

Abstract
Background and purpose:
Peroxisome proliferator-activated receptors (PPARs) are attractive targets for the treatment of type 2 diabetes and the metabolic syndrome. P633H (2-[4-(2-Fluoro-benzenesulphonyl)-piperazin-1-yl]-3-{4-[2-(5-methyl-2-phenyl-oxazol-4-yl)-ethoxy]-phenyl}-propionic acid), a novel PPARα/γ dual agonist, was investigated for its very different effects on insulin resistance and dyslipidemia in db/db and KK-Ay mice.
Experimental approach:
The action of P633H at PPARα/γ was characterized by using transactivation assays. Functional activation of PPARα/γin vitro was confirmed by pre-adipocyte differentiation and regulation of target gene expression. Anti-diabetic studies were performed in two different diabetic mice models in vivo.
Key results:
P633H activated both PPARα and PPAR γ, (with EC50 values of 0.012 µmol and 0.032 µmol respectively). Additionally, P633H promoted pre-adipocyte differentiation, up-regulated expression of adipose specific transport protein (aP2) mRNA (3T3-Ll cells) and acyl-CoA oxidase mRNA (LO2 cells). In db/db mice, P633H reduced serum glucose, insulin, triglycerides, non-esterified fatty acids and liver triglycerides. It also improved glucose intolerance without affecting food intake and body weight after 15 days of treatment. However in KK-Ay mice, hyperglycaemia, dyslipidemia and impaired glucose tolerance were not relieved even after a 25 day treatment with P633H. Further studies with real-time PCR and electron microscopy revealed that P633H promoted progression of diabetes in KK-Ay mice by increasing hepatic gluconeogenesis and exacerbating pancreatic pathology.
Conclusion and implications:
Although P633H was a high-potency PPARα/γ dual agonist, with good functional activity in vitro, it produced opposing anti-diabetic effects in db/db and KK-Ay mice.
Keywords: PPAR, type 2 diabetes mellitus, insulin resistance, pancreas
Introduction
Type 2 diabetes has become an epidemic and serious worldwide public health issue, characterized by insulin resistance, hyperglycaemia and often accompanied with dyslipidemia and obesity. Recently these diabetes-related symptoms were redefined as the metabolic syndrome (Tenenbaum et al., 2003), commonly present at earlier stages in type 2 diabetes. These conditions have affected the lives of more than 200 million people and constitute an enormous economic burden on society. Thus, effective treatment of type 2 diabetes and the closely associated metabolic syndrome would clearly be of great benefit to the individual and society.
Peroxisome proliferator-activated receptors (PPARs) modulate the expression of a series of target genes that play critical roles in regulating glucose, lipid and cholesterol metabolism (Ferre, 2004). This has made PPARs as attractive therapeutic targets for type 2 diabetes and metabolic syndrome (Desvergne et al., 2004). The currently used PPARα agonists (fibrates) are effective hypolipidemic agents, which decrease the progression of atherosclerosis while improving glucose intolerance in type 2 diabetic patients (Guerre-Millo et al., 2000; van Raalte et al., 2004). Activation of PPARγ by thiazolidinediones improved insulin sensitivity and decreased hyperglycaemia in diabetic patients, yet with limited beneficial lipid-lowering effects (Hung et al., 2005). Therefore, research in the last few years has been focusing on the activation of both PPARα and PPARγ, which might provide a broad spectrum of beneficial metabolic effects. As a result of this strategy, a number of structurally diverse PPARα/γ dual agonists have been developed, and some have progressed into phase III clinical trials (Murakami et al., 1998; Ljung et al., 2002; Flordellis et al., 2005). Even though the development of some compounds has been discontinued, due to a variety of safety issues, the pursuit of efficacious and safer medications through regulation of PPAR receptors has not lost its momentum and is ongoing (Ramachandran et al., 2006).
Currently used thiazolidinediones are insulin sensitizers and contain an asymmetric centre, of which only the (S)-enantiomer binds to the target receptor with high affinity. These compounds have been developed as racemates, as they rapidly undergo racemization in vivo (Henke et al., 1998). To overcome this problem, many novel compounds that are less prone to racemization have been developed through structural redesign. GW409544 and farglitazar, showing agonist activity towards PPARγ or/and PPARα, are two representatives of a series of L-tyrosine derivatives, and the chiral compounds with the S-configuration were conveniently synthesized from naturally occurring L-tyrosine. X-ray crystal structures of farglitazar and GW409544 bound to PPARγ demonstrated that the α-amino of tyrosine and the carbonyl, from benzophenone (farglitazar) or from the vinyl amide (GW409544), formed an intramolecular hydrogen bond, and the structural complex was then inserted into a lipophilic pocket (Gampe et al., 2000). On the basis of this six-membered ring structure, we designed a series of compounds containing the piperazine ring and ultimately discovered the novel structural lead compound P633H (2-[4-(2-Fluoro-benzenesulphonyl)-piperazin-1-yl]-3-{4-[2-(5-methyl-2-phenyl-oxazol-4-yl)-ethoxy]-phenyl}-propionic acid), a phenylpropionic acid derivative (Figure 1; Li et al., 2007). Preliminary studies in mice showed that P633H was well tolerated in mice, with favourable pharmacokinetics and excellent oral bioavailability (∼90%). Therefore, P633H displayed good drug-like characteristics.
Figure 1.
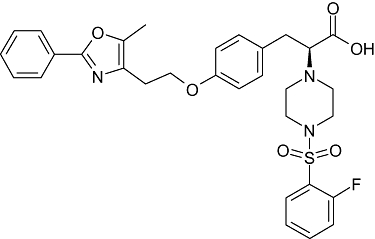
The structure of the dual peroxisome proliferator-activated receptors α and γ agonist P633H (MW, 593), 2-[4-(2-Fluoro-benzenesulphonyl)-piperazin-1-yl]-3-{4-[2-(5-methyl-2-phenyl-oxazol-4-yl)-ethoxy]-phenyl}-propionic acid.
From the experiments presented here, P633H was a potent PPARα/γ dual agonist in cell-based assays. However, unlike the well-known PPARα/γ dual agonists, such as GW409544 or ragaglitazar (Pickavance et al., 2005), P633H showed distinctly different pharmacological effects in two strains of diabetic mice, db/db and KK-Ay mice. Here we have investigated the mechanisms underlying this discrepancy.
Methods
In vitro transactivation assay
As previously described (Xu et al., 2006), cDNAs for human PPAR were obtained by RT-PCR from human liver or adipose tissues. Amplified cDNAs of the putative ligand-binding domain encoding amino acids 204–505 of human PPARγ, 154–421 of human PPARδ or 139–440 of human PPARα were cloned into the pM mammalian expression vector containing elements of GAL4 DNA-binding domain. GAL4-responsive reporter plasmid (pUAS (5x)-tk-luc) containing five copies of the GAL4 response element was placed adjacent to thymidine kinase (tk) minimal promoter and luciferase reporter gene. HEK-293 cells were co-transfected with GAL4-pM-hPPARα/pM-hPPARγ/pM-hPPARδ, pUAS (5x)-tk-luc receptor vector, and pRL-CMV-Rluc as an internal control for transactivation efficiency using the lipofectamine 2000 system (Invitrogen, Carlsbad, CA, USA). Then, after treatment with each compound for an additional 24 h, luciferase activity in cell extracts was analysed with an ML luminometer (Promega, Madison, WI, USA). All results were obtained from at least three independent experiments performed in triplicate.
Assessment of pre-adipocyte differentiation
3T3-L1 pre-adipocytes were seeded in 24-well culture plates and cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics at 37°C in 5% CO2. Two days post confluence, cells were treated with the tested compounds or vehicle [0.05% dimethyl sulphoxide (DMSO)] in the presence of 10 µg·mL−1 insulin for 3 days. Then the cells were further cultured for 6 days with renewed medium every other day. The effect on pre-adipocyte differentiation was represented as adipogenesis. Cells were washed, fixed and stained with Oil Red O (0.1 mg·mL−1). Medium was then removed, and 100 µL isopropyl alcohol was added to dissolve the precipitation. The absorbance (OD) at 490 nm was quantitated by microplate reader (Bio-Red 550) (Ramirez-Zacarias et al., 1992; Xu et al., 2006).
