cones into retraction-bulb like structures (Ertürk et al., 2007). These new results suggest that disrupting microtubules suffices to transform a growth cone into a retraction bulb which gives rise to the following two questions, ‘Does microtubule-stabilization lead to formation of new growth cones from retraction bulbs or does it reduce the development of retraction bulbs?’ Treating the central branches locally, immediately after dorsal column injury with the microtubule stabilizer Taxol, reduced the number of retraction bulbs from 71% to 23% and stimulated axon growth (Ertürk et al., 2007). These exciting data suggest that interfering with microtubule stability is a potential pharmacotherapeutic approach to preventing, reducing or reversing retraction bulb formation in CNS injury. Due to the potential side effect of systemic Taxol treatment with induction of peripheral neuropathies, local application is recommended or, alternatively, other ways of enhancing microtubule stability are worth evaluating. Target candidates would be proteins regulating microtubule-associated proteins such as Glycogen synthase kinase-3β, (GSK-3β), the synapses of amphids defective kinases (homologues of the partitioning defective-1, PAR-1, serine/threonine kinases), c-Jun N-terminal kinases or collapsin-response mediator protein 2 because their inhibition or deletion has been shown to affect the process of microtubule stabilization and thereby of axon formation (Witte et al., 2008). Anti-neoplastic agents, the taxanes, the epothilones, macrolide antibiotics with a distinct tubulin binding mode, laulimalide, a natural cytotoxic product isolated from marine sponges and structurally distinct from the taxanes, with a common working mechanism of binding to tubulin and stabilizing microtubules have been described in recent years (Cortes and Baselga, 2007; Fojo and Menefee, 2007; Gallagher, 2007; Liu et al., 2007) and might be candidates for local application in animal models of TBI to evaluate their potential ability in reversing the retraction bulb status of injured axons, thereby stimulating regenerative growth and functional recovery.
These approaches target the growth machinery of injured axons but another approach is to modify the micro-environment of the injured axons or their response to this micro-environment. Comparing transected central with transected peripheral axons suggests that the different behaviour of the severed axons in the peripheral nervous system (PNS) and CNS is not caused by a general inability to form a growth cone but that external influences, which differ in the PNS and CNS, are responsible for growth cone formation or retraction bulb induction. In the next paragraph, we will therefore focus on the repulsive or inhibitory micro-environment with which an injured or lesioned axon is confronted in the CNS.
The inhibitory environment of the injured axon in the central nervous system
It is now generally accepted that the damage caused by primary TBI is worsened by secondary pathological injury mechanisms (Gennarelli and Graham, 2005). Secondary pathology starts shortly after the traumatic insult and shares many features with spinal cord injury (Figure 1) such as ischemia, haemorrhaging, excitotoxicity, free radical formation, and necrotic and apoptotic cell death (Mueller et al., 2005). These secondary injury mechanisms worsen the primary insult and increase damage making any pharmacotherapeutic approach even more difficult. Due to the occurrence of multiple pathological injury mechanisms, any neuroprotective strategy focusing on only one of these mechanisms will probably not result in any significant therapeutic benefit for brain injury patients. However, it may support future, more promising restorative neuroregenerative therapy by limiting secondary injury-related damage to the brain.
Figure 1.
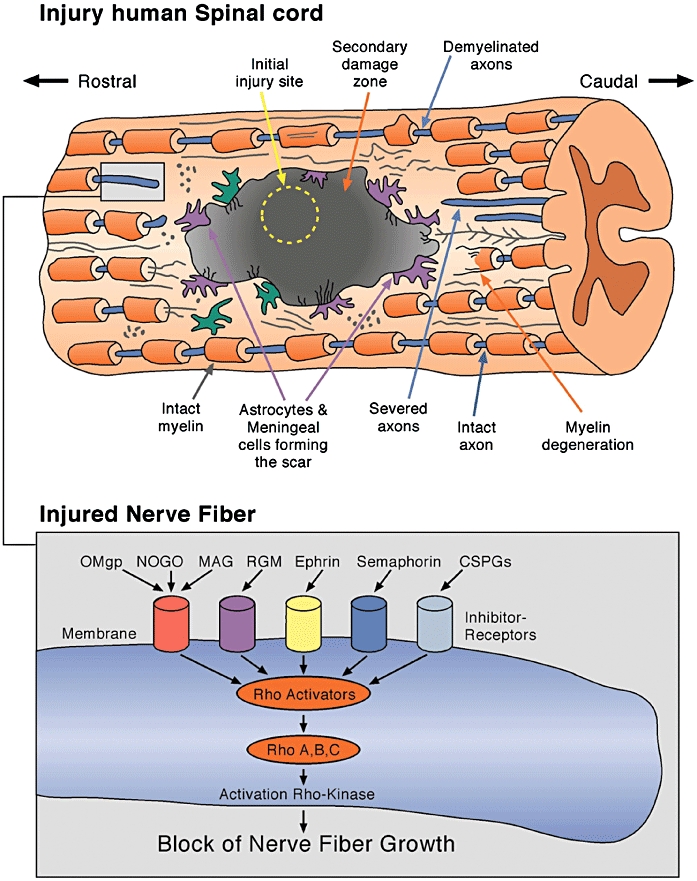
Diagram of human spinal cord injury. The primary injury site is usually small and secondary mechanisms such as excitotoxicity, inflammation and oedema formation contribute significantly to enlargement of the primary site of injury. An injured, transected and demyelinated nerve fibre is shown in the box. The transected nerve fibre expresses receptors for many neurite growth inhibitory proteins. Many of them activate the Rho-Rho-kinase pathway leading to irreversible growth arrest of the injured fibre.
