

Abstract
Background and purpose:
The transcription factor nuclear factor-κB (NF-κB) has been linked to the cell growth, apoptosis and cell cycle progression. NF-κB blockade induces apoptosis of cancer cells. Therefore, NF-κB is suggested as a potential therapeutic target for cancer. Here, we have evaluated the anti-cancer potential of a novel NF-κB inhibitor, quinoclamine (2-amino-3-chloro-1,4-naphthoquinone).
Experimental approach:
In a large-scale screening test, we found that quinoclamine was a novel NF-κB inhibitor. The global transcriptional profiling of quinoclamine in HepG2 cells was therefore analysed by transcriptomic tools in this study.
Key results:
Quinoclamine suppressed endogenous NF-κB activity in HepG2 cells through the inhibition of IκB-α phosphorylation and p65 translocation. Quinoclamine also inhibited induced NF-κB activities in lung and breast cancer cell lines. Quinoclamine-regulated genes interacted with NF-κB or its downstream genes by network analysis. Quinoclamine affected the expression levels of genes involved in cell cycle or apoptosis, suggesting that quinoclamine exhibited anti-cancer potential. Furthermore, quinoclamine down-regulated the expressions of UDP glucuronosyltransferase genes involved in phase II drug metabolism, suggesting that quinoclamine might interfere with drug metabolism by slowing down the excretion of drugs.
Conclusion and implications:
This study provides a comprehensive evaluation of quinoclamine by transcriptomic analysis. Our findings suggest that quinoclamine is a novel NF-κB inhibitor with anti-cancer potential.
Keywords: quinoclamine, nuclear factor-κB, microarray, cell cycle, UDP glucuronosyltransferases
Introduction
The transcription factor nuclear factor-κB (NF-κB) has been linked to the control of cell growth, apoptosis and cell cycle progression (Wu and Kral, 2005). Activated NF-κB promotes the expressions of over 150 downstream genes, which are involved in immune responses, stress responses, cellular proliferation, anti-apoptosis, cell migration and angiogenesis (Pahl, 1999). NF-κB is activated in many tumours, including breast cancers, liver epithelial tumours, pancreatic adenocarcinomas, cervical carcinomas, melanomas and haematological malignancies (Sovak et al., 1997; Wang et al., 1999; Arsura et al., 2000; Garg and Aggarwal, 2002; Nair et al., 2003; Ueda and Richmond, 2006). Roles of NF-κB in cancer cells include regulation of cell proliferation, control of apoptosis, promotion of angiogenesis and stimulation of invasion/metastasis (Karin, 2006; Kim et al., 2006). Because NF-κB blockade has been documented to induce apoptosis of cancer cells, NF-κB is suggested as a potential therapeutic target for cancer (Karin, 2006; Kim et al., 2006). As expected, NF-κB inhibitors exhibit therapeutic potentials in these tumours.
Many natural products, which are polyphenols, terpenes, alkaloids, flavonoids or phenolics, are potent NF-κB inhibitors (Aggarwal and Shishodia, 2006). They exhibit anti-proliferative effects by inhibiting IκB kinase, suppressing the phosphorylation or degradation of IκB, blocking the translocation of NF-κB into the nucleus, or blocking the DNA binding ability of NF-κB (Hsu et al., 2004a; 2004b; Rahman and Sarkar, 2005; Ichikawa and Aggarwal, 2006; Ishiguro et al., 2007). Naphthoquinones are also potent NF-κB inhibitors. For examples, plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) has been shown to suppress NF-κB activation through p65 nuclear translocation and IκB-α kinase activation (Sandur et al., 2006). Shikonin (5,8-dihydroxy-2-(1-hydroxy-4-methyl-3-pentenyl)-1,4-naphthoquinone) induces apoptosis of oral cancer cells through the inhibition of NF-κB and the subsequent activation of caspases (Min et al., 2008). PPM-18 (α-benoylamino-1,4-naphthoquinone) is a potent anti-inflammatory agent, which blocks the binding of NF-κB to promoter (Yu et al., 1997). Recently, in a large-scale screening test, we found that quinoclamine (2-amino-3-chloro-1,4-naphthoquinone), a chemically synthesized naphthoquinone compound, was a novel NF-κB inhibitor (see Figure 1). Quinoclamine has been shown to induce the differentiation of human leukemia HL-60 cells via protein kinase C activation (Bae and Choung, 1996; Kwon and Choung, 1997). Moreover, quinoclamine is thought to be a potent anti-malarial agent and its derivatives also display anti-platelet, anti-inflammatory and anti-allergic activities (Lien et al., 1997; Kapadia et al., 2001). In this study, we applied a comprehensive transcriptomic analysis to determine the NF-κB-downstream effects and particularly effects on drug metabolism of the novel NF-κB inhibitor, quinoclamine.
Figure 1.
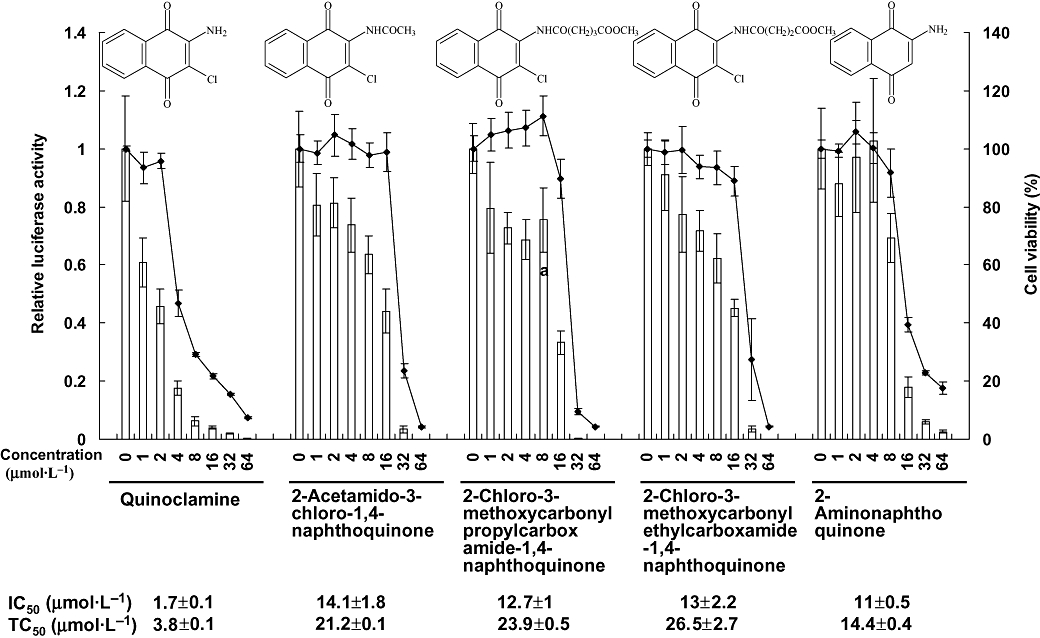
Effects of five naphthoquinone derivatives on nuclear factor-κB (NF-κB) activities and cell viabilities in HepG2 cells. Recombinant HepG2/NF-κB cells were treated with various amounts of naphthoquinone derivatives for 24 h. The luciferase activity and cell viability were then evaluated by luciferase assay and MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay, respectively. Bars represent the relative luciferase activity, and lines represent the cell viability. Structures of naphthoquinone derivatives are shown on the top. IC50 value and TC50 value of each compound are shown on the bottom. Values are mean ± standard error of four independent assays.
