

Abstract
Glycosylation is the most common form of post-translational modifications by which oligosaccharide side chains are covalently attached to specific residues of the core protein. Especially O-linked glycan structures like the glycosaminoglycans were found to contribute significantly to many (patho-)biological processes like inflammation, coagulation, cancer and viral infections. Glycans exert their function by interacting with proteins thereby changing the structure of the interacting proteins and consequently modulating their function. Given the complex nature of cell-surface and extracellular matrix glycan structures, this therapeutic site has been neglected for a long time, the only exception being the antithrombin III-glycan interaction which has been sucessfully targeted by unfractionated and low-molecular weight heparins for many decades. Due to the recent breakthrough in the ‘–ome’ sciences, among them proteomics and glycomics, protein–glycan interactions became more amenable for therapeutic approaches so that novel inhibitors of this interaction are currently in preclinical and clinical studies. An overview of current approaches, their advantages and disadvantages, is given and the promising potential of pharmacologically interfering with protein–glycan interactions is highlighted here.
Keywords: glycosaminoglycan, chemokine, growth factor
Structure–function considerations of glycans
Among post-translational modifications of eukaryotic proteins, glycosylation is the most common and the most diverse modification. It is therefore obvious, as each cell spends a tremedous biosynthetic effort on glycosylation, that these modifications are of general importance for many biological processes such as stability and folding assistance, embryonic development, antigenic and microbial recognition, inflammatory response and metastasis (Rudd et al., 2001; Middleton et al., 2002; Fears and Woods, 2006). The wide diversity of glycoforms found in nature, in addition to frequent dramatic consequences of altering these types of glycosylation – resulting in the loss of particular protein functions or in the degradation of the entire protein – has highlighted our limited understanding of structure–function relationships of complex glycans. Recently, tools to unravel glycan function on the basis of their saccharide building blocks have been developed as have methods for investigating the signals directing their biosynthetic enzymes (Varki, 1993; Prydz and Dalen, 2000; Taylor and Gallo, 2006). This opens ways for novel approaches to target this pharmacologically highly interesting site (Gesslbauer and Kungl, 2006).
Glycoproteins contain usually one or more oligosaccharide chains covalently attached to asparagine residues (termed N-linked) or to serine, threonine, hydroxylysine or hydroxyproline residues (termed O-linked). They are found ubiquitously in nature either as soluble (intracellular or extracellular) or as membrane bound molecules. There is a great structural variety based on the type, length and linkage of the carbohydrate components as well as on the degree of saturation of potential glycosylation sites on the protein itself (Berg et al., 2002). A major characteristic of most glycoproteins is that the carbohydrate component constitutes a much smaller percentage of the overall molecular weight of the glycoprotein in relation to the core protein. This is in clear contrast to the so-called proteoglycans (PGs) in which the protein core is modified to a much higher degree by long polysaccharide chains commonly of the glycosaminoglycan (GAG) type (Mulloy and Rider, 2006).
Protein glycosylation is an enzymatic process which takes place inside the lumen of the endoplasmic reticulum (ER) and the Golgi apparatus. N-linked glycosylation is a co-translational event accompanying protein synthesis in ribosomes located in the ER, whereas O-linked glycosylation is a post-translational process characterized by the stepwise addition of sugar residues directly to the protein. The most common type of O-linked glycans contain an initial N-acetylgalactosamine residue bound to the Ser/Thr residues (=mucine-type glycans). Other O-linked glycans are known to include glucosamine, xylose, galactose, fucose or mannose as the primary sugar. Furthermore, O-linked glycoproteins are very often large proteins (>200 kDa) with clusters of glycosylation chains which are comparatively less branching than N-glycans. Chain elongation and termination, if performed, are carried out by various glycosyltransferases localized in different regions of the Golgi. Termination of O-linked glycans usually includes Gal, GlcNAc, GalNAc, Fuc or sialic acid (Hang and Bertozzi, 2005; Lehle et al., 2006).
N-linked glycans are found among lysosomal enzymes, cytokines, hormones, growth factors and their receptors, plasma proteins, matrix proteins, membrane proteins and receptors, etc. (McKillop et al., 2000; Duchesne et al., 2006; Van Acker et al., 2006). O-Linked glycans are prevalent in most secretory cells and tissues. They are present in high concentrations in the zona pelucida surrounding mammalian eggs and may funtion as sperm receptors (ZP3 glycoprotein) (Shur et al., 2006). O-linked glycans are also involved in hematopoiesis, inflammation response mechanisms and the formation of ABO blood antigens (Rydberg, 2001). Nonelongated O-GlcNAc groups have been recently shown to be related to phosphorylation states and dynamic processing related to cell signalling events in the cell (Whelan and Hart, 2003). From a pharmacological and therapeutic point of view, O-linked glycans, and here especially the GAGs, have a much higher potential for targeted intervention. The reason for this being the molecular interaction framework in which GAGs are involved and which ultimately modulate the function of a large number of signalling molecules and extracellular enzymes (see below).
The structural distribution of glycans is strongly influenced by the tissue-specific expression of the biosynthetic enzymes, their activity and specificity as well as of sugar nucleotide pool composition (Dennis et al., 1999). Glycan patterns therefore reflect a certain biological/pathological state of a cell or a tissue. We have recently shown that human microvascular endothelial cells change their GAG sulfation pattern follwing an inflammatory (TNF-alpha) trigger (Krenn et al., 2008). As the methods for analysing cell-/tissue-derived glycans are still very limited, we have applied so-called glyco-genomic methods to infere the glycan expression pattern of endothelial cells from the expression pattern of GAG biosynthetic enzymes as determined by qPCR analysis.
Biosynthesis of O-linked glycans: heparan sulphate (HS) GAGs as a pharmacologically interesting case study
The protein core components of PGs are synthesized in ribosomes to be then translocated to the rough ER where a xylosyltransferase initiates the synthesis of the linker tetrasacharide by adding a xylose to a serine residue of the protein core. Two galactose residues are subsequently added in the cis or medial Golgi to the Xyl by galactosyltransferase I and galactosyltransferase II. The fourth residue, completing the linker tetrassacharide, is a GlcA added by glucuronyltransferase I and occurs in the trans-Golgi, the final location for all subsequent reactions. The addition of the fifth saccharide determines whether the GAG chain becomes chondroitin sulfate (CS)/DS or HS/heparin. GAG type, length of the chain(s), conformational flexibility and particularly the specific GAG sequence/structure determine the biological function of the glycan part of the PG. The unique structural design, which in turn determines the specific binding properties of the PG, is generated during biosynthesis by the concerted action of a complex set of enzymes (Esko and Selleck, 2002; Gesslbauer et al., 2007) which is supposed to be non-template dependent and therefore not related to a so-called glyco-code (Habuchi et al., 2004). During chain elongation, the nascent GAG chain is modified by an epimerase, converting GlcA into IdoA, and several sulfotransferases adding sulfate groups to distinct positions. Chain elongation and modification require an array of distinct enzymes for the HS and the CS pathway (see Figure 1). This conventional view of GAG biosynthesis, which is based on sequential modifications of the chain, has been recently challenged by the postulation of a so-called GAGososme (Ledin et al., 2006) in which the biosynthetic enzymes are topologically clustered within the Golgi membranes and can therefore act in a more concerted and regulated way. The mature HS chain can also be edited by the action of endosulfatases and heparanase. Especially, the enzymes involved in the generation of the sulfation pattern exist in several isoforms with divergent activities, substrate specificities and tissue distribution. Modulation in GAG structure is therefore likely to be achieved, at least to some extend, by the differential regulation of expression of a certain repertoire of modifying enzymes. In Figure 1, the overall scheme of HS synthesis is displayed indicating the shaping of AT-III and FGF2 binding sites.