One of the main reasons for the limited success of pharmacotherapeutic approaches in brain injury patients is the inability of the injured nerve fibres to regrow and establish new synaptic connections because the micro-environment in the CNS is hostile to the regeneration of nerve fibres with many molecular growth inhibitors appearing at the lesion site. As mentioned above, such a hostile micro-environment keeps the injured axons in a permanently inhibited growth mode, characterized by the occurrence of varicosities and retraction bulbs. The appearance of many different molecular neurite growth inhibitors at the lesion site, in the cicatrix and mature scar in brain injury patients is a formidable barrier for any function-restoring, neuroregenerative therapy and suggests that targeting these neurite growth inhibitors could be a way of overcoming the long-lasting, invariably permanent stalling of neurite growth.
A remarkable example of regenerative fibre growth in a human TBI patient
Before we focus on molecular neurite growth inhibitors, we would like to address the question of whether axonal regeneration is possible in patients with severe brain injury. In the adult mammalian CNS, axonal growth over a distance greater than 1 mm rarely occurs without therapeutic intervention (Cafferty et al., 2008) but in a recent publication, Voss and colleagues describe a 39-year-old patient, who was in a MCS for 19 years as a result of severe closed head injury caused by a motor-vehicle accident, and who spontaneously emerged from MCS after 19 years (Voss et al., 2006). In two DTI exams 8 and 18 months after recovery from MCS, the authors observed large increases in FA in the posterior white matter and correlated these anisotropy increases with the patient's clinical improvement suggesting that axonal regrowth was responsible for the late recovery of the patient (Voss et al., 2006). The fascinating study by Voss and colleagues suggests that significant axonal growth is possible, even in a patient with severe head injury, after years in MCS and that the axonal changes can be monitored or followed by DTI. It is not known what mechanisms induced axonal regrowth and the patient's recovery but it is assumed that treatment of TBI patients with neuroregeneration-stimulating drugs is a valuable strategy, no matter if the patient is in coma, a vegetative state, MCS or a locked-in state.
Neutralizing the molecular neurite growth inhibitors
Many neuroregenerative drug candidates have been evaluated in models of spinal cord injury (Table 1) and important lessons have been learned from these studies. Both spinal cord injury and brain injury share many similarities and a common feature of both injuries is the inhibition of regrowth after axonal injury. As mentioned above, neurite growth inhibitors present in CNS myelin and scar tissue are responsible for this growth inhibition (He and Koprivica, 2004; Schwab, 2004; Mueller et al., 2005; Schwab et al., 2005a; Liu et al., 2006). Table 2 lists well-known neurite growth inhibitors together with their receptors or the primary binding protein of their receptor complex. Scar- or lesion-associated inhibitors encompass CSPGs, RGMs and members of the ephrin and semaphorin families (Mueller et al., 2005). Well-characterized growth inhibitors of the CNS myelin are myelin-associated glycoprotein (MAG), NOGO A, and oligodendrocyte myelin glycoprotein, all binding to NgR1, a glycosylphosphatidyl-inositol-anchored (GPI) glycoprotein expressed by some neurons (Fournier et al., 2001; Domeniconi et al., 2002; Liu et al., 2002; Wang et al., 2002). The lipid-anchored NgR1 has no signal transduction domain and therefore requires transducing co-receptors. Three candidates have been identified, the low affinity neurotrophin receptor p75, LINGO 1 and TROY/TAJ, a p75-related member of the TNF receptor family (Wong et al., 2002; Mi et al., 2004; Mueller et al., 2005; Park et al., 2005; Shao et al., 2005). Neutralization of NOGO A using a monoclonal antibody resulted in induction of regenerative growth in spinal cord injury and enhancement of cognitive defects after experimental brain injury in rodents. Importantly, expression analysis in TBI patients gave a clear indication of Nogo A up-regulation or accumulation at the human lesion sites (Thallmair et al., 1998; Buss et al., 2005; Lenzlinger et al., 2005). Blocking MAG in an experimental TBI study resulted in neuroprotection and functional improvements (Thompson et al., 2006). Positive effects were also observed in brain injury models when NgR1 was neutralized by NgR antagonists (Lee et al., 2003). Inhibition of these myelin-associated inhibitors or of one of their receptors has so far resulted in neuroprotective, neuroregenerative effects with concomitant functional improvements. Targeting of other neurite growth inhibitors pathologically associated with TBI is therefore a possible strategy.
Table 2.
Neurite growth inhibitory biomolecules and their receptors
| Neurite growth inhibitor | Receptor |
|---|---|
| Nogo A | NgR1 (nogo66 receptor) |
| Myelin associated glycoprotein (MAG) | NgR1 (nogo66 receptor), NgR2 |
| Oligodendrocyte myelin glycoprotein (OMgp) | NgR1 (nogo66 receptor) |
| Semaphorin 3A | Neuropilin-1 (NP-1) |
| Semaphorin 4D | Plexin-B1 |
| Semaphorin 5A | Plexin-B3 |
| Repulsive guidance molecule A (RGM A) | Neogenin |
| Ephrin B3 | EphA recectors, class B Ephs |
| Chondroitin sulphate proteoglycans (CSPG) | Unknown, various candidates, e.g. L1, NCAM, tenascin |
| Netrin-1 | UNC5H |
| Wnt, Wnt 5a | RYK |
| Bone morphogenetic proteins (BMP-2, BMP-4) | BMP recs. I and II, RGM proteins as coreceptors |
Overview of neurite growth inhibitory molecules and their receptors. Many of these inhibitory proteins play an important role as guidance molecules during development of the vertebrate central nervous system.
Other myelin-associated molecules with neurite growth inhibitory activity play important roles in the early development of neuronal maps. They belong to the families of the ephrins, semaphorins and RGMs that are up-regulated or re-expressed in spinal cord injury sites and/or human brain injury sites (Miranda et al., 1999; Willson et al., 2002; Moreau-Fauvarque et al., 2003; Schwab et al., 2005a,b; Hata et al., 2006). We focus here on the family of the neurite growth inhibitors of the RGM family because the expression pattern of these GPI-anchored molecules has been described in human brain after TBI, in ischemic human brain and in rat spinal cord lesions (Monnier et al., 2002; Mueller et al., 2006).