Methods
Cell culture
Human hepatocellular carcinoma cell lines (HepG2 and Hep3B), normal liver cell line (Chang liver), breast adenocarcinoma cell line (MCF7) and lung epithelial cell line (A-549) were obtained from Bioresource Collection and Research Center (Hsinchu, Taiwan). Recombinant HepG2/NF-κB was constructed as described previously (Hsiang et al., 2007). Hep3B, Chang liver, MCF7 and A-549 cells were transiently transfected with 5 µg of pNF-κB-Luc plasmid DNA (Stratagene, La Jolla, CA, USA) by SuperFect® transfection reagent (Qiagen, Valencia, CA, USA). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies, Gaithersburg, MD, USA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (HyClone, Logan, UT, USA), 100 µg·mL−1 streptomycin and 100 units·mL−1 penicillin in 75 cm2 tissue culture flasks in a humidified incubator at 37°C with 5% CO2.
Naphthoquinone and 12-O-tetradecanoylphorbol-13-acetate (TPA) treatments
The chemically synthesized naphthoquinone derivatives were synthesized as described previously (Lien et al., 1997). These compounds were dissolved in dimethyl sulphoxide to a final concentration of 200 mmol·L−1 and stored at −30°C. TPA was purchased from Sigma (St. Louis, MO, USA) and dissolved in ethanol at 0.5 mg·mL−1. The cells were subcultured and transferred into 25 cm2 tissue culture flasks 24 h before compound treatments. We treated the cells with various amounts of compounds in the absence or presence of 100 ng·mL−1 TPA when cells reached a 100% confluence. The treated cells were then kept in a humidified incubator at 37°C with 5% CO2 for 24 h before total RNA extraction.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
MTT was purchased from Sigma (St. Louis, MO, USA) and dissolved in phosphate-buffered saline (137 mmol·L−1 NaCl, 1.4 mmol·L−1 KH2PO4, 4.3 mmol·L−1 Na2HPO4, 2.7 mmol·L−1 KCl, pH 7.2). Cell viability was monitored by MTT colorimetric assay as described previously (Hsiang et al., 2005). Cell viability (%) was calculated by OD of naphthoquinone-treated cells/OD of solvent-treated cells. The TC50 value was determined as the concentration of compound required to inhibit cell viability by 50%.
Luciferase assay
Cells were cultured in 24-well plates for 24 h, washed with DMEM and treated with different concentrations of naphthoquinone derivatives in the absence or presence of 100 ng·mL−1 TPA for an additional 24 h. The cells were then washed with ice-cold phosphate-buffered saline, lysed with 350 µL Triton lysis buffer (50 mmol·L−1 Tris-HCl, 1% Triton X-100, 1 mmol·L−1 dithiothreitol, pH 7.8) and collected with a cell scraper. The luciferase activity was measured as described previously (Ho et al., 2007). The IC50 value was determined as the concentration of compound required to inhibit NF-κB activity at 50%.
Western blot analysis
HepG2/NF-κB cells were treated with various amounts of quinoclamine in DMEM for 30 min and then lysed with 250 µL sample buffer (62.5 mmol·L−1 Tris-HCl, 2% sodium dodecyl sulphate, 10% glycerol, 50 mmol·L−1 dithiothreitol, 0.1% bromophenol blue, pH 6.8). The proteins (2 µg) were separated by 10% sodium dodecyl sulphate-polyacrylamide gel electrophoresis, and the protein bands were then transferred electrophoretically to nitrocellulose membranes. The membranes were probed with polyclonal antibodies against p65, IκB-α and phosphorylated IκB-α (Cell Signaling Technology, Beverly, MA, USA). The bound antibody was detected with peroxidase-conjugated anti-rabbit antibody followed by chemiluminescence (ECL system, Amersham, Buckinghamshire, UK) and exposed by autoradiography. The intensities of bands on the gels were calculated by Gel-Pro® Analyzer (Media Cybernetics Inc., Silver Spring, MD, USA)
Total RNA isolation
Total RNA was extracted from cells treated with or without quinoclamine by RNeasy Mini kit (Qiagen, Valencia, CA, USA). Total RNA was quantified by using the Beckman DU800 spectrophotometer (Beckman Coulter, Fullerton, CA, USA). Samples with A260/A280 ratios greater than 1.8 were further evaluated by using Agilent 2100 bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The RNA samples with a RNA integrity number greater than 8.0 were accepted for microarray analysis.
Microarray analysis
Microarray analysis was performed as described previously (Cheng et al., 2008). Briefly, fluorescent RNA targets were prepared from 5 µg of total RNA by using MessageAmp™ aRNA kit (Ambion, Austin, TX, USA) and Cy5 dye (Amersham Pharmacia, Piscataway, NJ, USA). Fluorescent targets were hybridized to the Human Whole Genome OneArray™ (Phalanx Biotech Group, Hsinchu, Taiwan) overnight, non-specific targets were washed away, and the slides were dried by centrifugation and scanned by an Axon 4000 scanner (Molecular Devices, Sunnyvale, CA, USA). The Cy5 fluorescent intensity of each spot was analysed by Genepix 4.1 software (Molecular Devices, Sunnyvale, CA, USA). The signal intensity of each spot was corrected by subtracting background signals in surroundings. We filtered out spots that signal-to-noise ratio was less than 0 or control probes. Spot intensities were then normalized by the Limma package of the R programme using ‘normalizeQuantiles’ function in single channel data and ‘normalizeWithinArray’ function in dual channel data (Smyth, 2005). Normalized data were tested for statistically significant genes according to the Limma package manual (Smyth, 2005). The statistically significant genes were considered as differentially expressed genes when the fold change was over twofold. These differentially expressed genes in both single and dual channel experiments were then analysed to find significant gene ontology (GO) categories by WebGestalt tool on Gene Ontology Tree Machine web site (Zhang et al., 2005). Number of replicates for single and dual channel experiments was three.
Network analysis
The target genes, which are regulated by NF-κB, were selected from the web site (http://rulai.cshl.edu/cgi-bin/TRED/tred.cgi?process=searchTFGeneForm). To estimate the overall regulatory effect of quinoclamine on these target genes, we used the ‘geneSetTest’ function implemented in the Limma package of the R programme to compare the absolute t-statistic values for these target genes with those for all genes in the single channel hybridization experiment. These target genes were then combined with the differentially expressed genes, which belong to the GO category of ‘regulation of biological process’, to investigate their relationship with NF-κB. We used BiblioSphere Pathway Edition software (Genomatix Applications, http://www.genomatix.de/index.html) to construct the interaction networks between NF-κB-downstream genes and part of the differentially expressed genes. To test the transcriptional significance for the first neighbour nodes of the NFKB1 and the other ones in this network, we used thee ‘geneSetTest’ function in the Limma package to compare the absolute t-statistic values for these two gene sets with those for all genes in the single channel hybridization experiment. Finally, we visualized the interaction network by Cytoscape software (Shannon et al., 2003).
Analysis of expression levels of genes associated with drug metabolism
Two hundred and nineteen genes associated with drug metabolism were selected from ‘The Pharmacogenetics and Pharmacogenomics Knowledge Base’ web site (https://www.pharmgkb.org/index.jsp). Among these genes, we analysed the expression levels of phase I drug metabolism genes, including alcohol dehydrogenases, aldehyde dehydrogenases and cytochrome P450 genes, and phase II drug metabolism genes, including glutathione S-transferases, sulphotransferase and UDP glucuronosyltransferase (UGT) genes.
Quantitative real-time polymerase chain reaction (qPCR)
The expression levels of UGTs and cancer progression-associated genes were further validated by qPCR. RNA samples were reverse-transcribed for 30 min at 42°C with High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). qPCR was performed using 1 µL of cDNA, 2X SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA) and 500 nmol·L−1 of forward and reverse primers. The reaction condition was followed: 2 min at 50°C, 10 min at 95°C and 40 cycles of 15 s at 95°C, 1 min at 60°C. Each assay was run on an Applied Biosystems 7300 Real-Time PCR system in triplicates. Fold changes were calculated using the comparative CT method. The primer set for each gene is shown in Table 1.