Figure 1.
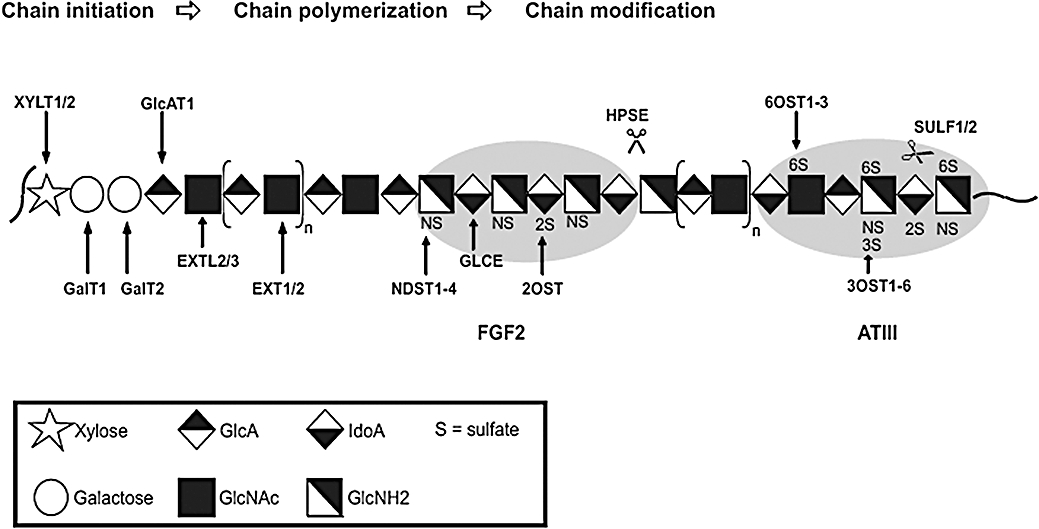
Scheme of HS-biosynthesis. Displayed is the consecutive action of the various biosynthetic enzymes durin chain initiation, polymerization and modification (the recently proposed model of a so-called GAGosome, in which the biosynthetic steps occur in a parallelized way (Ledin et al., 2006), has not been considered in this figure). Enzymes involved in HS biosynthesis are: XYLT1, xylosyltransferase-1; XYLT2, xylosyltransferase-2; GalT1, galactosyltransferase-1; GalT2, galactosyltransferase-2; GlcAT1, glucuronosyltransferase-1; EXT1, exostosin-1; EXT2, exostosin-2; EXTL2, exostosin-like-2; EXTL3, exostosin-like-3; NDST1-4, N-deacetylase/N-sulfotransferase-1, -2, -3, -4; GLCE, D-glucuronyl C5-epimerase; 2OST, 2-O-sulfotransferase; 3OST1-6, 3-O-sulfotransferase-1, -2, -3A, -3B, -4, -5, -6; 6OST1-3, 6-O-sulfotransferase-1, -2, -3; SULF1, extracellular sulfatase Sulf-1; SULF2, extracellular sulfatase Sulf-2; HPSE, heparanase. Two typical results of taylored HS biosynthesis are given in the form of the FGF2- and the ATIII-specific binding sites. HS, heparan sulphate.
Specific shaping of GAG structures was found to occur during pathogenesis, during development or in response to extracellular signals such as growth factors (Carter et al., 2003). Among the various physiological processes (like cell proliferation, differentiation, morphogenesis, angiogenesis, etc.) in which HS is involved, the interaction with extracellular signalling molecules deserves special attention. Among these signalling molecules are growth factors, and chemokines, lipid- and membrane-binding proteins, adhesions proteins, and certain proteases and esterases, as well as HS degrading enzymes (Capila and Linhardt, 2002; Handel et al., 2005; Vlodavsky et al., 2007). HS plays an essential role in the regulation of inflammatory processes, cell growth and differentiation, lipid transport and clearance, cell–cell and cell–matrix interactions and blood coagulation processes (Kolset and Salmivirta, 1999; Knox and Whitelock, 2006). The association of chemokines with HS helps to stabilize concentration gradients across the endothelial surface thereby providing directional clues for migrating leukocytes in inflammation (e.g. atherosclerosis, arthritis). HS was also shown to protect chemokines from proteolytic degradation and to induce their oligomerization, thus promoting local high concentrations in the vicinity of their G-coupled signalling receptors (Johnson et al., 2005). FGF1 and FGF2 were the first members of the fibroblast growth factor family to be discovered and their interaction with HS was intensively investigated. It has been established that HS is required for FGF to effectively activate its receptor and several models have been described trying to explain FGF/HS/FGF-receptor complex formation and its stoichiometry. For FGF2, to form the ternary signalling complex, there must be FGF-2, FGF receptor, and an HS chain having appropriately spaced 2-O and 6-O sulfation. FGF-2 released from HS chains in the extracellular matrix (ECM) by cleavage through mammalian heparanase has been shown to promote angiogenesis for tumour growth (Sanderson et al., 2005; Harmer, 2006).
Localization of O-linked glycans: they are part of PGs
Proteoglycans are a special class of glycanated proteins which consist of a core protein with one or more covalently attached GAG side chains. Their carbohydrate structure clearly distinguishes them from ‘ordinary’ glycoproteins: repeating disaccharide units composed of an hexosamine and an uronic acid are arranged to form the long linear GAG polysaccharide chains. Depending on the type of the disaccharide building blocks, the PG superfamily is subdivided into three major groups: (i) chondroitin/dermatan sulfate proteoglycans (CSPGs); (ii) heparin/heparin sulfate proteoglycans (HSPGs); and (iii) keratan sulfate proteoglycans (KSPGs) (Mulloy and Rider, 2006; Bishop et al., 2007). All three GAG structures are strongly negatively charged due to their high content of sulfate groups and uronic acids. Although there are intracellular types, most PGs are directed to the cell surface as well as to the ECM including cartilage, brain, skin, connective tissue, basement membranes and blood vessels. The biological functions of PGs are diverse ranging from purely structural function in the ECM, mechanical tissue function, influence in metastasis and their interaction with a variety of extracellular ligands, such as growth factors, chemokines and adhesions molecules (Vlodavsky et al., 2002; Kolset et al., 2004; Cattaruzza and Perris, 2005; Celie et al., 2005; Proudfoot, 2006). The common motif among the various functions is the interaction of the PG, in most cases only the GAG part, with other proteins (see below). The subsequent structural rearrangement of the interacting protein is usually the trigger for a specific biological function.
