

Abstract
The deleterious pathophysiological cascade induced after traumatic brain injury (TBI) is initiated by an excitotoxic process triggered by excessive glutamate release. Activation of the glutamatergic N-methyl-D-aspartate receptor, by increasing calcium influx, activates nitric oxide (NO) synthases leading to a toxic production of NO. Moreover, after TBI, free radicals are highly produced and participate to a deleterious oxidative stress. Evidence has showed that the major toxic effect of NO comes from its combination with superoxide anion leading to peroxynitrite formation, a highly reactive and oxidant compound. Indeed, peroxynitrite mediates nitrosative stress and is a potent inducer of cell death through its reaction with lipids, proteins and DNA. Particularly DNA damage, caused by both oxidative and nitrosative stresses, results in activation of poly(ADP-ribose) polymerase (PARP), a nuclear enzyme implicated in DNA repair. In response to excessive DNA damage, massive PARP activation leads to energetic depletion and finally to cell death. Since 10 years, accumulating data have showed that inactivation of PARP, either pharmacologically or using PARP null mice, induces neuroprotection in experimental models of TBI. Thus TBI generating NO, oxidative and nitrosative stresses promotes PARP activation contributing in post-traumatic motor, cognitive and histological sequelae. The mechanisms by which PARP inhibitors provide protection might not entirely be related to the preservation of cellular energy stores, but might also include other PARP-mediated mechanisms that needed to be explored in a TBI context. Ten years of experimental research provided rational basis for the development of PARP inhibitors as treatment for TBI.
Keywords: excitotoxicity, neuroprotection, nitric oxide, nitrosative stress, oxidative stress, peroxynitrite, poly(ADP-ribose)polymerase, cell death, traumatic brain injury
With a global incidence of traumatic brain injury (TBI) generally reported as ≈200 in 100 000 with a mortality of 20 per 100 000, neurotrauma is a major public health challenge. TBI remains one of the leading causes of death and disability in industrialized countries and its incidence increases in developing countries (Reilly, 2007). Over the last two decades, understanding of the complex pathobiology of TBI has improved significantly. However, despite numerous studies on animal models of TBI searching for therapeutic strategies, no neuroprotective therapy is currently available for human traumatized patients (Bramlett and Dietrich, 2004).
TBI promotes focal injuries resulting from direct loadings. Focal injuries to the brain account for one half of all severe head injuries. Diffuse injury also occurs after TBI and is most often caused by inertial forces such as translational and/or rotational acceleration (LaPlaca et al., 2007). The pathophysiology of TBI is complicated and involves both primary and secondary insults. Primary injury to the brain can be induced by numerous mechanisms, such as brain contusion, shearing and stretching of the brain tissue caused by motion of the brain structures relative to the skull and haematoma. Secondary injury development includes complex biochemical and physiological processes that are initiated by the primary insult and manifest over a period of hours to days and even months. Animal models commonly used in TBI research have been developed and used to experimentally mimic some aspects of the behavioural, tissular and cellular consequences of human TBI in order to understand post-traumatic pathophysiology (Cernak, 2005).
This report will outline the involvement of nitric oxide (NO), oxidative and nitrosative stresses, and poly(ADP-ribose)polymerase (PARP) in the pathophysiology of TBI. Because NO and oxidative stress-mediated toxicities are triggered by excitotoxicity, the role of glutamate will be briefly examined.
Excitotoxicity: initiator of the deleterious cascade of cell death
The deleterious pathophysiological cascade induced after TBI is initiated by an excitotoxic process triggered by excessive glutamate release. Glutamate is the major excitotoxic neurotransmitter in the mammalian central nervous system, but it is also a potent neurotoxin that can kill nerve cell. In traumatized patients, a correlation has been demonstrated between bad outcome and important increase in glutamate (Bullock et al., 1998; Zhang et al., 2001). Increased levels of extracellular glutamate following head injury causes overstimulation of both metabotropic and ionotropic receptors that may result in secondary events leading to neuronal cell death. Activation of metabotropic receptors causes mobilization of calcium (Ca2+) from internal stores. Three types of ionotropic glutamatergic receptors are activated: N-methyl-D-aspartate (NMDA) receptor, 2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl)propionic acid receptor (AMPAR) and kainate receptor. When activated, AMPAR leads to the entry of Na+ in cell contributing to membrane depolarization. In addition, intracellular water increases inducing a cell swelling also called cytotoxic oedema. When activated, NMDA receptor promotes the entry of Na+ and Ca2+ in cell contributing to membrane depolarization and to activation of Ca2+-dependent enzymes (Yi and Hazell, 2006). Studies using glutamatergic receptors antagonists showed neuroprotective effects in experimental models of TBI, demonstrating the deleterious role of excitotoxicity (Faden et al., 1989; McIntosh et al., 1990; Shapira et al., 1990; Faden, 1993; Allen et al., 1999). Calcium overload can trigger many downstream neurotoxic cascades, including the uncoupling mitochondrial electron transfer from ATP synthesis, and the activation and overstimulation of enzymes, such as calpains, protein kinases, endonucleases and NO synthases (NOS) (Farkas and Povlishock, 2007).
Involvement of NOS in TBI sequelae
Experimentally, TBI induces the activation of NOS leading to the production of NO in brain (Yamanaka et al., 1995; Sakamoto et al., 1997; Muralikrishna Rao et al., 1998; Cherian et al., 2000; Ahn et al., 2004). NO production has been also demonstrated in cerebrospinal fluid (CSF) and in brain tissue after human TBI (Clark et al., 1996; Uzan et al., 2001; Hlatky et al., 2002; 2003; Silberstein et al., 2002; Toczylowska et al., 2006) and the end–products of NO, nitrate/nitrite, levels are strongly correlated with TBI severity (Hlatky et al., 2002). Interestingly, injured patients who died had less nitrate/nitrite than the survivors. Particularly, it has been shown that NO can combine to cellular thiols compounds leading to the formation of S-nitrosothiols (Foster et al., 2003; Gow et al., 2004). Bayir et al. (2002; 2003) demonstrate that increase in S-nitrosothiols is correlated with intracranial pressure decrease, suggesting a neuroprotective role of S-nitrosothiols following TBI. NO can be synthetized by three isoforms of NOS: NOS1 (neuronal NOS) and NOS3 (endothelial NOS) that are both constitutive and Ca2+-dependent enzymes. The third one is NOS2 (inducible NOS) that produces large quantity of NO in inflammatory situations. The involvement of NOS has been quite well established in TBI pathophysiology, even if each isoform plays a different role. The first study demonstrating the role of NOS in TBI has been published by Mésenge et al., (1996). Indeed, post-treatment with Nω-nitro-L-arginine-methylester (L-NAME), a non-selective NOS inhibitor, decreases the post-traumatic neurological deficit with a therapeutic window of opportunity of 60 min. The same results have been observed with 7-nitroindazole, a selective NOS1 inhibitor (Mésenge et al., 1996). In addition, pretreatment with this inhibitor decreases the brain lesion volume following TBI (Wada et al., 1999). Intracerebral injection of NOS1 antiserum reduced blood–brain barrier permeability and cerebral oedema (Sharma et al., 2006). All these data demonstrate the deleterious role of NOS1.
Inhibition of all NOS isoforms is not desirable considering the vasodilator role of NO produced by NOS3 and involved in the regulation of vascular tone. Indeed, treatment with L-arginine, the substrate of NOS, reduces the brain lesion volume and this is associated with NOS3 activation and improved cerebral blood flow (CBF) (DeWitt et al., 1997; Wada et al., 1998a; 1999; Cherian et al., 1999). In addition, NOS3−/− mice have a decreased CBF compared with wild-type animals (Hlatky et al., 2003), and NOS3 is necessary to induce beneficial vascular effect of L-arginine after TBI (Hlatky et al., 2003). Considering these data, NO produced by NOS3 plays a beneficial role after TBI.