Table 1.
Primer sequences of genes for qPCR
| Gene | Primer sequences |
|---|---|
| UGT genes | |
| UGT1A5 | Sense: 5′-CAACGGGAAGCCACTATCTCAGG-3′ |
| Antisense: 5′-CCACAATTCCATGTTCTCCAGAAGC-3′ | |
| UGT2A1 | Sense: 5′-CATTGCTCACATGAAGGCCAAAGG-3′ |
| Antisense: 5′-GCGCTAAGCAAATCCACACTTGTC-3′ | |
| UGT2B4 | Sense: 5′-CCACTGCAAACCTGCCAAACC-3′ |
| Antisense: 5′-TCTTCTGACGTGTTACTGACCATCG-3′ | |
| UGT2B7 | Sense: 5′-TTGCCGATCAACCTGATAACATTGC-3′ |
| Antisense: 5′-AGCAAGTCTGTACTCGACATTGTG-3′ | |
| UGT2B15 | Sense: 5′-GGAAATGGAAGAGTTTGTGCAGAGC-3′ |
| Antisense: 5′-GGATCTGGGCAAGGGCTGATG-3′ | |
| UGT2B11 | Sense: 5′-TGCTTGTCATAAGGCAGACATCATC-3′ |
| Antisense: 5′-TGAGGTGACTGTACTGGCATCTTC-3′ | |
| Cancer progression-associated genes | |
| CCNA2 | Sense: 5′-AACAGCCAGACATCACTAACAG-3′ |
| Antisense: 5′-CTCAGCACTGACATGGAAGAC-3′ | |
| CDKN3 | Sense: 5′-GAGGGACTCCTGACATAGCC-3′ |
| Antisense: 5′-AGACAAGCAGCTACAAGACAAG-3′ | |
| ESCO2 | Sense: 5′-TCAGACTGAAGAGAAGAAAGCG-3′ |
| Antisense: 5′-TGGTGTTGGGTCAGAAAATGC-3′ | |
| EXT1 | Sense: 5′-AGGAGACAATGATGGGACAGAC-3′ |
| Antisense: 5′-AGTGGATCAGCGGCATGTAG-3′ | |
| LATS1 | Sense: 5′-GTGTGATTGGTGGAGTGTTGG-3′ |
| Antisense: 5′-TGTGGTGGAATGTGAAGAGATG-3′ | |
| PAFAH1B1 | Sense: 5′-TGCTGATGACAAGACCCTACG-3′ |
| Antisense: 5′-TGATCTACGCTGCCAGTGAC-3′ | |
| PLCB1 | Sense: 5′-GCTTAATCTTCGGCAAGAACAG-3′ |
| Antisense: 5′-CTGACACTCTTCTGCGACATC-3′ | |
| PRKCA | Sense: 5′-CCTATGGCGTCCTGTTGTATG-3′ |
| Antisense: 5′-CAGATAGAAACAGCCTCCTTGG-3′ | |
| Endogenous control | |
| GAPDH | Sense: 5′-ACACCCACTCCTCCACCTTT-3′ |
| Antisense: 5′-TAGCCAAATTCGTTGTCATACC-3′ | |
CCNA2, cyclin A2; CDKN3, cyclin-dependent kinase inhibitor 3; LATS1, large tumour suppressor homologue 1; PRKCA, protein kinase C alpha; qPCR, quantitative real-time polymerase chain reaction; UGT, UDP glucuronosyltransferase.
Statistical analysis
Data were presented as mean ± standard error. Student's t-test was used for comparisons between two experiments. A value of P < 0.05 was considered statistically significant.
Results
Naphthoquinone derivatives inhibited NF-κB activities in HepG2 cells
We tested the NF-κB activities and cell viabilities of HepG2/NF-κB cells, which were treated with 93 chemically synthesized naphthoquinone, chalcone, pyrazole or quinoline compounds. Among them, five compounds that belong to naphthoquinones were more effective than others. The IC50 values and TC50 values of these five compounds are listed in Figure 1. The most effective NF-κB inhibitor was quinoclamine, with the IC50 and TC50 values of 1.7 ± 0.1 and 3.8 ± 0.1 µmol·L−1 respectively. Therefore, we applied oligonucleotide microarray experiments to analyse the expression profiles of quinoclamine in HepG2 cells.
Quinoclamine suppressed TPA-induced NF-κB activities in different cancer cell lines
TPA is one of the well-accepted agents for studying the mechanism of carcinogenesis (Furstenberger et al., 1981). We then examined whether quinoclamine was able to suppress the TPA-induced NF-κB activities. Cells were treated with quinoclamine, and the luciferase assay was performed 24 h later. As shown in Figure 2, TPA induced NF-κB activations in HepG2 and four additional cell lines, whereas quinoclamine inhibited the TPA-induced NF-κB activities in these cell lines. However, we cannot discount the possibility that this loss of TPA-induced luciferase activity in the presence of 4 µmol·L−1 quinoclamine was due to a loss of cell viability in response to drug treatment. These findings suggested that quinoclamine suppressed induced NF-κB activities in liver, lung and breast cancer cell lines.
Figure 2.
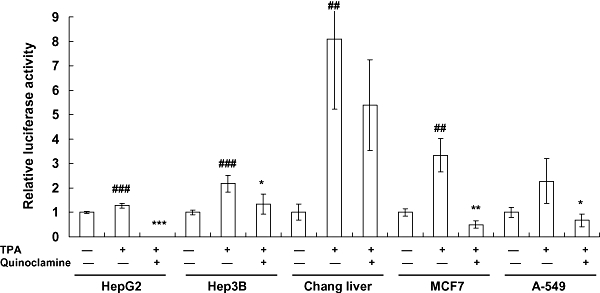
Effects of quinoclamine on 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced nuclear factor-κB (NF-κB) activities in HepG2, Hep3B, Chang liver, MCF7 and A-549 cells. Cells were cultured in 24-well plates for 24 h, washed with Dulbecco's modified Eagle's medium and treated with 4 µmol·L−1 of quinoclamine. Thirty minutes later, TPA (100 ng·mL−1) was added to the media. After a 24 h-incubation, luciferase activities were determined. Results are expressed as relative luciferase activity, which is presented as comparison with the relative luciferase unit relative to untreated cells. Values are mean ± standard error of four independent assays. ##P < 0.01, ###P < 0.001, compared with untreated cells. *P < 0.05, **P < 0.01, ***P < 0.001, compared with TPA-treated cells.
Quinoclamine inhibited IκB-α phosphorylation and p65 translocation in HepG2 cells
Nuclear factor-κB is a dimeric transcription factor that consists of p50, p52, p65, RelB and c-Rel. The activation of NF-κB is preceded by translocation of NF-κB to the nucleus following the phosphorylation and degradation of IκB-α (Barnes and Karin, 1997). To further investigate how the NF-κB signalling pathway was affected by quinoclamine, we determined the levels of p65 and IκB-α proteins in quinoclamine-treated HepG2 cells by Western blot analysis. As shown in Figure 3, quinoclamine suppressed p65 translocation to the nucleus in a dose-dependent manner. As IκB phosphorylation is a predominant pathway for NF-κB activation (Karin and Greten, 2005), we next determined the levels of IκB-α proteins in cellular extracts. The result showed that quinoclamine suppressed the phosphorylation of IκB-α in a dose-dependent manner. These findings demonstrated that quinoclamine suppressed NF-κB activation through the inhibition of IκB-α phosphorylation and p65 translocation.
Figure 3.