Cell surface HSPGs are subdivided into the glypican and the syndecan families, which are the major representatives of this class, betaglycan and CD44 being minor representatives. Glypicans are glycosylphosphatidylinositol (GPI)-linked molecules containing exclusively HS GAGs. The GPI anchor is responsible for a different turnover and involvement in different metabolic pathways compared with the integral PGs. It also affects their localization and distribution of the glypicans. There are six known members of the glypican family in mammals: glypican-1, glypican-2 (cerebroglycan), glypican-3 (OCI-5), glypican-4 (K-glypican), glypican-5 (GCP5) and glypican-6 (David, 1993; Fransson, 2003). The syndecan family contains four members: syndecan-1, syndecan-2 (fibroglycan), syndecan-3 (N-syndecan) and syndecan-4 (amphiglycan or ryudocan) which have been described in vertebrates. They are type I glycoproteins with a single membrane spanning protein core comprising a large ectodomain and a highly conserved transmembrane domain. Their short cytoplasmatic parts contain two conserved domains interrupted by a variable region, which is specific for each syndecan family member, thus allowing both common and core-specific interactions with various cytoplasmic molecules. Syndecans usually carry three HS chains close to their N-terminus, although substitutions with CS are possible (Gotte, 2003; Tkachenko et al., 2005).
Proteoglycans modified by CS chains include aggrecan, neurocan, brevican, bamacan, a CD44 isoform, syndecans, betaglycan and serglycin (Taylor and Gallo, 2006). DS is mainly found in skin and PGs bearing DS chains are versican, decorin, biglycan and endocan (Trowbridge and Gallo, 2002). Many of the PGs bearing CS chains are additionally modified by GAG chains of either HS or KS, for example, syndecans or aggrecan respectively. Decorin, however, a secretory product in the ECM, is one example of an exclusively CS/DS PG. Aggrecan has a molecular mass of about 2500 kDa and is a large KS/CSPG in cartilagenous tissue. Its distribution pattern is relatively restricted to a few tissues including brain, aorta and tendon, in addition to cartilage. The core protein of 210–250 kDa binds hyaluronic acid and forms a supramolecular complex together with the linker protein. Aggrecan provides a strongly hydrated space filling gel due to the large number (more than 100) of polyanionic GAG chains covalently attached to the protein core and thereby contributes to the physical strength of cartilagenous tissue (Roughley, 2006).
Pharmacological driving force: protein–GAG interactions and the glycan-protein network (GPN)
While PGs are involved in a plethora of physiological processes, much has been speculated about the individual roles of the core protein and the attached GAG chains. Although we have recently shown that the chemokine CXCL8 binds to both, the GAG sidechain and the core protein of syndecan-2 (Halden et al., 2004), and is therefore able to induce downstream signalling via its syndecan co-receptor into endothelial cells (S.F. Falsone et al., manuscript submitted), it is generally accepted that mainly the glycan side chains on PGs are responsible for the biological functions of PGs. This is achieved by immobilizing and by structurally influencing proteins which bind to these complex carbohydrate chains. Interestingly, although the affinity of proteins towards GAG ligands is commonly rather low, that is, in the µmol·L–1 range, a significant structural change can be induced in the protein (Goger et al., 2002). The function of the interacting protein is thereby modulated either by inducing a direct conformational change (Goger et al., 2002), sometimes by the induction of homo- or hetero-oligomerization (Hoogewerf et al., 1997; Weber and Koenen, 2006). Of special pharmacological interest in the context of protein–GAG interactions is the cell surface of endothelial cells. These cells, being the coating of the (micro)vasculature, are able to secrete, release or express growth factors and chemokines, adhesions proteins, proteases and esterases, as well as HS degrading enzymes into the serum which bind to HS or CS, the proto-typic and dominating glycan chains on most eukaryotic cells, thereby attracting and activating distant cells like leukocytes or metastatic cells or releasing cells in the course of metastasis or angiogenesis. Consequently, the surface of endothelial cells is characterized not only by the various membrane proteins and their short and branched N-linked glycan chains, the glycocalyx of a cell is rather constituted by a wealth of charged glycan (GAG) chains which, in addition, bind a multitude of proteins, thereby creating a dense matrix of membrane proteins, glycan chains and immobilized soluble proteins (see Figure 2). We define this matrix as the GPN which ultimately defines the active and passive signalling capacity of a given cell, as it relates secreted and presented proteins to the functional environment of the cell.
Figure 2.
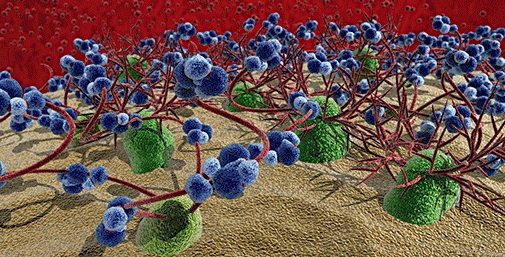
Cell surface of endothelial cells. In green, (trans-)membrane proteins are shown; branched and short red tubes represent N-linked glycans, long and unbranched red tubes represent O-linked (glycoasmino-)gycans; in blue, glycan-binding proteins are shown, different shades refer to different proteins and thus to hetero- or homo-oligomerization.
If the GPN is a signalling fingerprint of a cell, then the protein–glycan interactions are proposed to be of significant specificity. In their seminal work, Cardin and Weintraub (1989) have outlined two protein consensus sequences which are responsible for GAG binding. Their report describes defined arrays of basic amino acid clusters (lysine, arginine and histidine) characterized by the consensus sequences, XBBXBX and XBBBXXBX, ‘where B is the probability of a basic residue and X is a hydropathic residue’. More recently, Hileman et al. (Hileman et al., 1989) have extended this list by the new consensus sequence TXXBXXTBXXXTBB, where turns (T) are suggested to bring basic interacting amino acid residues into proximity. But what about protein-specific GAG sequences? Less is known about specific GAG sequences mainly because (bio)chemical methods for preparing and amplifying biologically relevant (i.e. cell- or tissue-derived) and protein-related GAGs are lacking, although highly sensitive technologies for sequencing these carbohydrates have been introduced by the Gallagher group in the late 1990s (Merry et al., 1999; Turnbull et al., 1999). However, no high throughput sequencing methods like for mass spectrometry (MS)/MS-based protein sequencing in proteomics have yet been established (see below). Table 1 shows specific GAG sequences which were to date found for proteins of various classes demonstrating the general principle of the ‘specificity’ within protein–GAG interactions. However, based on more general findings (Kreuger et al., 2006) but also based on findings in our own lab, we tend to use ‘selectivity’ rather than specificity for describing this interaction, which allows for a certain degree of degeneracy during recognition of proteins and glycan molecules.
Table 1.