In vitro gene expression studies
To test the functional activation of PPARα/γ by P633H, expression of respective direct target genes, acyl-CoA oxidase (ACO) mRNA in LO2 hepatocytes and the adipose specific transport protein (aP2) mRNA in 3T3-L1 pre-adipocytes, was measured. Human normal hepatic LO2 cells (provided by Professor Xiao-ming Yang in the Institute of Radioactive Medicine, Beijing) cultured in RPMI 1640 medium supplemented with 10% (v/v) FBS and were treated with 10 µmol·L−1 of P633H for 24 h. 3T3-L1 pre-adipocytes (obtained from Institute of Geriatrics, Beijing Hospital) cultured in DMEM medium with 10% (v/v) FBS and were treated with 10 µmol·L−1 of P633H for 72 h in the presence of 10 ng·mL−1 insulin. Cells were grown at 37°C in 5% CO2 in a humidified chamber and re-fed fresh media every 2 days. P633H were dissolved in DMSO and diluted with culture media. The final media concentration of DMSO was 0.1% in the dilutions. Cells treated with 0.1% DMSO in media served as vehicle controls. Then total RNA were isolated from these cells and analysed by semi-quantitative PCR by using primers specific for the indicated genes. Data were normalized to the expression of endogenous control gene (β-actin) and presented as fold of vehicle control.
Animals and treatment
All animal care and experimental procedures were strictly in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.
db/db mouse study
Male homozygous db/db mice (8 weeks old) were from Model Animal Research Center of Nanjing University, China. Animals were maintained on a 12 h day/night schedule with ad libitum access to standard mouse diet and water. The mice were housed four per cage and acclimatized for 2 weeks before the mice were randomized into five groups (n= 8 each) according to their initial body weight and levels of fasting blood glucose (FBG). Mice were treated by gavage once daily with rosiglitazone (10 mg·kg−1·day−1), P633H (1, 2.5 and 10 mg·kg−1·day−1 respectively), or vehicle (control). Compounds were dissolved in DMSO and suspended in 0.5% carboxymethylcellulose. Blood samples from mice fasted overnight were taken from the retro-orbital sinus after 15 days of drug treatment.
KK-Ay mouse study
Normal female C57BL/J mice (8 weeks old) and insulin-resistant female KK-Ay mice (8 weeks old) were purchased from the Experimental Animal Center, Chinese Academy of Medical Sciences. The KK-Ay mice were provided with a high-fat diet and water ad libitum. At the beginning of the study, KK-Ay mice were weighed, bled via the tail vein in the fasted state (4 h) and sorted into six groups (n= 6–7 each) based on their blood glucose levels and initial body weight. Rosiglitazone (10 mg·kg−1·day−1), P633H (1, 2.5, 5 and 10 mg·kg−1·day−1 respectively), or vehicle control were administered by gavage once a day for 25 days. Individual body weight and cage food consumption were measured every 2 days. At the end of the experimental period, blood samples were collected for immediate assessment of serum biochemical parameters.
Metabolic studies
Serum glucose, total cholesterol (TCHO), triglycerides (TG), non-esterified fatty acids (NEFA) and insulin levels were measured as previously described (Chen et al., 2008).The insulin sensitivity index (ISI) was calculated from the values of FBG and fasting blood insulin (FBI). ISI=1/(FBG*FBI)1000, where FBG is expressed as mg·dL−1 and FBI as mU·L−1.
Oral glucose tolerance test (OGTT)
OGTT was performed by giving a glucose bolus (2 g·kg−1) by gavage after overnight fasting. Glycaemia was measured through the tail tip before and after glucose load at the times indicated (Chen et al., 2008). The area under the curves (AUC) generated from the data collected during the OGTT was calculated.
Histopathological examination and electron microsocpy
Samples of liver, abdominal white adipose tissue (WAT), interscapular brown adipose tissue (BAT) and portions of splenic pancreata were resected and fixed with 10% formaldehyde phosphate buffered saline (pH 7.4), then embedded in paraffin, sectioned, stained with haematoxylin/eosin and analysed by either microscopy or morphometry. The ultrastructure of pancreas of KK-Ay mice was assessed by electron microscopy (Philips Em 400T).
Semi-quantitative and quantitative real-time PCR
Total RNA was isolated with the Trizol RNA preparation kit following the manufacturer's recommended procedures (GIBCO/BRL, Grand Island, NY, USA) and converted to cDNA with oligo dT primers by using a cDNA synthesis kit (GIBCO/BRL, Grand Island, NY, USA). The semi-quantitative RT-PCR analysis of gene expression subsequently amplified by PCR using the RNA PCR Kit Version 1.1(TaKaRa Biotechnology Co. Ltd. Dalian, China) in a thermocycler (Mastercycler, Eppendorff Germany). Real-time PCR with the ABI PRISM 7300 sequence detection system (Applied Biosystems) was performed by using the ABI Power SYBR green PCR Master Mix (catalog no.4367659; UK) as recommended by the manufacturer. All reactions were carried out in triplicate. The relative amount of all mRNAs was calculated by using the comparative CT method. Expression results of specific genes are always presented as relative to the expression of the control gene (β-actin).
Statistical analysis
All results are expressed as mean ± SD. For multiple comparisons, the statistical analysis was performed by using one-way anova followed by the Tukey's multiple comparison tests with spss. P < 0.05 was considered to be statistically significant.
Materials
Fenofibrate was procured from Sigma (St. Louis, MO, USA). P633H, rosiglitazone, GW501516 and GW409544 were synthesized by the New Drug Design Center of our Institute. The characteristics of the compounds synthesized matched the published data. The structure of P633H is shown in Figure 1. Melting points were determined on a capillary melting point apparatus without correction. 1H nuclear magnetic resonance (NMR) spectra were recorded by using a Varian Unity INOVA 400 MHz spectrometer. Mass spectrometry was carried out by using Zabspect mass spectrometer (Li et al., 2007).
Results
Cell-based reporter gene assays
By in silico screening of a virtual library and cell-based transcription assays using GAL4-hPPAR-LBD, a lead compound P633H was identified as a potent and selective PPARα/γ co-agonist. The maximal efficacies of GAL4-hPPARα-LBD induction by 10 µmol·L−1 rosiglitazone and 100 µmol·L−1 fenofibrate were only 3.5-fold and 8.5-fold respectively. However, the induction was 24.5-fold after treatment with 10 µmol·L−1 of P633H. P633H activated GAL4-hPPARγ-LBD in a dose-dependent manner, which produced 12.6-fold maximal luciferase activation. Treatment with the thiazolidinedione, rosiglitazone, resulted in only 8.3-fold activation under the same conditions. The EC50 of P633H for activation of hPPARγ was comparable to that of rosiglitazone (0.035 ± 0.003 µmol·L−1 and 0.032 ± 0.006 µmol·L−1 for P633H and rosiglitazone respectively). For hPPARα, P633H was nearly 1000 times more potent than fenofibrate and rosiglitazone (EC50, 0.012 ± 0.002 µmol·L−1, 14.5 ± 1.17 µmol·L−1 and 10.58 ± 1.36 µmol·L−1 for P633H, fenofibrate and rosiglitazone respectively). No significant PPARδ activity was observed at >30 µmol·L−1, while the EC50 of the specific PPARδ agonist GW501516 was found to be 0.015 ± 0.006 nmol·L−1.
Differentiation of 3T3-L1 pre-adipocytes
Stimulation of adipocyte differentiation is considered to be closely related to the insulin-sensitizing effect, and PPARγ agonists are well-known dominant regulators of adipocyte development and pre-adipocyte differentiation (Yang et al., 2007). As P633H activated PPARγ in the reporter assay, we then tested whether P633H was able to induce adipogenesis in vitro. As shown in Figure 2, P633H significantly promoted the differentiation of 3T3-L1 pre-adipocytes and intracellular adipogenesis (P < 0.01). At 10 µmol·L−1, P633H was about as active as 1 µmol·L−1 rosiglitazone.
Figure 2.