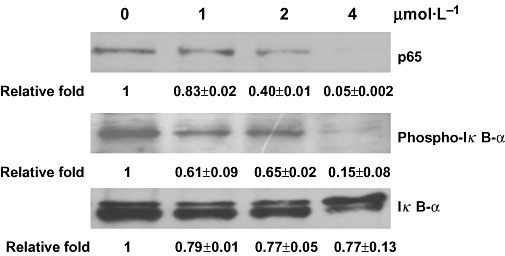
Signal transduction pathways contributing to the inhibition of nuclear factor-κB activity by quinoclamine in HepG2 cells. HepG2 cells were treated with 0, 1, 2 and 4 µmol·L−1 of quinoclamine. The phosphorylated IκB-α and non-phosphorylated IκB-α in cellular extracts were detected by Western blot. The p65 protein in nucleus was also determined by Western blot. Relative fold, which is presented as the comparison of the band intensity relative to untreated cells, is shown on the bottom. Values are mean ± standard error of two independent assays.
Quinoclamine-regulated genes might interact with NF-κB or its downstream genes
Approximately 100 target genes, which are regulated by NF-κB, were selected from web site, and the overall regulatory effect of quinoclamine on these target genes were estimated by the ‘geneSetTest’ function in the Limma package. These genes were significantly regulated (P= 0.004545). The relationship between target genes and differentially expressed genes belonging to the GO category of ‘regulation of biological process’ in single channel hybridization was analysed by Genomatix Applications software. As shown in Figure 4, the NFKB1 gene played a central role in the network. Moreover, quinoclamine-regulated genes interacted with NF-κB or its downstream genes. We further used Wilcoxon rank sum test (by the ‘geneSetTest’ function in the Limma package) to confirm the transcriptional significance for the first neighbour nodes of the NFKB1 and the other ones in this network. The down-regulated genes belonging to the GO category of ‘cell cycle’ in the network are shown in Table 2. Among these genes, those for cyclin A2 (CCNA2), cyclin-dependent kinase inhibitor 3 (CDKN3), growth arrest and DNA-damage-inducible protein alpha (GADD45A), large tumour suppressor homologue 1 (LATS1) and stromal antigen 1 (STAG1) are known to be involved in cell cycle, cell growth or apoptosis (Pagano et al., 1992; Gyuris et al., 1993; Jin et al., 2002; Xia et al., 2002; Anazawa et al., 2004).
Figure 4.
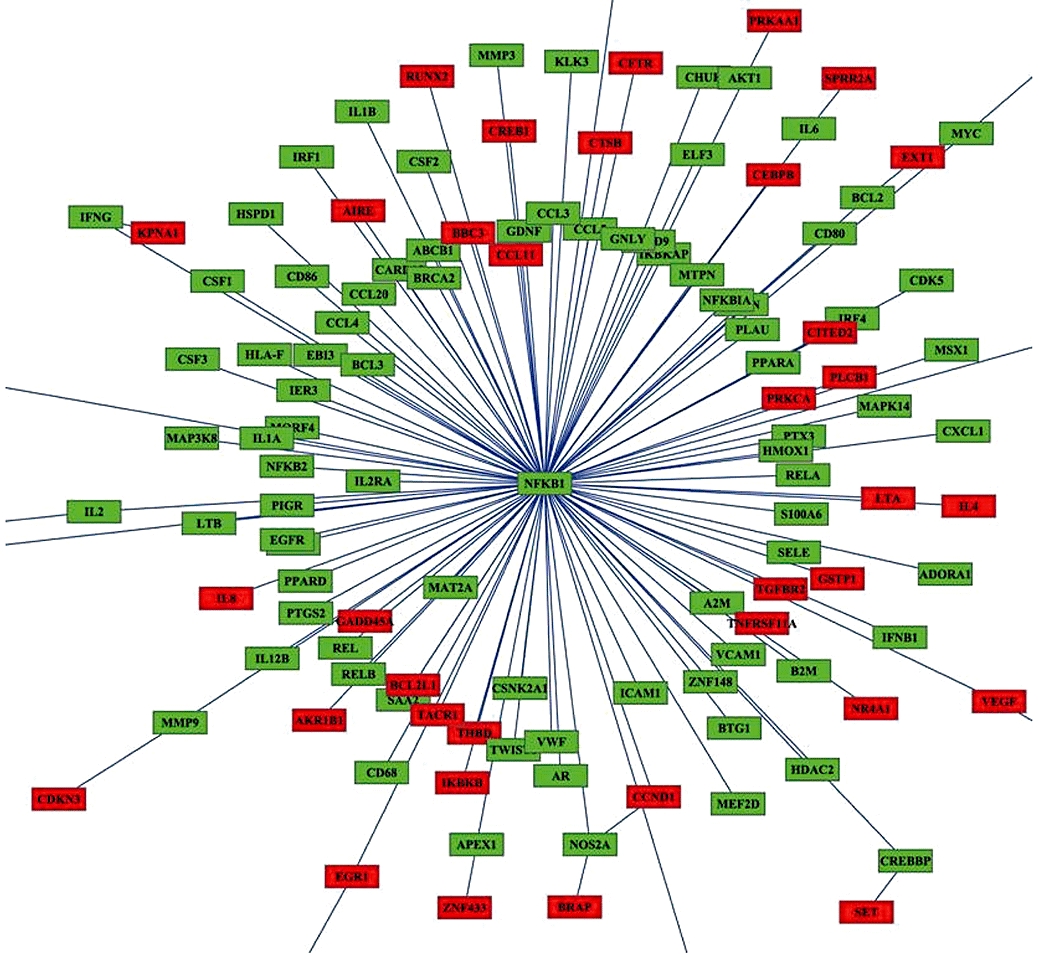
Network analysis of quinoclamine-regulated genes in HepG2 cells. The relationship between nuclear factor-κB (NF-κB) target genes and differentially expressed genes responsive to 4 µmol·L−1 quinoclamine was analysed by BiblioSphere Pathway Edition software. The connection between NF-κB and quinoclamine-regulated genes was visualized by cytoscape software. Nodes for over-expressed genes and NFKB1 are colour-coded according to their log2 expression values.
Table 2.
Expression levels of differentially expressed genes belonging to the gene ontology category of ‘cell cycle’
| Gene | Description | Fold changes | SD | P-value |
|---|---|---|---|---|
| CCNA2 | Cyclin A2 | −2.21 | 0.13 | 5.05E-06 |
| CDKN3 | Cyclin-dependent kinase inhibitor 3 (CDK2-associated dual specificity phosphatase) | −2.86 | 0.47 | 1.77E-05 |
| EXT1 | Exostoses (multiple) 1 | −2.74 | 0.37 | 5.94E-07 |
| GADD45A | Growth arrest and DNA-damage-inducible, alpha | 2.92 | 0.31 | 1.38E-08 |
| KNTC1 | Kinetochore-associated 1 | −2.01 | 0.30 | 1.16E-04 |
| LATS1 | LATS, large tumour suppressor, homolog 1 (Drosophila) | 3.66 | 1.35 | 8.87E-05 |
| MCM7 | MCM7 minichromosome maintenance deficient 7 (Saccharomyces cerevisiae) | −2.27 | 0.31 | 2.34E-06 |
| PAFAH1B1 | Platelet-activating factor acetylhydrolase, isoform Ib, alpha subunit 45 kDa | −2.44 | 0.40 | 3.82E-05 |
| PLCB1 | Phospholipase C, beta 1 (phosphoinositide-specific) | −2.45 | 0.30 | 5.94E-06 |
| PRKCA | Protein kinase C, alpha | 2.02 | 0.35 | 2.24E-05 |
| STAG1 | Stromal antigen 1 | 2.18 | 1.01 | 8.52E-05 |
| ESCO2 | Establishment of cohesion 1 homolog 2 (S. cerevisiae) | −2.00 | 0.25 | 1.18E-06 |
| GNL3 | Guanine nucleotide binding protein-like 3 (nucleolar) | 2.07 | 0.30 | 3.63E-06 |
Quinoclamine-regulated genes were associated with the cell cycle
The microarray data from single and dual channel hybridizations were analysed by the Limma package of the Bioconductor programme to get differentially expressed genes in cells responsive to quinoclamine treatments. Results showed that the transcripts of 220 genes were up-regulated and 147 genes were down-regulated in single channel hybridization experiment, while 128 genes were up-regulated and 175 genes were down-regulated in dual channel hybridization. Additionally, 123 genes were regulated in both single and dual channel hybridizations. We selected 123 genes for GO annotation by using the WebGestalt tool on Gene Ontology Tree Machine web site. Figure 5 shows that GO category associated with cell cycle was enriched. The expression levels of genes associated with cancer progression in the GO category of ‘cell cycle’ were further validated by qPCR (Table 3).