Protein-specific glycosaminoglycan sequences
| GAG-sequence | Interacting protein | References |
|---|---|---|
| GlcNAc/NS(6S)-GlcA-GlcNS(3,6S)-IdoA(2S)-GlcNS(6S) | Antithrombin III | Oscarsson LG et al. (1989). J Biol Chem264 (1): 296–304. |
| UA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S) | Annexin V | Capila I et al. (2001). Structure9: 57–64. |
| UA-GlcNS-IdoA(2S)-GlcNAc-UA(2S)-GlcNS-IdoA(2S) -GlcNH2(3,6S) | herpes simplex 1 envelope glycoprotein D (gD) | Liu J et al. (2002). J Biol Chem277 (36): 33456–33467. |
| IdoA-GlcNAc(6S)-GlcA-GlcNS(3,6-S)-IdoA(2S)- | Heparanase | Gong F et al. (2003). J Biol Chem278 (37): 35152–35158. |
| -IdoA2S-GlcNS6S- | HSulf1, HSulf2 | Morimoto-Tomita et al. (2002). J Biol Chem277 (51): 49175–49185. |
| GlcNS-IdoA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS | VEGF 165 | Robinson CJ et al. (2006). J Biol Chem281 (3): 1731–1740. |
| GlcA-GlcNS-[IdoA(2S)-GlcNS]5-IdoA-GlcNAc | FGF2 | Turnbull et al. (1992). J Biol Chem267 (15): 10337–10341. |
| -IdoA(2S)-GlcNS(6S)-IdoA(2S)- | FGF1 | Kreuger J et al. (2001). J Biol Chem276 (33): 30744–30752. |
| UA-GlcNS-[UA(2S)-GlcNS(6S)]3 | FGF4, FGF7 | |
| UA-GlcNS-[UA-GlcNS(6S)]3 | FGF10 | |
| UA-GlcNS-[UA(2S)-GlcNS]3, UA-GlcNS-[UA-GlcNS(6S)]3 | FGF18, HGF | Ashikari S et al. (2004). J Biol Chem279 (13): 12346–12354. |
| UA-GlcNS/NAc-IdoA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S)-UA-GlcNS/NAc- | HGF | Ashikari S et al. (1995). J Biol Chem270 (49): 29586–29593. |
| UA-GlcNS/NAc-UA-GlcNS/NAc-IdoA(2S)-GlcNS(6S)-IdoA(2S)-GlcNS(6S)- | ||
| UA-GlcNS/NAc-IdoA(2S)-GlcNS(6S)-UA-GlcNS/NAc-IdoA(2S)-GlcNS(6S)- | ||
| -GlcA-GlcNAc/S-[IdoA(2S)-GlcNS(6S)]3-[GlcA-GlcNAc/S]6-7-[IdoA(2S)-GlcNS(6S)]3-GlcA-GlcNAc/S | IL8 | Spillmann et al. (1998). J Biol Chem273 (25): 15487–15493. |
| (UA-GlcN)8containing 7S and 2Ac | MCP1, MCP2, MCP3 | Schenauer MR et al. (2007). J Biol Chem282 (35): 25182–25188. |
| IdoA(2S)-GlcNH2(3S ± 6S) within dp8 | CyPB | Vanpouille C et al. (2007). J Biol Chem282 (33): 24416–24429. |
GAG, glycosaminoglycan.
Identification of protein-specific GAG sequences
The pioneering work by Lindahl and co-workers has led to the identification of the first protein-specific GAG sequence, namely the heparin pentasaccharide structure required for antithrombin-III binding (Lindahl et al., 1989). Since then, only few more protein-specific GAG oligosaccharides have been added to this list (see Table 1). Traditionally, identification of protein-specific GAG oligosaccharides was accomplished in a ligand-biased manner, that is, by screening a naturally derived, diversified and size-defined GAG oligosaccharide library with respect to target protein binding. This approach, due to the limited size of the oligosaccharide library, is incomprehensive and therefore the very specific GAG ligand for a given protein may not (or not in sufficient amounts for detection) be contained in the screened library. The ‘classical’ methods used to obtain and to characterize protein-speficic GAG oligos include gel electrophoresis, affinity- and size-exclusion chromatography, filter binding and competition assays, microcalorimetry, and surface plasmon resonance. Recent developments in liquid chromatography and capillary electrophoresis coupled to MS in combination with subtle bioinformatic tools allow nowadays a more unbiased and efficient identification of protein-specific GAG oligos (Zamfir et al., 2004; Saad et al., 2005; Yu et al., 2005; Yu et al., 2006). Using tandem MS, the group of Leary was able to unravel the structure of a number of HS oligos and to identify oligos which bind to specific chemokines (Yu et al., 2005). This approach is still limited by origin and natural diversity of the GAG oligosaccharide library used to select for protein-specific ligands. However, because of the general sensitivity of MS methods, a number of oligosaccharide libraries can be screened with a much higher probability to pick the specific GAG ligand even if it occurs with low abundancy. This means the dawning of the glycomic age also for glycan analyses. In the group of Sasisekharan, an MS-based glycan fingerprinting method combined with bioinformatical integration of data sets – a method using the so-called property-encoded nomenclature which was developed to handle many data sets and to extract information on the investigated saccharide structure – was successfully applied to various linear glycans including the AT-III-specific heparin epitope (Shriver et al., 2000). Enzyme protection assays were shown to be especially useful for the identification of GAG ligands of dimeric and multimeric proteins such as PF4 and MIP-1α (Stringer and Gallagher, 1997; Stringer et al., 2002). By applying heparinase and/or chondroitinase to protein–ligand complexes, also distant protein binding sites on the GAG ligand have been identified like the split S-domains for PF4 and long sulfated regions for MIP-1α. A similar approach has been used to identify the HS motif for interferon-γ (Lortat-Jacob et al., 1995).
Therapeutic approaches
From Table 1, it becomes obvious that a multitude of pharmacologically highly interesting target proteins need a selective GAG partner to exert their physiological function which are therefore potential therapeutical targets (Gesslbauer and Kungl, 2006; Lindahl, 2007). In general, as outlined above, protein–glycan interactions are considered to occur with low affinity, that is, in the low micromolar range, although higher affinities (in the nanomolar range) especially for growth factors have been observed (Mohammadi and Olsen, 2005). There are consequently various ways to interfere with protein–glycan interactions (see Figure 3). The ‘classical’ approach to this problem is to chemically and/or enzymatically synthesize the target protein-specific glycan structure (see Figure 3B) or a mimetic thereof and to test for and to optimize inhibitory activity. Despite the initial success of Sanofi and Organon in co-developing various forms of the AT-III-specific heparin pentasaccharide as a very efficient anti-thromboticum, resulting in Fondaparinux (Arixtra®), an a priori GAG synthesizing approach is currently less pursued by large pharmaceutical companies but rather by biotech companies like Momenta Pharmaceuticals Inc. and Endotis Pharma. Momenta's M118 is a rationally engineered heparin which is modified and fractionated so to provide only sites with anti-coagulant activity. Endotis' EP37 was synthesized, using the company's unique oligosaccharide synthesis platform, for oral prevention of venous thromboembolism and stroke prevention in patients with atrial fibrillation. Upcoming on the glycomics horizon are miniaturized on-chip synthesis technologies which use combinatorial or parallel chemistry to generate a multitude of glycan oligosaccharides for biological activity screening (de Paz et al., 2007). These methods, however valuable they are for performing medium to highthroughput first screens and to find suitable hits, do not solve the problem of finally synthesizing the hit oligosaccharides in sufficient amounts needed for clinical trials or to feed the market for such drugs.
Figure 3.