Repulsive guidance molecule proteins are GPI-anchored glycoproteins that play a role in neurite growth inhibition (RGM A) and iron metabolism (RGM C/hemojuvelin), and exist in membrane-bound and soluble forms (Mueller et al., 2006). These proteins bind to their receptor Neogenin, thereby inducing neurite growth inhibition (Rajagopalan et al., 2004; Conrad et al., 2007). In addition, all RGM proteins have been shown to interact with bone morphogenetic proteins (BMP) 2 and 4 (Babitt et al., 2005; 2006; Samad et al., 2005) and early data suggest that BMP interaction with RGM A also results in neurite growth inhibition. RGM A therefore displays two different modes of neurite growth inhibition (Figure 2). In the trans-mode, RGM A on oligodendrocytes or microglial cells bind to Neogenin on axons thereby stimulating RhoA and Rho-kinase via LMO4 release (Hata et al., 2006; Conrad et al., 2007; Schaffar et al., 2008) whereas in the cis mode RGM A on axons interact with the BMP-2 or 4, promoting interaction of the RGM A/BMP-complex with the BMP-receptors, resulting in direct activation of LIM kinase 1 and axonal growth inhibition (Matsuura et al., 2007; 2008).
Figure 2.
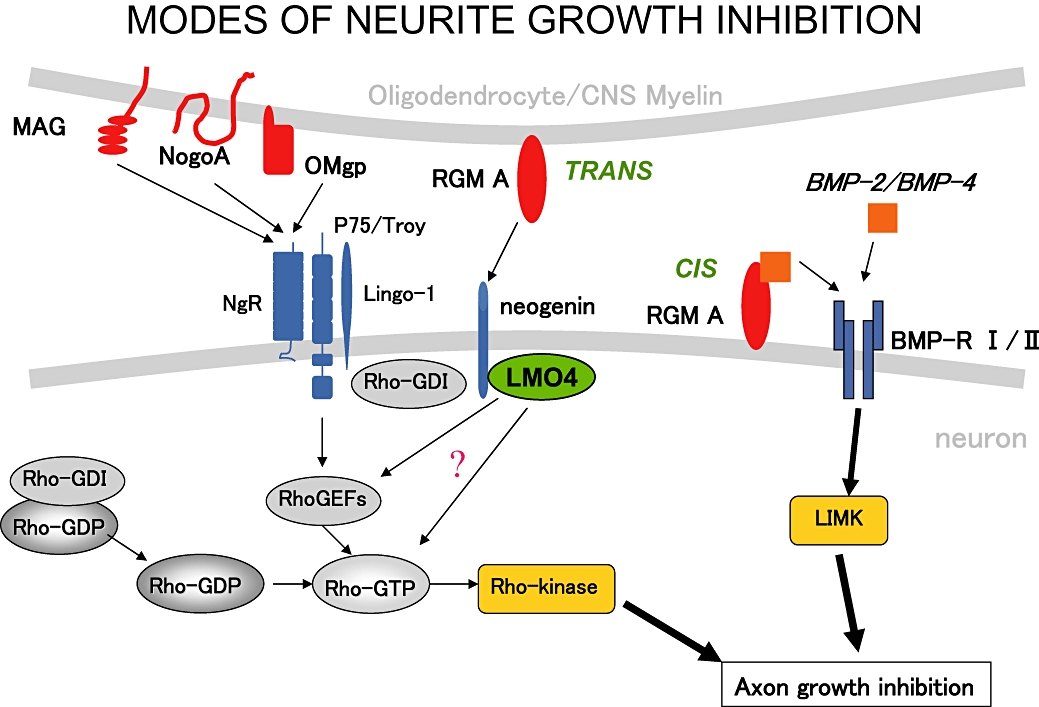
Modes of neurite growth inhibition of molecules expressed on CNS myelin and oligodendrocytes. RGM A has two modes of inducing growth inhibition: a trans-mode, with RGM A on myelin/oligodendrocytes binding to neuronal Neogenin and downstream activation of the Rho-Rho-kinase pathway. LMO4 is a transcriptional co-regulator, present in neurites and growth cones, stimulating the Rho pathway via an unknown mechanism. In the cis mode, RGM A expressed on neurites interacts with BMP-2/BMP-4. This complex then stimulates LIM-Kinase via activation of BMP-receptors I and II, leading to axonal growth inhibition (Figure 2 is a courtesy of Dr T. Yamashita, University of Osaka, Japan).
In healthy adult human brain, immunostaining with a pan-RGM antibody showed expression of RGM on some neuronal perikarya, oligodendrocytes and white matter fibre tracts as well as on smooth muscle cells of the vasculature and choroid plexus, and on endothelial cells (Schwab et al., 2005b). A very similar evolutionarily conserved expression pattern was observed in rat spinal cord (Schwab et al., 2005a), suggesting that these molecules contribute to the failure of regenerative neurite growth after traumatic brain and spinal cord injury. Injury to the human brain induces massive accumulation of RGM-positive cells at the injury site in rather rapid progress within 12 h of injury (Schwab et al., 2005b). For weeks after injury, much higher RGM-positive cell counts are found at the lesion site and, with the maturation of cicatrix and scar tissue, extracellular-like, pseudo-laminar RGM staining patterns are observed at the site of injury pointing to cleavage or secretion of the protein into the extracellular matrix of the forming scar (Schwab et al., 2005b). In one patient, a pseudo-laminar RGM staining pattern was observed in the scar tissue even 5 years after brain injury. This is a clear sign that the pathological up-regulation and deposition of such potent neurite growth inhibitors is a very stable process preventing any further restorative or regenerative growth of injured axons (Schwab et al., 2005b). Similar accumulation of RGM has been observed in the penumbral region of patients suffering from ischemic stroke and in spinal cord lesions in rats, suggesting that this molecule is an important inhibitor of regenerative growth of injured axons in the CNS of humans and rodents (Schwab et al., 2005a,b; Hata et al., 2006). Neutralizing RGM A in a rat spinal cord injury model using a small amount of a function-blocking RGM A-specific polyclonal antibody resulted in significant long distance regenerative growth of injured axons through the lesion and beyond, the formation of synapses and significant functional improvement (Hata et al., 2006; Kyoto et al., 2007), thereby proving that neutralization of the pathologically increased levels of RGM A stimulates regeneration and functional recovery (Mueller et al., 2006). Due to the similar RGM A molecular neuropathology of rat spinal cord injury and human TBI, it is therefore expected that RGM A neutralization will also stimulate the regrowth of injured axons and functional recovery in TBI models.
