

Abstract
Background and purpose:
Verapamil blocks current through the voltage-gated K+ channel Kv1.3 in the open and inactivated state of the channel but not the closed state. The binding site for verapamil was proposed to be close to the selectivity filter and the occupancy of the selectivity filter might therefore influence verapamil affinity.
Experimental“ approach:
We investigated the influence of intra- and extracellular K+ and Rb+ on the effect of verapamil by patch-clamp studies, in COS-7 cells transfected with hKv1.3 channels.
Key results:
Verapamil affinity was highest in high intracellular K+ concentrations ([K+]i) and lowest in low [Rb+]i, indicating an influence of intracellular cations on verapamil affinity. Experiments with a mutant channel (H399T), exhibiting a strongly reduced C-type inactivated state, demonstrated that part of this changed verapamil affinity in wild-type channels could be caused by altered C-type inactivation. External K+ and Rb+ could influence verapamil affinity by a voltage-dependent entry into the channel thereby modifying the verapamil off-rate and in addition causing a voltage-dependent verapamil off-rate.
Conclusions and implications:
Recovery from verapamil block was mainly due to the voltage-dependent closing of channels (state-dependent block), implying a second open state of the channel. This hypothesis was confirmed by the dependency of the tail current time course on duration of the prepulse. We conclude that the wild-type hKv1.3 channel undergoes at least two different conformational changes before finally closing with a low verapamil affinity in one open state and a high verapamil affinity in the other open state.
Keywords: K+ channel, electrophysiology, verapamil, second open state
Introduction
The voltage-gated K+ channel Kv1.3 (nomenclature follows Alexander et al., 2008) is a member of the Shaker-related K+ channel family (Grissmer et al., 1990) and plays an important role in the earliest stages of T-lymphocyte activation (DeCoursey et al., 1984). Kv1.3 channels can also be found for example in natural killer cells, macrophages, oligodendrocytes and adipocytes. The channel consists of four identical subunits and each subunit is composed of six transmembrane segments, termed S1-S6, with the amino and carboxy terminals located intracellularly. The S1 to S4 segments form the voltage sensor domain with the highly charged S4 as the major part. The segments S5 and S6 with the pore-helix between these segments shape the central pore. In the functional channel the highly conserved signature sequence, GYGD, localized in the pore-region of the identical four subunits form the selectivity filter which is responsible for the selective flux of K+ through the channel. The selectivity filter contains four ion binding sites, which are occupied under physiological conditions in two configurations either in the 1–3 configuration or in the 2–4 configuration (Zhou and MacKinnon, 2003). In addition, Zhou and MacKinnon (2003) also showed that in low concentrations of K+ the backbone carbonyl group of V76 in the KcsA K+ channel rotates away from the centre, the α-carbon atom of the residue G77 faces towards the pore and the ions are absent from positions 2 and 3. This ‘low-K+ structure’ is significantly different at the selectivity filter region and might therefore have different affinities for blockers in this region.
The intrinsic channel properties of Kv1.3 channels are well known (DeCoursey et al., 1984; Cahalan et al., 1985; DeCoursey, 1990; Grissmer et al., 1990). The channel has at least three states, the closed (C), open (O) and inactivated (I) state, all of which depend on voltage. At hyperpolarized potentials, Kv1.3 channels are closed. Depolarization leads to changes in the conformation of the channel resulting in channel opening. Prolonged depolarization leads to another conformational change of the channel resulting in a different non-conducting state of the channel separate from the closed state, called the ‘C-type inactivated state’. To avoid the C-type inactivated state, we have used the hKv1.3 H399T mutant channel. This mutation is located in the extracellular loop between the pore and the S6 helix of hKv1.3 (Dreker and Grissmer, 2005) and results in a strong reduction of the C-type inactivated state.
Another property of Kv1.3 channels that can be used to distinguish them from other Kv channels is their distinct pharmacology. For example, Kv1.3 channels are blocked by peptide inhibitors like scorpion toxins (margatoxin, agiotoxin, kaliotoxin) and sea anemone toxins (ShK). Margatoxin (MgTX), the most potent scorpion toxin, blocked current through Kv1.3 channels with an IC50 of 30 pmol·L−1, whereas currents through Kv1.6 or Shaker channels were blocked with IC50 values of 5 and 150 nmol·L−1 respectively. In addition, at 200 nmol·L−1, MgTX did not affect Kv1.5 and Kv3.1 channels (Garcia-Calvo et al., 1993). Similarly, the sea anemone toxin ShK blocks current through Kv1.3 channels with an IC50 of 16 pmol·L−1, whereas on Kv1.2 and Kv1.5 channels, the IC50 is >1 µmol·L−1 (Kalman et al., 1998).