If the roles of NOS1 and NOS3 are quite well understood, the one of NOS2 is still controversial in TBI. Some studies suggest that early after TBI, effects of NOS2 may be detrimental as aminoguanidine (AG), a selective NOS2 inhibitor, has been shown to reduce neuronal cell death following TBI (Wada et al., 1998b; Görlach et al., 2000; Stoffel et al., 2000; 2001). In addition, NOS2−/− mice have a reduced motor and cognitive deficits and brain lesion volume than those of wild-type animals after cryogenic TBI (Jones et al., 2004). More recently, three different selective NOS2 inhibitors, AG, L-N(6)-(1-iminoethyl) lysine hydrochloride (L-NIL) and 1400W reduce motor deficits and brain lesion 24 and 72 h after TBI (Jafarian-Tehrani et al., 2005; Louin et al., 2006). So these data argue in the deleterious role of NOS2 in the early phase after brain trauma. However, several reports have suggested potential beneficial effects of NOS2, particularly at delayed time points after injury. Indeed, Sinz et al. (1999) have shown that NOS2−/− mice present more important cognitive deficits than wild-type mice at 17–21 days after TBI. In addition, NOS2 knockout (KO) mice have been demonstrated to have greater loss of brain levels of ascorbate, an endogen antioxidant, compared with wild-type animals at 72 h after TBI (Bayir et al., 2005). Moreover, treatment with two selective inhibitors of NOS2, AG and L-NIL increases post-traumatic cognitive deficits at 21 days after injury (Sinz et al., 1999). Treatment with L-arginine 48 h after TBI, that is, when NOS2 is activated, induces neuroprotective effects (Cherian et al., 2003). As TBI decreases CBF (Bouma et al., 1991; Marion et al., 1991) and NO is a powerful cerebral vasodilator, two studies examined the role of NOS2-derived NO on CBF after TBI. Animals receiving antisense NOS2 oligodeoxynucleotides exhibited an exacerbation of the CBF reduction after TBI (Steiner et al., 2004) and NOS2 KO mice showed a reduced recovery of CBF 72 h after TBI in hippocampus, thalamus and amygdala/piriform cortex, which are structures outside the contusion (Foley et al., 2008). Taken together, NOS2 appears to mediate detrimental effects early after TBI while mediating beneficial actions at more delayed time points.
Involvement of oxidative and nitrosative stresses in TBI
The glutamate-mediated increase in intracellular Ca2+ also activates several free radical pathways, including the conversion of xanthine dehydrogenase to xanthine oxidase, NOS, the phospholipase A2-cycloxygenase pathway and mitochondria, leading to post-traumatic oxidative stress. The oxidative stress situation is defined as an imbalance between free radical production and endogen antioxidant systems. Brain is particularly vulnerable to oxidative stress as it contains low levels of antioxidant systems and high concentrations of iron that can catalyse the production of free radicals, and as it is rich in unsaturated fatty acids that are targets for lipid peroxidation (Margaill et al., 2005). In humans, TBI increases plasma and CSF levels of malonedialdehyde, a marker of lipid peroxidation, as early as 2–3 h that persists at least until 7 days after injury (Cernak et al., 2000; Cristofori et al., 2001; Bayir et al., 2002). In addition, superoxide dismutase (SOD) activity decreases at 24 h and during 7 days after severe TBI (Cernak et al., 2000).
After experimental TBI, hydroxyl radicals OH and superoxide anions O2− increase early after injury (Hall and Braughler, 1993; Fabian et al., 1998). In addition, production of OH is strongly correlated with lipid peroxidation and post-traumatic cerebral oedema (Nishio et al., 1997). Endogen antioxidants enzymatic systems, such as catalase and glutathione peroxidase (GPx), have increased activities between 3 and 7 days following TBI (Goss et al., 1997). If oxidative stress has been shown in brain after TBI, brain trauma also induces whole-body oxidative stress (Shohami et al., 1999; Pratico et al., 2002). There is compelling evidence supporting the role of oxidative stress in post-traumatic sequelae. Indeed, antioxidant strategies aiming at decreasing oxidative stress reduced post-traumatic neurological deficits, cerebral oedema and brain lesion volume: PEG-SOD, an antioxidant enzyme (Hamm et al., 1996); melatonine mimicking GPx (Mésenge et al., 1998a); OPC-14117 (7-hydroxy-1-[4-(3-methoxyphenyl)-1-piperazinyl]acetylamino-2,2,4,6-tetramethylindan), a scavenger of superoxide anions (Aoyama et al., 2002); phenyl-tert-butylnitrone, a free radical scavenger (Lewen et al., 2001; Marklund et al., 2001); mesylate tirilazad, an inhibitor of lipid peroxidation (Hall et al., 1988), and SOD1 (Mikawa et al., 1996) or SOD3 (Pineda et al., 2001) transgenic mice. In addition, KO mice for GPx1 present an increased neuronal cell death after TBI compared with wild-type animals (Flentjar et al., 2002).
NO-mediated toxicity has been shown to involve peroxynitrite. Indeed, superoxide anions could combine with NO to generate peroxynitrite anions. Peroxynitrite is a strong oxidant that can directly react with tyrosine of proteins producing nitrotyrosine (Pacher et al., 2007). The development of antibodies that recognize nitrotyrosine provided a major impetus to the study of peroxynitrite and nitrotyrosine is now widely used as a marker of nitrosative stress. Mésenge et al. (1998b) have shown an increase in nitrotyrosine in brain tissue between 4 and 24 h in a model of diffuse TBI. In a model of focal TBI, nitrotyrosine is present as early as 30 min after TBI and persists at least during 72 h (Besson et al., 2003a). Several studies have shown 3-nitrotyrosine staining in infiltrating polymorphonuclear neutrophils, microvascular endothelium and neurons throughout the cytoplasm of both cell bodies and the proximal portions of axons and dendrites (Whalen et al., 1999; Hall et al., 2004; Bayir et al., 2005). Treatment with L-NAME, a non-selective NOS inhibitor, decreases nitrotyrosine levels after TBI, demonstrating the involvement of NO in peroxynitrite formation (Mésenge et al., 1998b). The deleterious role of peroxynitrite is supported by reports demonstrating the beneficial effects of peroxynitrite scavengers such as penicillamine, penicillamine methyl-ester (Hall et al., 1999), and tempol (Deng-Bryant et al., 2008), and the peroxynitrite decomposition catalyst FP15 (Lacza et al., 2003) in reducing neuronal injury and improving neurological recovery following TBI.
Although not a free radical in nature, peroxynitrite is much more reactive than its parent molecules NO and O2− (Pacher et al., 2007). The half-life of peroxynitrite is short but sufficient to cross biological membranes and allow significant interactions with most critical biomolecules (Pacher et al., 2007). Peroxynitrite anions attack protein components, such as transition metal centres and amino acids with cysteine oxidation, tyrosine nitration and tryptophan, methionine and histidine oxidation. In many cases, nitration of antioxidant enzymes leads to their inactivation. In particular, it has been shown recently that SOD2 extracted from brains of mice and humans after TBI demonstrates both a significant increase in tyrosine nitration and a dramatic decrease in its enzymatic activity (Bayir et al., 2007). Furthermore, deficiency of NOS1 but not NOS2 and NOS3 attenuates SOD2 nitration after experimental TBI (Bayir et al., 2007). So nitration of SOD2 is likely a consequence of peroxynitrite within the intracellular milieu of neurons rather than activated inflammatory cells. Nitration and inactivation of SOD2 could lead to self-amplification of oxidative stress in the brain progressively enhancing peroxynitrite production and secondary damage. Peroxynitrite anions also trigger lipid peroxidation in membranes, liposomes and lipoproteins. Several intermediates products of lipid peroxidation including isoprostanes and 4-hydroxynonenal (4HNE) serves as an indirect marker of peroxynitrite effects on lipids. Lipid peroxidation has been demonstrated with 4HNE staining in experimental models of diffuse and focal TBI (Hall et al., 2004; Chen et al., 2007). Last but not the least, peroxynitrite can alter DNA by introducing oxidative damage on guanine producing 8-oxoguanine. It may also attack DNA generating strand breaks. The formation of peroxynitrite-mediated DNA breakage represents one of the most deleterious aspects of peroxynitrite anions as they represent the trigger for the activation of the nuclear DNA repair enzyme, PARP.
PARP: executioner of cell death
PARP is a constitutive nuclear enzyme present in eukaryotes. It belongs to an expanding family of 17 members implicated in such physiological processes as DNA repair, maintenance of genomic integrity, gene transcription, cell division and apoptosis (Schreiber et al., 2006). PARP-1 (EC 2.4.2.30), also known as poly(ADP-ribose) synthetase, the founding member of the superfamily, is the major isoform present in the nucleus. It has been the most extensively studied as it is responsible for most of the poly(ADP-ribose) formation in health and disease.