Figure 5.
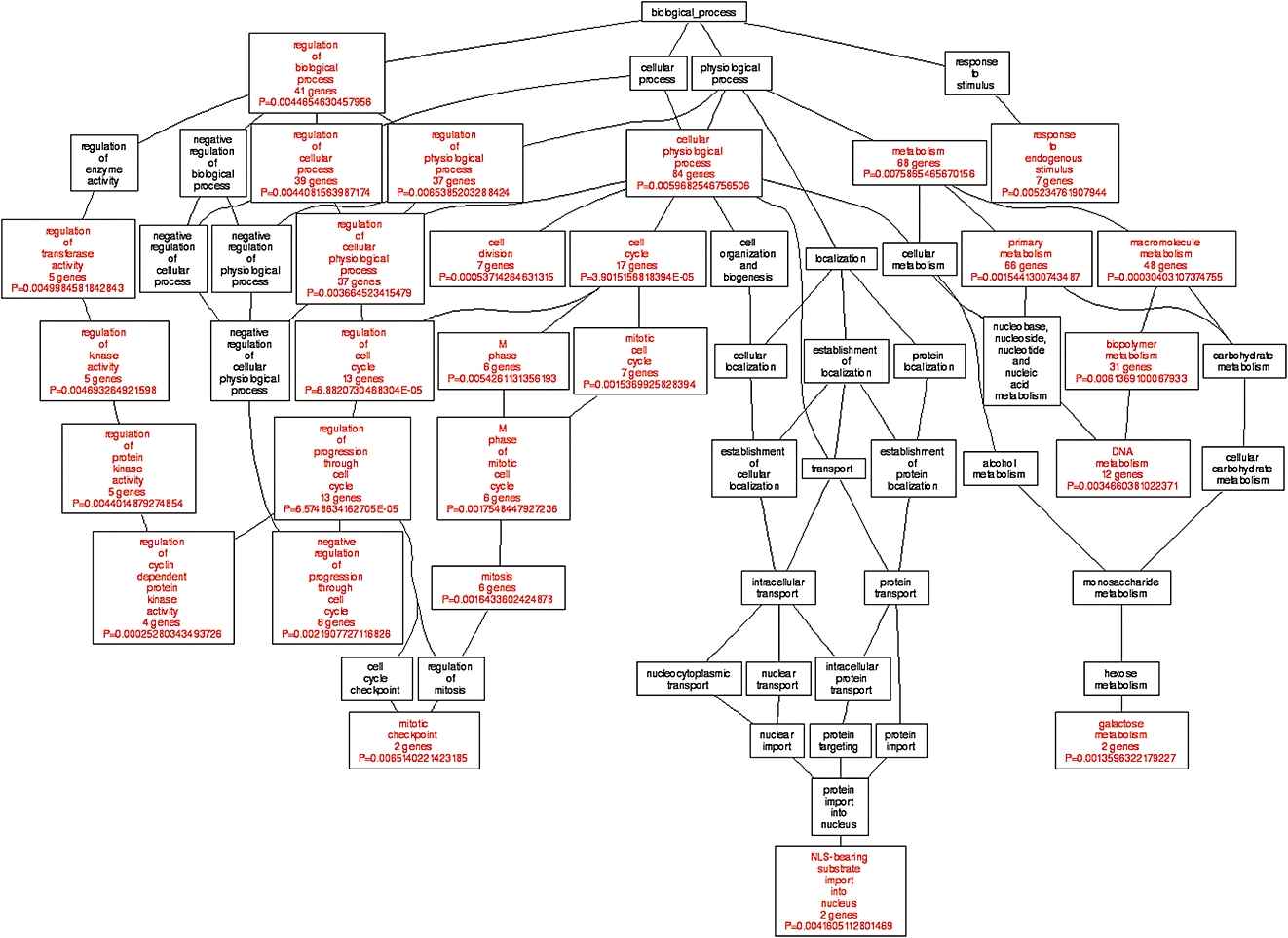
Gene ontology (GO) analysis of quinoclamine-regulated genes in HepG2 cells. Differentially expressed genes responsive to 4 µmol·L−1 quinoclamine in single and dual channel hybridization experiments were organized by using Gene Ontology Tree Machine. The tree indicates the GO structure. The enriched GO categories (P < 0.01 and at least two genes) are coloured red, and non-enriched ones are coloured black.
Table 3.
Expression levels of UGT and cancer progression-associated genes by qPCR
| Gene | Fold changes |
|---|---|
| UGT genes | |
| UGT1A10 | −1.55 |
| UGT2A1 | −2.13 |
| UGT2B4 | 1.23 |
| UGT2B7 | −117.78 |
| UGT2B11 | −1.49 |
| UGT2B15 | 3.05 |
| Cancer progression-associated genes | |
| CCNA2 | −1.52 |
| CDKN3 | −2.44 |
| ESCO2 | −1.22 |
| EXT1 | −7.14 |
| LATS1 | 1.11 |
| PAFAH1B1 | −2.63 |
| PLCB1 | −3.85 |
| PRKCA | 2.29 |
CCNA2, cyclin A2; CDKN3, cyclin-dependent kinase inhibitor 3; LATS1, large tumour suppressor homologue 1; PRKCA, protein kinase C alpha; qPCR, quantitative real-time polymerase chain reaction; UGT, UDP glucuronosyltransferase.
Quinoclamine affected the expression levels of genes involved in drug metabolism
To evaluate whether quinoclamine affected drug metabolism in hepatocytes, we analysed the expression levels of genes encoding phase I and II drug metabolism enzymes. Phase I drug metabolism genes encode alcohol dehydrogenases, aldehyde dehydrogenases and cytochrome P450 families, while phase II drug metabolism genes encode glutathione S-transferases, sulphotransferase and UGT families. Most genes encoding alcohol dehydrogenases were up-regulated, and all genes encoding UGTs were down-regulated (Figure 6). Moreover, genes encoding ADH7, ALDH1A2, ALDH3A2, CYP11B2, CYP1B1 and CYP4B1 in phase I drug metabolism, and genes encoding GSTP1, UGT2B7 and UGT2B11 in phase II drug metabolism were down-regulated or up-regulated >twofold compared with mock. We further applied qPCR to confirm the transcriptional expression levels of UGT genes. As shown in Table 3, the expression levels of four genes, including UGT1A10, UGT2A1, UGT2B11 and UGT2B7, were down-regulated in both microarray and qPCR analyses. However, the expression levels of UGT2B15 and UGT2B4 showed opposite results in these methods.
Figure 6.