The currents through Kv1.3 channels were also blocked by small molecule inhibitors, such as piperidines (UK-78282), psoralens (Psora-4, PAP-1) and phenylalkylamines (verapamil). In this present study we used verapamil, originally described as a classical calcium channel blocker. Verapamil, however, is also able to block current through Kv1.3 channels (IC50= 2.6 µmol·L−1). Earlier measurements demonstrated that verapamil blocked the open and the inactivated state of the channel but not the closed, deactivated state (DeCoursey, 1995; Rauer and Grissmer, 1996; 1999; see Figure 1). These studies suggested that the binding site was situated in the water-filled cavity below the selectivity filter. Several observations supported the proposed internal binding site for verapamil (Rauer and Grissmer, 1996), one of which was that intracellularly applied tetraethylammonium ions (TEA+) competed with verapamil for the same binding site. In this present study we wanted to see if intra- and extracellular K+ and Rb+ could influence verapamil affinity. Under physiological conditions the selectivity filter is occupied by two potassium ions (Zhou and MacKinnon, 2003) and the water-filled cavity also is occupied by a K+ (Zhou and MacKinnon, 2004). Changing intra- and extracellular K+ and Rb+ will change the occupancy of the selectivity filter which could modulate the effect of verapamil.
Figure 1.
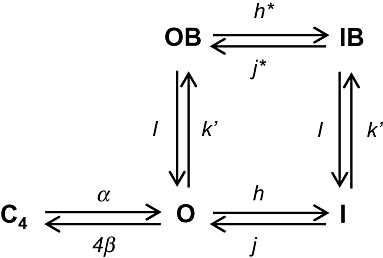
Simplified scheme of transitions of hKv1.3 channels by de- and hyperpolarizations in the absence and presence of verapamil. C4 is the last closed state, O is the open state, OB the open-blocked state, I the inactivated state and IB is the inactivated blocked state.
One interesting observation was the weak, voltage-dependent, verapamil block at positive potentials and the strong, voltage-dependent off-rate for verapamil at potentials more negative than −60 mV (Röbe and Grissmer, 2000). The authors concluded that the strong voltage dependence of the verapamil off-rate was due to the positively charged verapamil sensing the membrane electric field and leaving its binding site because of the negative voltage. The strong off-rate voltage dependence at negative potentials, however, could also be due to the closing of the channel itself and not due to the voltage sensed by verapamil. This interpretation was supported by our results, which also imply the existence of two different open states of the hKv1.3 channel, one of which is less sensitive to block by verapamil.
Methods
Cell culture
African green monkey kidney cells transformed with an origin-defective mutant of SV-40 (COS-7) were obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ, Braunschweig, Germany; No.: ACC 60). The cells were grown in Dulbecco's modified Eagle's medium with high glucose (Invitrogen, Carlsbad, CA, USA; Cat.-No. 41966) containing 10% fetal bovine serum (PAA Laboratories GmbH, Pasching, Austria; Cat.-No.: A15-041) in a humidified, 10% CO2 incubator at 37°C.
Chemicals and solutions
All measurements were performed in an external bath solution, high [K+]o, containing (in mmol·L−1): KCl 164.5, CaCl2 2, MgCl2 1, HEPES 5, or high [Rb+]o containing (in mmol·L−1): RbCl 164.5, CaCl2 2, MgCl2 1, HEPES 5 or high [Na+]o containing (in mmol·L−1): NaCl 160, KCl 4.5, CaCl2 2, MgCl2 1, HEPES 5. Osmolarity was 290–320 mOsm and the pH was adjusted to 7.4 with KOH, RbOH or NaOH respectively. The high [Na+]o solution was used as a low [K+]o condition. The internal pipette solution, high [K+]i, containing (in mmol·L−1): KF 145, MgCl2 2, EGTA 10, HEPES 10, or high [Rb+]i containing (in mmol·L−1): RbF 145, MgCl2 2, EGTA 10, HEPES 10, or in low [K+]i containing (in mmol·L−1): KF 10, tetramethylammonium fluoride (TMAF) 135, MgCl2 2, EGTA 10, HEPES 10, or in low [Rb+]i containing (in mmol·L−1): RbF 10, TMAF 135, MgCl2 2, EGTA 10, HEPES 10. Osmolarity was 290–320 mOsm and the pH was adjusted to 7.2 with KOH or RbOH.
The phenylalkylamine verapamil was purchased from Sigma-Aldrich Co. (Munich, Germany) as (+/−) verapamil hydrochloride, stock solutions made in DMSO and diluted to the appropriate concentration in the external bath solution before application. The proportion of DMSO in the final solution was always <0.3%.