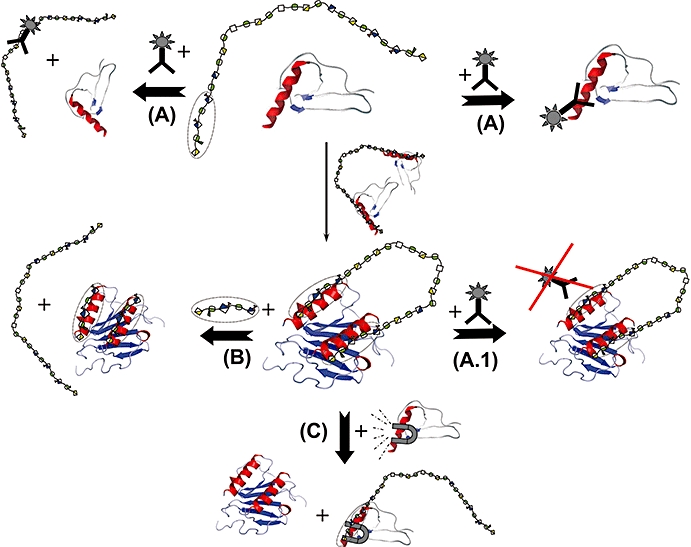
Different ways for therapeutic intervention in protein–GAG signalling.  sequence-specific GAG – Oligo within a proteoglycan side chain;
sequence-specific GAG – Oligo within a proteoglycan side chain;  antibody. (A) Antibody approach. The binding of a protein to a (specific) GAG chain can be prevented by an antibody to either the (proteo-)glycan or the protein interaction partner. (A.1) The GAG-bound protein may not be targetable by the antibody raised against the soluble protein as structural rearrangements of the protein upon GAG binding as well as the change of overall/surface charge influence/destroy the antibody binding epitope. (B) Glycan- or glycan mimetic-based approach. The protein–GAG interaction can be antagonized by the addition of the synthesized protein-specific GAG sequence (or a mimetic thereof) which displaces the target protein from the natural GAG ligand. (C) Protein-based approach. The protein–GAG interaction can be antagonized by the addition of a mutant form of the target protein which exhibits higher affinity towards the natural GAG ligand and thereby displaces the wild-type target protein from the GAG chain. GAG, glycosaminoglycan.
antibody. (A) Antibody approach. The binding of a protein to a (specific) GAG chain can be prevented by an antibody to either the (proteo-)glycan or the protein interaction partner. (A.1) The GAG-bound protein may not be targetable by the antibody raised against the soluble protein as structural rearrangements of the protein upon GAG binding as well as the change of overall/surface charge influence/destroy the antibody binding epitope. (B) Glycan- or glycan mimetic-based approach. The protein–GAG interaction can be antagonized by the addition of the synthesized protein-specific GAG sequence (or a mimetic thereof) which displaces the target protein from the natural GAG ligand. (C) Protein-based approach. The protein–GAG interaction can be antagonized by the addition of a mutant form of the target protein which exhibits higher affinity towards the natural GAG ligand and thereby displaces the wild-type target protein from the GAG chain. GAG, glycosaminoglycan.
The typical biotech approach, that is, raising monoclonal antibodies against the glycan binding target protein, seems not to be generally applicable for glycan-binding proteins. As glycan-binding proteins are supposed to act only (or only in the desired fashion) when bound to the glycan, any mAb raised against the soluble protein might fail to inactivate the glycan-bound protein simply because the structural change is prone to induce epitope loss and/or epitope masking due to the high charge of the GAG ligand (see Figure 3A.1). This was most probably the reason for the failure of the anti-IL-8 antibody ABX-IL8 from Abgenix Inc. in clinical phase II, as the authors stated themselves that ABX-IL8 was not able to bind IL-8 localized on endothelial cells, that is, bound to GAGs (Yang et al., 1999). On the other hand, it proved to be very difficult to raise monoclonal antibodies against protein-specific GAG sequences. Although the group of van Kuppevelt was successful in generating antibodies against various tissue-specific GAGs by phage-display (Lensen et al., 2005), no therapeutic antibody is yet available which inhibits specific protein–GAG interaction. This could be attributed to the low affinity of these phage display-derived antibodies and/or to the still low selectivity of these molecules. Antibodies against entire PGs have been available for quite some time but their potential for treatment of glycan-related diseases is not yet clearly foreseeable (Fjeldstad and Kolset, 2005). As these antibodies were raised either against only the recombinant core protein of PGs or against the entire molecule (core protein plus glycan chains), the effect on disrupting protein–glycan interactions remains elusive at this point. Another interesting therapeutic approach addressing protein–GAG interactions by knocking out the GAG interaction sites is the various chemokine mutants developed by Serono (Proudfoot et al., 2008; Shahrara et al., 2008). The group of Amanda Proudfoot engineered several chemokines towards lower or no GAG binding affinity with the aim to block the chemokines' GPCR receptors on neutrophils with these GAG knockout mutants, which consequently prevents leukocyte activation and firm adhesion by GAG-bound wild-type chemokines.
We have developed a protein-based technology to create a novel class of glycan-directed therapeutics which circumvents the above outlined problems for antagonizing protein/GAG interactions (Potzinger et al., 2006). Our CellJammer® (ProtAffin, Graz, Austria) approach takes advantage of the intrinsic specificity or selectivity (see above) of a GAG-binding protein for its ligand to create a protein-based GAG antagonist by engineering the protein towards higher GAG binding affinity (see Figure 3C). This is achieved by engineering basic amino acids into the GAG binding site of the selected protein in a way that the overall structure of the protein is conserved. This kind of affinity maturation is based on the fact that electrostatic interactions are mainly responsible for (high) affinity whereas hydrogen bonds and van der Vaals contacts determine specificity (as in the case of protein–DNA interactions). By this means, the precise nature of the glycan ligand does not need to be known as long as specific/selective GAG ligand recognition is conserved within the mutant protein. This can usually be verified by (i) competition experiments of mutant protein versus wild-type protein versus other (unspecific) GAG-binding proteins; and (ii) by comparative binding experiments of mutant versus wild-type protein using a (limited) library of synthetic and natural, fractionated GAG oligosaccharides. As a first proof of concept, we have developed an CXCL8-based therapeutic, PA401, which exhibits a 100-fold higher affinity towards HS while keeping selective GAG ligand recognition compared with the wild-type chemokine. PA401 was found to bind also to CS but the differences in selectivity with respect to wtCXCL8 need to be explored in more detail in the future. This mutant protein was shown to efficiently displace wtCXCL8 from HS chains on endothelial cells, whereas other chemokines are displaced only at much higher concentrations of PA401. To this engineered ‘dominant’ (higher GAG binding affinity) characteristics of the chemokine, we have added a ‘negative’ characteristic in order to create a potent anti-infammatory drug, otherwise a super-agonist of CXCL8 had been the result. For this purpose, the leukocyte attraction and activation domain of the chemokine have been knocked out by deleting six amino acids of the N-terminus. The dominant-negative CXCL8 mutant PA401 exhibited excellent inhibitory activity in a number of inflammatory animal models (A. Rek et al., manuscript in preparation) and is, among further proteins modified by the CellJammer® (ProtAffin) technology, currently in preclinical development. Our approach represents the first protein-based GAG antagonist which specifically inhibits information flow from the HS-presenting endothelium to the attracted leukocyte.
Acknowledgments
This work was supported by the Austrian Federal Ministry of Science and Research (GEN-AU/APP II project No. GZ 200.138/1-VI/1/2005) as well as by the Austrian Science Fund (FWF project No. P17872-B09).
Glossary
Abbreviations:
- CS
chondroitin sulfate
- GAG
glycosaminoglycan
- HS
heparan sulfate
- MS
mass spectrometry
Conflict of interest
A Rek and AJ Kungl are employees of ProtAffin Biotechnologie AG, Austria. The company has interest in the development of protein-based GAG antagonists.