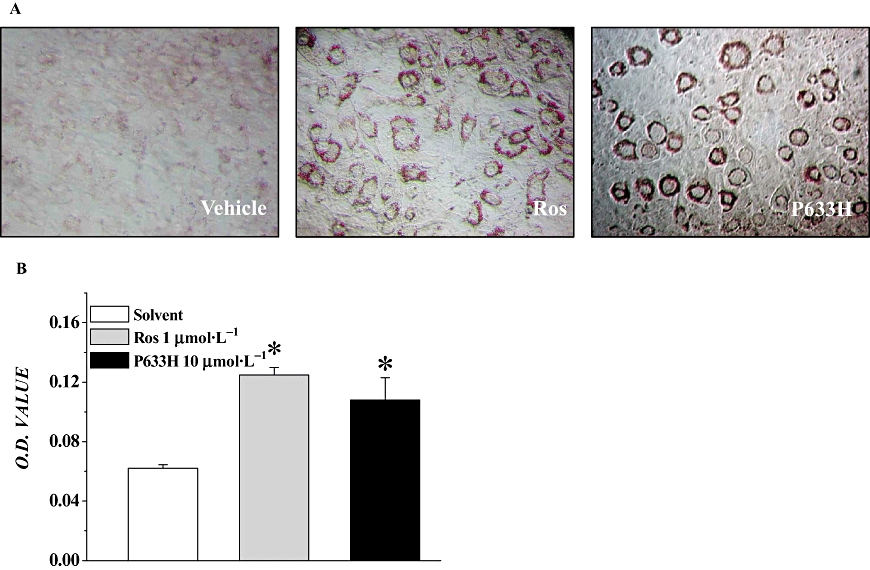
P633H, 2-[4-(2-Fluoro-benzenesulphonyl)-piperazin-1-yl]-3-{4-[2-(5-methyl-2-phenyl-oxazol-4-yl)-ethoxy]-phenyl}-propionic acid (10 µmol·L−1) promoted insulin-induced 3T3-L1 pre-adipocyte differentiation. (A) Images after staining with Oil Red O. (B) The OD490 of extracts of stained cells is shown as mean ± SD. *P < 0.01 versus vehicle control group. n= 4.
P633H up-regulated the expression of direct target genes of PPARα/γin vitro
Fatty ACO, the first and rate-limiting enzyme of the peroxisomal β-oxidation system and adipose specific transport protein (aP2) are direct target genes of PPARα/γ activation in liver and adipose tissue respectively. To test functional activation of the two PPAR subtypes by P633H, the expression of ACO mRNA in LO2 hepatocytes and aP2 mRNA in 3T3-L1 pre-adipocytes was investigated. P633H markedly increased the mRNA levels of ACO and aP2 by 3.1-fold (P < 0.01) and 2.3-fold (P < 0.05) as compared with vehicle respectively (data not shown).
Effects of P633H on metabolic parameters
All doses of P633H were without effect on food intake in both db/db and KK-Ay mice (data not shown) and body weight in db/db mice (Figure 3A). However, in KK-Ay mice, from the fourth day onwards, the group with 2.5 mg·kg−1 P633H showed a reduction in weight (Figure 3B). Even though rosiglitazone decreased food intake in KK-Ay mice (4.96 ± 0.43 and 3.67 ± 0.45 g·mice−1·day−1 for control and rosiglitazone respectively, P < 0.05), the body weight gain in this group was much higher than that of the control (Figure 3B). Rosiglitazone also significantly increased the body weight gain of db/db mice (Figure 3A) (Yajima et al., 2003).
Figure 3.
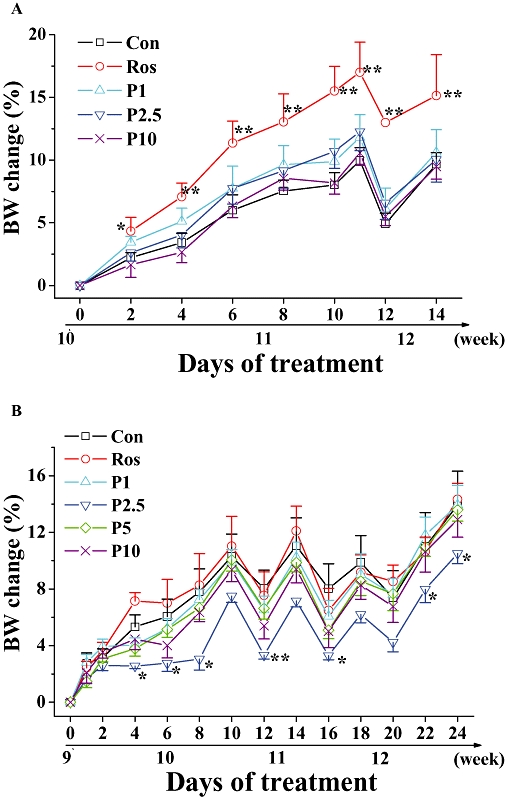
Effect of P633H on body weight in db/db and KK-Ay mice. Net body weight change in (A) db/db and (B) KK-Ay mice, calculated for individual mice and then averaged. Note that db/db mice were treated for 15 days, whereas the KK-Ay mice were given 25 days treatment. Values are mean ± SD (n= 8 for db/db mice and n= 6–7 for KK-Ay mice); *P < 0.05, **P < 0.01 versus control group. BW, body weight; Con, control; P633H, 2-[4-(2-Fluoro-benzenesulphonyl)-piperazin-1-yl]-3-{4-[2-(5-methyl-2-phenyl-oxazol-4-yl)-ethoxy]-phenyl}-propionic acid; P1, 1 mg·kg−1 P633H; P2.5, 2.5 mg·kg−1 P633H; P5, 5 mg·kg−1 P633H; P10, 10 mg·kg−1 P633H; Ros, rosiglitazone.
P633H did not affect liver weights in either mouse models and the adipose index in KK-Ay mice (Table 1). However, at doses of 2.5 and 10 mg·kg−1, P633H reduced the intra-abdominal adipose tissue in db/db mice (P < 0.05) (Table 1). On the other hand, rosiglitazone marked increased BAT weight in both mouse models and liver weight in the KK-Ay mice.
Table 1.
Phenotypic analysis in db/db and KK-Ay mice at the end of the treatment
|
db/db mice |
KK-Ay mice |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Control | Rosiglitazone | 1 mg·kg−1 | 2.5 mg·kg−1 | 10 mg·kg−1 | Normal | Control | Rosiglitazone | 1 mg·kg−1 | 2.5 mg·kg−1 | 5 mg·kg−1 | 10 mg·kg−1 | |
| TG (mmol·L−1) | 1.03 ± 0.29 | 0.59 ± 0.14** | 1.04 ± 0.08 | 0.60 ± 0.23** | 0.78 ± 0.09* | 0.97 ± 0.14** | 2.03 ± 0.43 | 0.90 ± 0.21** | 1.74 ± 0.22 | 1.31 ± 0.20* | 2.10 ± 0.64 | 2.16 ± 0.78 |
| TCHO (mmol·L−1) | 5.56 ± 0.60 | 4.45 ± 0.89 | 5.37 ± 0.43 | 4.98 ± 0.22* | 4.89 ± 0.36* | 2.28 ± 0.59** | 5.93 ± 0.80 | 5.60 ± 0.71 | 5.35 ± 0.50 | 5.17 ± 0.50 | 6.17 ± 1.20 | 5.64 ± 0.67 |
| HDLc (mmol·L−1) | 1.99 ± 0.13 | 1.46 ± 0.45 | 1.90 ± 0.20 | 2.05 ± 0.12 | 1.88 ± 0.19 | 0.77 ± 0.16** | 1.96 ± 0.13 | 1.92 ± 0.16 | 1.83 ± 0.12 | 1.80 ± 0.31 | 2.19 ± 0.45 | 1.92 ± 0.09 |
| TCHO/HDLc | 2.73 ± 0.11 | 2.90 ± 0.21 | 2.82 ± 0.10 | 2.50 ± 0.17* | 2.66 ± 0.16 | 3.23 ± 0.50 | 3.05 ± 0.50 | 2.91 ± 0.22 | 2.93 ± 0.13 | 2.93 ± 0.45 | 2.84 ± 0.20 | 2.93 ± 0.24 |
| NEFA (mmol·L−1) | 2.17 ± 0.23 | 1.53 ± 0.15** | 2.21 ± 0.30 | 1.75 ± 0.37* | 1.87 ± 0.19* | 2.35 ± 0.31** | 4.28 ± 0.45 | 2.70 ± 0.61** | 2.87 ± 0.89** | 3.39 ± 0.37* | 4.03 ± 1.06 | 4.01 ± 1.12 |
| Liver/BW (%) | 6.64 ± 0.68 | 7.25 ± 0.61 | 7.00 ± 0.44 | 6.59 ± 0.23 | 6.62 ± 0.60 | 4.18 ± 0.57** | 6.12 ± 0.20 | 7.51 ± 1.19* | 5.64 ± 0.61 | 5.11 ± 1.25 | 5.59 ± 0.60 | 6.32 ± 1.24 |
| BAT/BW (%) | 2.57 ± 0.33 | 4.36 ± 0.89** | 2.29 ± 0.17 | 2.64 ± 0.23 | 2.51 ± 0.36 | 0.72 ± 0.11** | 2.13 ± 0.34 | 2.86 ± 0.16** | 2.27 ± 0.32 | 2.16 ± 0.19 | 2.20 ± 0.15 | 2.11 ± 0.20 |
| WAT/BW (%) | 12.84 ± 0.94 | 11.97 ± 0.63 | 11.91 ± 0.50 | 11.67 ± 0.81* | 11.53 ± 0.85* | 2.43 ± 0.64** | 13.41 ± 1.60 | 14.23 ± 1.22 | 13.56 ± 1.22 | 13.24 ± 1.36 | 13.62 ± 1.01 | 12.91 ± 2.01 |
Compounds (rosiglitazone, 10 mg·kg−1 and P633H, 1, 2.5 or 10 mg·kg−1) were given once daily to db/db and KK-Ay mice by gavage for 15 and 25 days respectively as described in the Methods. The ratios Liver/BW, BAT/BW and WAT/BW are shown as %BW [(g liver, WAT, BAT/g body) ×100].