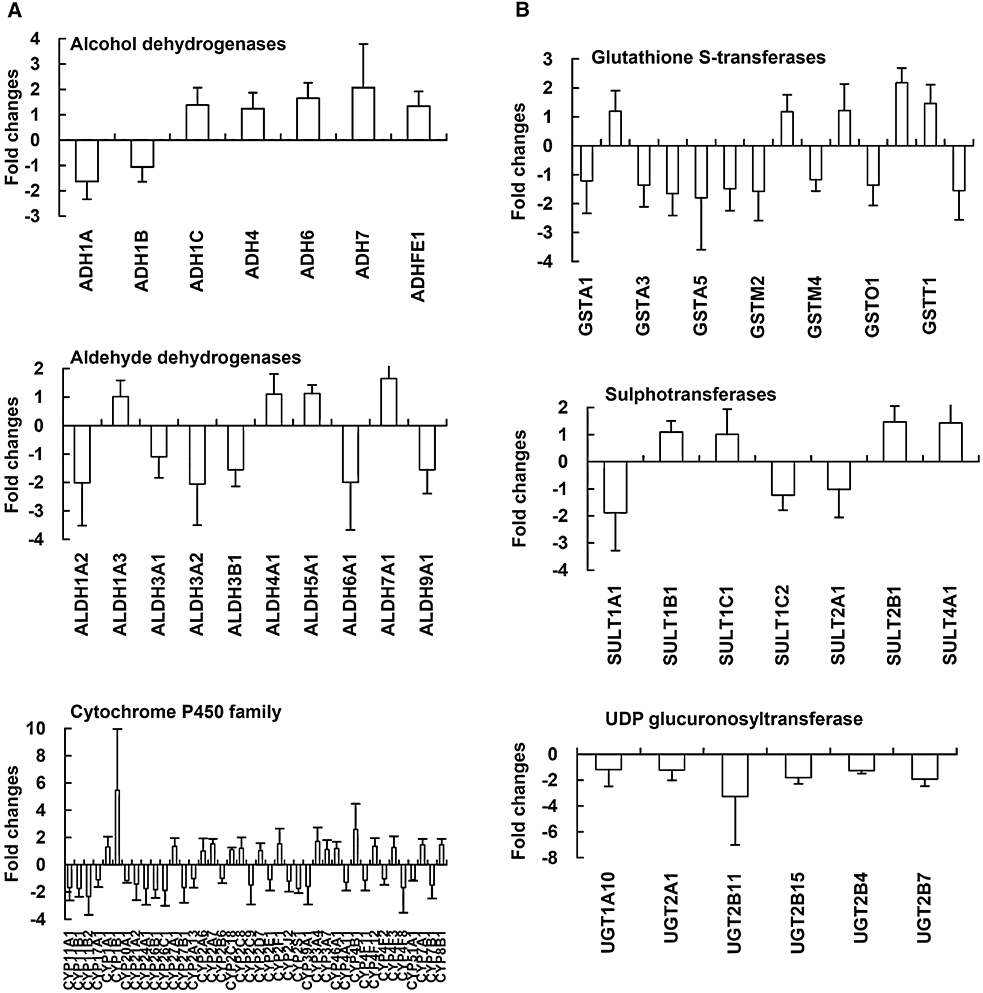
Expression levels of genes involved in drug metabolism in quinoclamine-treated HepG2 cells. (A) Phase I drug metabolism genes. (B) Phase II drug metabolism genes. Values are mean ± standard error of three independent assays.
Discussion and conclusions
The transcription factor NF-κB is known to be involved in many cell processes, such as cell cycle, apoptosis, inflammation and oncogenesis. Inhibition of NF-κB induces apoptosis of cancer cells, suggesting that NF-κB is a potential therapeutic target for cancer (Karin, 2006; Kim et al., 2006). Naphthoquinone derivatives are known to inhibit NF-κB activities and exhibit anti-proliferative potentials. In a large-scale screening test, we found that quinoclamine, a chemically synthesized naphthoquinone compound, was a novel NF-κB inhibitor. Transcriptomic analysis showed that quinoclamine regulated the expression levels of NF-κB-regulated genes. Additionally, these differentially expressed genes have been reported to be associated with cell cycle, cell growth, apoptosis, cell differentiation or anti-tumour effects. These results indicated that quinoclamine was a novel NF-κB inhibitor with anti-cancer potential. Furthermore, microarray analysis showed that all genes encoding UGTs were down-regulated by quinoclamine. These findings suggested that quinoclamine might interfere with drug metabolism by slowing down the excretion of drugs.
Transcriptomic analysis by DNA microarray tools has been applied to predict the effects of clinical drugs, to analyse the mechanisms of drugs and to evaluate the metabolism of drugs (Gerhold et al., 2001; Gunther et al., 2003; Sato et al., 2005). A comprehensive transcriptomic evaluation of the efficacies and metabolism of quinoclamine was done in this study. Compared with previous biochemical studies of naphthoquinones, we found that some genes regulated by quinoclamine are also regulated by other naphthoquinones. For examples, shikonin down-regulated Bcl-xL (BCL2L1) in human colorectal carcinoma cells (Hsu et al., 2004a). Our results showed that quinoclamine down-regulated BCL2L1 (fold change =−2.12, P= 0.02) in HepG2 cells. 5-Hydroxy-2-methyl-1,4-naphthoquinone down-regulates the expression of product cyclin D1 (CCND1) (Sandur et al., 2006). In our study, CCND1 was also down-regulated (fold change =−6.89, P= 0.13) by quinoclamine in HepG2 cells. Additionally, quinoclamine induces differentiation of leukemia cells via protein kinase C alpha activation (Kwon and Choung, 1997). Protein kinase C alpha was also affected by quinoclamine in our experiments. Therefore, these results suggested that quinoclamine might regulate apoptosis or cell proliferation partially through the genes affected by other naphthoquinones.
By using network analysis, we demonstrated that quinoclamine-regulated genes might interact with NF-κB or its downstream genes. Several quinoclamine-affected genes, including CCNA2, CDKN3, GADD45A, LATS1 and STAG1, are known to be involved in cell cycle, cell growth or apoptosis (Pagano et al., 1992; Gyuris et al., 1993; Jin et al., 2002; Xia et al., 2002; Anazawa et al., 2004). It has been implicated that proteins of the NF-κB family participate in cell cycle regulation via cyclin, cyclin-dependent kinase and cyclin-dependent kinase inhibitors (Perkins et al., 1997; Joyce et al., 2001). Quinoclamine-regulated CCNA2 and CDKN3 genes were associated with NF-κB by the knowledge-based network analysis, indicating that quinoclamine might regulate cell cycle through NF-κB activity. It has been reported that inhibition of NF-κB in cancer cells results in GADD-dependent induction of apoptosis and inhibition of tumour growth (Zerbini et al., 2004). The LATS1 tumour suppressor may play an important role in the control of human tumour development and suppress tumorigenesis by negatively regulating cell proliferation and modulating cell survival (Xia et al., 2002). STAG1 is suggested to mediate the p53-dependent apoptosis and may be a good candidate for cancer therapy (Anazawa et al., 2004). CDKN3 is considered to be potential therapeutic targets against cancer (Okamoto et al., 2006). Quinoclamine affected the expression levels of GADD45A, LATS1, STAG1 and CDKN3 genes, suggesting that quinoclamine might have anti-cancer activity.