Electrophysiology
All experiments were performed in the whole-cell or outside-out recording mode of the patch-clamp technique (Hamill et al., 1981) at room temperature (18–22°C). Electrodes were pulled from glass capillaries (Science Products, Hofheim, Germany) in three stages and fire-polished to resistances of 2–4 MΩ. Data were acquired with an EPC-9 patch-clamp amplifier (HEKA elektronik, Lambrecht, Germany) connected to a computer running Patchmaster/Fitmaster v2.00 data acquisition and analysis software (HEKA elektronik, Lambrecht, Germany). All currents were filtered by a 2.9 kHz Bessel Filter and recorded with a sampling frequency of 4.00 kHz. Capacitative and leak currents were subtracted and series resistance compensation (70–80%) was used in all measurements. The mean voltage error was in all measurements 6 ± 4 mV (n= 53). Holding potential was always −120 mV. To determine the verapamil affinity in the wild-type channel we used the reduction of the peak current amplitude after several depolarizing pulses in order to allow the use-dependent block of verapamil to reach steady state. Due to the lack of C-type inactivation in the H399T mutant channel a simpler approach to determine verapamil affinity to this channel was chosen, namely, the reduction of the current amplitude at the end of a 200 ms depolarizing pulse. Further data analysis was performed using the Igor Pro 3.12 (WaveMetrics, Oregon, USA) software package or Microsoft® Office Excel 2003. Time constants for tail currents were determined by an exponential fit using the computer fitting routine of Fitmaster.
Simulation of the tail currents
To evaluate the time course of recovery from verapamil block of the hKv1.3 channels, the tail currents were simulated using the following equation:
| (1) |
where 4β describes the transition from the open (O) to the closed (C) state, k' is the transition from O to the open-blocked (OB) state and l from OB to O (Figure 1), and all rate constant are in 1/s. From k' we deduced k as k=k'/[verapamil]. For the simulation during hyperpolarization, we assumed a voltage dependence of k of 138 mV per e-fold (DeCoursey, 1995).
Molecular biology
The hKv1.3 wild-type plasmid (gene: KCNA3) was a generous gift from Prof Dr O. Pongs (Institut für Neurale Signalverarbeitung, Zentrum für Molekulare Neurobiologie, Hamburg, Germany). It contained the human Kv1.3 K+ channel gene in a pRc/CMV vector (Invitrogen, Carlsbad, CA, USA) with a CMV promoter for protein expression in mammalian cells. The H399T mutant channel was originally generated in our laboratory (Dreker and Grissmer, 2005) by introducing the corresponding point mutation in the cloned hKv1.3 gene with the QuikChange site-directed mutagenesis kit (Stratagene, Amsterdam, Netherlands). COS-7 cells were transfected using the Fugene 6 transfection reagent (Roche Molecular Biochemicals, Mannheim, Germany). Cells were grown to ∼80% confluence and co-transfected with ∼1 µg hKv1.3 and ∼0.5 µg eGFP-N1 (CLONTECH, California, USA) DNA. One day after transfection, sufficient protein for electrophysiological measurements was expressed.
Results
In this present study we investigated the influence of intracellular cations on the effect of verapamil. For this purpose we used different cations each at two concentrations in the wild type and a mutant channel with a strongly reduced C-type inactivated state, the H399T channel (Rauer and Grissmer, 1996; Dreker and Grissmer, 2005).
Influence of intracellular cations on the effect of verapamil to block current through the wild-type channel
At first we performed experiments in the wild-type channel under physiological control conditions, with 145 mmol·L−1[K+]i and 4.5 mmol·L−1[K+]o (Figure 2A). We elicited currents through hKv1.3 channels in the whole-cell recording mode of the patch-clamp technique by depolarizing pulses to +40 mV for 200 ms from a holding potential of −120 mV in the absence and presence of different concentrations of verapamil. After an application of, for example 3 µmol·L−1 verapamil, the peak current in high [K+]i was blocked more than 50% compared with the control current without verapamil (Figure 2A). From the decrease of the peak current with different verapamil concentrations, we calculated the IC50 value of verapamil to block the peak outward current. In high [K+]i the IC50 for verapamil was 2.6 µmol·L−1 (Figure 2E) which agrees well with published values (Dreker and Grissmer, 2005).
Figure 2.