Values are mean ± SD (n= 8 for db/db mice and n= 6–7 for KK-Ay mice);
P < 0.05,
P < 0.01 versus control group.
BAT, brown adipose tissue; BW, body weight; HDLc, high-density lipoprotein cholesterol; NEFA, non-esterified fatty acids; TCHO, total cholesterol; TG, triglyceride; WAT, white adipose tissue.
Effect of P633H on blood glucose and OGTT
In db/db mice, sequential monitoring of blood glucose showed that rosiglitazone and P633H (2.5 and 10 mg·kg−1 groups) decreased the fasted blood glucose (Figure 4A), serum insulin levels (Figure 4C) and dose-dependently increased the ISI (Figure 4E). However, rosiglitazone and the higher dose of P633H significantly decreased the blood glucose at each test point after glucose load and the AUC for the glucose response during the OGTT (Figure 5A and B).
Figure 4.
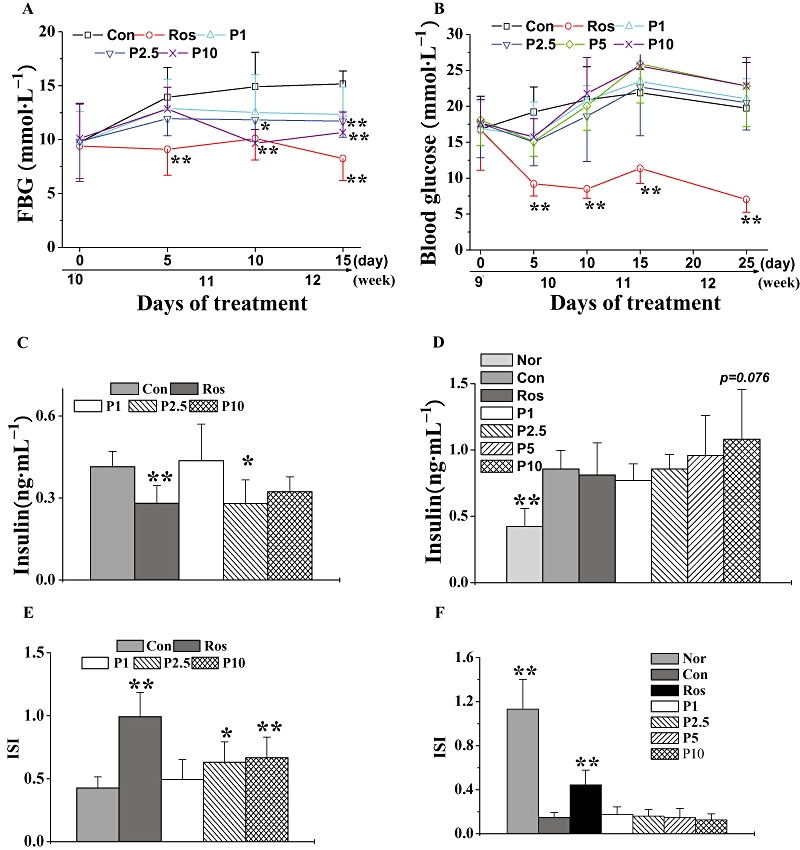
Effect of P633H on blood glucose, serum insulin levels and ISI. (A) Sequential monitoring of blood glucose in db/db mice after overnight fasting and (B) in KK-Ay mice after 4 h fasting. Blood glucose was tested at the same time point on the indicated day. Serum insulin in db/db mice (C) and (D) KK-Ay mice. (E) ISI in db/db mice and (F) KK-Ay mice. Values are mean ± SD (n= 8 for db/db mice and n= 6–7 for KK-Ay mice); *P < 0.05, **P < 0.01 versus control group. Con, control; FBG, fasting blood glucose; ISI, insulin sensitivity index; Nor, normal; P633H, 2-[4-(2-Fluoro-benzenesulphonyl)-piperazin-1-yl]-3-{4-[2-(5-methyl-2-phenyl-oxazol-4-yl)-ethoxy]-phenyl}-propionic acid; P1, 1 mg·kg−1 P633H; P2.5, 2.5 mg·kg−1 P633H; P5, 5 mg·kg−1 P633H; P10, 10 mg·kg−1 P633H; Ros, rosiglitazone.
Figure 5.
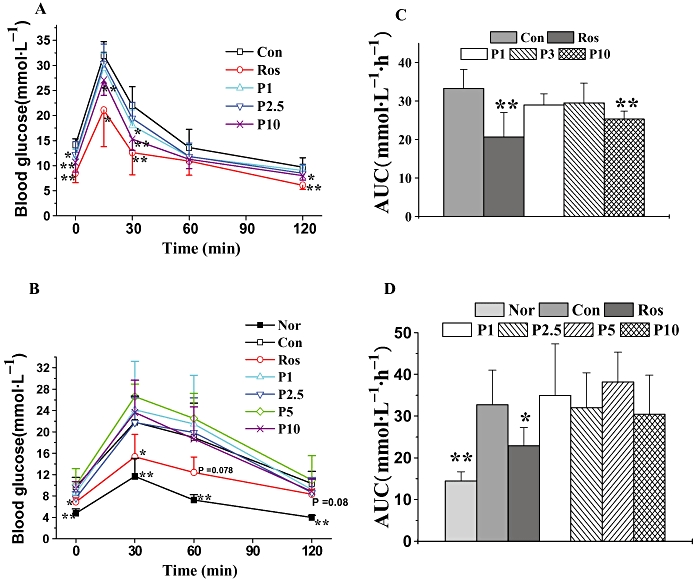
Effects of P633H on glucose tolerance in db/db and KK-Ay mice. Mice were fasted overnight and then subjected to OGTT. (A) OGTT and (B) AUC of the OGTT on the twelfth day of treatment in db/db mice. (C) OGTT and (D) AUC of the OGTT on the twentieth day of treatment in KK-Ay mice. Values are mean ± SD (n= 8 for db/db mice and n= 6–7 for KK-Ay mice); *P < 0.05, **P < 0.01 versus control group. Con, control; Nor, normal; OGTT, oral glucose tolerance test; P633H, 2-[4-(2-Fluoro-benzenesulphonyl)-piperazin-1-yl]-3-{4-[2-(5-methyl-2-phenyl-oxazol-4-yl)-ethoxy]-phenyl}-propionic acid; P1, 1 mg·kg−1 P633H; P2.5, 2.5 mg·kg−1 P633H; P3, 3 mg·kg−1 P633H; P5, 5 mg·kg−1 P633H; P10, 10 mg·kg−1 P633H; Ros, rosiglitazone.
The KK-Ay mice displayed apparent hyperinsulinemia (Figure 4D), and the blood glucose levels increased with the progress of disease (Figure 4B). Rosiglitazone significantly reduced blood glucose from the fifth day onwards and lasting until the end of the experiment, but it did not affect the fasting serum insulin (Figure 4D). The most unexpected finding was that these mice were refractory to P633H. Moreover, at the last two test points, the blood glucose in most of the treated groups exhibited a non-significant trend towards levels higher than those in the untreated animals (Figure 4B). P633H, at any dose, did not increase serum insulin levels relative to that in untreated KK-Ay mice. There was a similar lack of response of ISI in KK-Ay mice to P633H treatment, although ISI was raised by rosiglitazone (Figure 4D and F). As shown in Figure 5, rosiglitazone effectively improved glucose intolerance after 20 days of treatment, while P633H had no beneficial effects, even at the highest dose (10 mg·kg−1).