At the beginning of PARP research, strategies aiming at studying approaches to inhibit PARP have been carried out almost exclusively by scientist working on cancer research. Enhanced PARP expression and/or activity have been demonstrated in several tumour cell lines. This may allow tumour cells to withstand genotoxic stress and increase their resistance to DNA-damaging agents. Interestingly, inhibition of PARP sensitizes tumour cells to cytotoxic therapy, including temozolomide, platinums, topoisomerase I inhibitors and radiation (Ratnam and Low, 2007). Thus inhibition of PARP could enhance the anti-tumour effect of radiation or chemotherapy. In this order, PARP inhibitors are being developed for clinical use both alone and as chemosensitizers in combination with chemotherapy. Several clinical studies are conducted to evaluate the safety and effectiveness of the combination of PARP inhibitor with chemotherapy (de la Lastra et al., 2007; Lewis and Low, 2007; Ratnam and Low, 2007).
PARP is also known to have an essential role in other various diseases (Pacher and Szabo, 2008). When activated in response to oxidative and nitrosative stress-induced DNA strand breaks, PARP initiates an energy-consuming cycle by transferring ADP-ribose units from NAD to a set of nuclear acceptor proteins, including histones, several chromatin-binding proteins and PARP itself (Hassa et al., 2006). In order to study the role of PARP in pathological conditions, numerous inhibitors of PARP have been synthetized (Southan and Szabó, 2003; Jagtap & Szabó, 2005) and three different KO mice for PARP-1 have been generated (Wang et al., 1995; de Murcia et al., 1997; Masutani et al., 2000). Using these pharmacological tools, it has been shown that PARP mediates necrotic cell death in response to excessive DNA damage under pathological conditions. This process results in rapid depletion of intracellular NAD, in a loss of ATP as it is used to synthesize new NAD, and finally to cell death. It was first suggested by Nathan Berger more than two decades ago (Berger, 1985) and this ‘PARP suicide hypothesis’ was demonstrated in vitro. Oxidative stress induced in vivo in brain has been demonstrated to promote PARP activation, which contributes to neuronal cell death (Besson et al., 2003b). Additionally, PARP inhibitors have been shown to prevent in vitro neuronal cell injury from glutamate (Cosi et al., 1994), NMDA (Mandir et al., 2000), direct NO (Wallis et al., 1993) and NO donors (Zhang et al., 1994). So these results demonstrate a role of PARP in glutamate-mediated excitotoxicity. As this deleterious cascade occurs in TBI, massive DNA breakage has been reported after TBI (Rink et al., 1995; Colicos and Dash, 1996; LaPlaca et al., 1999; Satchell et al., 2003), and peroxynitrite and poly(ADP-ribosyl)ation co-localize in areas of cell death in traumatic brain tissues (Besson et al., 2003a). PARP is markedly activated as early as 30 min after TBI and its activation persists for 72 h after TBI (LaPlaca et al., 1999; Besson et al., 2003a; Satchell et al., 2003) Tyrosine nitration and PARP activation are both found to be persistently increased compared with normal brain, with relative peaks seen at 8 and 72 h (Satchell et al., 2003). As poly(ADP-ribose) glycohydrolase rapidly degrades polymers of ADP-ribose (Davidovic et al., 2001), this suggests that ADP-ribosylation, that is, PARP activation, is a prolonged phenomenon. This pattern of PARP activation is likely related to the continuing presence of peroxynitrite in the lesioned brain tissue. It is also conceivable that a massive early DNA breakage, which remains unrepaired for prolonged periods of time, is responsible for the prolonged pattern of PARP activation. Cleavage of PARP by caspase-3, occurring only 7 days after TBI (LaPlaca et al., 1999), is one of the mechanism to inactivate PARP, thus preserving cell energy stores required during apoptosis. Finally taken together, these data indicate that, following brain trauma, NO leads to peroxynitrite formation, via its combination with superoxide anion, which in turn induces DNA strand breaks rendering PARP active.
After TBI, PARP is present in human brain pericontusional tissue in neurons either in the nucleus exclusively or in both nuclear and cytoplasmic compartments (Ang et al., 2003). Moreover, PARP is activated by brain trauma as poly(ADP-ribose)-modified proteins are increased at 24 h and until 48 h in CSF of pediatric TBI patients (Fink et al., 2008). Mitochondrial proteins belonging to electron transport chain complexes, such as cytochrome c reductase and oxidase, F1F0ATPase β subunit, HSP60, were found to be proteins poly-ADP-ribosylated (Lai et al., 2008). Post-traumatic PARP activation is also associated to tissue NAD decrease in cortex and hippocampus (Lai et al., 2008). These data suggest that poly(ADP-ribosyl)ation on electron transport chain function may impair mitochondrial respiration contributing, at least partly, to PARP-mediated energy failure. Two PARP inhibitors, INH2BP (5-iodo-6-amino-1,2-benzopyrone) (Satchell et al., 2003) and INO-1001 (Clark et al., 2007), prevent NAD depletion and improve post-TBI deficits, demonstrating PARP-mediated energy failure as a contributor to the pathological sequelae of TBI. In vitro PARP inhibition protects hippocampal slices against percussion-induced loss of CA1 pyramidal cell-evoked response (Wallis et al., 1996). Whalen et al. (1999) showed that motor and cognitive deficits of mice submitted to TBI are less severe when the PARP-1 gene is inactivated. The prototypical PARP inhibitor, 3-aminobenzamide (3-AB), and other benzamide derivatives induce neuroprotective effects on the neurological deficit and the brain lesion after closed head injury in mice (Mésenge et al., 1999) and after TBI induced by fluid percussion (Besson et al., 2003a). Delaying the treatment of PARP inhibition relative to the TBI produces a therapeutic window of opportunity of 2 h (Mésenge et al., 1999). GPI-6150 (1,11b-dihydro-[2H]benzopyrano[4,3,2-de]isoquinolin-3-one), another PARP inhibitor, reduces the injured area in the very early phase (24 h) after TBI (LaPlaca et al., 2001). Two water-soluble PARP inhibitors, PJ34 [N-(6-oxo-5,6-dihydrophenanthridin-2-yl)-2-(N,N-dimethylamino)acetamide, HCl] and INO-1001 induce neurological recovery-promoting effects in a model of TBI caused by fluid percussion in rat (Besson et al., 2005). Indeed, repeated treatment with PJ34 and INO-1001 decreases the neurological deficit 3 days after injury and these protective effects still persist at 7 days post injury (Besson et al., 2005). It is important to emphasize that the protective effect of PARP inhibition on neurological function is a lasting one, which remains significant even 7 (Besson et al., 2003a; 2005) or 21 days (Satchell et al., 2003) after TBI. These data strengthen the hypothesis that PARP activation promotes deleterious effects on the neurological consequences in the early and late phases after TBI. Finally, a NOS1 inhibitor, 3-bromo-7-nitroindazole, significantly decreases the production of poly(ADP-ribose) in damaged cerebral cortex after cryogenic lesion, demonstrating that TBI-induced PARP activation depends, at least in part, on prior activation of NOS1 (Hortobagyi et al., 2003).
Pharmacological inhibition of PARP activation may also be a viable approach for improving the outcome of stem cell transplantation. This can be achieved by two ways. First, it may make the host tissue environment more receptive for a graft. Second, it may directly protect the transplanted cells from necrosis induced by peroxynitrite. Indeed, treatment with the PARP inhibitor PJ34 improves the neurological score after cryogenic lesion, and increases it even further following stem cell transplantation. High peroxynitrite production coincides with the loss of the majority of the grafted cells, and inhibition of the PARP activation cascade increases the number of surviving cells (Lacza et al., 2003). This PARP inhibition approach may ultimately lead to an optimized grafting strategy.