DNA microarray has been applied to evaluate drug metabolism (Gerhold et al., 2001). Here, we have analysed the expression levels of phase I and II drug metabolism genes during quinoclamine treatment. It is interested to find that all genes encoding UGTs were down-regulated by quinoclamine. The gene expression levels of these genes were further validated by qPCR. Although the gene expression tendencies of UGT2B4 and UGT2B15 were in the reverse direction by microarray and qPCR analyses, the global view at the gene set level for these genes displayed the same tendency. In addition to quinoclamine, menadione (2-methyl-1,4-naphthoquinone) has been reported to induce phase I and II enzymes for the metabolism of xenobiotics, drugs and procarcinogens (Sidorova and Grishanova, 2004). The human UGT superfamily is comprised of two families (UGT1 and UGT2) and three subfamilies (UGT1A, UGT2A and UGT2B) (Mackenzie et al., 1997). A glucuronidation reaction catalysed by UGT enzymes makes the drug molecule more hydrophilic and thus the drug molecule tends to be excreted easily (Ouzzine et al., 2003). Glucuronidation usually abolishes the pharmacological activity in most cases. Various drugs have been identified in vitro as inhibitors for UGT-mediated glucuronidation reactions, and immunosuppressants appear to be particularly potent UGT inhibitors (Kiang et al., 2005). The suppression of UGTs by quinoclamine might inhibit the UGT-mediated glucuronidation and increase the difficulty of drug excretion. Therefore, our findings suggested that drug metabolism might be affected by quinoclamine.
In conclusion, we comprehensively evaluated the effects and metabolism of a novel potent NF-κB inhibitor, quinoclamine, by transcriptomic analysis. Quinoclamine regulated genes involved in cell cycle or apoptosis, suggesting that quinoclamine exhibited the anti-cancer potential by the regulation of cell cycle and apoptosis. Furthermore, quinoclamine down-regulated UGT genes, suggesting that quinoclamine might interfere with drug metabolism by slowing down the excretion of drugs. Our results demonstrated the feasibility of microarray data-, GO- and knowledge-driven analysis for new drug development. Additionally, our findings suggested that quinoclamine was a novel NF-κB inhibitor with the anti-cancer potential.
Acknowledgments
We thank Mr Wei-Shuen Shen for his technical assistance. This work was supported by grants from National Research Program for Genomic Medicine, National Science and Technology Program for Agricultural Biotechnology, National Science Council, Committee on Chinese Medicine and Pharmacy, Department of Health (CCMP96-RD-201 and CCMP97-RD-201), and China Medical University (CMU97-064 and CMU97-CMC-004), Taiwan.
Glossary
Abbreviations:
- CCNA2
cyclin A2
- CCND1
cyclin D1
- CDKN3
cyclin-dependent kinase inhibitor 3
- DMEM
Dulbecco's modified Eagle's medium
- GADD45A
growth arrest and DNA-damage-inducible protein alpha
- GO
gene ontology
- LATS1
large tumour suppressor homologue 1
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NF-κB
nuclear factor-κB
- PRKCA
protein kinase C alpha
- qPCR
quantitative real-time polymerase chain reaction
- STAG1
stromal antigen 1
- TPA
12-O-tetradecanoylphorbol-13-acetate
- UGT
UDP glucuronosyltransferase
Conflict of interest
The authors state no conflict of interest.
References
- Aggarwal BB, Shishodia S. Molecular targets of dietary agents for prevention and therapy of cancer. Biochem Pharmacol. 2006;71:1397–1421. doi: 10.1016/j.bcp.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Anazawa Y, Arakawa H, Nakagawa H, Nakamura Y. Identification of STAG1 as a key mediator of a p53-dependent apoptotic pathway. Oncogene. 2004;23:7621–7627. doi: 10.1038/sj.onc.1207270. [DOI] [PubMed] [Google Scholar]
- Arsura M, Mercurio F, Oliver AL, Thorgeirsson SS, Sonenshein GE. Role of the IκB kinase complex in oncogenic Ras- and Raf-mediated transformation of rat liver epithelial cells. Mol Cell Biol. 2000;20:5381–5391. doi: 10.1128/mcb.20.15.5381-5391.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae KA, Choung SY. Differentiation inducing effects of 2-chloro-3-amino-1,4-naphthoquinone on human leukemia HL-60. Biol Pharm Bull. 1996;19:824–827. doi: 10.1248/bpb.19.824. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Karin M. Nuclear factor-κB: a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1071. doi: 10.1056/NEJM199704103361506. [DOI] [PubMed] [Google Scholar]
- Cheng WY, Wu SL, Hsiang CY, Li CC, Lai TY, Lo HY, et al. Relationship between San-Huang-Xie-Xin-Tang and its herbal components on the gene expression profiles in HepG2 cells. Am J Chin Med. 2008;36:783–797. doi: 10.1142/S0192415X08006235. [DOI] [PubMed] [Google Scholar]
- Furstenberger G, Berry DL, Sorg B, Marks F. Skin tumor promotion by phorbol ester is a two-stage process. Proc Natl Acad Sci USA. 1981;78:7722–7726. doi: 10.1073/pnas.78.12.7722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A, Aggarwal BB. Nuclear transcription factor-κB as a target for cancer drug development. Leukemia. 2002;16:1053–1068. doi: 10.1038/sj.leu.2402482. [DOI] [PubMed] [Google Scholar]
- Gerhold D, Lu M, Xu J, Austin C, Caskey CT, Rushmore T. Monitoring expression of genes involved in drug metabolism and toxicology using DNA microarrays. Physiol Genomics. 2001;5:161–170. doi: 10.1152/physiolgenomics.2001.5.4.161. [DOI] [PubMed] [Google Scholar]
- Gunther EC, Stone DJ, Gerwien RW, Bento P, Heyes MP. Prediction of clinical drug efficacy by classification of drug-induced genomic expression profiles in vitro. Proc Natl Acad Sci USA. 2003;100:9608–9613. doi: 10.1073/pnas.1632587100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyuris J, Golemis E, Chertkov H, Brent R. Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell. 1993;75:791–803. doi: 10.1016/0092-8674(93)90498-f. [DOI] [PubMed] [Google Scholar]
- Ho TY, Chen YS, Hsiang CY. Noninvasive nuclear factor-κB bioluminescence imaging for the assessment of host-biomaterial interaction in transgenic mice. Biomaterials. 2007;28:4370–4377. doi: 10.1016/j.biomaterials.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Hsiang CY, Wu SL, Ho TY. Morin inhibits 12-O-tetradecanoylphorbol-13-acetate-induced hepatocellular transformation via activator protein 1 signaling pathway and cell cycle progression. Biochem Pharmacol. 2005;69:1603–1611. doi: 10.1016/j.bcp.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Hsiang CY, Wu SL, Chen JC, Lo HY, Li CC, Chiang SY, et al. Acetaldehyde induces matrix metalloproteinase-9 gene expression via nuclear factor-κB and activator protein 1 signaling pathways in human hepatocellular carcinoma cells: association with the invasive potential. Toxicol Lett. 2007;171:78–86. doi: 10.1016/j.toxlet.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Hsu PC, Huang YT, Tsai ML, Wang YJ, Lin JK, Pan MH. Induction of apoptosis by shikonin through coordinative modulation of the Bcl-2 family, p27, and p53, release of cytochrome c, and sequential activation of caspases in human colorectal carcinoma cells. J Agric Food Chem. 2004a;52:6330–6337. doi: 10.1021/jf0495993. [DOI] [PubMed] [Google Scholar]
- Hsu YL, Kuo PL, Lin CC. Proliferative inhibition, cell-cycle dysregulation, and induction of apoptosis by ursolic acid in human non-small cell lung cancer A549 cells. Life Sci. 2004b;75:2303–2316. doi: 10.1016/j.lfs.2004.04.027. [DOI] [PubMed] [Google Scholar]