Effect of P633H on serum TG, TCHO and NEFA values
After treatment for 15 days, serum TG (P < 0.01 for 2.5 mg·kg−1 group and P < 0.05 for 10 mg·kg−1 group), TCHO (P < 0.05 for 2.5 and 10 mg·kg−1 group), TCHO/HDLc (high-density lipoprotein cholesterol) (P < 0.05 for 2.5 mg·kg−1 group) and NEFA (P < 0.05 for 2.5 and 10 mg·kg−1 group) were all decreased in db/db mice treated with P633H, compared with the values in the control, untreated db/db mice (Table 1). However, rosiglitazone only reduced the serum TG and NEFA. Furthermore, TG content in the liver was lowered by treatments (Figure 6A)
Figure 6.
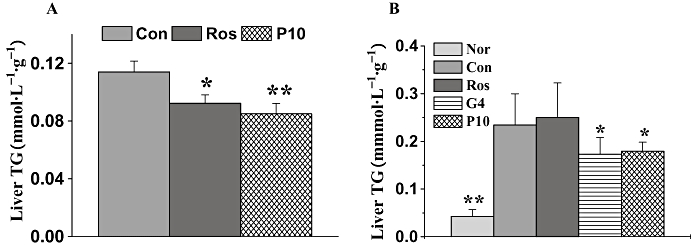
Effects of P633H on liver TG in db/db mice (A) and (B) KK-Ay mice. Values are mean ± SD (n= 8 for db/db mice and n= 6–7 for KK-Ay mice); *P < 0.05, **P < 0.01 versus control group. Con, control; G4, 10 mg·kg−1 GW409544; Nor, normal; P633H, 2-[4-(2-Fluoro-benzenesulphonyl)-piperazin-1-yl]-3-{4-[2-(5-methyl-2-phenyl-oxazol-4-yl)-ethoxy]-phenyl}-propionic acid; P10, 10 mg·kg−1 P633H; Ros, rosiglitazone; TG, triglycerides.
KK-Ay mice had significantly elevated levels of serum TG, TCHO and NEFA compared with that of normal mice (Table 1). Rosiglitazone and 2.5 mg·kg−1 P633H significantly decreased the serum TG (P < 0.01 and P < 0.05 respectively) and NEFA levels (P < 0.01 and P < 0.05 respectively). In this strain of diabetic mice, only GW409544 and P633H and not rosiglitazone lowered liver TG (Figure 6B).
Gene expression analysis in mice
To evaluate the molecular mechanism of P633H, insulin-sensitive target organs (i.e. the liver, skeletal muscle) were chosen to detect the levels of representative mRNA-encoding genes involved in glucose utilization and fatty acid oxidation. Rosiglitazone and P633H both up-regulated the expression of mRNA for the glucose transporter 4 (GLUT4) in skeletal muscle and down-regulated phosphoenolpyruvate carboxykinase (PEPCK) mRNA expression in liver of db/db mice (Figure 7A). However, only P633H markedly induce the expression of carnitine palmitoyl transferase Ia (CPTIa) and ACO mRNA in liver, which were consistent with the results from LO2 hepatocytes. It is possible that P633H decreased plasma and liver lipid content in db/db mice at least partially by activating PPARα.
Figure 7.
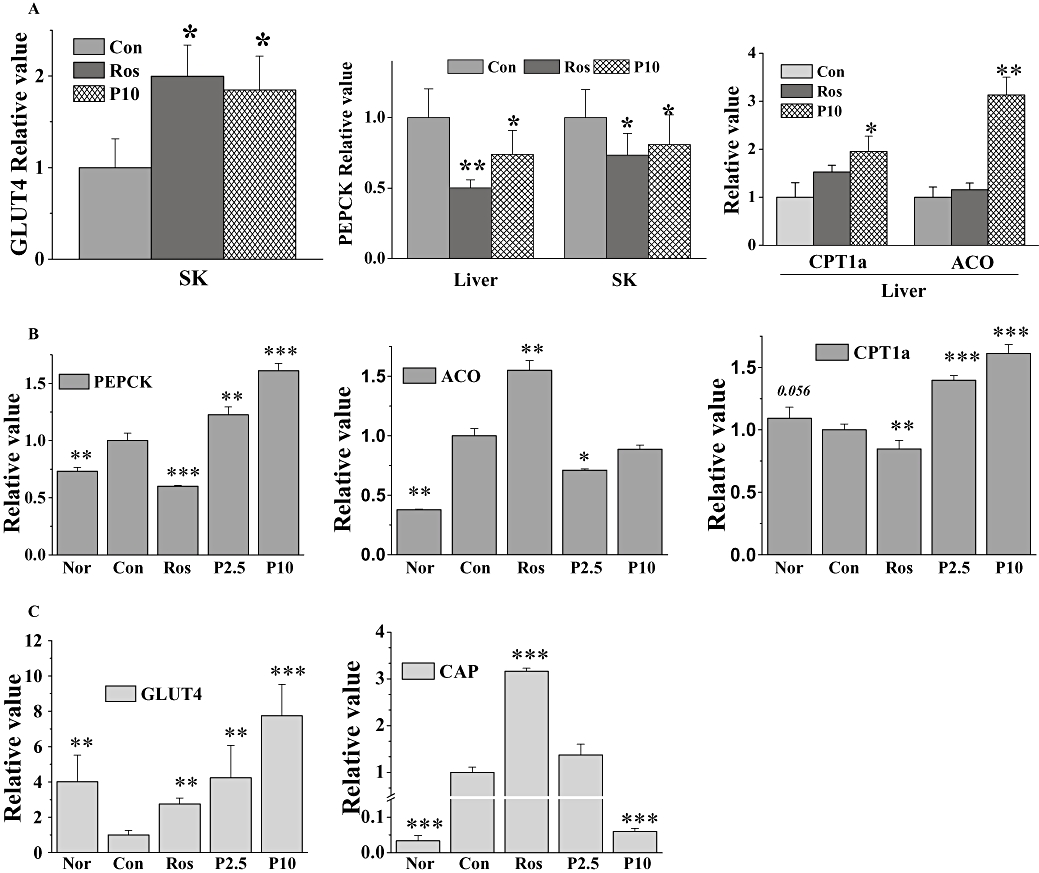
Effect of P633H on expression of representative target genes in db/db and KK-Ay mice. Total RNA was isolated from liver and skeletal muscle (SK) and then subjected to PCR to detect expression of the indicated genes. (A) Relative quantitation of gene expression in db/db mice. (B) Relative quantitation of gene expression in the liver and (C) skeletal muscle of KK-Ay mice. Results are normalized to β-actin and expressed as mean fold increase of mRNA ±SD compared with control group. *P < 0.05, **P < 0.01, ***P < 0.001 versus control group. ACO, acyl-CoA oxidase; CAP, c-cbl-associated protein; Con, control; CPTI, carnitine palmitoyl transferase I; GLUT4, glucose transporter 4; Nor, normal; P633H, 2-[4-(2-Fluoro-benzenesulphonyl)-piperazin-1-yl]-3-{4-[2-(5-methyl-2-phenyl-oxazol-4-yl)-ethoxy]-phenyl}-propionic acid; P2.5, 2.5 mg·kg−1 P633H; P10, 10 mg·kg−1 P633H; PEPCK, phosphoenolpyruvate carboxykinase; Ros, rosiglitazone.
As revealed by real-time PCR, P633H significantly and dose-dependently up-regulated the expression of PEPCK and CPTIa mRNA in the liver of KK-Ay mice (Figure 7B). Meanwhile ACO mRNA expression was suppressed, but was still greater than that in non-diabetic mice. Expression of mRNA for GLUT4 and c-cbl-associated protein (CAP), the adapter protein in the insulin-signalling cascade, was increased by rosiglitazone in skeletal muscle (Figure 7C). In the same system, P633H dose-dependently increased GLUT4 expression but, at the highest dose (10 mg·kg−1), significantly diminished CAP expression.