References
- Ashikari S, Habuchi H, Kimata K. Characterization of heparan sulfate oligosaccharides that bind to hepatocyte growth factor. J Biol Chem. 1995;270:29586–29593. doi: 10.1074/jbc.270.49.29586. [DOI] [PubMed] [Google Scholar]
- Ashikari-Hada S, Habuchi H, Kariya Y, Itoh N, Reddi AH, Kimata K. Characterization of growth factor-binding structures in heparin/heparan sulfate using an octasaccharide library. J Biol Chem. 2004;279:12346–12354. doi: 10.1074/jbc.M313523200. [DOI] [PubMed] [Google Scholar]
- Berg JM, Tymodczko JL, Stryer L. Biochemistry. New York: W. H. Freeman and Company; 2002. [Google Scholar]
- Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- Capila I, Linhardt RJ. Heparin-protein interactions. Angew Chem Int Ed Engl. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Capila I, Hernáiz MJ, Mo YD, Mealy TR, Campos B, Dedman JR, et al. Annexin V–heparin oligosaccharide complex suggests heparan sulfate–mediated assembly on cell surfaces. Structure. 2001;9:57–64. doi: 10.1016/s0969-2126(00)00549-9. [DOI] [PubMed] [Google Scholar]
- Cardin AD, Weintraub HJ. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis. 1989;9:21–32. doi: 10.1161/01.atv.9.1.21. [DOI] [PubMed] [Google Scholar]
- Carter NM, Ali S, Kirby JA. Endothelial inflammation: the role of differential expression of N-deacetylase/N-sulphotransferase enzymes in alteration of the immunological properties of heparan sulphate. J Cell Sci. 2003;116:3591–3600. doi: 10.1242/jcs.00662. [DOI] [PubMed] [Google Scholar]
- Cattaruzza S, Perris R. Proteoglycan control of cell movement during wound healing and cancer spreading. Matrix Biol. 2005;24:400–417. doi: 10.1016/j.matbio.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Celie JW, Keuning ED, Beelen RH, Drager AM, Zweegman S, Kessler FL, et al. Identification of L-selectin binding heparan sulfates attached to collagen type XVIII. J Biol Chem. 2005;280:26965–26973. doi: 10.1074/jbc.M502188200. [DOI] [PubMed] [Google Scholar]
- David G. Integral membrane heparan sulfate proteoglycans. FASEB J. 1993;7:1023–1030. doi: 10.1096/fasebj.7.11.8370471. [DOI] [PubMed] [Google Scholar]
- Dennis JW, Granovsky M, Warren CE. Protein glycosylation in development and disease. Bioessays. 1999;21:412–421. doi: 10.1002/(SICI)1521-1878(199905)21:5<412::AID-BIES8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Duchesne L, Tissot B, Rudd TR, Dell A, Fernig DG. N-glycosylation of fibroblast growth factor receptor 1 regulates ligand and heparan sulfate co-receptor binding. J Biol Chem. 2006;281:27178–27189. doi: 10.1074/jbc.M601248200. [DOI] [PubMed] [Google Scholar]
- Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- Fears CY, Woods A. The role of syndecans in disease and wound healing. Matrix Biol. 2006;25:443–456. doi: 10.1016/j.matbio.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Fjeldstad K, Kolset SO. Decreasing the metastatic potential in cancers – targeting the heparan sulfate proteoglycans. Curr Drug Targets. 2005;6:665–682. doi: 10.2174/1389450054863662. [DOI] [PubMed] [Google Scholar]
- Fransson LA. Glypicans. Int J Biochem Cell Biol. 2003;35:125–129. doi: 10.1016/s1357-2725(02)00095-x. [DOI] [PubMed] [Google Scholar]
- Gesslbauer B, Kungl AJ. Glycomic approaches toward drug development: therapeutically exploring the glycosaminoglycanome. Curr Opin Mol Ther. 2006;8:521–528. [PubMed] [Google Scholar]
- Gesslbauer B, Rek A, Falsone F, Rajkovic E, Kungl AJ. Proteoglycanomics tools to unravel the biological function of glycosamino-glycans. Proteomics. 2007;7:2870–2880. doi: 10.1002/pmic.200700176. [DOI] [PubMed] [Google Scholar]
- Goger B, Halden Y, Rek A, Moesl R, Pye D, Gallagher J, et al. Different affinities of glycosaminoglycan oligosaccharides for monomeric and dimeric interleukin-8: a model for chemokine regulation at inflammatory sites. Biochemistry. 2002;41:1640–1646. doi: 10.1021/bi011944j. [DOI] [PubMed] [Google Scholar]
- Gong F, Jemth P, Escobar Galvis ML, Vlodavsky I, Horner A, Lindahl U, et al. Processing of macromolecular heparin by heparanase. J Biol Chem. 2003;278:35152–35158. doi: 10.1074/jbc.M300925200. [DOI] [PubMed] [Google Scholar]
- Gotte M. Syndecans in inflammation. FASEB J. 2003;17:575–591. doi: 10.1096/fj.02-0739rev. [DOI] [PubMed] [Google Scholar]
- Habuchi H, Habuchi O, Kimata K. Sulfation pattern in glycosaminoglycan: does it have a code? Glycoconj J. 2004;21:47–52. doi: 10.1023/B:GLYC.0000043747.87325.5e. [DOI] [PubMed] [Google Scholar]
- Halden Y, Rek A, Atzenhofer W, Szilak L, Wabnig A, Kungl AJ. Interleukin-8 binds to syndecan-2 on human endothelial cells. Biochem J. 2004;377:533–538. doi: 10.1042/BJ20030729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel TM, Johnson Z, Crown SE, Lau EK, Proudfoot AE. Regulation of protein function by glycosaminoglycans – as exemplified by chemokines. Annu Rev Biochem. 2005;74:385–410. doi: 10.1146/annurev.biochem.72.121801.161747. [DOI] [PubMed] [Google Scholar]
- Hang HC, Bertozzi CR. The chemistry and biology of mucin-type O-linked glycosylation. Bioorg Med Chem. 2005;13:5021–5034. doi: 10.1016/j.bmc.2005.04.085. [DOI] [PubMed] [Google Scholar]
- Harmer NJ. Insights into the role of heparan sulphate in fibroblast growth factor signalling. Biochem Soc Trans. 2006;34:442–445. doi: 10.1042/BST0340442. [DOI] [PubMed] [Google Scholar]
- Hileman RE, Fromm JR, Weiler JM, Linhardt RJ. Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. Bioessays. 1989;20:156–167. doi: 10.1002/(SICI)1521-1878(199802)20:2<156::AID-BIES8>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Hoogewerf AJ, Kuschert GSV, Proudfoot AEI, Borlat F, Clark-Lewis I, Power CA, et al. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry. 1997;36:13570–13578. doi: 10.1021/bi971125s. [DOI] [PubMed] [Google Scholar]
