

Abstract
Background and purpose:
The transcription factor peroxisome proliferator-activated receptor γ (PPARγ) is essential for glucose homeostasis. PPARγ ligands reducing insulin levels in vivo are used as drugs to treat type 2 diabetes mellitus. Genes regulated by PPARγ have been found in several tissues including insulin-producing pancreatic islet β-cells. However, the role of PPARγ at the insulin gene was unknown. Therefore, the effect of PPARγ and PPARγ ligands like rosiglitazone on insulin gene transcription was investigated.
Experimental approach:
Reporter gene assays were used in the β-cell line HIT and in primary mature pancreatic islets of transgenic mice. Mapping studies and internal mutations were carried out to locate PPARγ-responsive promoter regions.
Key results:
Rosiglitazone caused a PPARγ-dependent inhibition of insulin gene transcription in a β-cell line. This inhibition was concentration-dependent and had an EC50 similar to that for the activation of a reporter gene under the control of multimerized PPAR binding sites. Also in normal primary pancreatic islets of transgenic mice, known to express high levels of PPARγ, rosiglitazone inhibited glucose-stimulated insulin gene transcription. Transactivation and mapping experiments suggest that, in contrast to the rat glucagon gene, the inhibition of the human insulin gene promoter by PPARγ/rosiglitazone does not depend on promoter-bound Pax6 and is attributable to the proximal insulin gene promoter region around the transcription start site from −56 to +18.
Conclusions and implications:
The human insulin gene represents a novel PPARγ target that may contribute to the action of thiazolidinediones in type 2 diabetes mellitus.
Keywords: β-cells, thiazolidindiones, human insulin gene transcription, PPARγ
Introduction
The nuclear hormone receptor, peroxisome proliferator-activated receptor γ (PPARγ), is, among other functions, an important regulator of glucose homeostasis (Desvergne and Wahli, 1999). While the endogenous ligand of PPARγ is not known, there are several synthetic compounds that bind PPARγ with high affinity and activate the receptor. These include the thiazolidinedione class of oral antidiabetic drugs, which are in use for the treatment of type 2 diabetes mellitus (Desvergne and Wahli, 1999). Thiazolidinediones like rosiglitazone and pioglitazone are effective agents for the control of glycaemia in patients with type 2 diabetes (Natali and Ferranini, 2006). In addition, in a randomized controlled clinical trial including 5238 patients, pioglitazone significantly reduced by 16% a composite end point. This included death from any cause, nonfatal myocardial infarction and stroke (Dormandy et al., 2005). The overall clinical benefit from use of thiazolidinediones, however, remains to be defined (Lago et al., 2007; Lincoff et al., 2007; Nissen and Wolski, 2007; Singh et al., 2007).
When compared with other oral antidiabetic drugs like the sulphonylureas and metformin, thiazolidinediones exhibit a unique antidiabetic effect. Through PPARγ, they reduce hepatic glucose output and increase insulin sensitivity (Natali and Ferranini, 2006). Furthermore, decreases in fasting plasma insulin levels have been reported in most trials performed with thiazolidinediones (Walter and Lübben, 2005; Wajchenberg, 2007). Reductions in fasting plasma insulin levels have been in the range of 10.7–31.8 pmol·L−1 with pioglitazone monotherapy and 6.6–27.2 pmol·L−1 with rosiglitazone monotherapy (Walter and Lübben, 2005). Thiazolidinediones also reverse the decline in the function of insulin-producing pancreatic islet β-cells in type 2 diabetes mellitus with improvements in islet architecture, insulin content, proinsulin to total immunoreactive insulin ratio and glucose-stimulated insulin secretion (Zeender et al., 2004; Walter and Lübben, 2005; Wajchenberg, 2007). Although these effects on insulin could be explained as the beneficial sequelae of reducing hyperglycaemia and peripheral insulin resistance, direct effects of PPARγ on β-cells may contribute as well. PPARγ serves as transcription factor for a great number of genes involved in fatty acid uptake and storage, inflammation and glucose homoeostasis in adipose tissue, skeletal muscle and liver (Kostadinova et al., 2005; Gervois et al., 2007). Furthermore, PPARγ is expressed also in pancreatic islet β-cells (Braissant and Wahli, 1998). Nevertheless, the role of PPARγ in β-cells in the regulation of the insulin gene has received little attention so far.
Therefore, the direct effect of PPARγ and thiazolidinediones on insulin gene transcription in pancreatic islet β-cells was examined in the present study. Using transfection studies in the β-cell line HIT and in primary islets of mature transgenic mice, this study demonstrates that PPARγ ligands inhibit insulin gene transcription. This study suggest that, in contrast to the glucagon gene, the inhibition of the insulin gene does not depend on promoter-bound Pax6 and is conferred by the proximal insulin gene promoter region around the transcription start site.
Methods
Plasmid constructs
The plasmids pT81Luc, −350GluLuc (Schwaninger et al., 1993), −410rInsLuc (Siemann et al., 1999), −336hInsLuc, −258hInsLuc, −222hInsLuc, −193hInsLuc, −140hInsLuc, −93hInsLuc, −56hInsLuc, −336/+18hInsLuc (Oetjen et al., 2007), PPRE-Luc, pPPARγ (Schinner et al., 2002) and pcDNA3-retinoid X receptor (RXR)α, pcDNA3-PPARγ1-475N, pcDNA3-PPARγ175-475N (Krätzner et al., 2008) have been described previously. The expression vector pBAT14.mPax6 was kindly provided by Dr M. German (University of California, San Francisco, CA) (Sander et al., 1997). The plasmid pCMV-GFPtpz was purchased from Canberra-Packard (Dreieich, Germany). To generate −336/−31P-Luc, the fragment from −336 to −31 of the human insulin gene promoter was amplified by PCR using −336hInsLuc as template and cloned in front of the heterologous minimal promoter −85rInsLuc (P) (Siemann et al., 1999). All constructs were verified by sequencing using the enzymic method.
Cell culture and transfection of DNA
The insulin-producing pancreatic islet β-cell line HIT-T15 (Santerre et al., 1981) was grown in RPMI 1640 medium supplemented with 10% foetal calf serum, 5% horse serum, penicillin (100 U·mL−1) and streptomycin (100 µg·mL−1). JEG-3 human choriocarcinoma cells (Oetjen et al., 2007) were grown in DMEM supplemented with 10% foetal calf serum, penicillin (100 U·mL−1) and streptomycin (100 µg·mL−1). HIT cells were trypsinized and transfected in suspension by the DEAE-dextran method (Schwaninger et al., 1993) with 2 µg of reporter gene plasmids and, when indicated, 1 µg of expression vector per 6 cm dish. Twenty-four hours after transfection, cells were incubated in RPMI 1640 containing 0.5% bovine serum albumin and antibiotics as described above. Thiazolidinediones were added 24 h before harvest. JEG cells were transfected by the calcium phosphate precipitation method (Oetjen et al., 2007) with 3 µg of reporter gene plasmid and, when indicated, 207 ng of expression vector per 6 cm dish. In all experiments (HIT and JEG), cotransfections were carried out with a constant amount of DNA, which was maintained by adding Bluescript (Stratagene, La Jolla, CA). In all experiments 1 µg (HIT) or 50 ng (JEG) of cytomegalovirus–GFP (plasmid pCMV-GFPtpz) per 6 cm dish was cotransfected to check for transfection efficiency. Cell extracts (Schwaninger et al., 1993) were prepared 48 h after transfection. The luciferase assay was performed as described previously (Schwaninger et al., 1993). For the transfection of small interference RNA (siRNA) against Pax6, cells were transfected by metafectene (Biontex, Munich, Germany) with 50 or 100 pmole per 2 cm dish of siRNA as indicated according to the manufacturer's recommendations. The following sequences were employed: siRNA 1 – GGGACCACUUCAACAGGACUCAUUU, siRNA 2 – GGAGUGAACCUGACAUGUCUCAGUA, siRNA 3 – ACCACACCUGUCUCCUCCUUCACAU and their respective complementary strands. The efficiency of Pax6 knock down was tested by immunoblot using an antibody against Pax6 (Santa Cruz, Heidelberg, Germany). Green fluorescent protein was measured in the cell extracts using the FluoroCount™ microplate fluorometer (Packard) with a 485 nm (excitation)/530 nm (emission) filter pair.