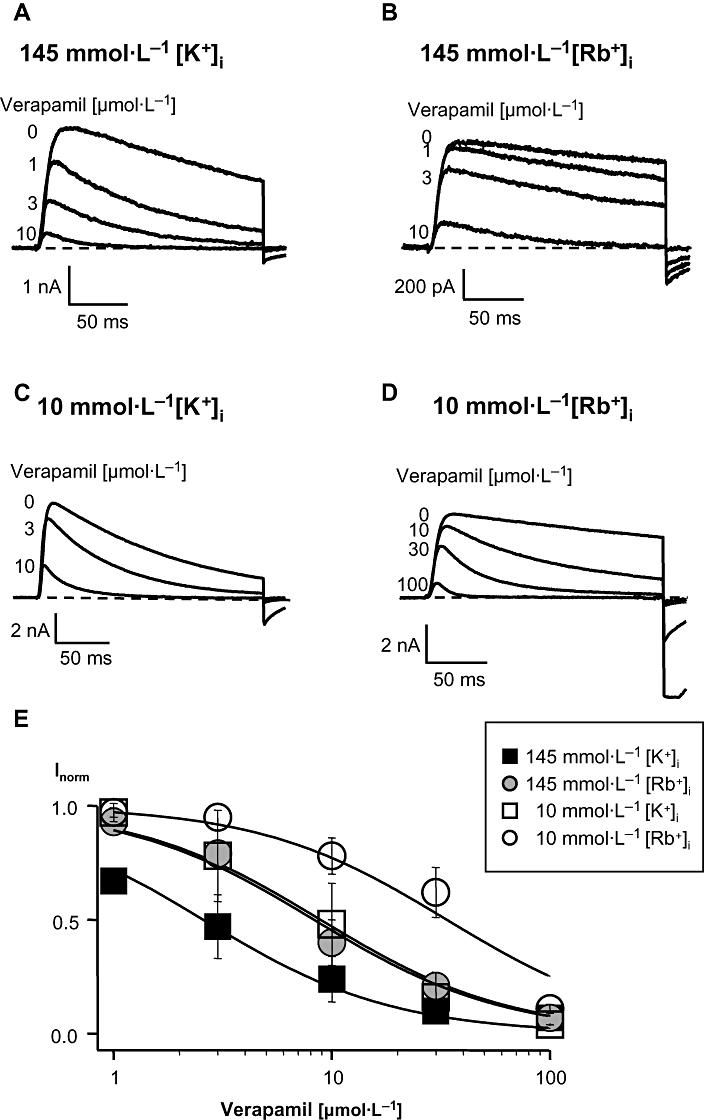
Block of current through wild-type hKv1.3 channels by verapamil in the presence of different [K+]i or [Rb+]i with low [K+]o. Currents were elicited in the whole-cell configuration by a 200-ms depolarizing pulse to +40 mV from a holding potential of −120 mV every 30 s and were obtained in high, 145 mmol·L−1, [K+]i (A) or [Rb+]i (B) and in low, 10 mmol·L−1, [K+]i (C) or [Rb+]i (D) before and after application of different concentrations of verapamil. (E) Dose-response curves in the wild-type channel. IC50 values were calculated by fitting a Hill function (Idrug/I0= 1/[1 + (IC50/Cdrug)]) to the normalized peak current data points obtained after application of different verapamil concentrations. I0 represents the peak current before and Idrug the peak current after verapamil application, Cdrug is the concentration of verapamil in the external bath solution. All data points were obtained from at least three identical experiments and are given as mean ± SD.
In another set of experiments, we used 145 mmol·L−1 Rb+ as the intracellular cation (Figure 2B), which resulted in a slower decay of the outward current due to a modified C-type inactivation. The block of the peak currents through hKv1.3 channels by 3 µmol·L−1 verapamil in the presence of 145 mmol·L−1 Rb+, resulted in a 20% reduction of current compared with the control current (Figure 2B). The IC50 value under these conditions was 8.3 µmol·L−1 (Figure 2E), which is about 3 times higher than the value with high [K+]i, suggesting either an influence of the intracellular cation on the verapamil affinity or a modified inactivation leading to a modified verapamil effect.
The use of a 10 mmol·L−1[K+]i solution (Figure 2C) resulted in a faster transition to the C-type inactivated state. After application of 3 µmol·L−1 verapamil approximately 20% of the peak current was blocked, compared with the current without verapamil. The affinity of verapamil decreased by a factor of 3.5 to an IC50 of 9.1 µmol·L−1 (Figure 2E) compared with high [K+]i (2.6 µmol·L−1). This reduced affinity suggests that K+ ions are involved in the effect of verapamil on the channel. Low K+ in the intracellular solution resulted in a reduced verapamil affinity which might be at least in part due to the stronger inactivation in 10 mmol·L−1[K+]i (Figure 2C) than in 145 mmol·L−1[K+]i. In 10 mmol·L−1[Rb+]i the C-type inactivated state is not affected, as it is in low [K+]i. The peak current in low [Rb+]i was blocked about 40% by 30 µmol·L−1 verapamil compared with the control current (Figure 2D). Therefore, with 10 mmol·L−1[Rb+]i solution the affinity of verapamil was lowered by a factor of 4 to an IC50 of 33.7 µmol·L−1 (Figure 2E), relative to that in 145 mmol·L−1[Rb+]i. Thus, a decreased cation concentration in the intracellular solution resulted in a reduction of the verapamil affinity in the wild-type channel, which could be at least in part due to the faster inactivation (Figure 2C).
Influence of intracellular cations on the effect of verapamil to block current through the mutant channel H399T
To exclude the influence of the C-type inactivation on verapamil affinity, we carried out the same experiments, as described above, in the mutant channel H399T, which exhibits a strongly reduced C-type inactivated state (Dreker and Grissmer, 2005). The currents through H399T channels were elicited in the whole-cell recording mode with the same voltage pulse protocol as well as solutions as described for the wild-type channel. For the H399T channels we calculated the IC50 after the application of different verapamil concentrations by the decrease of the steady-state current at the end of the 200 ms depolarizing pulse (Röbe and Grissmer, 2000; Dreker and Grissmer, 2005) and not by the decrease of the peak current as in the wild-type channel. Under physiological control conditions in H399T channels with 145 mmol·L−1[K+]i and 4.5 mmol·L−1[K+]o 42% of the steady-state current was blocked by 1 µmol·L−1 verapamil compared with the control current without verapamil (Figure 3A). The affinity of verapamil was 2.1 µmol·L−1 (Figure 3E) which was similar to what we determined for the wild-type channel (2.6 µmol·L−1, Figure 2E). The currents through H399T channels with 145 mmol·L−1[Rb+]i were blocked about 33% with 1 µmol·L−1 verapamil (Figure 3B), resulting in a slightly lower verapamil affinity with an IC50 of 3.3 µmol·L−1 (Figure 3E). In the mutant channel the verapamil affinity was less affected by the different intracellular cation compared with the wild-type channel indicating that part of the change in verapamil affinity by using different intracellular cations was presumably due to a modified C-type inactivation in the wild-type channel.