In cerebral ischaemia studies in which different PARP inhibitors were used and full dose–response curves were obtained, the protection provided by PARP inhibitors diminishes when the dose of agent is increased; that is, a bell-shaped dose–response is observed (Takahashi et al., 1997). Consistent with this observation, similar data have been noticed with 3-AB in a model of TBI (Mésenge et al., 1999). The beneficial effects of PJ34 and INO-1001, two potent PARP inhibitors, on the neurological score have been seen despite no significant benefit on brain lesion volume, suggesting that the protective effect by PARP inhibition could not be attributed to salvaging significant amounts of lesioned tissue (Besson et al., 2005). This observation has been also noticed in both pharmacological inhibition and genetic intervention in models of TBI induced by controlled cortical impact (Whalen et al., 1999; Satchell et al., 2003) and cold injury (Lacza et al., 2003), indicating that functional improvement does not always correlate with the extent of brain damage. In addition, more complete inhibition of PARP with INH2BP impairs spatial memory acquisition independent of injury, and is associated with ribosylation of 14-3-3gamma, a protein implicated in learning and memory (Satchell et al., 2003). As the catalytic domain is very similar in PARP members, PARP inhibitors are not isoenzyme-specific but block total PARP activity, thus demonstrating the overall role of PARP in TBI (Figure 1). These observations suggest that complete and non-selective inhibition of all PARP isoforms may produce some potential adverse effects, and that partial inhibition may be a more desirable approach. All PARP family members ribosylate target proteins and use NAD, but PARP-1 and -2 are the only family members known to be activated by DNA strand breaks (Schreiber et al., 2002). Even if both enzymes have overlapping and redundant functions (Ménissier de Murcia et al., 2003), PARP-2 has been thought to be less active isoform contributing only 5–10% of total PARP activity in response to DNA damage (Améet al., 1999; Schreiber et al., 2002). PARP-2 KO mice have been generated (Ménissier de Murcia et al., 2003) and very recently, Pellicciari et al. (2008) have synthetized selective PARP-2 inhibitors. These two pharmacological approaches will further help in the understanding of the role of PARP-2 in TBI. In addition, it will be very interesting to further evaluate the only effect of PARP-1 inhibition.
Figure 1.
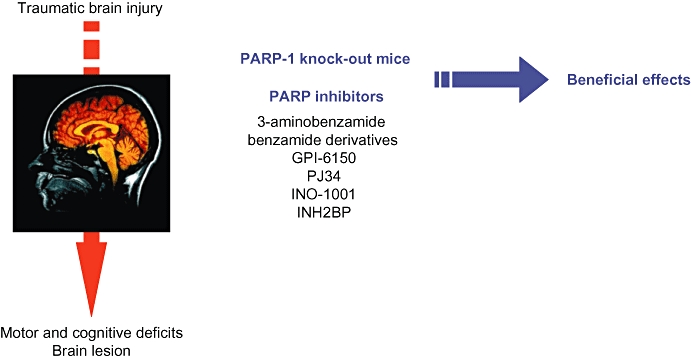
Poly(ADP-ribose) polymerase (PARP) strategies evaluated on deleterious consequences induced by traumatic brain injury. GPI-6150, 1,11b-dihydro-[2H]benzopyrano[4,3,2-de]isoquinolin-3-one; INH2BP, 5-iodo-6-amino-1,2-benzopyrone; PJ34 N-(6-oxo-5,6-dihydrophenanthridin-2-yl)-2-(N,N-dimethylamino)acetamide, HCl.
Deleterious mechanisms of PARP activation toxicity in brain are multiple (Figure 2). First, PARP activation mediates cell death. When DNA is severely damaged, PARP is massively activated resulting in high consumption of NAD and ATP and finally necrosis. Second, PARP regulates inflammation as it acts also as a co-activator of the transcription factor nuclear factor-kappa B resulting in the synthesis of pro-inflammatory mediators. In addition, PARP is able to directly poly-ADP-ribosylate other transcription factors including STAT and activator protein-1 and -2 (Kauppinen, 2007). By this way, inhibition of PARP has been shown to mediate many anti-inflammatory effects in various inflammatory diseases (Szabó, 2006) and acute brain injuries including stroke (Koh et al., 2004; Haddad et al., 2006). Moreover, PAR synthesis induced by PARP promotes translocation of apoptosis-inducing factor (AIF) from mitochondria to the nucleus, subsequent DNA fragmentation and caspase-independent programmed cell death (Andrabi et al., 2006; Yu et al., 2006). As TBI induces neuroinflammation (Ray et al., 2002) and AIF translocation (Zhang et al., 2002), one can ask whether PARP inhibition may promote beneficial effects by interacting theses mechanisms.
Figure 2.
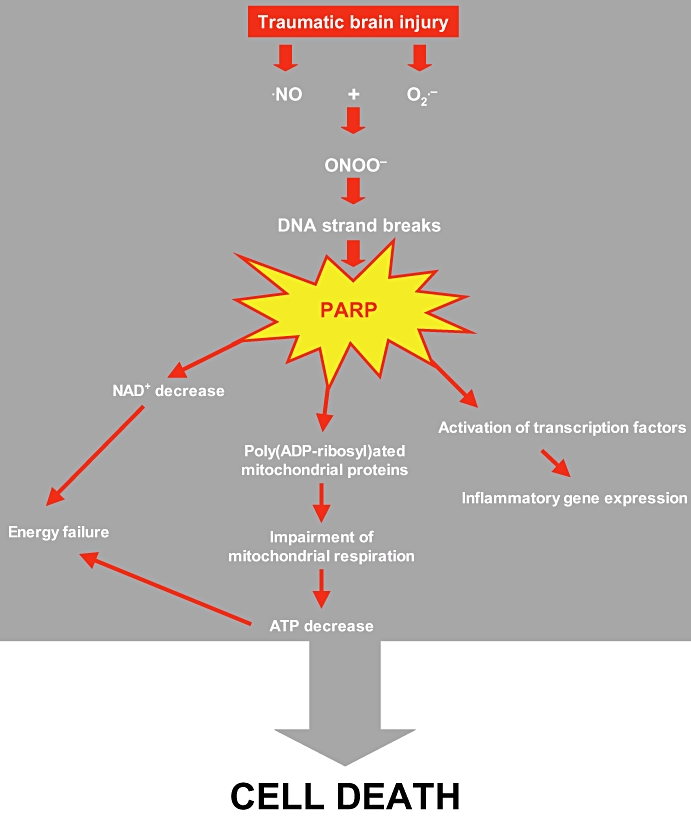
The nitric oxide-oxidative and nitrosative stress–Poly(ADP-ribose) polymerase (PARP) pathway in traumatic brain injury.
General conclusion
Research in the area of NO-oxidative and nitrosative stresses–PARP cascade has led to a better understanding of the pathophysiology of TBI. The mechanisms by which PARP inhibitors provide protection might not entirely be related to the preservation of cellular energy stores, but might also include other PARP-mediated mechanisms that needed to be explored in the context of TBI. The marked beneficial effects of PARP inhibitors in different animal models of TBI suggest that PARP inhibitors can be exploited to treat this important cause of mortality. It is necessary to point out that PARP participates in DNA repair, many global cellular functions (Hassa et al., 2006) and memory formation (Satchell et al., 2003). Partial inhibition may be a safer approach for the TBI treatment than complete PARP inhibition. The advantage of PARP inhibitors as agents for the treatment of TBI includes the short duration of treatment, which is an important safety concern when inhibiting an enzyme that regulates nuclear integrity. Thus the future clinical use of PARP inhibitors for acute life-threatening indications, such as TBI, seems to be well justified and very promising.
Glossary
Abbreviations:
- 3-AB
3-aminobenzamide
- 4HNE
4-hydroxynonenal
- AG
aminoguanidine
- AIF
apoptosis-inducing factor
- AMPAR
2-amino-3-(3-hydroxy-5-methylisoxazol-4-yl)propionic acid receptor
- Ca2+
calcium
- CBF
cerebral blood flow
- CSF
cerebrospinal fluid
- GPx
glutathione peroxidase
- KO
knockout
- L-NAME
Nω-nitro-L-arginine-methylester
- L-NIL
L-N(6)-(1-iminoethyl) lysine hydrochloride
- Na+
sodium
- NMDAR
N-methyl-D-aspartate receptor
- NOS
nitric oxide synthase
- PARP
poly(ADP-ribose)polymerase
- SOD
superoxide dismutase
- TBI
traumatic brain injury
Conflics of interest
None.