Electrophoretic mobility shift assay
Synthetic complementary oligonucleotides (hIns PPRE wt: 5′-GGCCCAGCAGCCCTCAGCCCTCCAGGACAGGCT-3′, hIns PPRE mut: 5′-GGCCCAGCAGCAAGCATCTTGCCAGGACAGGCT-3′) were annealed and labelled by a fill-in reaction using [α-32P]dCTP and Klenow enzyme. Fifteen microlitres of islet extracts was pre-incubated with 2 µg poly dI/dC in binding buffer (20 mmol·L−1 HEPES, 1 mmol·L−1 EDTA) and when indicated with a 200-fold molar excess of the competitors (PPRE wt: 5′-GGTAAAGGTCAAAGGTCAAT-3′, PPRE mut: 5′-GGTAAAGAACAAAGAACAAT-3′) for 10 min at room temperature, followed by a 15 min incubation with the labelled oligonucleotides at room temperature. The binding reaction was subjected to electrophoresis on a 5% non-denaturing poly acrylamide gel.
Generation and analysis of transgenic mice
All animal studies were conducted according to the National Institutes of Health's guidelines for care and use of experimental animals and were approved by the Committee on Animal Care and Use of the local institution and state. The generation and analysis of transgenic mice carrying a transgene with the luciferase reporter gene under the control of the human insulin gene promoter from −336 to +112 have been described before (Oetjen et al., 2003a).
Isolation and culture of islets
Pancreatic islets from mice or transgenic mice were isolated and incubated as described previously (Oetjen et al., 2003a). In short, isolated islets were pre-incubated in a humified atmosphere of 95% air/5% CO2 for 12 h in RPMI 1640 medium containing 5 mmol·L−1 glucose and supplemented with 10% foetal calf serum, penicillin (100 U·mL−1) and streptomycin (100 µg·mL−1). Rosiglitazone was added 7 h, glucose (final concentration 20 mmol·L−1) 6 h before harvest. Islet collection and extraction, as well as the measurement of luciferase activity and protein content were performed as has been described (Oetjen et al., 2003a). Following this protocol, the human insulin promoter has been shown to confer a normal, physiological glucose response to reporter gene expression in isolated islets (Oetjen et al., 2003a). For the electrophoretic mobility shift assay, approximately 800 islets were lysed in 150 µL lysis buffer (50 mmol·L−1 HEPES, 150 mmol·L−1 NaCl, 1.5 mmol·L−1 MgCl2, 1 mmol·L−1 EGTA, 10% glycerol, 0.5% NP-40, 1 mmol·L−1 NaVO4, 50 mmol·L−1 NaF, 20 mmol·L−1β-glycerophosphate), passed five times through a 20 G needle, incubated on ice for 30 min and centrifuged at 4°C, 20 800×g for 5 min. The supernatant was used for the binding reaction.
Statistical analysis
All results are expressed as means ± SEM. Statistical significance was calculated with anova, followed by Student's t-test. A value of P < 0.05 was considered significant.
Materials
Rosiglitazone was kindly provided by GlaxoSmith-Kline (Welwyn Garden City, Hertfordshire, UK). Darglitazone and englitazone (CP-72,467-02, sodium salt) were provided by Pfizer Inc (Groton, CT). RNAi was obtained from Invitrogen (Karlsruhe, Germany). Luciferin was purchased from Promega (Mannheim, Germany).
Results
Effect of PPARγ and thiazolidinediones on human insulin gene transcription in HIT β-cells
The expression of PPARγ is very high in normal pancreatic islets, approximately two-thirds of the expression level in white adipose tissue (Braissant and Wahli, 1998; Rosen et al., 2003; Lupi et al., 2004). In contrast, HIT cells express low levels of PPARγ as indicated by the observation that activation of a PPAR-dependent promoter (PPRE-Luc) by the thiazolidinedione rosiglitazone required transfection of a PPARγ expression vector (Figure 1). Consequently, this cell line allowed a direct assessment of the role of PPARγ in insulin gene transcription. Similarly, low-level expression of PPARγ in cell lines derived from tissues with high-level expression has been reported previously (Ricote et al., 1998) including the islet α-cell line InR1-G9 (Schinner et al., 2002) and β-cell line MIN6 (Nakamichi et al., 2003). To study the effect of PPARγ and thiazolidinediones on insulin gene transcription, a fragment from −336 to +112 of the human insulin gene was fused to the luciferase reporter gene (construct −336hInsLuc) (Oetjen et al., 2007; 2003a). This insulin promoter fragment is sufficient to confer tissue-specific gene expression and regulation of gene transcription by cAMP-, calcium-, glucose-, calcineurin- and mitogen-activated protein kinase-induced signalling pathways (Melloul et al., 2002; Hay and Docherty, 2006; Oetjen et al., 2003a,b; 2007). In the absence of cotransfected PPARγ expression plasmid, treatment of HIT cells with rosiglitazone at concentrations of up to 100 µmol·L−1 had no effect on insulin gene transcription (data not shown). This is consistent with the reported lack of effect of rosiglitazone on insulin gene transcription in the MIN6 β-cell line (Richardson et al., 2006). However, when a PPARγ expression plasmid was cotransfected, rosiglitazone inhibited insulin gene transcription (Figure 2). Consistently, rosiglitazone activated a PPRE-driven reporter gene only when PPARγ was cotransfected (Figure 1). Thus, rosiglitazone inhibits insulin gene transcription by a PPARγ-dependent mechanism. Inhibition of insulin gene transcription by rosiglitazone was concentration-dependent with an IC50 value of about 1 µmol·L−1 (Figure 2). Cotransfection of an expression vector encoding the RXRα together with PPARγ did not alter the concentration–response curve for inhibition of insulin gene transcription by rosiglitazone (data not shown). The concentrations of rosiglitazone that inhibit insulin gene transcription (Figure 2) are similar to those that activate the PPARγ-dependent promoter (Figure 1). The maximum inhibition of insulin gene transcription by rosiglitazone was about 60% (Figure 2). In addition to rosiglitazone, two other thiazolidinediones, darglitazone and englitazone, also inhibited insulin gene transcription (Figure 3). The specificity of the effect of PPARγ/rosiglitazone on insulin gene transcription is further supported by the lack of effect of PPARγ/rosiglitazone on CMV-promoter activity (not shown). These data indicate that PPARγ inhibits insulin gene transcription in response to the binding of thiazolidinediones.