Figure 3.
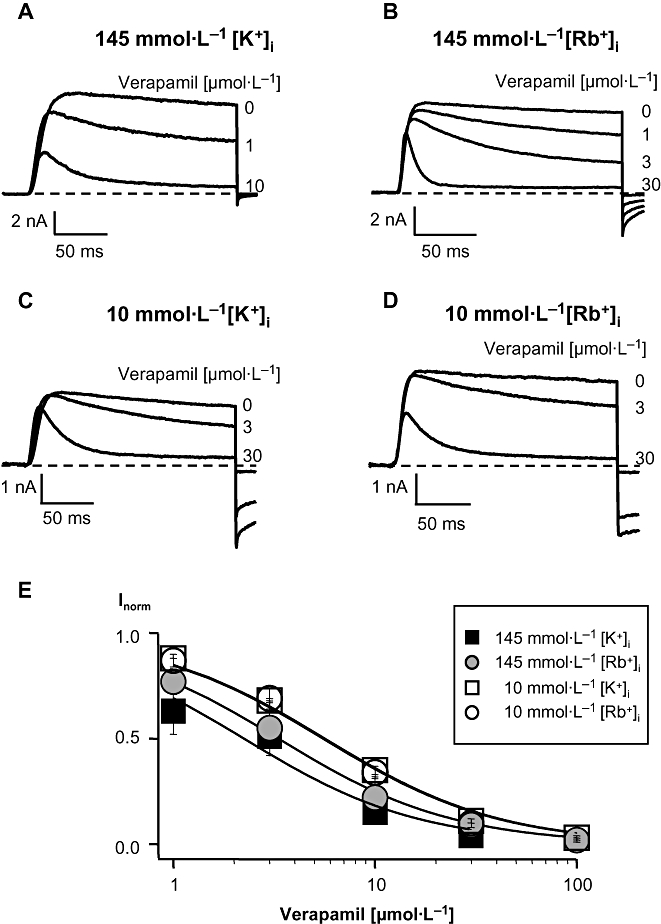
Block of current through the mutant channel H399T by verapamil in the presence of different [K+]i or [Rb+]i with low [K+]o. Currents were elicited in the whole-cell configuration as described in the legend to Figure 2 and were obtained in high, 145 mmol·L−1, [K+]i (A) or [Rb+]i (B) and in low, 10 mmol·L−1, [K+]i (C) or [Rb+]i (D) before and after application of different concentrations of verapamil. (E) Dose-response curves in the mutant channel. IC50 values were calculated by fitting a Hill function (Idrug/I0= 1/[1 + (IC50/Cdrug)]) to the normalized steady-state current data points obtained after application of different verapamil concentrations. I0 represents the steady-state current before and Idrug the steady-state current after verapamil application, Cdrug is the concentration of verapamil in the external bath solution. All data points were obtained from at least three identical experiments and are given as mean ± SD.
In 10 mmol·L−1[K+]i the application of 3 µmol·L−1 verapamil blocked more than 30% of the steady-state current (Figure 3C). The low [K+]i decreased the verapamil affinity on the H399T channel to an IC50 of 5.6 µmol·L−1 (Figure 3E), a factor of 2.6 compared with its value in high [K+]i. With low Rb+ (Figure 3D) in the intracellular solution 30% of the steady-state current was blocked by 3 µmol·L−1 verapamil as in 10 mmol·L−1[K+]i whereas in high [Rb+]i∼50% of the steady-state current was blocked by 3 µmol·L−1 verapamil. The IC50 for verapamil in 10 mmol·L−1[Rb+]i was 5.5 µmol·L−1 (Figure 3E).
Thus, in the mutant channel H399T the internal cations did have less influence on verapamil affinity compared with the wild-type channel and a part of the cation effect on the verapamil affinity for the wild-type channel could be due to change in C-type inactivation brought about by the cations.
This C-type inactivation is strongly reduced in the mutant H399T channel and therefore the effect of the internal cations to modulate verapamil affinity is less compared with the wild-type channel.