References
- Ahn MJ, Sherwood ER, Prough DS, Lin CY, De Witt DS. The effects of traumatic brain injury on cerebral blood flow and brain tissue nitric oxide levels and cytokine expression. J Neurotrauma. 2004;21:1431–1442. doi: 10.1089/neu.2004.21.1431. [DOI] [PubMed] [Google Scholar]
- Allen JW, Ivanova SA, Fan L, Espey MG, Basile AS, Faden AI. Group II metabotropic glutamate receptor activation attenuates traumatic neuronal injury and improves neurological recovery after traumatic brain injury. J Pharmacol Exp Ther. 1999;290:112–120. [PubMed] [Google Scholar]
- Amé JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, et al. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem. 1999;274:17860–17868. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, Sasaki M, et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci USA. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang BT, Yap E, Lim J, Tan WL, Ng PY, Ng I, et al. Poly(adenosine diphosphate-ribose) polymerase expression in human traumatic brain injury. J Neurosurg. 2003;99:125–130. doi: 10.3171/jns.2003.99.1.0125. [DOI] [PubMed] [Google Scholar]
- Aoyama N, Katayama Y, Kawamata T, Maeda T, Mori T, Yamamoto T, et al. Effects of antioxidant, OPC-14117, on secondary cellular damage and behavioral deficits following cortical contusion in the rat. Brain Res. 2002;934:117–124. doi: 10.1016/s0006-8993(02)02366-1. [DOI] [PubMed] [Google Scholar]
- Bayir H, Kagan VE, Tyurina YY, Tyurin V, Ruppel RA, Adelson PD, et al. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatr Res. 2002;51:571–578. doi: 10.1203/00006450-200205000-00005. [DOI] [PubMed] [Google Scholar]
- Bayir H, Kochanek PM, Liu SX, Arroyo A, Osipov A, Jiang J, et al. Increased S-nitrosothiols and S-nitrosoalbumin in cerebrospinal fluid after severe traumatic brain injury in infants and children: indirect association with intracranial pressure. J Cereb Blood Flow Metab. 2003;23:51–61. doi: 10.1097/01.WCB.0000040399.30600.E3. [DOI] [PubMed] [Google Scholar]
- Bayir H, Kagan VE, Borisenko GG, Tyurina YY, Janesko KL, Vagni VA, et al. Enhanced oxidative stress in iNOS-deficient mice after traumatic brain injury: support for a neuroprotective role of iNOS. J Cereb Blood Flow Metab. 2005;25:673–684. doi: 10.1038/sj.jcbfm.9600068. [DOI] [PubMed] [Google Scholar]
- Bayir H, Kagan VE, Clark RSB, Janseko-Feldman K, Rafikov R, Huang Z, et al. Neuronal NOS-mediated nitration and inactivation of manganese superoxide dismutase in brain after experimental and human brain injury. J Neurochem. 2007;101:168–181. doi: 10.1111/j.1471-4159.2006.04353.x. [DOI] [PubMed] [Google Scholar]
- Berger NA. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat Res. 1985;101:4–15. [PubMed] [Google Scholar]
- Besson VC, Croci N, Plotkine M, Marchand-Verrecchia C. Deleterious poly(ADP-ribose)polymerase-1 pathway activation in traumatic brain injury in rat. Brain Res. 2003a;989:58–66. doi: 10.1016/s0006-8993(03)03362-6. [DOI] [PubMed] [Google Scholar]
- Besson VC, Margaill I, Plotkine M, Marchand-Verrecchia C. Deleterious activation of poly(ADP-ribose)polymerase-1 in brain after in vivo oxidative stress. Free Radic Res. 2003b;37:1201–1208. doi: 10.1080/10715760310001612568. [DOI] [PubMed] [Google Scholar]
- Besson VC, Zsengellér Z, Plotkine M, Szabó C, Marchand-Verrecchia C. Beneficial effects of PJ34 and INO-1001, two novel water-soluble poly(ADP-ribose) polymerase inhibitors, on the consequences of traumatic brain injury in rat. Brain Res. 2005;1041:149–156. doi: 10.1016/j.brainres.2005.01.096. [DOI] [PubMed] [Google Scholar]
- Bouma GJ, Muizelaar JP, Choi SC, Newlon PG, Young HF. Cerebral circulation and metabolism after severe traumatic brain injury: the elusive role of ischemia. J Neurosurg. 1991;75:685–693. doi: 10.3171/jns.1991.75.5.0685. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab. 2004;24:133–150. doi: 10.1097/01.WCB.0000111614.19196.04. [DOI] [PubMed] [Google Scholar]
- Bullock R, Zauner A, Woodward JJ, Myseros J, Choi SC, Ward JD, et al. Factors affecting excitatory amino acid release following severe human head injury. J Neurosurg. 1998;89:507–518. doi: 10.3171/jns.1998.89.4.0507. [DOI] [PubMed] [Google Scholar]
- Cernak I. Animal models of head trauma. NeuroRX. 2005;2:410–422. doi: 10.1602/neurorx.2.3.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cernak I, Savic VJ, Kotur J, Prokic V, Veljovic M, Grbovic D. Characterization of plasma magnesium concentration and oxidative stress following graded traumatic brain injury in humans. J Neurotrauma. 2000;17:53–68. doi: 10.1089/neu.2000.17.53. [DOI] [PubMed] [Google Scholar]
- Chen XR, Besson VC, Palmier B, Garcia Y, Plotkine M, Marchand-Leroux C. Neurological recovery-promoting, anti-inflammatory and anti-oxidative effects afforded by fenofibrate, a PPAR alpha agonist, in traumatic brain injury. J Neurotrauma. 2007;24:1119–1131. doi: 10.1089/neu.2006.0216. [DOI] [PubMed] [Google Scholar]
- Cherian L, Chacko G, Goodman JC, Robertson CS. Cerebral hemodynamic effects of phenylephrine and L-arginine after cortical impact injury. Crit Care Med. 1999;27:2512–2517. doi: 10.1097/00003246-199911000-00031. [DOI] [PubMed] [Google Scholar]
- Cherian L, Goodman JC, Robertson CS. Brain nitric oxide changes after controlled cortical impact injury in rats. J Neurophysiol. 2000;83:2171–2178. doi: 10.1152/jn.2000.83.4.2171. [DOI] [PubMed] [Google Scholar]
- Cherian L, Chacko G, Goodman C, Robertson CS. Neuroprotective effects of L-arginine administration after cortical impact injury in rats: dose response and time window. J Pharmacol Exp Ther. 2003;304:617–623. doi: 10.1124/jpet.102.043430. [DOI] [PubMed] [Google Scholar]
- Clark RS, Kochanek PM, Obrist WD, Wong HR, Billiar TR, Wisniewski SR, et al. Cerebrospinal fluid and plasma nitrite and nitrate concentrations after head injury in humans. Crit Care Med. 1996;24:1243–1251. doi: 10.1097/00003246-199607000-00030. [DOI] [PubMed] [Google Scholar]
- Clark RSB, Vagni VA, Nathaniel PD, Jenkins LW, Dixon CE, Szabo C. Local administration of the poly(ADP-ribose)polymerase inhibitor INO-1001 prevents NAD depletion and improves water-maze performance after traumatic brain injury. J Neurotrauma. 2007;24:1399–1405. doi: 10.1089/neu.2007.0305. [DOI] [PubMed] [Google Scholar]
- Colicos MA, Dash PK. Apoptotic morphology of dentate gyrus granule cells following experimental cortical impact injury in rats: possible role in spatial memory deficits. Brain Res. 1996;739:120–131. doi: 10.1016/s0006-8993(96)00824-4. [DOI] [PubMed] [Google Scholar]
- Cosi C, Suzuki H, Milani D, Facci L, Menegazzi M, Vantini G, et al. Poly(ADP-ribose) polymerase: early involvement in glutamate-induced neurotoxicity in cultured cerebellar granule cells. J Neurosci Res. 1994;39:38–46. doi: 10.1002/jnr.490390106. [DOI] [PubMed] [Google Scholar]
- Cristofori L, Tavazzi B, Gambin R, Vagnozzi R, Vivenza C, Amorini AM, et al. Early onset of lipid peroxydation after human traumatic brain injury: a fatal limitation for the free radical scavenger pharmacological therapy? J Investig Med. 2001;49:450–458. doi: 10.2310/6650.2001.33790. [DOI] [PubMed] [Google Scholar]