Histopathological examination of KK-Ay mice
Histological analysis of liver
Coincident with increased hepatic TG content (P < 0.05), the untreated KK-Ay mice showed hypertrophy of hepatocytes and hepatic steatosis (Figure 8A). Unlike rosiglitazone, which had no obvious impact on hepatic TG and TCHO contents in KK-Ay mice (Bedoucha et al., 2001), GW409544 and P633H ameliorated the hepatocyte hypertrophy and significantly lowered the lipid droplet accumulation, in accordance with the decreased hepatic TG (Figure 6B),
Figure 8.
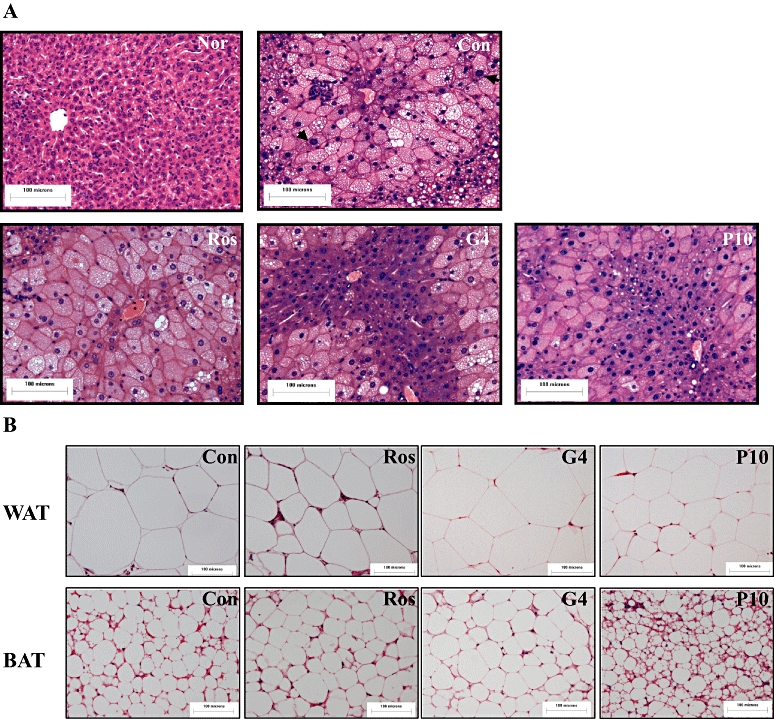
Photomicrographs of liver, WAT and BAT sections of the KK-Ay mice treated with different PPAR agonists. (A) Liver. Hypertrophy of hepatocytes and hepatic steatosis are evident. Black arrow in ‘Con’ indicates the hypertrophy of nuclear. (B) WAT and BAT. The bar indicates 100 µm in the panels (HE: 40×). BAT, brown adipose tissue; Con, control; Nor, normal; G4, 10 mg·kg−1 GW409544; P633H, 2-[4-(2-Fluoro-benzenesulphonyl)-piperazin-1-yl]-3-{4-[2-(5-methyl-2-phenyl-oxazol-4-yl)-ethoxy]-phenyl}-propionic acid; P10, 10 mg·kg−1 P633H; PPAR, peroxisome proliferator-activated receptor; Ros, rosiglitazone; WAT, white adipose tissue.
Histological analysis of adipose tissue
Rosiglitazone and P633H both decreased the size of the adipocytes in WAT (adipocyte area, 7.57 ± 6.10, 4.87 ± 3.63, 5.17 ± 3.65 µm2 for control, rosiglitazone and 10 mg·kg−1 P633H, P < 0.05 and 0.01 respectively) (Figure 8B). However, the WAT/body weight ratios were unaltered, indicating increased cell numbers (Table 1). GW409544 also increased the size of the WAT adipocytes (adipocyte area, 9.42 ± 5.55 µm2) and the WAT/body weight ratio (15.32 ± 1.45).
Although rosiglitazone and GW409544 augmented the size of BAT adipocytes based on the increased BAT/body weight ratios (Figure 8B; Table 1), P633H markedly reduced the diameter of BAT adipocytes and increased the total cell numbers with unchanged BAT/body weight ratios (Table 1). These results were in consistent with promotion by P633H of pre-adipocyte differentiation in vitro.
Histological analysis of pancreatic morphology
Histological results demonstrated that rosiglitazone and GW409544 ameliorated some pathological changes of the pancreas in KK-Ay mice, such as anomalous nuclear or pycnosis. However P633H made the islet more hyperplastic accompanied by some abnormal nuclear changes, which were further confirmed by electron microscopy.
As previously reported, the islets of untreated KK-Ay mice showed hyperplastic changes, degranulation, proliferation and infrequent hypertrophy of mitochondria and rough endoplasmic reticulum (Figure 9). Rosiglitazone-treated islets of KK-Ay mice had unremarkable organelles, except for some rare cases with modest proliferation of mitochondria. GW409544 also ameliorated the abnormal changes and increased the amount of insulin secretory organelles. On the contrary, P633H seemed to accelerate the degeneration of β-cells, as suggested by the further damaged structure of pancreatic β-cells, the increased anomalous nuclei and the swollen mitochondria with lost ridges (Figure 9).
Figure 9.
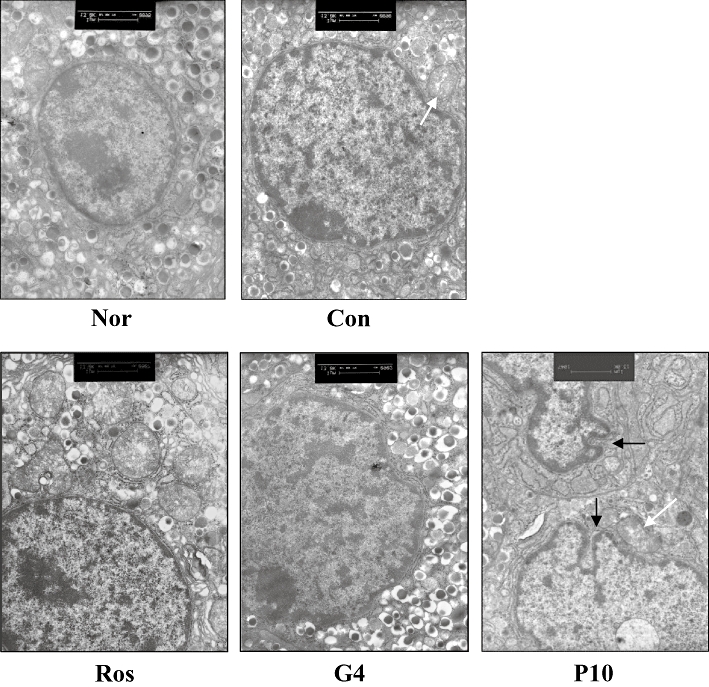
Electron microscopic analysis of pancreas in KK-Ay mice. White arrow, hypertrophy of mitochondria. Black arrow, deformed nucleus. The bar indicates 1 µm in the panels (13 000×). Con, control; Nor, normal; G4, 10 mg·kg−1 GW409544; P633H, 2-[4-(2-Fluoro-benzenesulphonyl)-piperazin-1-yl]-3-{4-[2-(5-methyl-2-phenyl-oxazol-4-yl)-ethoxy]-phenyl}-propionic acid; P10, 10 mg·kg−1 P633H; Ros, rosiglitazone.
Discussion
By transactivation assays P633H was shown to be an effective PPARα/γ dual agonist. The in vitro activation by P633H was confirmed by its promotion of 3T3-L1 pre-adipocyte differentiation. P633H also concomitantly up-regulated the expression of ACO and aP2, target genes of PPARα in liver and PPARγ in adipose tissue respectively. Additionally, preliminary experiments found that P633H (10 mg·kg−1) decreased the FBG of diet-induced obese mice by 58%.
However, further pharmacological results were completely unexpected. Unlike the well-known PPARα/γ dual agonist G409544 and the PPARγ agonist rosiglitazone, P633H showed clearly opposing activities in db/db and KK-Ay mice. In db/db mice, P633H significantly ameliorated FBG, impaired glucose tolerance, hyperinsulinemia, ISI, as well as increased TCHO levels. However in KK-Ay mice, these variables were either resistant to, or exacerbated by, treatment with P633H. Sequential blood glucose monitoring indicated that blood glucose level showed initial descending tendency on the fifth day, but relapsed on the tenth day and rose above that of control animals from the fifteenth day on. The increased blood glucose level showed no sign of falling, relative to that of controls, until the 25th day.