Tail currents with high K+ in the extracellular solution
External cations also influence the rate of C-type inactivation (López-Barneo et al., 1993) and extracellular K+ displaced, at hyperpolarized potentials, intracellular TEA (tetraethylammonium) ions from their blocking site due to the voltage-dependent entry of external K+ into the channel (Armstrong, 1971). Therefore, the voltage-dependent entry of K+ or Rb+ could also influence the dissociation of verapamil from its binding site (verapamil off-rate). The simplest way to assess the voltage dependence of verapamil dissociation would be to create conditions where the channels stay open longer at potentials more negative than −60 mV and to achieve this we performed experiments using 164.5 mmol·L−1 K+ in the extracellular solution and 145 mmol·L−1 K+ in the intracellular solution. We used high K+ in the extracellular solution to prevent the transition to the C-type inactivated state and to have a more precise measurement of the tail current.
The results of these experiments are shown in Figure 4. We elicited currents (Figure 4A) through hKv1.3 channels in the outside-out recording mode from a holding potential of −120 mV with 50 ms depolarizing pulses to +60 mV followed by different hyperpolarized potentials in high [K+]o. In the absence of verapamil, the depolarizing pulses activated and opened the channels (C → O) giving rise to a peak outward current within 15–20 ms that declined minimally during depolarization, i.e. did not show inactivation due to the high [K+]o. Hyperpolarization deactivated and closed the channels (O → C) resulting in an instantaneous inward tail current that declined mono-exponentially. The average time constant for this decline was 22 ± 9 ms (mean ± SD, n= 4) at −160 mV. From the deactivation time constants at different hyperpolarized potentials we calculated the deactivation rate constants and plotted those against the applied voltage (Figure 4B). We did not include the values obtained at −80 and −60 mV because, at those potentials, channels become activated and open and therefore the time course of the tail current decay is not only due to the transition from the open to the closed, deactivated state of the channel, but also from the closed, deactivated state to the open state. The straight line in Figure 4B represents a mono-exponential fit to the data with a slope of −0.014 mV−1 indicating a voltage dependence of deactivation of hKv1.3 channels in [K+]o of e-fold/−71 mV.
Figure 4.
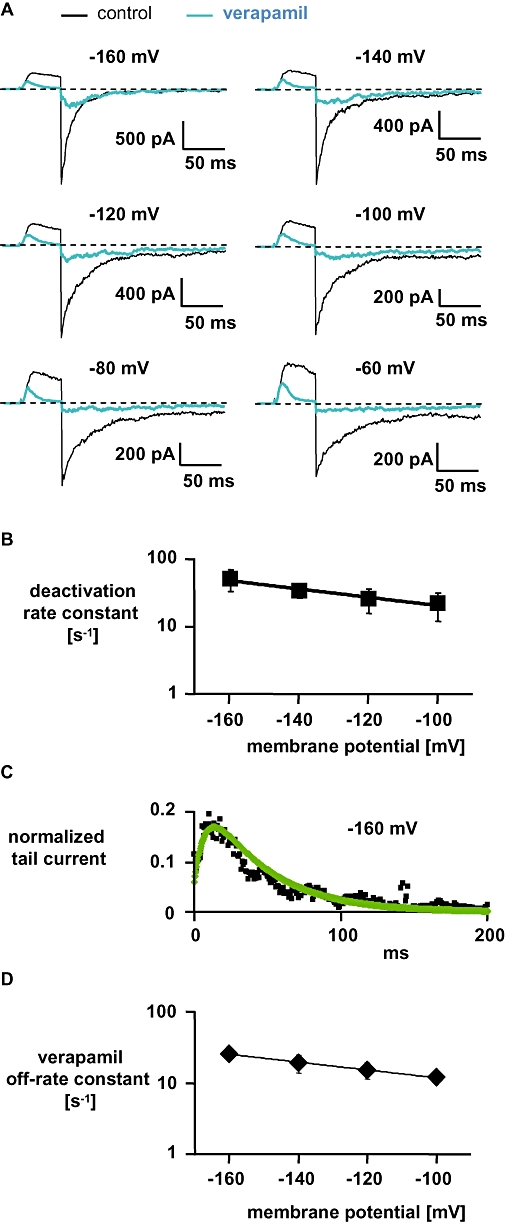
Characteristics of verapamil-induced block of currents through wild-type hKv1.3 channels in high [K+]o and high [K+]i. (A) Currents were elicited in the outside-out configuration from a holding potential of −120 mV with 50-ms depolarizing pulses to +60 mV followed by various hyperpolarizing pulses. (B) Deactivation rate constants (mean ± SD, n= 4) were obtained by single-exponential fits to the tail currents shown in (A) in the absence of verapamil and plotted against the applied membrane potential. The straight line represents a mono-exponential fit to the data with a slope of −0.014 mV−1 indicating a voltage dependence of deactivation in [K+]o of e-fold/−71 mV. (C) Tail currents at −160 mV in the presence of 50 µmol·L−1 verapamil were normalized to the instantaneous tail current in the absence of verapamil (data from A; black squares). The continuous green line represents the simulation of the current based on Equation 1 yielding off-rate constants (mean ± SD, n= 4) for verapamil which are plotted in (D) against the applied membrane potential. The straight line represents a mono-exponential fit to the data with a slope of −0.0126 mV−1 indicating a voltage dependence of the verapamil off-rate in [K+]o of e-fold/−79 mV. Error bars are shown when they exceed the size of the symbols.