Releasing the molecular brake in stalled neurites
Targeting external inhibitors of the micro-environment like RGM A, NOGO A, MAG or Nogo receptor 1, is one mechanism for stimulating regrowth of the injured axon. Another mechanism separate from stimulation of microtubule assembly but probably converging on it, focuses on elements of the signal pathway of injured axons, the mechanism underlying the irreversible brake. The inhibitors of neurite growth mentioned above have all been shown to stimulate the serine/threonine kinase, Rho kinase, within the injured nerve (Mueller et al., 2005; Kubo and Yamashita, 2007). Neutralization of this kinase with small molecule (e.g. Fasudil, Y-27632) or peptidic inhibitors (e.g. p21-tat) promoted functional recovery through neuroprotective and neuroregenerative mechanisms in rodent spinal cord injury models (Hara et al., 2000; Dergham et al., 2002; Fournier et al., 2003; Sung et al., 2003; Tanaka et al., 2004). In addition, Nogo-66, a potent neurite growth inhibitory fragment of NOGO A and MAG, was shown to inhibit microtubule assembly by activating Rho kinase, thereby linking the molecular braking mechanism with the motor driving force, the microtubule assembly (Mimura et al., 2006). Other interference points for drug candidates to stimulate regenerative growth of injured axons are certain isoforms of protein kinase C (Hasegawa et al., 2004), the epidermal growth factor (EGF) receptor (Koprivica et al., 2005) and GSK-3β (Burgaya et al., 2006; Ito et al., 2006). Inhibitors blocking these elements of neural inhibitory signalling pathways may be good candidates for testing in brain injury models.
Conclusion
The results obtained over the last 30 years from lesion paradigms such as spinal cord injury have taught us important lessons. One lesson deals with regrowing axons and their ability to re-establish broken connections. This process is not at all stochastic but precisely follows guidance-molecule-decorated pathways in the damaged CNS. Aberrant behaviour resulting from the formation of faulty connections is therefore not necessarily a critical issue. The transected axon is not inherently immobile but is in a latent state of growth which is suppressed by factors of the local environment. Attenuation of inhibition by antagonists neutralizing single inhibitory factors can therefore reverse the permanent growth inhibitory mode. With the development of new in vitro and in vivo assays focused on the axonal retraction bulb, the development of small molecules and antibodies, new imaging techniques enabling visualization of axonal damage not only in animal models of brain injury but also in humans suffering from brain injury, we are in a position to develop new strategies for the development of regeneration-promoting therapeutics which will help to overcome the silent epidemic of TBI.
Acknowledgments
The authors thank Dr Toshihide Yamashita, Osaka University, Japan, for his kind gift of Figure 2. We owe many thanks to our colleague Bridget Grout for critically reading and improving our manuscript.
Glossary
Abbreviations:
- APP
amyloid precursor protein
- BMP
bone morphogenetic protein
- CDC
Centres for Disease Control and Prevention
- CNS
central nervous system
- CSPG
chondroitinsulfate proteoglycan
- DAI
diffuse axonal injury
- DTI
diffusion tensor imaging
- FA
fractional anisotropy
- GFP
green fluorescent protein
- GPI-anchor
glycosylphosphatidyl-inositol-anchor
- GSK-3β
Glycogen synthase kinase – 3β
- LMO4
LIM-only protein 4
- MAG
myelin-associated glycoprotein
- MCS
minimally conscious state
- MPTP
mitochondrial permeability transition pore
- NgR1
Nogo-Receptor 1
- PAR-1 kinase
partitioning defective-1 kinase
- RGM
repulsive guidance molecule
- TBI
traumatic brain injury
- YFP
yellow fluorescent protein
Conflict of interest
None declared.