- Johnson Z, Proudfoot AE, Handel TM. Interaction of chemokines and glycosaminoglycans: a new twist in the regulation of chemokine function with opportunities for therapeutic intervention. Cytokine Growth Factor Rev. 2005;16:625–636. doi: 10.1016/j.cytogfr.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Knox SM, Whitelock JM. Perlecan: how does one molecule do so many things? Cell Mol Life Sci. 2006;63:2435–2445. doi: 10.1007/s00018-006-6162-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolset SO, Salmivirta M. Cell surface heparan sulfate proteoglycans and lipoprotein metabolism. Cell Mol Life Sci. 1999;56:857–870. doi: 10.1007/s000180050031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolset SO, Prydz K, Pejler G. Intracellular proteoglycans. Biochem J. 2004;379:217–227. doi: 10.1042/BJ20031230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenn EC, Wille I, Gesslbauer B, Poteser M, van Kuppevelt TH, Kungl AJ. Glycanogenomics: a qPCR-approach to investigate biological glycan function. Biochem Biophys Res Commun. 2008;375:297–302. doi: 10.1016/j.bbrc.2008.07.144. [DOI] [PubMed] [Google Scholar]
- Kreuger J, Spillmann D, Li JP, Lindahl U. Interactions between heparan sulfate and proteins: the concept of specificity. J Cell Biol. 2006;174:323–327. doi: 10.1083/jcb.200604035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuger J, Salmivirta M, Sturiale L, Giménez-Gallego G, Lindahl U. Sequence analysis of heparan sulfate epitopes with graded affinities for fibroblast growth factors 1 and 2. J Biol Chem. 2001;276:30744–30752. doi: 10.1074/jbc.M102628200. [DOI] [PubMed] [Google Scholar]
- Ledin J, Ringvall M, Thuveson M, Eriksson I, Wileén M, Kusche-Gullberg M, et al. Enzymatically active N-deacetylase/N-sulfotransferase-2 is present in liver but does not contribute to heparan sulfate N-sulfation. J Biol Chem. 2006;281:35727–35734. doi: 10.1074/jbc.M604113200. [DOI] [PubMed] [Google Scholar]
- Lehle L, Strahl S, Tanner W. Protein glycosylation, conserved from yeast to man: a model organism helps elucidate congenital human diseases. Angew Chem Int Ed Engl. 2006;45:6802–6818. doi: 10.1002/anie.200601645. [DOI] [PubMed] [Google Scholar]
- Lensen JF, Rops AL, Wijnhoven TJ, Hafmans T, Feitz WF, Oosterwijk E, et al. Localization and functional characterization of glycosaminoglycan domains in the normal human kidney as revealed by phage display-derived single chain antibodies. J Am Soc Nephrol. 2005;16:1279–1288. doi: 10.1681/ASN.2004050413. [DOI] [PubMed] [Google Scholar]
- Lindahl U. Heparan sulfate-protein interactions – a concept for drug design? Thromb Haemost. 2007;98:109–115. [PubMed] [Google Scholar]
- Lindahl U, Backstrom G, Thunberg L, Leder IG. Evidence for a 3-O-sulfated D-glucosamine residue in the antithrombin-binding sequence of heparin. Proc Natl Acad Sci USA. 1989;77:6551–6555. doi: 10.1073/pnas.77.11.6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Shriver Z, Pope RM, Thorp SC, Duncan MB, Copeland RJ, et al. Characterization of a heparin sulfate octasaccharide that binds to herpes simplex virus type 1 glycoprotein D. J Biol Chem. 2002;277:33456–33467. doi: 10.1074/jbc.M202034200. [DOI] [PubMed] [Google Scholar]
- Lortat-Jacob H, Turnbull JE, Grimaud JA. Molecular organization of the interferon y-binding domain in heparan sulphate. Biochem J. 1995;310:497–505. doi: 10.1042/bj3100497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKillop AM, McCluskey JT, Boyd AC, Mooney MH, Flatt PR, O'Harte FP. Production and characterization of specific antibodies for evaluation of glycated insulin in plasma and biological tissues. J Endocrinol. 2000;167:153–163. doi: 10.1677/joe.0.1670153. [DOI] [PubMed] [Google Scholar]
- Merry CLM, Lyon M, Deakin JA, Hopwood JJ, Gallagher JT. Highly sensitive sequencing of the sulfated domains of heparan sulfate. J Biol Chem. 1999;274:18455–18462. doi: 10.1074/jbc.274.26.18455. [DOI] [PubMed] [Google Scholar]
- Middleton J, Patterson AM, Gardner L, Schmutz C, Ashton BA. Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood. 2002;100:3853–3860. doi: 10.1182/blood.V100.12.3853. [DOI] [PubMed] [Google Scholar]
- Mohammadi M, Olsen SK, Goetz R. A protein canyon in the FGF–FGF receptor dimer selects from an a` la carte menu of heparan sulfate motifs. Curr Opinion Struct Biol. 2005;15:1–11. doi: 10.1016/j.sbi.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Morimoto-Tomita M, Uchimura K, Werb Z, Hemmerich S, Rosen SD. Cloning and characterization of two extracellular heparin-degrading endosulfatases in mice and humans. J Biol Chem. 2002;277:49175–49185. doi: 10.1074/jbc.M205131200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulloy B, Rider CC. Cytokines and proteoglycans: an introductory overview. Biochem Soc Trans. 2006;34:409–413. doi: 10.1042/BST0340409. [DOI] [PubMed] [Google Scholar]
- Oscarsson LG, Pejler G, Lindahl U. Location of the antithrombin-binding sequence in the heparin chain. J Biol Chem. 1989;264:296–304. [PubMed] [Google Scholar]
- de Paz JL, Moseman EA, Noti C, Polito L, von Andrian UH, Seeberger PH. Profiling heparin-chemokine interactions using synthetic tools. ACS Chem Biol. 2007;2:735–744. doi: 10.1021/cb700159m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potzinger H, Geretti E, Brandner B, Wabitsch V, Piccinini AM, Rek A, et al. Developing chemokine mutants with improved proteoglycan affinity and knocked-out GPCR activity as anti-inflammatory recombinant drugs. Biochem Soc Trans. 2006;34:435–437. doi: 10.1042/BST0340435. [DOI] [PubMed] [Google Scholar]
- Proudfoot AE. The biological relevance of chemokine-proteoglycan interactions. Biochem Soc Trans. 2006;34:422–426. doi: 10.1042/BST0340422. [DOI] [PubMed] [Google Scholar]
- Proudfoot AEI, Souza ALS, Muzio V. The use of chemokine antagonists in EAE models. J Neuroimmunol. 2008;198:27–30. doi: 10.1016/j.jneuroim.2008.04.007. [DOI] [PubMed] [Google Scholar]
- Prydz K, Dalen KT. Synthesis and sorting of proteoglycans. J Cell Sci. 2000;113:193–205. doi: 10.1242/jcs.113.2.193. [DOI] [PubMed] [Google Scholar]
- Robinson CJ, Mulloy B, Gallagher JT, Stringer SE. VEGF165-binding sites within heparan sulfate encompass two highly sulfated domain and can be liberated by K5 lyase. J Biol Chem. 2006;281:1731–1740. doi: 10.1074/jbc.M510760200. [DOI] [PubMed] [Google Scholar]
- Roughley PJ. The structure and function of cartilage proteoglycans. Eur Cell Mater. 2006;12:92–101. doi: 10.22203/ecm.v012a11. [DOI] [PubMed] [Google Scholar]
- Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Glycosylation and the immune system. Science. 2001;291:2370–2376. doi: 10.1126/science.291.5512.2370. [DOI] [PubMed] [Google Scholar]