Figure 1.
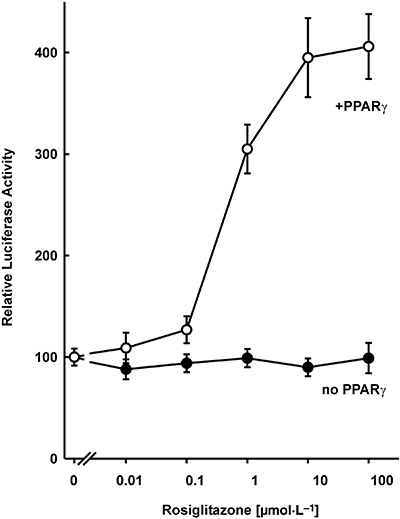
Activation of a peroxisome proliferator-activated receptor (PPAR)-dependent promoter by rosiglitazone and PPARγ in HIT β-cells. A luciferase reporter gene under the control of three copies of a PPARγ response element (plasmid PPRE-Luc) was transfected into HIT cells together with and without an expression vector encoding PPARγ. Increasing concentrations of rosiglitazone were added 24 h before harvest. Luciferase activity is expressed as percentage of the mean value of the activity measured in the untreated controls. Values are means ± SEM of three independent experiments, each in duplicate.
Figure 2.
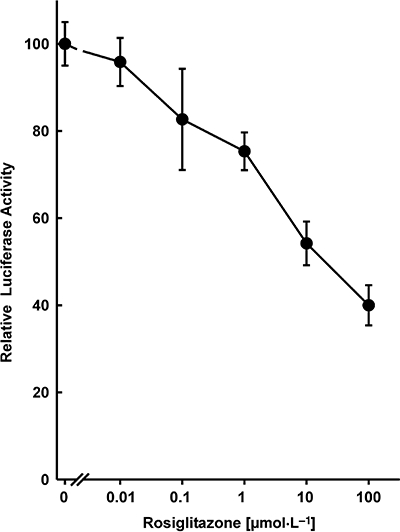
Inhibition of human insulin gene promoter activity by rosiglitazone and peroxisome proliferator-activated receptor γ (PPARγ). Plasmid −336hInsLuc was transfected into HIT β-cells together with pPPARγ. Rosiglitazone was added 24 h before harvest. Luciferase activity is expressed as percentage of the mean value of the activity measured in the untreated controls. Values are means ± SEM of three independent experiments, each in duplicate.
Figure 3.
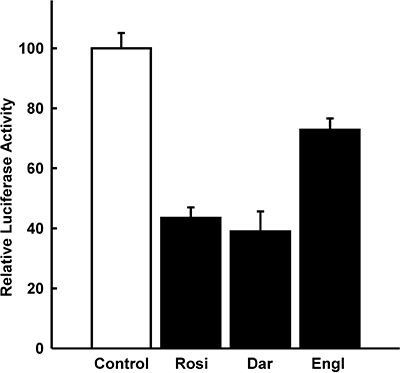
Inhibition of insulin gene transcription by the thiazolidinediones rosiglitazone, darglitazone and englitazone. HIT β-cells were transfected with −336hInsLuc and pPPARγ. They were treated with rosiglitazone (Rosi, 10 µmol·L−1), darglitazone (Dar, 30 µmol·L−1) or englitazone (Engl, 100 µmol·L−1) 24 h before harvest as indicated. Luciferase activity is expressed as percentage of the mean value of the activity measured in the untreated controls. Values are means ± SEM of three independent experiments, each in duplicate.
Effect of rosiglitazone on glucose-stimulated insulin gene transcription in primary pancreatic islets
HIT cells are a well-established β-cell line and very useful in studies of insulin gene transcription (Santerre et al., 1981; Melloul et al., 2002; Hay and Docherty, 2006). As a tumour cell line, they may differ in critical aspects from normal β-cells. To investigate the effect of rosiglitazone in normal primary β-cells and under stimulation by glucose as the major physiological stimulus of insulin gene transcription (Melloul et al., 2002; Hay and Docherty, 2006), islets of adult mice carrying a luciferase reporter transgene under the control of the human insulin gene promoter (from −336 to +112) were used. The expression of the human insulin gene within the islets of these transgenic mice has previously been shown to be regulated by glucose within the physiological concentration range (Oetjen et al., 2003a). In the present study, we show that the treatment of the isolated islets with rosiglitazone abolished glucose-induced human insulin gene transcription (Figure 4).
Figure 4.
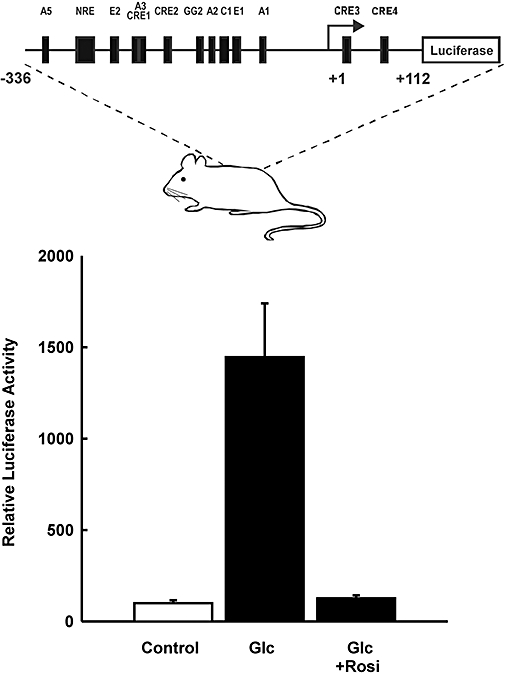
Effect of rosiglitazone on glucose-stimulated insulin gene transcription in normal primary pancreatic islets of transgenic mice. The upper panel depicts a scheme of the reporter gene used to generate the transgenic mice. Islets of transgenic mice were isolated and treated with glucose (Glc, 20 mmol·L−1) 6 h prior to harvest with and without rosiglitazone (Rosi, 30 µmol·L−1; 7 h prior to harvest). The control received 5 mmol·L−1 glucose only. Luciferase activity is expressed as percentage of the activity measured in the untreated controls. Values are means ± SEM of four experiments.
Mapping of the PPARγ-responsive segment in the human insulin gene promoter
The transcription factor Pax6 has been shown to interact with PPARγ through its transactivation domain and to mediate the inhibition by PPARγ and thiazolidinediones of rat glucagon gene transcription (Schinner et al., 2002; Krätzner et al., 2008). Pax6 also binds to the rat insulin I gene promoter (Knepel et al., 1991; Sander et al., 1997). To examine whether Pax6 may bind to and activate also the human insulin gene promoter, hInsLuc was transfected into the heterologous cell line JEG, with and without cotransfection of an expression vector encoding Pax6. The luciferase reporter gene under the control of the rat insulin I (rInsLuc) or rat glucagon gene promoter (GluLuc) was transfected as a control. As shown in Figure 5A, Pax6 activated the rat glucagon gene promoter 117-fold and, less so, the rat insulin I gene promoter. In contrast, the activation of the human insulin gene promoter by Pax6 was only fivefold (Figure 5A). This activation appears to be non-specific, as similar slight increases were produced by Pax6 using the luciferase reporter gene under the control of promoters that lack a Pax6 binding site such as the truncated viral thymidine kinase promoter (pT81Luc) (not shown). Reduction of the cellular Pax6 content of HIT cells by siRNA did not decrease the transcriptional activity of the human insulin gene promoter (100 ± 5.6%, control: 92.3 ± 1.8% in the presence of siRNA, n = 6; P < 0.05) nor did it interfere with the inhibitory effect of rosiglitazone on human insulin gene transcription (Figure 5B). In addition, rosiglitazone inhibited human insulin gene transcription only when the overexpressed PPARγ contained its DNA binding domain, indicating that binding of PPARγ to the insulin gene promoter is necessary for the inhibitory effect of the thiazoldindione (Figure 5C). These data do not support the view that PPARγ and thiazolidinediones may inhibit the human insulin gene promoter through inhibition of the transcriptional activity of promoter-bound Pax6, as is the case with the rat glucagon gene promoter (Schinner et al., 2002; Krätzner et al., 2008).