Why did P633H, a PPARα/γ dual agonist, produce such different effects in KK-Ay mice? There are several possible explanations for the lack of response to P633H: (i) insulin secretion was impaired; (ii) damage of pancreatic islets was aggravated; (iii) peripheral insulin resistance was exacerbated; and (iv) hepatic glucose output was increased. In KK-Ay mice, we found P633H did not affect serum insulin levels or ISI, a result that differed significantly from that obtained in db/db mice. At the initial diagnosis of type 2 diabetes, most patients show higher levels of fasting and postprandial plasma insulin, presumably to compensate for a loss of peripheral sensitivity to insulin. This increase is taken to indicate normally functional pancreatic β-cells. If untreated, most patients will soon exhibit evidence of β-cell dysfunction, leading eventually to complete loss of insulin secretion. Dysfunction of β-cells, insulin resistance and hyperglycaemia constitute a vicious cycle. Desensitized peripheral tissues keep demanding higher insulin output, which leads to a decline in β-cells function and further tissue damage. Histopathological analysis of the pancreas in KK-Ay mice treated with 10 mg·kg−1 P633H revealed pathological changes in islet hyperplasticity. Analysis by transmission electron microscopy found that the structure of the pancreatic β-cells was disrupted, with more increased anomalous nuclei and swollen mitochondria with lost ridges. Such toxicity does not seem to be specifically related to PPARα/γ dual agonists, as GW409544 effectively ameliorated the diabetic symptoms of KK-Ay mice, without obvious damage to the pancreas. However, the damage caused by P633H did not seem either to be a toxic effect of P633H directed towards pancreatic cells, as P633H, up to 10 mmol·L−1, did not promote proliferation or apoptosis of INS-1 pancreatic cells, over 2 days exposure and the IC50 value for P633H in HLF fibroblasts was lower than that of GW409544. Nevertheless, analysis of the effects of P633H on primary cultures of pancreatic cells from diabetic animals might contribute to an understanding of this unexpected toxicity.
Apart from impaired insulin-mediated glucose utilization, primarily in muscle, the major underlying mechanism of insulin resistance is impaired insulin-dependent down-regulation of hepatic glucose production, which determines the level of FBG. The key enzyme involved in gluconeogenesis is PEPCK. P633H, at the same time, promoted gene expression of the insulin-sensitive GLUT4 in skeletal muscle and decreased that of PEPCK in liver and muscle of db/db mice, changes responsible for improving insulin resistance in db/db mice. However, in KK-Ay mice, P633H dose-dependently increased the expression of PEPCK mRNA in liver and down-regulated mRNA of CAP and insulin receptor substrate in skeletal muscle (10 mg·kg−1 P633H). CAP, a multifunctional adapter protein, facilitates insulin receptor signalling and insulin sensitivity (Saltiel and Kahn, 2001). Thus, although GLUT4 expression was dose-dependently increased in muscle of P633H-treated KK-Ay mice, its membrane translocation under stimulation by insulin was affected. Meanwhile, the differential regulation of CAP and insulin receptor substrate by different doses of P633H explains why 2.5 mg·kg−1 P633H showed some reduction in the serum lipid and body weight, relative to the effect of the higher dose (10 mg·kg−1). Furthermore, from the response curve of OGTT in P633H-treated KK-Ay mice, we concluded that the impaired first and second phase insulin secretion from β-cells were exacerbated under acute glucose stress, which further suggested impairment of pancreatic function and coincided with the observed structural deterioration of pancreatic islets in KK-Ay mice.
At the same time, from the histological analysis, we found that P633H reduced liver TG content, improved hepatic steatosis and the hypertrophy in hepatocytes. This profile of effects in KK-Ay mice treated with 10 mg·kg−1 P633H suggested a decrease of ectopic hepatic lipid accumulation (Boden, 2002), which was related to the increased hepatic fatty acid oxidation upon PPARα activation. On the other hand, morphological analysis of adipose tissue also revealed that P633H treatment increased small adipocytes in both WAT and BAT, without significant increase in fat storage, which was different from the effect of rosiglitazone and GW409544. The effects of P633H were consistent with promotion of pre-adipocyte differentiation in vitro and did not suggest exacerbation of insulin resistance in adipose tissue. Overall, it is possible that P633H promoted gluconeogenesis in liver and exacerbated the burden on the pancreas, under pre-existing conditions of insulin resistance. Such a scheme might partially explain the discrepant results in KK-Ay mice. But the reasons for the contrary regulation of PEPCK need further investigation.
Even though the exact mechanism remains to be elucidated, a strain-specific effect cannot be excluded. As demonstrated in rats and mice, the specific PPARα agonist fenofibrate also showed model-specific differences in pharmacological effects, that is, it relieved insulin resistance and hyperglycaemia in ZDF rats and KK-Ay mice (Tsuchida et al., 2005), yet it was ineffective in monosodium L-glutamate-induced obese rats and mice, a model of insulin resistance (Li et al., 2006; Chen et al., 2008). The KK-Ay mice are a congenic strain in which the Ay allele at the agouti locus (initially from the C57BL/6J-Ay strain) has been transferred to the inbred KK strain by repetitive backcrossing, as a polygenic model for human type 2 diabetes (Huang et al., 2006; Jeppesen et al., 2007). This strain of mice exhibit marked hyperglycaemia, hyperinsulinemia and elevated TG levels at 10 weeks of age (Iwatsuka, et al., 1970; Zhang et al., 2007) and has a complex aetiology involving multiple genetic and environmental factors. Although db/db and KK-Ay mice both displayed the typical symptoms of type 2 diabetes, the polygenetic background combined with a high-caloric diet made KK-Ay mice a better model for human type 2 diabetes than db/db mice. The latter, with the loss of leptin receptors, are homozygous mice from a diabetic spontaneous mutation (Leprdb) and are prone to hyperleptinemia. Additionally, there are differences in the expression and content of PPAR subtypes and diverse nuclear co-activator or co-repressor recruitment styles among different species when treated with different agents (Qi et al., 2000).
Relatively small differences in chemical structure may lead to completely different pharmacological effects. Previously, molecular docking simulations with P633H using Flex-X module of Sylbyl 6.9 had identified that, unlike GW409544 and rosiglitazone, P633H did not share the same access to the ligand-binding pocket of PPAR, because of the polarity and the resistance of the sulphonyl group in P633H. It is therefore possible that P633H may exhibit different ways of combination or binding modes (orientation or sites) towards the PPAR subtypes. Whether such binding differences constitute the basis of the different pharmacodynamic effects described here will require further investigation.
In conclusion, this is the first report of an effective PPARα/γ dual agonist, P633H, promoting hepatic gluconeogenesis and increasing blood glucose level in KK-Ay diabetic mice. This unexpected finding may derive from the details of the compound's structure. Our findings also emphasize the importance of using a wide range of pharmacological tests when assessing a lead compound, whose activity is based on a specific therapeutic target.
Acknowledgments
We acknowledge, with thanks, Liangzeng Yan, for proofreading of the manuscript (Lilly Research Laboratories, Eli Lilly and Company), Professor Benli Yuan (Pathological Laboratory in our Institute) for pathological analysis. This project was supported by Chinese National Hi-tech Research and Development Grants ‘863’ (no. 2003AA235010) and Chinese National Science Foundation (no. 30600782).
Glossary
Abbreviations:
- ACO
acyl-CoA oxidase
- CPTI
carnitine palmitoyl transferase I
- GLUT4
glucose transporter 4
- ISI
insulin sensitivity index
- NEFA
non-esterified fatty acids
- PEPCK
phosphoenolpyruvate carboxykinase
- PPAR
peroxisome proliferator-activated receptor
- TCHO
total cholesterol
- TG
triglycerides
Conflict of interest
None.