- Davidovic L, Vodenicharov M, Affar EB, Poirier GG. Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp Cell Res. 2001;268:7–13. doi: 10.1006/excr.2001.5263. [DOI] [PubMed] [Google Scholar]
- Deng-Bryant Y, Singh IN, Carrico KM, Hall ED. Neuroprotective effects of tempol, a catalytic cavenger of peroxynitrite-derived free radicals, in a mouse traumatic brain injury model. J Cereb Blood Flow Metab. 2008;28:1114–1126. doi: 10.1038/jcbfm.2008.10. [DOI] [PubMed] [Google Scholar]
- DeWitt DS, Smith TG, Deyo DJ, Miller KR, Uchida T, Prough DS. L-arginine and superoxide dismutase prevent or reverse cerebral hypoperfusion after fluid-percussion traumatic brain injury. J Neurotrauma. 1997;14:223–233. doi: 10.1089/neu.1997.14.223. [DOI] [PubMed] [Google Scholar]
- Fabian RH, DeWitt DS, Kent TA. The 21-aminosteroid U-74389G reduces cerebral superoxide anion concentration following fluid percussion injury of the brain. J Neurotrauma. 1998;15:433–440. doi: 10.1089/neu.1998.15.433. [DOI] [PubMed] [Google Scholar]
- Faden AI. Comparison of single and combination drug treatment strategies in experimental brain trauma. J Neurotrauma. 1993;10:91–100. doi: 10.1089/neu.1993.10.91. [DOI] [PubMed] [Google Scholar]
- Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- Farkas O, Povlishock JT. Cellular and subcellular change evoked by diffuse traumatic brain injury: a complex web of change extending far beyond focal damage. Prog Brain Res. 2007;161:43–59. doi: 10.1016/S0079-6123(06)61004-2. [DOI] [PubMed] [Google Scholar]
- Fink EL, Lai Y, Zhang X, Janesko-Feldman K, Adelson PD, Szabo C, et al. Quantification of poly(ADP-ribose)-modified proteins in cerebrospinal fluid from infants and children after traumatic brain injury. J Cereb Blood Flow Metab. 2008;28:1523–1529. doi: 10.1038/jcbfm.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flentjar NJ, Crack PJ, Boyd R, Malin M, de Haan JB, Hertzog P, et al. Mice lacking glutathione peroxidase-1 activity show increased TUNEL staining and an accelerated inflammatory response in brain following a cold-induced injury. Exp Neurol. 2002;177:9–20. doi: 10.1006/exnr.2002.7927. [DOI] [PubMed] [Google Scholar]
- Foley LM, Hitchens TK, Melick JA, Bayir H, Ho C, Kochanek PM. Effect of inducible nitric oxide synthase on cerebral blood flow after experimental traumatic brain injury in mice. J Neurotrauma. 2008;25:299–310. doi: 10.1089/neu.2007.0471. [DOI] [PubMed] [Google Scholar]
- Foster MW, McMahon TJ, Stamler JS. S-nitrosylation in health and disease. Trends Mol Med. 2003;9:160–168. doi: 10.1016/s1471-4914(03)00028-5. [DOI] [PubMed] [Google Scholar]
- Görlach C, Hortobágyi T, Benyó Z, Wahl M. Aminoguanidine reduces brain lesion volume after cold injury in the rat. Pflugers Arch. 2000;440:309–314. doi: 10.1007/s004240000293. [DOI] [PubMed] [Google Scholar]
- Goss JR, Taffe KM, Kochanek PM, Dekosky ST. The antioxidant enzymes glutathione peroxidase and catalase increase following traumatic brain injury in the rat. Exp Neurol. 1997;146:291–294. doi: 10.1006/exnr.1997.6515. [DOI] [PubMed] [Google Scholar]
- Gow AJ, Farkouh CR, Munson DA, Posencheg MA, Ischiropoulos H. Biological significance of nitric oxide-mediated protein modifications. Am J Physiol Lung Cell Mol Physiol. 2004;287:L262–L268. doi: 10.1152/ajplung.00295.2003. [DOI] [PubMed] [Google Scholar]
- Haddad M, Rhinn H, Bloquel C, Coqueran B, Szabó C, Plotkine M, et al. Anti-inflammatory effects of PJ34, a poly(ADP-ribose) polymerase inhibitor, in transient focal cerebral ischemia in mice. Br J Pharmacol. 2006;149:23–30. doi: 10.1038/sj.bjp.0706837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Braughler JM. Free radicals in CNS injury. Res Publ Assoc Res Nerv Ment Dis. 1993;71:81–105. [PubMed] [Google Scholar]
- Hall ED, Yonkers PA, McCall JM, Braughler JM. Effects of the 21-aminosteroid U74006F on experimental head injury in mice. J Neurosurg. 1988;68:456–661. doi: 10.3171/jns.1988.68.3.0456. [DOI] [PubMed] [Google Scholar]
- Hall ED, Kupina NC, Althaus JS. Peroxynitrite scavengers for the acute treatment of traumatic brain injury. Ann NY Acad Sci. 1999;890:462–468. doi: 10.1111/j.1749-6632.1999.tb08025.x. [DOI] [PubMed] [Google Scholar]
- Hall ED, Detloff MR, Johnson K, Kupina NC. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J Neurotrauma. 2004;21:9–20. doi: 10.1089/089771504772695904. [DOI] [PubMed] [Google Scholar]
- Hamm RJ, Temple MD, Pike BR, Ellis EF. The effect of postinjury administration of polyethylene glycol-conjugated superoxide dismutase (pegorgotein, Dismutec) or lidocaine on behavioral function following fluid-percussion brain injury in rats. J Neurotrauma. 1996;13:325–332. doi: 10.1089/neu.1996.13.325. [DOI] [PubMed] [Google Scholar]
- Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlatky R, Furuya Y, Valadka AB, Goodman JC, Robertson CS. Microdialysate nitrate/nitrite levels following severe head injury. Acta Neurochir Suppl. 2002;81:331–333. doi: 10.1007/978-3-7091-6738-0_84. [DOI] [PubMed] [Google Scholar]
- Hlatky R, Goodman JC, Valadka AB, Robertson CS. Role of nitric oxide in cerebral blood flow abnormalities after traumatic brain injury. J Cereb Blood Flow Metab. 2003;23:582–588. doi: 10.1097/01.WCB.0000059586.71206.F3. [DOI] [PubMed] [Google Scholar]
- Hortobagyi T, Gorlach C, Benyo Z, Lacza Z, Hortobagyi S, Wahl M, et al. Inhibition of neuronal nitric oxide synthase-mediated activation of poly(ADP-ribose) polymerase in traumatic brain injury: neuroprotection by 3-aminobenzamide. Neuroscience. 2003;121:983–990. doi: 10.1016/s0306-4522(03)00482-2. [DOI] [PubMed] [Google Scholar]
- Jafarian-Tehrani M, Louin G, Royo NC, Besson VC, Bohme GA, Plotkine M, et al. 1400W, a potent selective inducible NOS inhibitor, improves histopathological outcome following traumatic brain injury in rats. Nitric Oxide. 2005;12:61–69. doi: 10.1016/j.niox.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Jagtap P, Szabó C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- Jones NC, Constantin D, Gibson CL, Prior MJ, Morris PG, Marsden CA, et al. A detrimental role for nitric oxide synthase-2 in the pathology resulting from acute cerebral injury. J Neuropathol Exp Neurol. 2004;63:708–720. doi: 10.1093/jnen/63.7.708. [DOI] [PubMed] [Google Scholar]
- Kauppinen TM. Multiple roles for poly(ADP-ribose)polymerase-1 in neurological disease. Neurochem Int. 2007;50:954–958. doi: 10.1016/j.neuint.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Koh SH, Park Y, Song CW, Kim JG, Kim K, Kim J, et al. The effect of PARP inhibitor on ischemic cell death, its related inflammation and survival signals. Eur J Neurosci. 2004;20:1461–1472. doi: 10.1111/j.1460-9568.2004.03632.x. [DOI] [PubMed] [Google Scholar]
- Lacza Z, Horvath EM, Komjati K, Hortobagyi T, Szabó C, Busija DW. PARP inhibition improves the effectiveness of neural stem cell transplantation in experimental brain trauma. Int J Mol Med. 2003;12:153–159. [PubMed] [Google Scholar]