Figure 5.
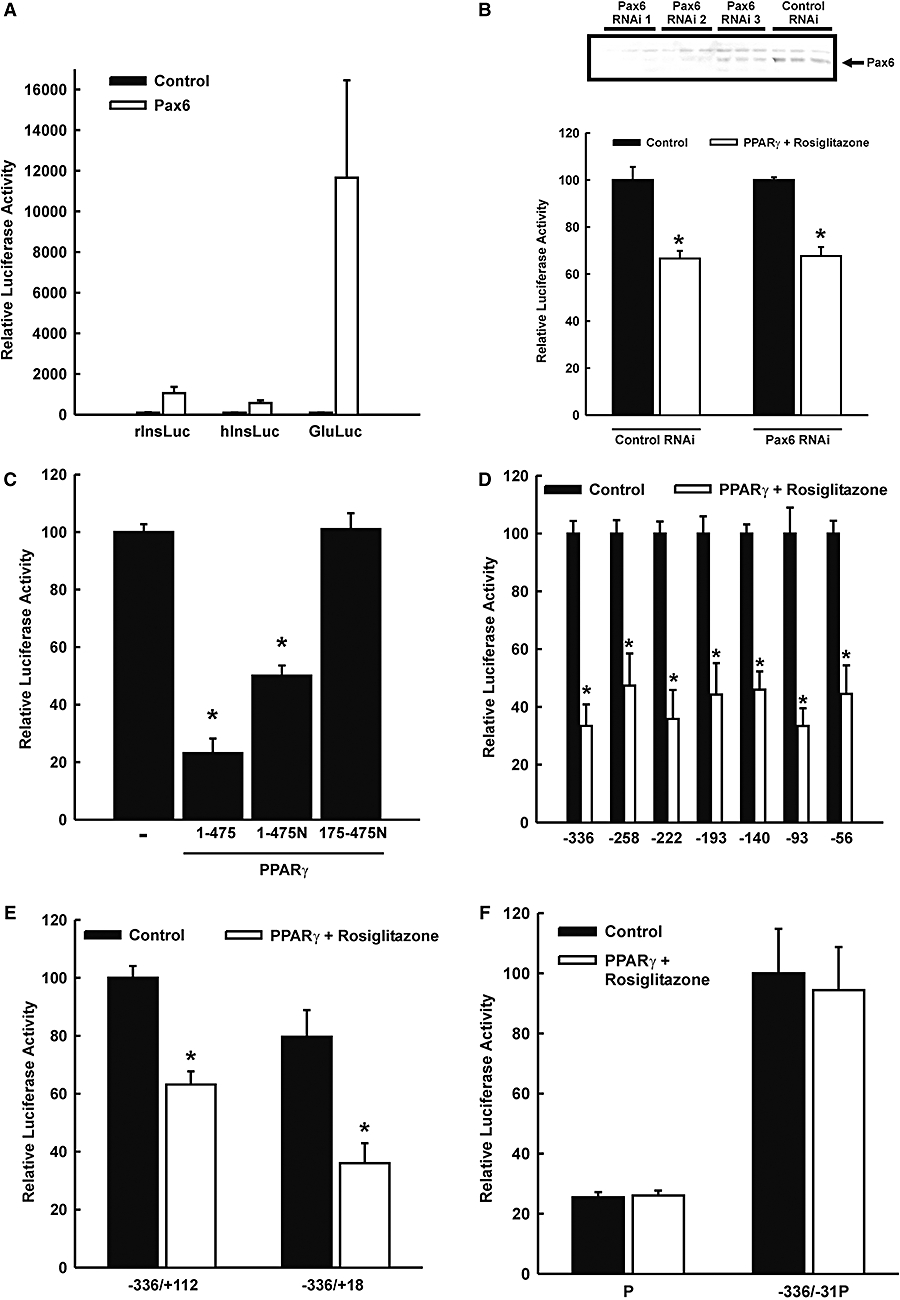
Mapping of the segment of the human insulin gene promoter that mediates the responsiveness to peroxisome proliferator-activated receptor γ (PPARγ)/rosiglitazone. (A) The transcription factor Pax6 is known to activate the rat glucagon gene and to confer responsiveness to PPARγ. This figure shows the effect of Pax6 on promoter activity of the rat insulin I, rat glucagon and human insulin gene. Luciferase reporter genes under the control of the rat insulin I (rInsLuc), rat glucagon (GluLuc) or human insulin gene promoter (hInsLuc) were transfected into heterologous JEG cells with and without cotransfection of an expression vector encoding Pax6. Luciferase activity is expressed as percentage of the activity measured in the controls (no Pax6). Values are means ± SEM of three experiments, each done in duplicate. (B) Reduction of cellular Pax6 does not interfere with rosiglitazone-induced inhibition of insulin gene transcription. Upper panel, HIT cells were transiently transfected with three different small interference RNA (shown as RNAi 1–3, see Methods; 50 and 100 pmole per dish) against Pax6. Cells were harvested after 48 h and an immunoblot using a Pax6 antibody was performed. RNAi 1 and 2 decrease the content of Pax6 in HIT cells. Lower panel, HIT cells were transiently transfected with RNAi 1 (50 pmole per dish), the luciferase reporter gene under control of the humane insulin gene promoter and the expression vector for PPARγ. Cells were treated with rosiglitazone (30 µmol·L−1) for 24 h or left untreated. Luciferase activity is expressed as percentage of the mean value measured in the control (without PPARγ cotransfection, without rosiglitazone). Values are means ± SEM of two independent experiments, each in triplicate; *P < 0.05. (C) Inhibition of insulin gene transcription by rosiglitazone depends on the DNA binding domain of PPARγ. A luciferase reporter gene under control of the human insulin gene promoter from −336 to +112 bp was transiently cotransfected into HIT cells with expression vectors for PPARγ wild type (1–475 PPARγ), PPARγ with extended carboxyl terminus by a nuclear localization signal (1–475N PPARγ), PPARγ lacking the AF-1 and the DNA binding domain and carrying at the carboxyl terminal a nuclear localization signal (175–475N PPARγ). Rosiglitazone (30 µmol·L−1) was added 24 h before harvest. Luciferase activity is expressed as percentage of the mean value measured in the control (without PPARγ cotransfection, without rosiglitazone). Values are means ± SEM of two independent experiments, each in triplicate. *P < 0.01 versus PPARγ wild type. (D) 5′-Deletion analysis. Human insulin gene promoter-luciferase reporter genes with 5′ ends as indicated were transfected into HIT β-cells with and without cotransfection of pPPARγ and rosiglitazone treatment (30 µmol·L−1). Luciferase activity is expressed as percentage of the activity measured in the respective untreated controls. Values are means ± SEM of three independent experiments, each in duplicate, *P < 0.05. (E) Effect of a 3′-deletion of the human insulin gene promoter from +112 to +18 on PPARγ/rosiglitazone responsiveness. The plasmids −336hInsLuc (−336/+112) or −336/+18hInsLuc were transfected into HIT β-cells with or without cotransfection of pPPARγ and rosiglitazone treatment (30 µmol·L−1). Luciferase activity is expressed as percentage of the activity measured in the untreated −336hInsLuc controls. Values are means ± SEM of four independent experiments, each in duplicate, *P < 0.05. (F) The fragment of the human insulin gene promoter from −336 to −31 was placed in front of a heterologous nonresponsive promoter (P). The plasmids were transfected into HIT β-cells with and without cotransfection of pPPARγ and rosiglitazone treatment (30 µmol·L−1). Values are means ± SEM of three independent experiments, each in duplicate.