References
- Aguayo AJ, Rasminsky M, Bray GM, Carbonetto S, McKerracher L, Villegas-Pérez MP, et al. Degenerative and regenerative responses of injured neurons in the central nervous system of adult mammals. Philos Trans R Soc Lond B Biol Sci. 1991;331:337–343. doi: 10.1098/rstb.1991.0025. [DOI] [PubMed] [Google Scholar]
- Babitt JL, Zhang Y, Samad TA, Xia Y, Tang J, Campagna JA, et al. Repulsive guidance molecule (RGMa), a DRAGON homologue, is a bone morphogenetic protein co-receptor. J Biol Chem. 2005;280:29820–29827. doi: 10.1074/jbc.M503511200. [DOI] [PubMed] [Google Scholar]
- Babitt JL, Huang FW, Wrighting DM, Xia Y, Sidis Y, Samad TA, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38:531–539. doi: 10.1038/ng1777. [DOI] [PubMed] [Google Scholar]
- Baptiste DC, Fehlings MG. Emerging drugs for spinal cord injury. Expert Opin Emerg Drugs. 2008;13:63–80. doi: 10.1517/14728214.13.1.63. [DOI] [PubMed] [Google Scholar]
- Bazarian JJ, Zhong J, Blyth B, Zhu T, Kavcic V, Peterson D. Diffusion Tensor Imaging detects clinically important axonal damage after mild traumtic brain injury: a pilot study. J Neurotrauma. 2007;2:1447–1459. doi: 10.1089/neu.2007.0241. [DOI] [PubMed] [Google Scholar]
- Berry M. Post-injury myelin-breakdown products inhibit axonal growth: an hypothesis to explain the failure of axonal regeneration in the mammalian central nervous system. Bibl Anat. 1982;23:1–11. [PubMed] [Google Scholar]
- Bruns J, Hauser WA. The epidemiology of traumatic brain injury: a review. Epilepsia. 2003;44(Suppl. 10):2–10. doi: 10.1046/j.1528-1157.44.s10.3.x. [DOI] [PubMed] [Google Scholar]
- Büki A, Povlishock JT. All roads lead to disconnection? – Traumatic axonal injury revisited. Acta Neurochir (Wien) 2006;148:181–194. doi: 10.1007/s00701-005-0674-4. [DOI] [PubMed] [Google Scholar]
- Büki A, Siman R, Trojanowski JQ, Povlishock JT. The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol. 1999;58:365–375. doi: 10.1097/00005072-199904000-00007. [DOI] [PubMed] [Google Scholar]
- Büki A, Okonkwo DO, Wang KK, Povlishock J. Cytochrome c release and caspase activation in traumatic axonal injury. J Neurosci. 2000;20:2825–2834. doi: 10.1523/JNEUROSCI.20-08-02825.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgaya F, Fontana X, Martínez A, Montolio M, Mingorance A, Simó S, et al. Semaphorin 6C leads to GSK-3-dependent growth cone collapse and redistributes after entorhino-hippocampal axotomy. Mol Cell Neurosci. 2006;33:321–334. doi: 10.1016/j.mcn.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Buss A, Sellhaus B, Wolmsley A, Noth J, Schwab ME, Brook GA. Expression pattern of NOGO-A protein in the human nervous system. Acta Neuropathol. 2005;110:113–119. doi: 10.1007/s00401-004-0942-z. [DOI] [PubMed] [Google Scholar]
- Cafferty WBJ, McGee AW, Strittmatter SM. Axonal growth therapeutics: regeneration or sprouting or plasticity. Trends Neurosci. 2008;31:215–220. doi: 10.1016/j.tins.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. Traumatic Brain Injury. Available at: http://www.cdc.gov/ncipc/tbi/TBI.htm.
- Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci. 2005;6:889–898. doi: 10.1038/nrn1788. [DOI] [PubMed] [Google Scholar]
- Colicos MA, Dash PK. Apoptotic morphology of dentate gyrus granule cells following experimental cortical impact injury in rats: possible role in spatial memory deficits. Brain Res. 1996;739:120–131. doi: 10.1016/s0006-8993(96)00824-4. [DOI] [PubMed] [Google Scholar]
- Conrad S, Genth H, Hofmann F, Just I, Skutella T. Neogenin-RGMa signaling at the growth cone is bone morphogenetic protein-independent and involves RhoA, ROCK, and PKC. J Biol Chem. 2007;282:16423–16433. doi: 10.1074/jbc.M610901200. [DOI] [PubMed] [Google Scholar]
- Corso P, Finkelstein E, Miller T, Fiebelkorn I, Zaloshnja E. Incidence and lifetime costs of injuries in the United States. Inj Prev. 2006;12:212–218. doi: 10.1136/ip.2005.010983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes J, Baselga J. Targeting the microtubules in breast cancer beyond taxanes: the epothilones. Oncologist. 2007;12:271–280. doi: 10.1634/theoncologist.12-3-271. [DOI] [PubMed] [Google Scholar]
- David S, Aguayo AJ. Axonal elongation into peripheral nervous system ‘bridges’ after central nervous system injury in adult rats. Science. 1981;214:931–933. doi: 10.1126/science.6171034. [DOI] [PubMed] [Google Scholar]
- Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci. 2002;22:6570–6577. doi: 10.1523/JNEUROSCI.22-15-06570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson TC, Chung RS, McCormack GH, Staal JA, Vickers JC. Acute reactive and regenerative changes in mature cortical axons following injury. Neuroreport. 2007;18:283–288. doi: 10.1097/WNR.0b013e3280143cdb. [DOI] [PubMed] [Google Scholar]
- Domeniconi M, Cao Z, Spencer T, Sivasankaran R, Wang K, Nikulina E, et al. Myelin-associated glycoprotein interacts with the Nogo66 receptor to inhibit neurite outgrowth. Neuron. 2002;35:283–290. doi: 10.1016/s0896-6273(02)00770-5. [DOI] [PubMed] [Google Scholar]
- Donovan WH. Spinal cord injury – past, present and future. J Spinal Cord Med. 2007;30:85–100. doi: 10.1080/10790268.2007.11753918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertürk A, Hellal F, Enes J, Bradke F. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J Neurosci. 2007;27:9169–9180. doi: 10.1523/JNEUROSCI.0612-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fojo T, Menefee M. Mechanisms of multidrug resistance: the potential role of microtubule-stabilizing agents. Ann Oncol. 2007;18(Suppl. 5):v3–v8. doi: 10.1093/annonc/mdm172. [DOI] [PubMed] [Google Scholar]