References
- Bedoucha M, Atzpodien E, Boelsterli UA. Diabetic KKAy mice exhibit increased hepatic PPARgamma1 gene expression and develop hepatic steatosis upon chronic treatment with antidiabetic thiazolidinediones. J Hepatol. 2001;35:17–23. doi: 10.1016/s0168-8278(01)00066-6. [DOI] [PubMed] [Google Scholar]
- Boden G. Interaction between free fatty acids and glucose metabolism. Curr Opin Clin Nutr Metab Care. 2002;5:545–549. doi: 10.1097/00075197-200209000-00014. [DOI] [PubMed] [Google Scholar]
- Chen W, Wang LL, Liu HY, Long L, Li S. Peroxisome proliferator-activated receptor delta-agonist, GW501516, ameliorates insulin resistance, improves dyslipidaemia in monosodium L-glutamate metabolic syndrome mice. Basic Clin Pharmacol Toxicol. 2008;103:240–246. doi: 10.1111/j.1742-7843.2008.00268.x. [DOI] [PubMed] [Google Scholar]
- Desvergne B, Michalik L, Wahli W. Be fit or be sick: peroxisome proliferator-activated receptors are down the road. Mol Endocrinol. 2004;18:1321–1332. doi: 10.1210/me.2004-0088. [DOI] [PubMed] [Google Scholar]
- Ferre P. The biology of peroxisome proliferator-activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes. 2004;53(Suppl. 1):S43–S50. doi: 10.2337/diabetes.53.2007.s43. [DOI] [PubMed] [Google Scholar]
- Flordellis CS, Ilias I, Papavassiliou AG. New therapeutic options for the metabolic syndrome: what's next? Trends Endocrinol Metab. 2005;16:254–260. doi: 10.1016/j.tem.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Gampe RT, Jr, Montana VG, Lambert MH, Miller AB, Bledsoe RK, Milburn MV, et al. Asymmetry in the PPARgamma/RXRalpha crystal structure reveals the molecular basis of heterodimerization among nuclear receptors. Mol Cell. 2000;5:545–555. doi: 10.1016/s1097-2765(00)80448-7. [DOI] [PubMed] [Google Scholar]
- Guerre-Millo M, Gervois P, Raspe E, Madsen L, Poulain P, Derudas B, et al. Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity. J Biol Chem. 2000;275:16638–16642. doi: 10.1074/jbc.275.22.16638. [DOI] [PubMed] [Google Scholar]
- Henke BR, Blanchard SG, Brackeen MF, Brown KK, Cobb JE, Collins JL, et al. N-(2-Benzoylphenyl)-L-tyrosine PPARgamma agonists. 1. Discovery of a novel series of potent antihyperglycemic and antihyperlipidemic agents. J Med Chem. 1998;41:5020–5036. doi: 10.1021/jm9804127. [DOI] [PubMed] [Google Scholar]
- Huang H, Iida KT, Sone H, Yokoo T, Yamada N, Ajisaka R. The effect of exercise training on adiponectin receptor expression in KKAy obese/diabetic mice. Journal of. Endocrinology. 2006;189:643–653. doi: 10.1677/joe.1.06630. [DOI] [PubMed] [Google Scholar]
- Hung YJ, Hsieh CH, Pei D, Kuo SW, Lee JT, Wu LY, et al. Rosiglitazone improves insulin sensitivity and glucose tolerance in subjects with impaired glucose tolerance. Clin Endocrinol (Oxf) 2005;62:85–91. doi: 10.1111/j.1365-2265.2004.02178.x. [DOI] [PubMed] [Google Scholar]
- Iwatsuka H, Shino A, Suzuoki Z. General survey of diabetic features of yellow KK mice. Endocrinol Jpn. 1970;17:23–35. doi: 10.1507/endocrj1954.17.23. [DOI] [PubMed] [Google Scholar]
- Jeppesen PB, Nordentoft I, Abudula R, Hong J, Hermansen K. Isosteviol as novel oral anti-diabetic agent for treatment of type 2 diabetes and the metabolic syndrome – a in vivo study on diabetic KKAy-mice. Diabetologia. 2007;50:S95. [Google Scholar]
- Li PP, Shan S, Chen YT, Ning ZQ, Sun SJ, Liu Q, et al. The PPARalpha/gamma dual agonist chiglitazar improves insulin resistance and dyslipidemia in MSG obese rats. Br J Pharmacol. 2006;148:610–618. doi: 10.1038/sj.bjp.0706745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Zhou XB, Wang LL, Xu C, Ruan CM, Lin CF, et al. Substituted a-piperazinglyphenylpropionic acid derivatives as hPPAR alpha and/or hPPAR gamma agonists. US2007259883A1.
- Ljung B, Bamberg K, Dahllof B, Kjellstedt A, Oakes ND, Ostling J, et al. AZ 242, a novel PPARalpha/gamma agonist with beneficial effects on insulin resistance and carbohydrate and lipid metabolism in ob/ob mice and obese Zucker rats. J Lipid Res. 2002;43:1855–1863. doi: 10.1194/jlr.m200127-jlr200. [DOI] [PubMed] [Google Scholar]
- Murakami K, Tobe K, Ide T, Mochizuki T, Ohashi M, Akanuma Y, et al. A novel insulin sensitizer acts as a coligand for peroxisome proliferator-activated receptor-alpha (PPAR-alpha) and PPAR-gamma: effect of PPAR-alpha activation on abnormal lipid metabolism in liver of Zucker fatty rats. Diabetes. 1998;47:1841–1847. doi: 10.2337/diabetes.47.12.1841. [DOI] [PubMed] [Google Scholar]
- Pickavance LC, Brand CL, Wassermann K, Wilding JPH. The dual PPAR alpha/gamma agonist, ragaglitazar, improves insulin sensitivity and metabolic profile equally with pioglitazone in diabetic and dietary obese ZDF rats. Br J Pharmacol. 2005;144:308–316. doi: 10.1038/sj.bjp.0706041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi C, Zhu Y, Reddy JK. Peroxisome proliferator-activated receptors, coactivators, and downstream targets. Cell Biochem Biophys. 2000;32:187–204. doi: 10.1385/cbb:32:1-3:187. Spring. [DOI] [PubMed] [Google Scholar]
- van Raalte DH, Li M, Pritchard PH, Wasan KM. Peroxisome proliferator-activated receptor (PPAR)-alpha: a pharmacological target with a promising future. Pharm Res. 2004;21:1531–1538. doi: 10.1023/b:pham.0000041444.06122.8d. [DOI] [PubMed] [Google Scholar]
- Ramachandran U, Kumar R, Mittal A. Fine tuning of PPAR ligands for type 2 diabetes and metabolic syndrome. Mini Rev Med Chem. 2006;6:563–573. doi: 10.2174/138955706776876140. [DOI] [PubMed] [Google Scholar]
- Ramirez-Zacarias JL, Castro-Munozledo F, Kuri-Harcuch W. Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with Oil red O. Histochemistry. 1992;97:493–497. doi: 10.1007/BF00316069. [DOI] [PubMed] [Google Scholar]
- Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- Tenenbaum A, Fisman EZ, Motro M. Metabolic syndrome and type 2 diabetes mellitus: focus on peroxisome proliferator activated receptors (PPAR) Cardiovasc Diabetol. 2003;2:4. doi: 10.1186/1475-2840-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida A, Yamauchi T, Takekawa S, Hada Y, Ito Y, Maki T, et al. Peroxisome proliferator-activated receptor (PPAR)alpha activation increases adiponectin receptors and reduces obesity-related inflammation in adipose tissue: comparison of activation of PPARalpha, PPARgamma, and their combination. Diabetes. 2005;54:3358–3370. doi: 10.2337/diabetes.54.12.3358. [DOI] [PubMed] [Google Scholar]
- Xu C, Wang LL, Liu HY, Zhou XB, Cao YL, Li S. C333H, a novel PPARalpha/gamma dual agonist, has beneficial effects on insulin resistance and lipid metabolism. Acta Pharmacol Sin. 2006;27:223–228. doi: 10.1111/j.1745-7254.2006.00263.x. [DOI] [PubMed] [Google Scholar]
- Yajima K, Hirose H, Fujita H, Seto Y, Fujita H, Ukeda K, et al. Combination therapy with PPARgamma and PPARalpha agonists increases glucose-stimulated insulin secretion in db/db mice. Am J Physiol Endocrinol Metab. 2003;284:E966–E971. doi: 10.1152/ajpendo.00149.2002. [DOI] [PubMed] [Google Scholar]
- Yang Y, Shang W, Zhou L, Jiang B, Jin H, Chen M. Emodin with PPARgamma ligand-binding activity promotes adipocyte differentiation and increases glucose uptake in 3T3-Ll cells. Biochem Biophys Res Commun. 2007;353:225–230. doi: 10.1016/j.bbrc.2006.11.134. [DOI] [PubMed] [Google Scholar]
- Zhang ZY, Jung DY, Ola MS, Linden J, Lanoue KF, Kim JK. Short-term treatment with adenosine A(2B) receptor antagonist improves insulin sensitivity in diabetic KKAy mice. Diabetes. 2007;56:A64. [Google Scholar]