To define the cis-acting DNA sequences within the human insulin gene promoter that mediate transcriptional repression by PPARγ, a deletion analysis was performed. As shown in Figure 5D, expression of 5′-deleted mutant plasmids in HIT cells revealed that the repression by PPARγ and rosiglitazone was unimpaired when the 5′ end was shortened from −336 to −56. Basal activity was eventually reduced by 5′-deletion; for −56hInsLuc it was low but still detectable (2.4 ± 0.2% of −336hInsLuc). These results indicate that DNA sequences that allow repression by PPARγ may reside 3′ to −56. When the 3′ end of the human insulin gene promoter was shortened from +112 to +18 (construct −336/+18hInsLuc), the inhibition by PPARγ and rosiglitazone was fully preserved (Figure 5E). Bearing in mind the results of the 5′-deletion analysis, these data suggest that DNA sequences that are critical for repression by PPARγ to the human insulin gene may be located between −56 and +18. This conclusion was further supported by the lack of inhibition by PPARγ and rosiglitazone when the DNA sequences around the transcription start site of the human insulin gene promoter (sequences 3′ to −31) were replaced by a heterologous non-responsive minimal promoter (Figure 5F). The electrophoretic mobility shift assay showed that protein(s) of primary islet extracts bind in a mutation-sensitive way to the proximal sequence of the human insulin gene promoter (Figure 6). The protein complex was competed for by the consensus PPRE sequence (Desvergne and Wahli, 1999) but less so by a mutated PPRE sequence (Figure 6B), indicating that PPARγ is among the binding proteins.
Figure 6.
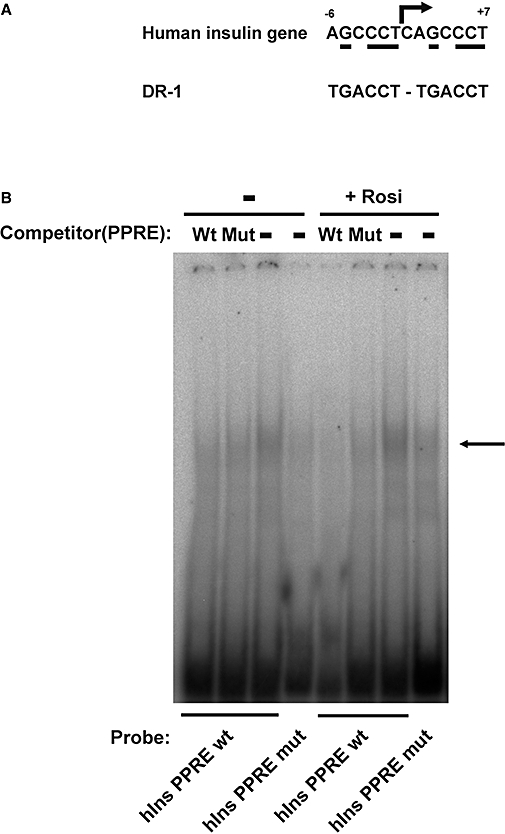
Protein binding to the proximal human insulin gene promoter around the transcription start site. (A) Sequence comparison between the proximal human insulin gene promoter around the transcription start site and a typical peroxisome proliferator-activated receptor γ (PPARγ) DNA binding site. PPARγ binds as a heterodimer with retinoid X receptor to response elements, which consist of a direct repeat of a hexamer half-site, spaced by one nucleotide (DR-1). The arrow indicates the transcription start site. The numbers give the first and last nucleotide relative to the transcriptional start site. The bases that match the consensus sequence are underlined. (B) Electrophoretic mobility shift assay. Islet extracts were incubated with radioactively labelled hIns PPARγ response element (PPRE) wt or hIns PPRE mut. For competition, PPRE consensus sequence (Wt) or its mutation (Mut) was used as indicated. The arrow indicates the PPARγ containing complex.
Discussion
As in adipose tissue, skeletal muscle and liver (Gervois et al., 2007), PPARγ and thiazolidinediones also exert important glucose regulatory functions within pancreatic islets (Walter and Lübben, 2005; Wajchenberg, 2007). In α-cells, they inhibit glucagon gene transcription (Schinner et al., 2002; Krätzner et al., 2008), which may contribute to the thiazolidinediones' antidiabetic effect by reducing glucagon-dependent hepatic glucose production (Schinner et al., 2002). In β-cells, PPARγ and thiazolidinediones activate the genes encoding GLUT2 (Kim et al., 2000), glucokinase (Kim et al., 2002) and ABCA1 cholesterol efflux transporter (Brunham et al., 2007). These effects seem to allow thiazolidinediones to establish a more timely insulin secretion, as is indicated by the findings that 13 week treatment with rosiglitazone increases the ability of an oscillatory glucose infusion to programme high-frequency pulsatile insulin secretion in patients with type 2 diabetes mellitus (Juhl et al., 2003), and that pioglitazone and rosiglitazone restore the first-phase insulin response to an intravenous glucose tolerance test in patients with impaired glucose tolerance and with frank type 2 diabetes mellitus (Ovalle and Bell, 2004). The present study now identifies the insulin gene as a new PPARγ target gene in pancreatic islet β-cells.
The early work on characterizing insulin gene transcription focused on the rat insulin I gene (Melloul et al., 2002; Hay and Docherty, 2006). The early perception that human insulin promoter constructs would not function in transfected rodent cells proved to be unfounded (Melloul et al., 2002; Hay and Docherty, 2006), and the human insulin promoter exhibited the expected pattern of activity in transgenic mice (Fromont-Racine et al., 1990). Furthermore, it became apparent that rodent insulin promoters differ considerably from the human promoter, leading to the conclusion that extreme care should be taken when extrapolating rodent-based data to the human insulin gene (Melloul et al., 2002; Hay and Docherty, 2006). In order to avoid these problems, the human insulin gene promoter was used in the present study.
In this study, we found human insulin gene transcription to be inhibited by PPARγ and thiazolidinediones in HIT β-cells. Rosiglitazone inhibited human insulin promoter activity also in normal primary pancreatic islets as revealed by the use of islets from transgenic mice carrying a human insulin promoter-luciferase transgene, which has been described before (Oetjen et al., 2003a; 2007). The effect of thiazolidiones on insulin gene expression is still a matter for debate: a recent study showed no effect of 1 µmol·L−1 of rosiglitazone on insulin gene expression in human pancreatic islets after chronic fatty acid exposure (Vandewalle et al., 2008). However, consistent with the present study, troglitazone reduced preproinsulin mRNA levels in primary islets (Bollheimer et al., 2003), and PPARγ and pioglitazone decreased proinsulin biosynthesis as indicated by [3H]leucine labelling in MIN6 cells (Nakamichi et al., 2003).