- Lai Y, Chen Y, Watkins SC, Nathaniel PD, Guo F, Kochanek PM, et al. Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J Neurochem. 2008;104:1700–1711. doi: 10.1111/j.1471-4159.2007.05114.x. [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Raghupathi R, Verma A, Pieper AA, Saatman KE, Snyder SH, et al. Temporal patterns of poly(ADP-ribose) polymerase activation in the cortex following experimental brain injury in the rat. J Neurochem. 1999;73:205–213. doi: 10.1046/j.1471-4159.1999.0730205.x. [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Zhang J, Raghupathi R, Li JH, Smith F, Bareyre FM, et al. Pharmacologic inhibition of poly(ADP-ribose) polymerase is neuroprotective following traumatic brain injury in rats. J Neurotrauma. 2001;18:369–376. doi: 10.1089/089771501750170912. [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Simon CM, Prado GR, Cullen DK. CNS injury biomechanics and experimental models. Prog Brain Res. 2007;161:13–26. doi: 10.1016/S0079-6123(06)61002-9. [DOI] [PubMed] [Google Scholar]
- de la Lastra CA, Villegas I, Sanchez-Fidalgo S. Poly(ADP-ribose)polymerase inhibitors: new pharmacological functions and potential clinical applications. Curr Pharm Des. 2007;13:933–962. doi: 10.2174/138161207780414241. [DOI] [PubMed] [Google Scholar]
- Lewen A, Skoglosa Y, Clausen F, Marklund N, Chan PH, Lindholm D, et al. Paradoxical increase in neuronal DNA fragmentation after neuroprotective free radical scavenger treatment in experimental traumatic brain injury. J Cereb Blood Flow Metab. 2001;21:344–350. doi: 10.1097/00004647-200104000-00003. [DOI] [PubMed] [Google Scholar]
- Lewis C, Low JA. Clinical poly(ADP-ribose) polymerase inhibitors for the treatment of cancer. Curr Opin Investig Drugs. 2007;8:1051–1056. [PubMed] [Google Scholar]
- Louin G, Marchand-Verrecchia C, Palmier B, Plotkine M, Jafarian-Tehrani M. Selective inhibition of inducible nitric oxide synthase reduces neurological deficit but not cerebral edema following traumatic brain injury. Neuropharmacology. 2006;50:182–190. doi: 10.1016/j.neuropharm.2005.08.020. [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Vink R, Soares H, Hayes R, Simon R. Effect of noncompetitive blockade of N-methyl-D-aspartate receptors on the neurochemical sequelae of experimental brain injury. J Neurochem. 1990;55:1170–1179. doi: 10.1111/j.1471-4159.1990.tb03122.x. [DOI] [PubMed] [Google Scholar]
- Mandir AS, Poitras MF, Berliner AR, Herring WJ, Guastella DB, Feldman A, et al. NMDA but not non-NMDA excitotoxicity is mediated by poly(ADP-ribose) polymerase. J Neurosci. 2000;20:8005–8011. doi: 10.1523/JNEUROSCI.20-21-08005.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margaill I, Plotkine M, Lerouet D. Antioxidant strategies in the treatment of stroke. Free Radic Biol Med. 2005;39:429–443. doi: 10.1016/j.freeradbiomed.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Marion DW, Darby J, Yonas H. Acute regional cerebral blood flow changes caused by severe head injuries. J Neurosurg. 1991;74:407–414. doi: 10.3171/jns.1991.74.3.0407. [DOI] [PubMed] [Google Scholar]
- Marklund N, Clausen F, McIntosh TK, Hillered L. Free radical scavenger posttreatment improves functional and morphological outcome after fluid percussion injury in the rat. J Neurotrauma. 2001;18:821–832. doi: 10.1089/089771501316919184. [DOI] [PubMed] [Google Scholar]
- Masutani M, Nozaki T, Nakamoto K, Nakagama H, Suzuki H, Kusuoka O, et al. The response of PARP knockout mice against DNA damaging agents. Mutat Res. 2000;462:159–166. doi: 10.1016/s1383-5742(00)00033-8. [DOI] [PubMed] [Google Scholar]
- Ménissier de Murcia J, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, et al. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003;22:2255–2263. doi: 10.1093/emboj/cdg206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mésenge C, Verrecchia C, Allix M, Boulu RG, Plotkine M. Reduction of the neurological deficit in mice with traumatic brain injury by nitric oxide synthase inhibitors. J Neurotrauma. 1996;13:209–214. [PubMed] [Google Scholar]
- Mésenge C, Margaill I, Verrecchia C, Allix M, Boulu RG, Plotkine M. Protective effect of melatonin in a model of traumatic brain injury in mice. J Pineal Res. 1998a;25:41–46. doi: 10.1111/j.1600-079x.1998.tb00384.x. [DOI] [PubMed] [Google Scholar]
- Mésenge C, Charriaut-Marlangue C, Verrecchia C, Allix M, Boulu RG, Plotkine M. Reduction of tyrosine nitration after N(ω)-nitro-L-arginine-methylester treatment of mice with traumatic brain injury. Eur J Pharmacol. 1998b;353:53–57. doi: 10.1016/s0014-2999(98)00432-4. [DOI] [PubMed] [Google Scholar]
- Mésenge C, Verrecchia C, Charriaut-Marlangue C, Boulu RG, Plotkine M. Peroxynitrite-PARS pathway activation following traumatic brain injury in mice. J Cereb Blood Flow Metab. 1999;19:S390. [Google Scholar]
- Mikawa S, Kinouchi H, Kamii H, Gobbel GT, Chen SF, Carlson E, et al. Attenuation of acute and chronic damage following traumatic brain injury in copper, zinc-superoxide dismutase transgenic mice. J Neurosurg. 1996;85:885–891. doi: 10.3171/jns.1996.85.5.0885. [DOI] [PubMed] [Google Scholar]
- Muralikrishna Rao A, Dogan A, Hatcher JF, Dempsey RJ. Fluorometric assay of nitrite and nitrate in brain tissue after traumatic brain injury and cerebral ischemia. Brain Res. 1998;793:265–270. doi: 10.1016/s0006-8993(98)00183-8. [DOI] [PubMed] [Google Scholar]
- de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc Natl Acad Sci USA. 1997;94:7303–7307. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishio S, Yunoki M, Noguchi Y, Kawauchi M, Asari S, Ohmoto T. Detection of lipid peroxidation and hydroxyl radicals in brain contusion of rats. Acta Neurochir Suppl (Wien) 1997;70:84–86. doi: 10.1007/978-3-7091-6837-0_26. [DOI] [PubMed] [Google Scholar]
- Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173:2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicciari R, Camaioni E, Costantino G, Formentini L, Sabbatini P, Venturoni F, et al. On the way to selective PARP-2 inhibitors. Design, synthesis, and preliminary evaluation of a series of isoquinolinone derivatives. Chem Med Chem. 2008;3:914–923. doi: 10.1002/cmdc.200800010. [DOI] [PubMed] [Google Scholar]
- Pineda JA, Aono M, Sheng H, Lynch J, Wellons JC, Laskowitz DT, et al. Extracellular superoxide dismutase overexpression improves behavioral outcome from closed head injury in the mouse. J Neurotrauma. 2001;18:625–634. doi: 10.1089/089771501750291864. [DOI] [PubMed] [Google Scholar]
- Pratico D, Reiss P, Tang LX, Sung S, Rokach J, McIntosh TK. Local and systemic increase in lipid peroxidation after moderate experimental traumatic brain injury. J Neurochem. 2002;80:894–898. doi: 10.1046/j.0022-3042.2002.00777.x. [DOI] [PubMed] [Google Scholar]
- Ratnam K, Low JA. Current development of clinical inhibitors of poly(ADP-ribose) polymerase in oncology. Clin Cancer Res. 2007;13:1383–1388. doi: 10.1158/1078-0432.CCR-06-2260. [DOI] [PubMed] [Google Scholar]
- Ray SK, Dixon CE, Banik NL. Molecular mechanisms in the pathogenesis of traumatic brain injury. Histol Histopathol. 2002;17:1137–1152. doi: 10.14670/HH-17.1137. [DOI] [PubMed] [Google Scholar]
- Reilly P. The impact of neurotrauma on society: an international perspective. Prog Brain Res. 2007;161:3–9. doi: 10.1016/S0079-6123(06)61001-7. [DOI] [PubMed] [Google Scholar]