- Fournier AE, GrandPre T, Strittmatter SM. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature. 2001;409:341–346. doi: 10.1038/35053072. [DOI] [PubMed] [Google Scholar]
- Fournier AE, Takizawa BT, Strittmatter SM. Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci. 2003;23:1416–1423. doi: 10.1523/JNEUROSCI.23-04-01416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher BM., Jr Microtubule-stabilizing natural products as promising cancer therapeutics. Curr Med Chem. 2007;14:2959–2967. doi: 10.2174/092986707782794014. [DOI] [PubMed] [Google Scholar]
- Gennarelli TA, Graham DI. In: Neuropathology in Textbook of Traumatic Brain Injury. Silver JM, McAllister TW, Yudofsky SC, editors. Washington DC: American Psychiatric Publishing Inc.; 2005. pp. 27–50. Chapter 2. [Google Scholar]
- Goldstein M. Traumatic Brain Injury: A Silent Epidemic. Editorial. Ann Neurology. 1990;27:327. doi: 10.1002/ana.410270315. [DOI] [PubMed] [Google Scholar]
- Gonzenbach RR, Schwab ME. Disinhibition of neurite growth to repair the injured adult CNS: focusing on Nogo. Cell Mol Life Sci. 2008;65:161–176. doi: 10.1007/s00018-007-7170-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham D, Gennarelli T, McIntosh T. Trauma. In: Graham D, Lantos P, editors. Greenfield's Neuropathology. Vol. 1. London: Arnold, Hodder Headline Group; 2002. pp. 823–898. [Google Scholar]
- Han BS, Kim SH, Kim OL, Cho SH, Kim YH, Jang SH. Recovery of corticospinal tract with diffuse axonal injury: a diffusion tensor image study. NeuroRehabilitation. 2007;22:151–155. [PubMed] [Google Scholar]
- Hara M, Takayasu M, Watanabe K, Noda A, Takagi T, Suzuki Y, et al. Protein kinase inhibition by fasudil hydrochloride promotes neurological recovery after spinal cord injury in rats. J Neurosurg. 2000;93(1) Suppl.:94–101. doi: 10.3171/spi.2000.93.1.0094. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y, Fujitani M, Hata K, Tohyama M, Yamagishi S, Yamashita T. Promotion of axon regeneration by myelin-associated glycoprotein and Nogo through divergent signals downstream of Gi/G. J Neurosci. 2004;24:6826–6832. doi: 10.1523/JNEUROSCI.1856-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata K, Fujitani M, Yasuda Y, Doya H, Saito T, Yamagishi S, et al. RGMa inhibition promotes axonal growth and recovery after spinal cord injury. J Cell Biol. 2006;173:47–58. doi: 10.1083/jcb.200508143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Koprivica V. The Nogo signaling pathway for regeneration block. Annu Rev Neurosci. 2004;27:341–368. doi: 10.1146/annurev.neuro.27.070203.144340. [DOI] [PubMed] [Google Scholar]
- Ito Y, Oinuma I, Katoh H, Kaibuchi K, Negishi M. Sema4D/plexin-B1 activates GSK-3beta through R-Ras GAP activity, inducing growth cone collapse. EMBO Rep. 2006;7:704–709. doi: 10.1038/sj.embor.7400737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley BJ, Farkas O, Lifshiz J, Povlishock JT. Traumatic axonal injury in the perisomatic domain triggers ultrarapid secondary axotomy and Wallerian degeneration. Exp Neurol. 2006;198:350–360. doi: 10.1016/j.expneurol.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat Med. 2005;11:572–577. doi: 10.1038/nm1229. [DOI] [PubMed] [Google Scholar]
- Koprivica V, Cho KS, Park JB, Yiu G, Atwal J, Gore B, et al. EGFR activation mediates inhibition of axon regeneration by myelin and chondroitin sulfate proteoglycans. Science. 2005;310:106–110. doi: 10.1126/science.1115462. [DOI] [PubMed] [Google Scholar]
- Kraus JF, Chu LD. In: Epidemiology in Textbook of Traumatic Brain Injury. Silver JM, McAllister TW, Yudofsky SC, editors. Washington DC: American Psychiatric Publishing Inc.; 2005. pp. 3–26. Chapter 1. [Google Scholar]
- Kraus MF, Susmaras T, Caughlin P, Walker CJ, Sweeney JA, Little DM. White mater integrity and cognition in chronic traumaticbrain injury: a diffusion tensor imaging study. Brain. 2007;130:2508–2519. doi: 10.1093/brain/awm216. [DOI] [PubMed] [Google Scholar]
- Kubo T, Yamashita T. Rho-ROCK inhibitors for the treatment of CNS injury. Recent Pat CNS Drug Discov. 2007;2:173–179. doi: 10.2174/157488907782411738. [DOI] [PubMed] [Google Scholar]
- Kyoto A, Hata K, Yamashita T. Synapse formation of the cortico-spinal axons is enhanced by RGMa inhibition after spinal cord injury. Brain Res. 2007;1186:74–86. doi: 10.1016/j.brainres.2007.10.038. [DOI] [PubMed] [Google Scholar]
- Lee DH, Strittmatter SM, Sah DW. Targeting the Nogo receptor to treat central nervous system injuries. Nat Rev Drug Discov. 2003;2:872–878. doi: 10.1038/nrd1228. [DOI] [PubMed] [Google Scholar]
- Lenzlinger PM, Shimizu S, Marklund N, Thompson HJ, Schwab ME, Saatman KE, et al. Delayed inhibition of Nogo-A does not alter injury-induced axonal sprouting but enhances recovery of cognitive function following experimental traumatic brain injury in rats. Neuroscience. 2005;134:1047–1056. doi: 10.1016/j.neuroscience.2005.04.048. [DOI] [PubMed] [Google Scholar]
- Li Y, Raisman G. Sprouts from cut corticospinal axons persist in the presence of astrocytic scarring in long-term lesions of the adult rat spinal cord. Exp Neurol. 1995;134:102–111. doi: 10.1006/exnr.1995.1041. [DOI] [PubMed] [Google Scholar]
- Liu BP, Fournier A, GrandPré T, Strittmatter SM. Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science. 2002;297:1190–1193. doi: 10.1126/science.1073031. [DOI] [PubMed] [Google Scholar]