The fact that rosiglitazone inhibited insulin gene transcription over the same range of concentrations as it stimulated, through PPARγ, the expression of a reporter gene directed by a well-defined PPARγ DNA binding site suggests that inhibition of insulin gene transcription may accompany other PPARγ-mediated effects. All findings, taken together, support the conclusion that inhibition of insulin gene transcription may be relevant both physiologically and therapeutically for the action of thiazolidinediones. Consistent with this view, the insulin content was significantly elevated in pancreatic islets from mice in which the expression of the PPARγ gene in β-cells was eliminated (βγKOmice), in spite of normal glucose and insulin levels in their sera (Rosen et al., 2003).
PPARγ is well known to bind as a heterodimer with RXR to response elements in target genes and to activate transcription (Desvergne and Wahli, 1999; Natali and Ferranini, 2006). With the human insulin gene promoter, the present study provides another example that PPARγ can also inhibit gene transcription (Ricote et al., 1998; Schinner et al et al., 2002; Krätzner et al., 2008). As has been first established for the glucocorticoid receptor (Reichardt et al., 1998; Tuckermann et al., 1999), nuclear receptors including PPARγ may mediate transrepression in a DNA binding-independent manner (Li et al., 2000). Thus, the PPARγ/RXR heterodimer binds through protein–protein interaction to the transcription factor Pax6 and thereby represses the activity of this transcriptional activator, leading to inhibition of rat glucagon gene transcription in α-cells (Schinner et al., 2002; Krätzner et al., 2008). Pax6 is expressed also in β-cells (St-Onge et al., 1997) and binds to the rat insulin I gene promoter at about −310 (Knepel et al., 1991; Sander et al., 1997). This Pax6 binding site may mediate the activation of the rat insulin I gene promoter when Pax6 is expressed in the heterologous JEG cells (this study). The rat glucagon gene promoter was more markedly activated by Pax6 (this study), most likely because it contains two Pax6 binding sites that interact synergistically (Knepel et al., 1991; 1990; Sander et al., 1997; Beimesche et al., 1999; Grzeskowiak et al., 2000). Nuclear protein binding to the rat insulin I gene at about −310 (Knepel et al., 1991), later to be identified as Pax6 (Sander et al., 1997; (Beimesche et al., 1999), is not conserved in the human insulin gene (Yildiz et al., 1996). The human promoter may also not contain other Pax6 binding sites, as is indicated by the low Pax6-induced activation of the human insulin gene promoter when compared with that of the glucagon gene promoter (this study). The finding that the inhibitory effect of rosiglitazone depends on the DNA-binding domain of PPARγ indicates that PPAR might directly bind to the promoter (this study). Mapping experiments revealed that DNA sequences between −56 and +18 may confer PPARγ responsiveness to the human insulin gene promoter. Importantly, this fragment contains a sequence motif from −6 to +7 with similarity to a typical PPARγ response element (Figure 6A), which consists of a direct repeat of hexamer half-sites, TGACCT, spaced by one nucleotide (DR-1) (Desvergne and Wahli, 1999). This raises the possibility that PPARγ may inhibit human insulin promoter activity by competing for binding to the region around the transcription start site with the general transcription machinery. Indeed, a complex of primary islet proteins bound to the sequence of the proximal promoter element, and this binding was competed for by additional incubation with a typical PPRE sequence but not to the same extent with the mutated PPRE sequence. Thus, although the mode of inhibition remains to be verified, the present study suggests that the mechanism through which PPARγ inhibits human insulin gene transcription differs from the one at the rat glucagon gene and may target the proximal insulin promoter around the transcription start site.
Thiazolidinediones efficiently improve glycaemic control and may reduce the risk of death from any cause in type 2 diabetic patients, with significant side effects (Dormandy et al., 2005; Lago et al., 2007; Lincoff et al., 2007; Nissen and Wolski, 2007; Singh et al., 2007). Inhibition of insulin gene transcription appears to be detrimental to these patients, who already suffer from insulin deficiency, and to be in opposition to several lines of evidence suggesting that treatment with thiazolidinediones may preserve and even improve β-cell function (Walter and Lübben, 2005; Wajchenberg, 2007). However, repression by thiazolidinediones of the insulin gene may in fact be in keeping with known thiazolidinedione effects and even be beneficial for the patients. Consequently, inhibition of insulin gene transcription is in line with and may contribute to the reductions in fasting plasma insulin levels found in most clinical trials performed with rosiglitazone, pioglitazone or troglitazone in patients with type 2 diabetes mellitus (Walter and Lübben, 2005; Wajchenberg, 2007). These reductions have so far been attributed solely to the increase in insulin sensitivity and the decrease in blood glucose concentrations (Walter and Lübben, 2005; Wajchenberg, 2007). Furthermore, insulin gene repression may protect β-cells from some of the damage induced by chronic overstimulation. Firm genetic and other lines of evidence indicate that β-cells may be especially sensitive to adverse effects of perturbed endoplasmic reticulum function (Wajchenberg, 2007). Glucose-mediated stimulation of proinsulin biosynthesis promotes some endoplasmic reticulum stress because it imposes a load on the folding and protein processing machinery of the endoplasmic reticulum. In hypersecretory states, such as insulin resistance, glucose intolerance and frank diabetes mellitus, proinsulin and other client proteins are translocated into the lumen of the endoplasmic reticulum in excess of the folding capacity of the organelle, inducing a state of severe endoplasmic reticulum stress with induction of programmed cell death. This mechanism may well contribute to the decline in β-cell function and mass in the insulin-resistant patient (Wajchenberg, 2007). Inhibition of insulin gene transcription by thiazolidinediones, we presume, decreases protein flux through the endoplasmic reticulum of the β-cell and thus reduces endoplasmic reticulum stress. Evidence of endoplasmic reticulum stress such as the accumulation of electron-dense material in the endoplasmic reticulum and distorted organelle morphology in diabetic islets are indeed reduced by thiazolidinedione treatment (Diani et al., 1984; 2004; Walter and Lübben, 2005; Wajchenberg, 2007). Similarly, induction of β-cell rest by KATP-channel openers has been shown to improve β-cell function (Ritzel et al., 2004). In conclusion, in this study we show an inhibition of insulin gene transcription by thiazolidinediones. Our results suggest that this inhibition is mediated by the proximal promoter region.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (SFB402/A3, GRK 335).
Glossary
Abbreviations:
- PPARγ
peroxisome proliferator-activated receptor γ
- RXRα
retinoid X receptor α
Statement of conflicts of interest
The authors state no conflict of interest.