- Rink A, Fung KM, Trojanowski JQ, Lee VM, Neugebauer E, McIntosh TK. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am J Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- Sakamoto KI, Fujisawa H, Koizumi H, Tsuchida E, Ito H, Sadamitsu D, et al. Effects of mild hypothermia on nitric oxide synthesis following contusion trauma in the rat. J Neurotrauma. 1997;14:349–353. doi: 10.1089/neu.1997.14.349. [DOI] [PubMed] [Google Scholar]
- Satchell MA, Zhang X, Kochanek PM, Dixon CE, Jenkins LW, Melick J, et al. A dual role for poly-ADP-ribosylation in spatial memory acquisition after traumatic brain injury in mice involving NAD+ depletion and ribosylation of 14-3-3gamma. J Neurochem. 2003;85:697–708. doi: 10.1046/j.1471-4159.2003.01707.x. [DOI] [PubMed] [Google Scholar]
- Schreiber V, Amé JC, Dollé P, Schultz I, Rinaldi B, Fraulob V, et al. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem. 2002;277:23028–23036. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- Schreiber V, Dantzer F, Amé JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- Shapira Y, Yadid G, Cotev S, Niska A, Shohami E. Protective effect of MK801 in experimental brain injury. J Neurotrauma. 1990;7:131–139. doi: 10.1089/neu.1990.7.131. [DOI] [PubMed] [Google Scholar]
- Sharma HS, Wiklund L, Badgaiyan RD, Mohanty S, Alm P. Intracerebral administration of neuronal nitric oxide synthase antiserum attenuates traumatic brain injury-induced blood-brain barrier permeability, brain edema formation, and sensory motor disturbances in the rat. Acta Neurochir Suppl. 2006;96:288–294. doi: 10.1007/3-211-30714-1_62. [DOI] [PubMed] [Google Scholar]
- Shohami E, Gati I, Beit-Yannai E, Trembovler V, Kohen R. Closed head injury in the rat induces whole body oxidative stress: overall reducing antioxidant profile. J Neurotrauma. 1999;16:365–376. doi: 10.1089/neu.1999.16.365. [DOI] [PubMed] [Google Scholar]
- Silberstein M, Lane D, Dodd S, Opeskin K. Identification of a by-product of nitric oxide synthase activity in human acute brain injury with in vivo proton magnetic resonance spectroscopy. AJNR Am J Neuroradiol. 2002;23:389–392. [PMC free article] [PubMed] [Google Scholar]
- Sinz EH, Kochanek PM, Dixon CE, Clark RS, Carcillo JA, Schiding JK, et al. Inducible nitric oxide synthase is an endogenous neuroprotectant after traumatic brain injury in rats and mice. J Clin Invest. 1999;104:647–656. doi: 10.1172/JCI6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan GJ, Szabó C. Poly(ADP-ribose) polymerase inhibitors. Curr Med Chem. 2003;10:321–340. doi: 10.2174/0929867033368376. [DOI] [PubMed] [Google Scholar]
- Steiner J, Rafols D, Park HK, Katar MS, Rafols JA, Petrov T. Attenuation of iNOS mRNA exacerbates hypoperfusion and upregulates endothelin-1 expression in hippocampus and cortex after brain trauma. Nitric Oxide. 2004;10:162–169. doi: 10.1016/j.niox.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Stoffel M, Rinecker M, Plesnila N, Eriskat J, Baethmann A. Attenuation of secondary lesion growth in the brain after trauma by selective inhibition of the inducible NO-synthase. Acta Neurochir Suppl. 2000;76:357–358. doi: 10.1007/978-3-7091-6346-7_74. [DOI] [PubMed] [Google Scholar]
- Stoffel M, Rinecker M, Plesnila N, Eriskat J, Baethmann A. Role of nitric oxide in the secondary expansion of a cortical brain lesion from cold injury. J Neurotrauma. 2001;18:425–434. doi: 10.1089/089771501750171010. [DOI] [PubMed] [Google Scholar]
- Szabó C. Poly(ADP-ribose) polymerase activation by reactive nitrogen species – relevance for the pathogenesis of inflammation. Nitric Oxide. 2006;14:169–179. doi: 10.1016/j.niox.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Greenberg JH, Jackson P, Maclin K, Zhang J. Neuroprotective effects of inhibiting poly(ADP-ribose) synthetase on focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1997;17:1137–1142. doi: 10.1097/00004647-199711000-00001. [DOI] [PubMed] [Google Scholar]
- Toczylowska B, Chalimoniuk M, Wodowska M, Mayzner-Zawadzk E. Changes in concentration of cerebrospinal fluid components in patients with traumatic brain injury. Brain Res. 2006;1104:183–189. doi: 10.1016/j.brainres.2006.05.057. [DOI] [PubMed] [Google Scholar]
- Uzan M, Tanriover N, Bozkus H, Gumustas K, Guzel O, Kuday C. Nitric oxide (NO) metabolism in the cerebrospinal fluid of patients with severe head injury. Inflammation as a possible cause of elevated NO metabolites. Surg Neurol. 2001;56:350–356. doi: 10.1016/s0090-3019(01)00633-4. [DOI] [PubMed] [Google Scholar]
- Wada K, Chatzipanteli K, Busto R, Dietrich WD. Role of nitric oxide in traumatic brain injury in the rat. J Neurosurg. 1998a;89:807–818. doi: 10.3171/jns.1998.89.5.0807. [DOI] [PubMed] [Google Scholar]
- Wada K, Chatzipanteli K, Kraydieh S, Busto R, Dietrich WD. Inducible nitric oxide synthase expression after traumatic brain injury and neuroprotection with aminoguanidine treatment in rats. Neurosurgery. 1998b;43:1427–1436. doi: 10.1097/00006123-199812000-00096. [DOI] [PubMed] [Google Scholar]
- Wada K, Chatzipanteli K, Busto R, Dietrich WD. Effects of L-NAME and 7-NI on NOS catalytic activity and behavioral outcome after traumatic brain injury in the rat. J Neurotrauma. 1999;16:203–212. doi: 10.1089/neu.1999.16.203. [DOI] [PubMed] [Google Scholar]
- Wallis RA, Panizzon KL, Henry D, Wasterlain CG. Neuroprotection against nitric oxide injury with inhibitors of ADP-ribosylation. Neuroreport. 1993;5:245–248. [PubMed] [Google Scholar]
- Wallis RA, Panizzon KL, Girard JM. Traumatic neuroprotection with inhibitors of nitric oxide and ADP-ribosylation. Brain Res. 1996;710:169–177. doi: 10.1016/0006-8993(95)01278-8. [DOI] [PubMed] [Google Scholar]
- Wang ZQ, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, et al. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995;9:509–520. doi: 10.1101/gad.9.5.509. [DOI] [PubMed] [Google Scholar]
- Whalen MJ, Clark RS, Dixon CE, Robichaud P, Marion DW, Vagni V, et al. Reduction of cognitive and motor deficits after traumatic brain injury in mice deficient in poly(ADP-ribose) polymerase. J Cereb Blood Flow Metab. 1999;19:835–842. doi: 10.1097/00004647-199908000-00002. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Kumura E, Iwatsuki K, Yoshimine T, Masana Y, Hayakawa T, et al. Increase in plasma nitric oxide end products following rat cortical injury. Neurosci Lett. 1995;194:124–126. doi: 10.1016/0304-3940(95)11715-9. [DOI] [PubMed] [Google Scholar]
- Yi JH, Hazell AS. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem Int. 2006;48:394–403. doi: 10.1016/j.neuint.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, Dawson TM, et al. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci USA. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhang X, Zhang T, Chen L. Excitatory amino acids in cerebrospinal fluid of patients with acute head injuries. Clin Chem. 2001;47:1458–1462. [PubMed] [Google Scholar]
- Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chen J, Graham SH, Du L, Kochanek PM, Draviam R, et al. Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem. 2002;82:181–191. doi: 10.1046/j.1471-4159.2002.00975.x. [DOI] [PubMed] [Google Scholar]