- Rydberg L. ABO-incompatibility in solid organ transplantation. Transfus Med. 2001;11:325–342. doi: 10.1046/j.1365-3148.2001.00313.x. [DOI] [PubMed] [Google Scholar]
- Saad OM, Myers RA, Castleton DL, Leary JA. Analysis of hyaluronan content in chondroitin sulfate preparations by using selective enzymatic digestion and electrospray ionization mass spectrometry. Anal Biochem. 2005;344:232–239. doi: 10.1016/j.ab.2005.06.041. [DOI] [PubMed] [Google Scholar]
- Sanderson RD, Yang Y, Kelly T, MacLeod V, Dai Y, Theus A. Enzymatic remodeling of heparan sulfate proteoglycans within the tumor microenvironment: growth regulation and the prospect of new cancer therapies. J Cell Biochem. 2005;96:897–905. doi: 10.1002/jcb.20602. [DOI] [PubMed] [Google Scholar]
- Schenauer MR, Yu Y, Sweeney MD, Leary JA. CCR2 chemokines bind selectively to acetylated heparan sulfate octasaccharides. J Biol Chem. 2007;282:25182–25188. doi: 10.1074/jbc.M703387200. [DOI] [PubMed] [Google Scholar]
- Shahrara S, Proudfoot AE, Park CC, Volin MV, Haines GK, Woods JM, et al. Inhibition of monocyte chemoattractant protein-1 ameliorates rat adjuvant-induced arthritis. J Immunol. 2008;180:3447–3456. doi: 10.4049/jimmunol.180.5.3447. [DOI] [PubMed] [Google Scholar]
- Shriver Z, Raman R, Venkataraman G, Drummond K, Turnbull J, Toida T, et al. Sequencing of 3-O sulfate containing heparin decasaccharides with a partial antithrombin III binding site. Proc Natl Acad Sci USA. 2000;97:10359–10364. doi: 10.1073/pnas.97.19.10359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shur BD, Rodeheffer C, Ensslin MA, Lyng R, Raymond A. Identification of novel gamete receptors that mediate sperm adhesion to the egg coat. Mol Cell Endocrinol. 2006;250:137–148. doi: 10.1016/j.mce.2005.12.037. [DOI] [PubMed] [Google Scholar]
- Spillmann D, Witt D, Lindahl U. Defining the interleukin-8-binding domain of heparan sulfate. J Biol Chem. 1998;273:15487–15493. doi: 10.1074/jbc.273.25.15487. [DOI] [PubMed] [Google Scholar]
- Stringer SE, Gallagher JT. Specific binding of the chemokine platelet factor 4 to heparan sulfate. J Biol Chem. 1997;272:20508–20514. doi: 10.1074/jbc.272.33.20508. [DOI] [PubMed] [Google Scholar]
- Stringer SE, Forster MJ, Mulloy B, Bishop CR, Graham GJ, Gallagher JT. Characterization of the binding site on heparan sulfate for macrophage inflammatory protein 1α. Blood. 2002;100:1543–2703. [PubMed] [Google Scholar]
- Taylor KR, Gallo RL. Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J. 2006;20:9–22. doi: 10.1096/fj.05-4682rev. [DOI] [PubMed] [Google Scholar]
- Tkachenko E, Rhodes JM, Simons M. Syndecans: new kids on the signaling block. Circ Res. 2005;96:488–500. doi: 10.1161/01.RES.0000159708.71142.c8. [DOI] [PubMed] [Google Scholar]
- Trowbridge JM, Gallo RL. Dermatan sulfate: new functions from an old glycosaminoglycan. Glycobiology. 2002;12:117R–125R. doi: 10.1093/glycob/cwf066. [DOI] [PubMed] [Google Scholar]
- Turnbull JE, Hopwood JJ, Gallagher JT. A strategy for rapid sequencing of heparan sulfate and heparin saccharides. Proc Natl Acad Sci USA. 1999;96:2698–2703. doi: 10.1073/pnas.96.6.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull JE, Fernig DG, Ke Y, Wilkinson MC, Gallagher JT. Identification of the basic fibroblast growth factor binding sequence in fibroblast heparan sulfate. J Biol Chem. 1992;267:10337–10341. [PubMed] [Google Scholar]
- Van Acker GJ, Perides G, Steer ML. Co-localization hypothesis: a mechanism for the intrapancreatic activation of digestive enzymes during the early phases of acute pancreatitis. World J Gastroenterol. 2006;12:1985–1990. doi: 10.3748/wjg.v12.i13.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanpouille C, Deligny A, Delehedde M, Denys A, Melchior A, Liénard X, et al. The heparin/heparan sulfate sequence that interacts with cyclophilin B contains a 3-O-sulfated N-unsubstituted glucosamine residue. J Biol Chem. 2007;282:24416–24429. doi: 10.1074/jbc.M701835200. [DOI] [PubMed] [Google Scholar]
- Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlodavsky I, Goldshmidt O, Zcharia E, Atzmon R, Rangini-Guatta Z, Elkin M, et al. Mammalian heparanase: involvement in cancer metastasis, angiogenesis and normal development. Semin Cancer Biol. 2002;12:121–129. doi: 10.1006/scbi.2001.0420. [DOI] [PubMed] [Google Scholar]
- Vlodavsky I, Ilan N, Naggi A, Casu B. Heparanase: structure, biological functions, and inhibition by heparin-derived mimetics of heparan sulfate. Curr Pharm Des. 2007;13:2057–2073. doi: 10.2174/138161207781039742. [DOI] [PubMed] [Google Scholar]
- Weber C, Koenen RR. Fine-tuning leukocyte responses: towards a chemokine ‘interactome’. Trends Immunol. 2006;27:268–273. doi: 10.1016/j.it.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Whelan SA, Hart GW. Proteomic approaches to analyze the dynamic relationships between nucleocytoplasmic protein glycosylation and phosphorylation. Circ Res. 2003;93:1047–1058. doi: 10.1161/01.RES.0000103190.20260.37. [DOI] [PubMed] [Google Scholar]
- Yang XD, Corvalan JRF, Wang P, Roy CMN, Davis CG. Fully human anti-interleukin-8 monoclonal antibodies: potential therapeutics for the treatment of inflammatory disease states. J Leukoc Biol. 1999;66:401–410. doi: 10.1002/jlb.66.3.401. [DOI] [PubMed] [Google Scholar]
- Yu Y, Sweeney MD, Saad OM, Crown SE, Hsu AR, Handel TM, et al. Chemokine-glycosaminoglycan binding: specificity for CCR2 ligand binding to highly sulfated oligosaccharides using FTICR mass spectrometry. J Biol Chem. 2005;280:32200–32208. doi: 10.1074/jbc.M505738200. [DOI] [PubMed] [Google Scholar]
- Yu Y, Sweeney MD, Saad OM, Leary JA. Potential inhibitors of chemokine function: analysis of noncovalent complexes of CC chemokine and small polyanionic molecules by ESI FT-ICR mass spectrometry. J Am Soc Mass Spectrom. 2006;17:524–535. doi: 10.1016/j.jasms.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Zamfir A, Seidler DG, Schonherr E, Kresse H, Peter-Katalinic J. On-line sheathless capillary electrophoresis/nanoelectrospray ionization-tandem mass spectrometry for the analysis of glycosaminoglycan oligosaccharides. Electrophoresis. 2004;25:2010–2016. doi: 10.1002/elps.200405925. [DOI] [PubMed] [Google Scholar]