References
- Beimesche S, Neubauer A, Herzig S, Grzeskowiak R, Diedrich T, Cierny I, et al. Tissue-specific transcriptional activity of a pancreatic islet cell specific enhancer sequence/Pax6-binding site determined in normal adult tissues in vivo using transgenic mice. Mol Endocrinol. 1999;13:718–728. doi: 10.1210/mend.13.5.0273. [DOI] [PubMed] [Google Scholar]
- Bollheimer LC, Troll S, Landauer H, Wrede CE, Schölmerich J, Buettner R. Insulin sparing effects of troglitazone in rat pancreatic islets. J Mol Endocrinol. 2003;31:61–69. doi: 10.1677/jme.0.0310061. [DOI] [PubMed] [Google Scholar]
- Braissant O, Wahli W. Differential expression of peroxisome proliferator-activated receptor-alpha, -beta, and -gamma during rat embryonic development. Endocrinology. 1998;139:2748–2754. doi: 10.1210/endo.139.6.6049. [DOI] [PubMed] [Google Scholar]
- Brunham LR, Kruit JK, Pape TD. Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinediones. Nat Med. 2007;13:340–347. doi: 10.1038/nm1546. [DOI] [PubMed] [Google Scholar]
- Desvergne B, Wahli B. Peroxisome proliferators-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–688. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- Diani AR, Peterson T, Sawada GA, Wyse BM, Gilchrist BJ, Hearron AE, et al. Ciglitazone, a new hypoglycaemic agent. 4. Effect on pancreatic islets of C57BL/6J ob/ob and C57BL/KsJ-db/db mice. Diabetologia. 1984;27:225–234. doi: 10.1007/BF00273811. [DOI] [PubMed] [Google Scholar]
- Diani AR, Sawada G, Wyse B, Murray FT, Khan M. Pioglitazone preserves pancreatic islets structure and insulin secretory function in three murine models of type 2 diabetes. Am J Physiol Endocrinol Metab. 2004;286:E116–E122. doi: 10.1152/ajpendo.00331.2003. [DOI] [PubMed] [Google Scholar]
- Dormandy JA, Charbonnel B, Eckland DJ, Erdmann E, Massi-Benedetti M, Moules IK, et al. SEcondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised trial. Lancet. 2005;366:1279–1289. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- Fromont-Racine M, Bucchini D, Madsen O, Desbois P, Line S, Nielsen JH, et al. Effect of 5′-flanking sequence deletions on expression of the human insulin gene in transgenic mice. Mol Endocrinol. 1990;4:669–677. doi: 10.1210/mend-4-5-669. [DOI] [PubMed] [Google Scholar]
- Gervois P, Fruchart JC, Staels B. Drug insight: mechanisms of action and therapeutic applications for agonist of peroxisome proliferator-activated receptors. Nat Clin Pract Endocrinol Metab. 2007;3:145–156. doi: 10.1038/ncpendmet0397. [DOI] [PubMed] [Google Scholar]
- Grzeskowiak R, Amin J, Oetjen E, Knepel W. Insulin responsiveness of the glucagon gene conferred by interactions between proximal promoter and more distal enhancer like elements involving the paired-domain transcription factor Pax6. J Biol Chem. 2000;275:30037–30045. doi: 10.1074/jbc.M000984200. [DOI] [PubMed] [Google Scholar]
- Hay CW, Docherty K. Comparative analysis of insulin gene promoters implications for diabetes research. Diabetes. 2006;50:3201–3213. doi: 10.2337/db06-0788. [DOI] [PubMed] [Google Scholar]
- Juhl CB, Hollingdal M, Porksen N, Prange A, Lönnqvist F, Schmitz O. Influence of rosiglitazone treatment on beta-cell function in type 2 diabetes: evidence of an increased ability of glucose to entrain high-frequency insulin pulsatility. J Clin Endocrinol Metab. 2003;88:3794–3800. doi: 10.1210/jc.2002-021181. [DOI] [PubMed] [Google Scholar]
- Kim HI, Kim JW, Kim SH, Cha JY, Kim KS, Ahn YH. Identification and functional characterization of the peroxisomal proliferators response element in rat GLUT2 promoter. Diabetes. 2000;49:1517–1524. doi: 10.2337/diabetes.49.9.1517. [DOI] [PubMed] [Google Scholar]
- Kim HI, Cha JY, Kim SY, Kim JW, Roh KJ, Seong JK, et al. Peroxisomal proliferator activated receptor-γ upregulates glucokinase gene expression in β-cells. Diabetes. 2002;51:676–685. doi: 10.2337/diabetes.51.3.676. [DOI] [PubMed] [Google Scholar]
- Knepel W, Jepeal L, Habener JF. A pancreatic islet cell-specific enhancer-like element in the glucagon gene contains two domains binding distinct cellular proteins. J Biol Chem. 1990;265:8725–8735. [PubMed] [Google Scholar]
- Knepel W, Vallejo M, Chafitz JA, Habener JF. The pancreatic islets-specific glucagon G3 transcription factors recognize control elements in the rat somatostatin and insulin 1 gene. Mol Endocrinol. 1991;5:1457–1466. doi: 10.1210/mend-5-10-1457. [DOI] [PubMed] [Google Scholar]
- Kostadinova R, Wahli W, Michalik L. PPARs in diseases: control mechanisms of inflammation. Curr Med Chem. 2005;12:2995–3009. doi: 10.2174/092986705774462905. [DOI] [PubMed] [Google Scholar]
- Krätzner R, Fröhlich F, Lepler K, Schröder M, Röher K, Dickel C, et al. A Peroxisome proliferator-activated receptor γ-retinoid X receptor heterodimer physically interacts with the transcriptional activator PAX6 to inhibit glucagon gene transcription. Mol Pharmacol. 2008;73:509–517. doi: 10.1124/mol.107.035568. [DOI] [PubMed] [Google Scholar]
- Lago RM, Singh PP, Nesto RW. Congestive heart failure and cardiovascular death in patients with prediabetes and type 2 diabetes given thiazolidindiones: a meta-analysis of randomised clinical trials. Lancet. 2007;370:1129–1136. doi: 10.1016/S0140-6736(07)61514-1. [DOI] [PubMed] [Google Scholar]
- Li M, Pascual G, Glass CK. Peroxisome proliferators-activated receptor gamma dependent repression of the inducible nitric oxide synthase gene. Mol Cell Biol. 2000;20:4699–4707. doi: 10.1128/mcb.20.13.4699-4707.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA. 2007;298:1180–1188. doi: 10.1001/jama.298.10.1180. [DOI] [PubMed] [Google Scholar]
- Lupi R, Del Guerra S, Marselli L, Bugliani M, Boggi U, Mosca F, et al. Rosiglitazone prevents the impairment of human islets function induced by fatty acids: evidence for a role of PPARgamma2 in the modulation of insulin secretion. Am J Physiol Endocrinol Metab. 2004;286:E560–E567. doi: 10.1152/ajpendo.00561.2002. [DOI] [PubMed] [Google Scholar]
- Melloul D, Marshak S, Cerasi E. Regulation of insulin gene transcription. Diabetologia. 2002;45:309–326. doi: 10.1007/s00125-001-0728-y. [DOI] [PubMed] [Google Scholar]
- Nakamichi Y, Kikuta T, Ito E, Ohara-Imaizumi M, Nishikawa C, Ishida H, et al. PPAR-gamma overexpression suppresses glucose-induced proinsulin biosynthesis and insulin release synergistically in MIN6 cells. Biochem Biophys Res Commun. 2003;306:832–836. doi: 10.1016/s0006-291x(03)01045-3. [DOI] [PubMed] [Google Scholar]