- Ichikawa H, Aggarwal BB. Guggulsterone inhibits osteoclastogenesis induced by receptor activator of nuclear factor-kappaB ligand and by tumor cells by suppressing nuclear factor-kappaB activation. Clin Cancer Res. 2006;12:662–668. doi: 10.1158/1078-0432.CCR-05-1749. [DOI] [PubMed] [Google Scholar]
- Ishiguro K, Ando T, Maeda O, Ohmiya N, Niwa Y, Kadomatsu K, et al. Ginger ingredients reduce viability of gastric cancer cells via distinct mechanisms. Biochem Biophys Res Commun. 2007;362:218–223. doi: 10.1016/j.bbrc.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Jin S, Tong T, Fan W, Fan F, Antinore MJ, Zhu X, et al. GADD45-induced cell cycle G2-M arrest associates with altered subcellular distribution of cyclin B1 and is independent of p38 kinase activity. Oncogene. 2002;21:8696–8704. doi: 10.1038/sj.onc.1206034. [DOI] [PubMed] [Google Scholar]
- Joyce D, Albanese C, Steer J, Fu M, Bouzahzah B, Pestell RG. NF-κB and cell-cycle regulation: the cyclin connection. Cytokine Growth Factor Rev. 2001;12:73–90. doi: 10.1016/s1359-6101(00)00018-6. [DOI] [PubMed] [Google Scholar]
- Kapadia GJ, Azuine MA, Balasubramanian V, Sridhar R. Aminonaphthoquinones – a novel class of compounds with potent antimalarial activity against Plasmodium falciparum. Pharmacol Res. 2001;43:363–367. doi: 10.1006/phrs.2000.0791. [DOI] [PubMed] [Google Scholar]
- Karin M. Nuclear factor-κB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Kiang TK, Ensom MH, Chang TK. UDP-glucuronosyltransferases and clinical drug-drug interactions. Pharmacol Ther. 2005;106:97–132. doi: 10.1016/j.pharmthera.2004.10.013. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Hawke N, Baldwin AS. NF-κB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006;13:738–747. doi: 10.1038/sj.cdd.4401877. [DOI] [PubMed] [Google Scholar]
- Kwon H, Choung SY. Induction of differentiation of U-937 cells by 2-chloro-3-amino-1,4-naphthoquinone. Res Commun Mol Pathol Pharmacol. 1997;97:215–227. [PubMed] [Google Scholar]
- Lien JC, Huang LJ, Wang JP, Teng CM, Lee KH, Kuo SC. Synthesis and antiplatelet, antiinflammatory, and antiallergic activities of 2-substituted 3-chloro-1,4-naphthoquinone derivatives. Bioorg Med Chem. 1997;5:2111–2120. doi: 10.1016/s0968-0896(97)00133-8. [DOI] [PubMed] [Google Scholar]
- Mackenzie PI, Owens IS, Burchell B, Bock KW, Bairoch A, Bélanger A, et al. The UDP glycosyltransferase gene superfamily: recommended nomenclature update based on evolutionary divergence. Pharmacogenetics. 1997;7:255–269. doi: 10.1097/00008571-199708000-00001. [DOI] [PubMed] [Google Scholar]
- Min R, Tong J, Wenjun Y, Wenhu D, Xiaojian Z, Jiacai H, et al. Growth inhibition and induction of apoptosis in human oral squamous cell carcinoma Tca-8113 cell lines by Shikonin was partly through the inactivation of NF-kappaB pathway. Phytother Res. 2008;22:407–415. doi: 10.1002/ptr.2340. [DOI] [PubMed] [Google Scholar]
- Nair A, Venkatraman M, Maliekal TT, Nair B, Karunagaran D. NF-κB is constitutively activated in high-grade squamous intraepithelial lesions and squamous cell carcinomas of the human uterine cervix. Oncogene. 2003;22:50–58. doi: 10.1038/sj.onc.1206043. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Kitabayashi I, Taya Y. KAP1 dictates p53 response induced by chemotherapeutic agents via Mdm2 interaction. Biochem Biophys Res Commun. 2006;351:216–222. doi: 10.1016/j.bbrc.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Ouzzine M, Barré L, Netter P, Magdalou J, Fournel-Gigleux S. The human UDP-glucuronosyltransferases: structural aspects and drug glucuronidation. Drug Metab Rev. 2003;35:287–303. doi: 10.1081/dmr-120026397. [DOI] [PubMed] [Google Scholar]
- Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992;11:961–971. doi: 10.1002/j.1460-2075.1992.tb05135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl HL. Activators and target genes of Rel/NF-κB transcription factors. Oncogene. 1999;18:6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Perkins ND, Felzien LK, Betts JC, Leung K, Beach DH, Nabel GJ. Regulation of NF-κB by cyclin-dependent kinases associated with the p300 coactivator. Science. 1997;275:523–527. doi: 10.1126/science.275.5299.523. [DOI] [PubMed] [Google Scholar]
- Rahman KW, Sarkar FH. Inhibition of nuclear translocation of nuclear factor-kappaB contributes to 3,3′-diindolylmethane-induced apoptosis in breast cancer cells. Cancer Res. 2005;65:364–371. [PubMed] [Google Scholar]
- Sandur SK, Ichikawa H, Sethi G, Ahn KS, Aggarwal BB. Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone) suppresses NF-κB activation and NF-κB-regulated gene products through modulation of p65 and IκB-α kinase activation, leading to potentiation of apoptosis induced by cytokine and chemotherapeutic agents. J Biol Chem. 2006;281:17023–17033. doi: 10.1074/jbc.M601595200. [DOI] [PubMed] [Google Scholar]
- Sato H, Ishida S, Toda K, Matsuda R, Hayashi Y, Shigetaka M, et al. New approaches to mechanism analysis for drug discovery using DNA microarray data combined with KeyMolnet. Curr Drug Discov Technol. 2005;2:89–98. doi: 10.2174/1570163054064701. [DOI] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidorova YA, Grishanova AY. Dose- and time-dependent effects of menadione on enzymes of xenobiotic metabolism in rat liver. Bull Exp Biol Med. 2004;137:231–234. doi: 10.1023/b:bebm.0000031556.47763.04. [DOI] [PubMed] [Google Scholar]
- Smyth GK. Limma: linear models for microarray data. In: Gentleman R, Carey V, Dudoit S, Irizarry R, Huber W, editors. Bioinformatics and Computational Biology Solutions Using R and Bioconductor. New York: Springer; 2005. pp. 397–420. [Google Scholar]
- Sovak MA, Bellas RE, Kim DW, Zanieski GJ, Rogers AE, Traish AM, et al. Aberrant nuclear factor-κB/Rel expression and the pathogenesis of breast cancer. J Clin Invest. 1997;100:2952–2960. doi: 10.1172/JCI119848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y, Richmond A. NF-κB activation in melanoma. Pigment Cell Res. 2006;19:112–124. doi: 10.1111/j.1600-0749.2006.00304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ. The nuclear factor-κB RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res. 1999;5:119–127. [PubMed] [Google Scholar]
- Wu JT, Kral JG. The NF-κB/IκB signaling system: a molecular target in breast cancer therapy. J Surg Res. 2005;123:158–169. doi: 10.1016/j.jss.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Xia H, Qi H, Li Y, Pei J, Barton J, Blackstad M, et al. LATS1 tumor suppressor regulates G2/M transition and apoptosis. Oncogene. 2002;21:1233–1241. doi: 10.1038/sj.onc.1205174. [DOI] [PubMed] [Google Scholar]
- Yu SM, Wu JF, Lin TL, Kuo SC. Inhibition of nitric oxide synthase expression by PPM-18, a novel anti-inflammatory agent, in vitro and in vivo. Biochem J. 1997;328:363–369. doi: 10.1042/bj3280363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbini LF, Wang Y, Czibere A, Correa RG, Cho JY, Ijiri K, et al. NF-κB-mediated repression of growth arrest- and DNA-damage-inducible proteins 45alpha and gamma is essential for cancer cell survival. Proc Natl Acad Sci USA. 2004;101:13618–13623. doi: 10.1073/pnas.0402069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Kirov S, Snoddy J. WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 2005;33:W741–W748. doi: 10.1093/nar/gki475. [DOI] [PMC free article] [PubMed] [Google Scholar]