In the presence of 50 µmol·L−1 verapamil during the depolarizing pulses, hKv1.3 channels opened and currents through the channels were almost completely blocked by verapamil at the end of the depolarizing pulses. During the following hyperpolarizing pulses, a clear ‘hook’ in the tail currents was observed, that is, an initial increase in inward current reaching a peak followed by a decrease in inward current. This initial increase in tail inward current resembled recovery from verapamil block. We then determined the rate constants for recovery from verapamil block at different potentials, from the hooked tail current by a simulation of the tail current based on Equation 1. Figure 4C represents the normalized tail current in [K+]o at −160 mV after the application of verapamil. At the beginning of the hyperpolarizing pulse, most of the channels are in the open-blocked state and undergo a transition from the open-blocked to the open state (OB → O) until they reach a peak after ∼13 ms. After the peak, the channels finally go to the deactivated, closed state (O → C). We simulated this time course of the tail current and obtained in this example (Figure 4C) an off-rate constant for verapamil of 30 s−1, with similar deactivation rate constants as determined in the absence of verapamil. The off-rate constants for verapamil from the simulation at different potentials were plotted against the applied membrane potential (Figure 4D). The straight line in Figure 4D represents a mono-exponential fit to the data with a slope of −0.0126 mV−1 indicating a voltage dependence of the verapamil off-rate of e-fold/−79 mV in [K+]o. The similarity between the voltage dependence of deactivation of hKv1.3 channels (e-fold/−71 mV), that is, the voltage dependence of channel closing and the verapamil off-rate (e-fold/−79 mV) suggested to us that the voltage dependence of the verapamil off-rate may be caused by the voltage dependence of channel closure.
Tail currents with high Rb+ in the extracellular solution
We also used Rb+ in the extracellular solution to keep the channels open longer at potentials more negative than −60 mV. This approach had been described to slow down deactivation by a factor of ∼5 compared with using K+ in the extracellular solution (Cahalan et al., 1985).
The currents through hKv1.3 channels were elicited in the outside-out recording mode as described before only in high [Rb+]o and in high [K+]i and the results of these experiments are shown in Figure 5. In the absence of verapamil, the currents elicited in high [Rb+]o were similar to those observed in high [K+]o (Figure 4) except that deactivation in high [Rb+]o was much slower than in high [K+]o. For example, at −160 mV the deactivation time constant was 112 ± 41 ms (mean ± SD, n= 4) compared with 22 ms in high [K+]o indicating that deactivation in high [Rb+]o was about 5 times slower than in high [K+]o. In Figure 5B the deactivation rate constants were plotted against the applied membrane potential as described for Figure 4B. With the mono-exponential fit (straight line) we obtained a slope of −0.0286 mV−1 indicating a voltage dependence of deactivation of hKv1.3 channels in high [Rb+]o of e-fold/−35 mV.
Figure 5.
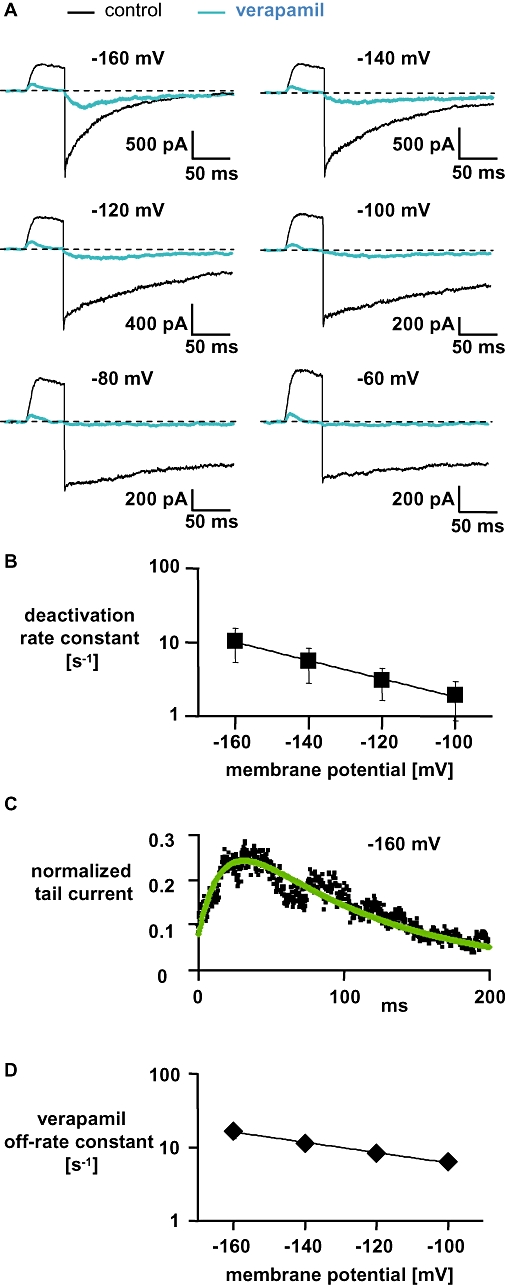
Characteristics of verapamil-induced block of currents through wild-type hKv1.3 in high [Rb+]o and high [K+]i. (A) Currents were elicited in the outside-out configuration as described in the legend to Figure 4A. (B) Deactivation rate constants (mean ± SD, n= 4) were obtained as described in the legend to Figure 4B and plotted against the applied membrane potential. The straight line represents a mono-exponential fit to the data with a slope of −0.0286 mV−1 indicating a voltage dependence of deactivation in [Rb+]o of e-fold/−35 mV. (C) Normalized tail currents at −160 mV (data from A; black squares) and the simulation of the current (green line) were obtained as described in the legend to Figure 4C. (D) Off-rate constants (mean ± SD, n= 3) for verapamil were obtained by the simulation and plotted against the applied membrane potential. The straight line represents a mono-exponential fit to the data with a slope of −0.012 indicating a voltage dependence of the verapamil off-rate in [Rb+]o of e-fold/−83 mV. Error bars are shown when they exceed the size of the symbols.