- Natali A, Ferranini E. Effects of metformin and thiazolidindiones on suppression of hepatic glucose production and stimulation of glucose uptake in type 2 diabetes: a systematic review. Diabetologia. 2006;49:434–441. doi: 10.1007/s00125-006-0141-7. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- Oetjen E, Baun D, Beimesche S, Krause D, Cierny I, Blume R, et al. Inhibition of human insulin gene transcription by the immunosuppressive drugs cyclosporin A and tacrolimus in primary, mature islets of transgenic mice. Mol Pharmacol. 2003a;63:1289–1295. doi: 10.1124/mol.63.6.1289. [DOI] [PubMed] [Google Scholar]
- Oetjen E, Grapentin D, Blume R, Seeger M, Krause D, Eggers A, et al. Regulation of human insulin gene transcription by the immunosuppressive drugs cyclosporin A and tacrolimus at concentrations that inhibit calcineurin activity and involving the transcription factor CREB. Naunyn-Schmiedeberg's. Arch Pharmacol. 2003b;367:227–236. doi: 10.1007/s00210-003-0694-7. [DOI] [PubMed] [Google Scholar]
- Oetjen E, Blume R, Cierny I, Schlag C, Kutschenko A, Krätzner R, et al. Inhibition of MafA transcriptional activity and human insulin gene transcription by interleukin-1β and mitogen-activated protein kinase kinase kinase in pancreatic islet beta cells. Diabetologia. 2007;50:1678–1687. doi: 10.1007/s00125-007-0712-2. [DOI] [PubMed] [Google Scholar]
- Ovalle F, Bell DS. Effect of rosiglitazone versus insulin on the pancreatic beta-cell function of subjects with type 2 diabetes. Diabetes Care. 2004;27:2585–2589. doi: 10.2337/diacare.27.11.2585. [DOI] [PubMed] [Google Scholar]
- Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Weesely O, Bock R, et al. DNA binding of the glucocortiocid receptor is not essential for survival. Cell. 1998;93:531–541. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- Richardson H, Campbell SC, Smith SC, Macfarlane WM. Effects of rosiglitazone on pancreatic beta cell gene expression. Diabetologia. 2006;49:685–696. doi: 10.1007/s00125-006-0155-1. [DOI] [PubMed] [Google Scholar]
- Ricote M, Huang J, Fajas L, Welch J, Najib J, Witzum JL, et al. Expression of the peroxisome proliferator activated receptor gamma (PPARγ) in human artherosclerosis and regulation in macrophages by colony stimulating factors and oxidized low density lipoprotein. Proc Natl Acad Sci USA. 1998;95:7614–7619. doi: 10.1073/pnas.95.13.7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzel RA, Hansen JB, Veldhuis JD, Butler PC. Induction of beta-cell rest by a Kir6.2/SUR1-selective K(ATP)-channel opener preserves beta-cell insulin stores and insulin secretion in human islets cultured at high (11 mmol·L−1) glucose. J Clin Endocrinol Metab. 2004;89:795–805. doi: 10.1210/jc.2003-031120. [DOI] [PubMed] [Google Scholar]
- Rosen ED, Kulkarni RN, Sarraf P, Ozcan U, Okada T, Hsu T, et al. Targeted elimination of peroxisome proliferator-activated receptor γ in β cells leads to abnormalities in islet mass without compromising glucose homeostasis. Mol Cell Biol. 2003;23:7222–7229. doi: 10.1128/MCB.23.20.7222-7229.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander M, Neubüser A, Kalamaras J, Ee HC, Martin GR, German MS. Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev. 1997;11:1662–1673. doi: 10.1101/gad.11.13.1662. [DOI] [PubMed] [Google Scholar]
- Santerre RF, Cook RA, Crisel RM, Sharp JD, Schmidt RJ, Williams DC, et al. Insulin synthesis in a clonal cell line of simian virus 40-transformed hamster pancreatic β cells. Proc Natl Acad Sci USA. 1981;78:4339–4343. doi: 10.1073/pnas.78.7.4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinner S, Dellas C, Schröder M, Heinlein CA, Chang C, Fischer J, et al. Repression of glucagon gene transcription by peroxisome proliferator-activated receptor γ through inhibition of Pax6 transcriptional activity. J Biol Chem. 2002;277:1941–1948. doi: 10.1074/jbc.M109718200. [DOI] [PubMed] [Google Scholar]
- Schwaninger M, Lux G, Blume R, Oetjen E, Hidaka H, Knepel W. Membrane depolarisation and calcium influx induce glucagon gene transcription in pancreatic islet cells through cyclic AMP-responsive element. J Biol Chem. 1993;268:5168–5177. [PubMed] [Google Scholar]
- Siemann G, Blume R, Grapentin D, Oetjen E, Knepel W. Inhibition of cyclic AMP response element-binding protein/cyclic AMP response element-mediated transcription by the immunosuppressive drugs cyclosporin A and FK506 depends on the promoter context. Mol Pharmacol. 1999;55:1094–1100. doi: 10.1124/mol.55.6.1094. [DOI] [PubMed] [Google Scholar]
- Singh S, Lke YK, Furberg CD. Long-term risk of cardiovascular events with rosiglitazone: a meta-analysis. JAMA. 2007;298:1189–1195. doi: 10.1001/jama.298.10.1189. [DOI] [PubMed] [Google Scholar]
- St-Onge L, Sosa-Pineda B, Chowdhury K, Mansouri A, Gruss P. Pax6 is required for differentiation of glucagon-producing α-cells in mouse pancreas. Nature. 1997;387:406–409. doi: 10.1038/387406a0. [DOI] [PubMed] [Google Scholar]
- Tuckermann JP, Reichardt HM, Arribas R, Richter KH, Schütz G, Angel P. The DNA binding-independent function of the glucocorticoid receptor mediates repression of AP-1-dependent genes in skin. J Cell Biol. 1999;147:1365–1370. doi: 10.1083/jcb.147.7.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandewalle B, Moerman E, Lefebvre B, Defrance F, Gmyr V, Lukowiak B, et al. PPARγ-dependent and–independent effects of rosiglitazone on lipotoxic human pancreatic islets. Biochem Biophys Res Commun. 2008;366:1096–1101. doi: 10.1016/j.bbrc.2007.12.088. [DOI] [PubMed] [Google Scholar]
- Wajchenberg BL. β-cell failure in diabetes and preservation by clinical treatment. Endocr Rev. 2007;28:187–218. doi: 10.1210/10.1210/er.2006-0038. [DOI] [PubMed] [Google Scholar]
- Walter H, Lübben G. Potential role of oral thiazolidindione therapy in preserving beta cell function in type 2 diabetes mellitus. Drugs. 2005;65:1–13. doi: 10.2165/00003495-200565010-00001. [DOI] [PubMed] [Google Scholar]
- Yildiz N, Diedrich T, Knepel W. Nuclear protein binding and functional activity of a variant insulin gene found in non-insulin-dependent diabetes mellitus. Exp Clin Endocrinol Diabetes. 1996;104:218–227. doi: 10.1055/s-0029-1211446. [DOI] [PubMed] [Google Scholar]
- Zeender E, Maedler K, Bosco D, Berney T, Donath MY, Halban PA. Pioglitazone and sodium salicylate protect human beta-cells against apoptosis and impaired function induced by glucose and interleukin-1beta. J Clin Endocrinol Metab. 2004;89:5059–5066. doi: 10.1210/jc.2004-0446. [DOI] [PubMed] [Google Scholar]