- Liu BP, Cafferty WB, Budel SO, Strittmatter SM. Extracellular regulators of axonal growth in the adult central nervous system. Philos Trans R Soc Lond B Biol Sci. 2006;361:1593–1610. doi: 10.1098/rstb.2006.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Towle MJ, Cheng H, Saxton P, Reardon C, Wu J, et al. In vitro and in vivo anticancer activities of synthetic (-)-laulimalide, a marine natural product microtubule stabilizing agent. Anticancer Res. 2007;27:1509–1518. [PubMed] [Google Scholar]
- Mac Donald CL, Dikranian K, Bayly P, Holtzman D, Brody D. Diffusion Tensor Imaging reliably detects experimental traumatic axonal injury and indicates approximate time of injury. J Neurosci. 2007;27:11869–11876. doi: 10.1523/JNEUROSCI.3647-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura I, Endo M, Hata K, Kubo T, Yamaguchi A, Saeki N, et al. BMP inhibits neurite growth by a mechanism dependent on LIM-kinase. Biochem Biophys Res Commun. 2007;360:868–873. doi: 10.1016/j.bbrc.2007.06.157. [DOI] [PubMed] [Google Scholar]
- Matsuura I, Taniguchi J, Hata K, Saeki N, Yamashita T. BMP inhibition enhances axonal growth and functional recovery after spinal cord injury. J Neurochem. 2008;105(4):1471–1479. doi: 10.1111/j.1471-4159.2008.05251.x. [DOI] [PubMed] [Google Scholar]
- Mi S, Lee X, Shao Z, Thill G, Ji B, Relton J, et al. LINGO-1 is a component of the Nogo-66 receptor/p75 signaling complex. Nat Neurosci. 2004;7:221–228. doi: 10.1038/nn1188. [DOI] [PubMed] [Google Scholar]
- Mi S, Hu B, Hahm K, Luo Y, Kam Hui ES, Yuan Q, et al. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat Med. 2007;13:1228–1233. doi: 10.1038/nm1664. [DOI] [PubMed] [Google Scholar]
- Mimura F, Yamagishi S, Arimura N, Fujitani M, Kubo T, Kaibuchi K, et al. Myelin-associated glycoprotein inhibits microtubule assembly by a Rho-kinase-dependent mechanism. J Biol Chem. 2006;281:15970–15979. doi: 10.1074/jbc.M510934200. [DOI] [PubMed] [Google Scholar]
- Miranda JD, White LA, Marcillo AE, Willson CA, Jagid J, Whittemore SR. Induction of Eph B3 after spinal cord injury. Exp Neurol. 1999;156:218–222. doi: 10.1006/exnr.1998.7012. [DOI] [PubMed] [Google Scholar]
- Monnier PP, Sierra A, Macchi P, Deitinghoff L, Andersen JS, Mann M, et al. RGM is a repulsive guidance molecule for retinal axons. Nature. 2002;419:392–395. doi: 10.1038/nature01041. [DOI] [PubMed] [Google Scholar]
- Moreau-Fauvarque C, Kumanogoh A, Camand E, Jaillard C, Barbin G, Boquet I, et al. The transmembrane semaphorin Sema4D/CD100, an inhibitor of axonal growth, is expressed on oligodendrocytes and upregulated after CNS lesion. J Neurosci. 2003;23:9229–9239. doi: 10.1523/JNEUROSCI.23-27-09229.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori S, Zhang J. Principles of Diffusion Tensor Imaging and its applications to basic neuroscience research. Neuron. 2006;51:527–539. doi: 10.1016/j.neuron.2006.08.012. [DOI] [PubMed] [Google Scholar]
- Mueller BK. Growth cone guidance: first steps towards a deeper understanding. Annu Rev Neurosci. 1999;22:351–388. doi: 10.1146/annurev.neuro.22.1.351. [DOI] [PubMed] [Google Scholar]
- Mueller BK, Mack H, Teusch N. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov. 2005;4:387–398. doi: 10.1038/nrd1719. [DOI] [PubMed] [Google Scholar]
- Mueller BK, Yamashita T, Schaffar G, Mueller R. The role of repulsive guidance molecules in the embryonic and adult vertebrate central nervous system. Philos Trans R Soc Lond B Biol Sci. 2006;361:1513–1529. doi: 10.1098/rstb.2006.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonkwo D, Povlishock JT. An intrathecal bolus of cyclosporin A before injury preserves mitochondrial integrity and attenuates axonal disruption in traumatic brain imjury. J Cereb Blood Flow Metab. 1998;19:443–451. doi: 10.1097/00004647-199904000-00010. [DOI] [PubMed] [Google Scholar]
- Park JB, Yiu G, Kaneko S, Wang J, Chang J, He XL, et al. A TNF receptor family member, TROY, is a coreceptor with Nogo receptor in mediating the inhibitory activity of myelin inhibitors. Neuron. 2005;45:345–351. doi: 10.1016/j.neuron.2004.12.040. [DOI] [PubMed] [Google Scholar]
- Pettus EH, Povlishock JT. Characterization of a distinct set of intra-axonal ultrastructural changes associated with traumatically induced alteration in axolemmal permeability. Brain Res. 1996;722:1–11. doi: 10.1016/0006-8993(96)00113-8. [DOI] [PubMed] [Google Scholar]
- Pettus EH, Christman CW, Giebel ML, Povlishock JT. Traumatically induced altered membrane permeability: its relationship to traumatically induced reactive axonal change. J Neurotrauma. 1994;11:507–522. doi: 10.1089/neu.1994.11.507. [DOI] [PubMed] [Google Scholar]
- Posmantur R, Kampfl A, Siman R, Liu J, Zhao X, Clifton GL, et al. A calpain inhibitor attenuates cortical cytoskeletal protein loss after experimental traumatic brain injury in the rat. Neuroscience. 1997;77:875–888. doi: 10.1016/s0306-4522(96)00483-6. [DOI] [PubMed] [Google Scholar]
- Rajagopalan S, Deitinghoff L, Davis D, Conrad S, Skutella T, Chedotal A, et al. Neogenin mediates the action of repulsive guidance molecule. Nat Cell Biol. 2004;6:756–762. doi: 10.1038/ncb1156. [DOI] [PubMed] [Google Scholar]
- Richardson PM, McGuinness UM, Aguayo AJ. Axons from CNS neurons regenerate into PNS grafts. Nature. 1980;284:264–265. doi: 10.1038/284264a0. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Bozycko-Coyne D, Siman R, Marcy V, Gennarelli TA, McIntosh TK. Prolonged calpain mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol. 1996;55:852–862. doi: 10.1097/00005072-199607000-00010. [DOI] [PubMed] [Google Scholar]
- Samad TA, Rebbapragada A, Bell E, Zhang Y, Sidis Y, Jeong SJ, et al. DRAGON, a bone morphogenetic protein co-receptor. J Biol Chem. 2005;280:14122–14129. doi: 10.1074/jbc.M410034200. [DOI] [PubMed] [Google Scholar]
- Schaffar G, Taniguchi J, Brodbeck T, Meyer AH, Schmidt M, Yamashita T, et al. LIM-Only-Protein 4 (LMO4) interacts directly with the RGM A Receptor Neogenin. J Neurochem. 2008;107:418–431. doi: 10.1111/j.1471-4159.2008.05621.x. 2008 Aug 12 [Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Schnell L, Schwab ME. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature. 1990;343:269–272. doi: 10.1038/343269a0. [DOI] [PubMed] [Google Scholar]
- Schwab ME. Nogo and axon regeneration. Curr Opin Neurobiol. 2004;14:118–124. doi: 10.1016/j.conb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Schwab ME, Thoenen H. Dissociated neurons regenerate into sciatic but not optic nerve explants in culture irrespective of neurotrophic factors. J Neurosci. 1985;5:2415–2423. doi: 10.1523/JNEUROSCI.05-09-02415.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab ME, Bartholdi D. Degeneration and regeneration of axons in the lesioned spinal cord. Physiol Rev. 1996;76:319–370. doi: 10.1152/physrev.1996.76.2.319. [DOI] [PubMed] [Google Scholar]
- Schwab JM, Conrad S, Monnier PP, Julien S, Mueller BK, Schluesener HJ. Spinal cord injury-induced lesional expression of the repulsive guidance molecule (RGM) Eur J Neurosci. 2005a;21:1569–1576. doi: 10.1111/j.1460-9568.2005.03962.x. [DOI] [PubMed] [Google Scholar]
- Schwab JM, Monnier PP, Schluesener HJ, Conrad S, Beschorner R, Chen L, et al. Central nervous system injury-induced repulsive guidance molecule expression in the adult human brain. Arch Neurol. 2005b;62:1561–1568. doi: 10.1001/archneur.62.10.1561. [DOI] [PubMed] [Google Scholar]
- Shao Z, Browning JL, Lee X, Scott ML, Shulga-Morskaya S, Allaire N, et al. TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66 receptor 1 and regulates axonal regeneration. Neuron. 2005;45:353–259. doi: 10.1016/j.neuron.2004.12.050. [DOI] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Stone JR, Okonkwo DA, Dialo AO, Rubin DG, Mutlu LK, Povlishock JT, et al. Impaired axonal transport and altered axolemmal permeability occur in distinct populations of damaged axons following traumatic brain injury. Exp Neurol. 2004;190:59–69. doi: 10.1016/j.expneurol.2004.05.022. [DOI] [PubMed] [Google Scholar]
- Sung JK, Miao L, Calvert JW, Huang L, Louis Harkey H, Zhang JH. A possible role of RhoA/Rho-kinase in experimental spinal cord injury in rat. Brain Res. 2003;959:29–38. doi: 10.1016/s0006-8993(02)03717-4. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Yamashita T, Yachi K, Fujiwara T, Yoshikawa H, Tohyama M. Cytoplasmic p21(Cip1/WAF1) enhances axonal regeneration and functional recovery after spinal cord injury in rats. Neuroscience. 2004;127:155–164. doi: 10.1016/j.neuroscience.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Thallmair M, Metz GA, Z'Graggen WJ, Raineteau O, Kartje GL, Schwab ME. Neurite growth inhibitors restrict plasticity and functional recovery following corticospinal tract lesions. Nat Neurosci. 1998;1:124–131. doi: 10.1038/373. Erratum in Nat Neurosci1 (4): 329. [DOI] [PubMed] [Google Scholar]
- Thompson HJ, Marklund N, LeBold DG, Morales DM, Keck CA, Vinson M, et al. Tissue sparing and functional recovery following experimental traumatic brain injury is provided by treatment with an anti-myelin-associated glycoprotein antibody. Eur J Neurosci. 2006;24:3063–3072. doi: 10.1111/j.1460-9568.2006.05197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tom VJ, Steinmetz MP, Miller JH, Doller CM, Silver J. Studies on the development and behavior of the dystrophic growth cone, the hallmark of regeneration failure, in an in vitro model of the glial scar and after spinal cord injury. J Neurosci. 2004;24:6531–6539. doi: 10.1523/JNEUROSCI.0994-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss HU, Uluç AM, Dyke JP, Watts R, Kobylarz EJ, McCandliss BD, et al. Possible axonal regrowth in late recovery from the minimally conscious state. J Clin Invest. 2006;116:2005–2011. doi: 10.1172/JCI27021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chun SJ, Treloar H, Vartanian T, Greer CA, Strittmatter SM. Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J Neurosci. 2002;22:5505–5515. doi: 10.1523/JNEUROSCI.22-13-05505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willson CA, Irizarry-Ramírez M, Gaskins HE, Cruz-Orengo L, Figueroa JD, Whittemore SR, et al. Upregulation of EphA receptor expression in the injured adult rat spinal cord. Cell Transplant. 2002;11:229–239. [PubMed] [Google Scholar]
- Witte H, Neukirchen D, Bradke F. Microtubule stabilization specifies initial neuronal polarization. J Cell Biol. 2008;180:619–632. doi: 10.1083/jcb.200707042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M, Poo MM. A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat Neurosci. 2002;5:1302–1308. doi: 10.1038/nn975. [DOI] [PubMed] [Google Scholar]
- Xie F, Zheng B. White matter inhibitors in CNS axon regeneration failure. Exp Neurol. 2008;209:302–312. doi: 10.1016/j.expneurol.2007.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Jin LQ, Sul JY, Haydon PG, Selzer ME. Live imaging of regenerating lamprey spinal axons. Neurorehabil Neural Repair. 2005;9:46–57. doi: 10.1177/1545968305274577. [DOI] [PubMed] [Google Scholar]