The traces obtained in the presence of 50 µmol·L−1 verapamil in high [Rb+]o are also shown in Figure 5A. Currents through the hKv1.3 channels elicited in high [Rb+]o were like those with high [K+]o, that is, a complete block by verapamil at the end of the depolarizing pulses and the appearance of a hook in the tail current. From this hook we determined the time constants to the peak of the inward tail current in high [Rb+]o, at −160 mV (17 ± 7 ms; mean ± SD, n= 4) and at −100 mV (91 ± 41 ms; mean ± SD, n= 5). These values were ∼4–6 times greater in high [Rb+]o compared with in high [K+]o (Figure 4A), indicating a correlation between the speed of deactivation and the speed of verapamil leaving its binding site. Besides this correlation we also determined from the hooked tail currents the rate constants for recovery from verapamil block at different potentials by a simulation of the current based on Equation 1. Figure 5C shows and represents the observed values for the normalized tail current in [Rb+]o at −160 mV after the application of verapamil and the line represents the simulation. At the beginning of the hyperpolarizing potential, most of the channels are in the open-blocked state and undergo a transition to the open state (OB → O) until they reach a peak after ∼27 ms. After the peak the channel finally closes (O → C). With the simulation we obtained in this example (Figure 5C) an off-rate constant for verapamil of 20 s−1, with similar deactivation rate constants as determined in the absence of verapamil in [Rb+]o. In Figure 5D the verapamil off-rate constants at different hyperpolarizing potentials in high [Rb+]o obtained by the simulation were plotted against the applied membrane potential. The straight line in Figure 5D represents the mono-exponential fit with a slope of −0.012 mV−1 indicating a voltage dependence of e-fold/−83 mV. The voltage dependence of the verapamil off-rate in high [Rb+]o (e-fold/−83 mV) was comparable with the voltage dependence of the verapamil off-rate in high [K+]o (e-fold/−79 mV) in spite of differences in the absolute values of the off-rate constants l. The off-rate constants (l; mean ± SD) for verapamil were in high [K+]o, 26 ± 3 s−1 (n= 4) at −160 mV and 12 ± 2 s−1 (n= 4) at −100 mV, compared with 17 ± 3 s−1 (n= 3) and 7 ± 1 s−1 (n= 3) in high [Rb+]o.
Influence of the voltage-dependent entry of K+ on the voltage dependence of the verapamil off-rate
One interpretation of the voltage dependence of the recovery from verapamil block (verapamil off-rate) is that there was voltage-dependent entry of potassium ions into the channel which displaced verapamil from its binding sites, analogous to the observation that increasing external K+ increased the rate constant of dissociation of TEA+ (tetraethylammonium) ion from its blocking sites (Armstrong, 1971).
The simplest method of testing if dissociation of verapamil was due to the entry of K+ into the channel was to repeat the measurements described in Figure 4, in low [K+]o and in high [K+]i and in the whole-cell recording mode. Due to the C-type inactivation in wild-type hKv1.3 channels in high [Na+]o we used the mutant channel H399T (Rauer and Grissmer, 1996; Dreker and Grissmer, 2005). Figure 6A shows the currents through H399T channels in the absence and presence of 100 µmol·L−1 verapamil. We used 100 µmol·L−1 verapamil to block almost all currents through the H399T channels in order to be able to visualize the presence of a hook in the tail current. In the enlarged part of the current (Figure 6B), a clear hook can be observed. From the hooked tail current we determined the verapamil off-rate by a simulation of the tail current based on Equation 1. In Figure 6C the verapamil off-rate constants at different hyperpolarizing potentials in low [K+]o obtained by the simulation were plotted against the applied membrane potential. The straight line in Figure 6C represents the mono-exponential fit with a slope of −0.015 mV−1 indicating a voltage dependence of the verapamil off-rate of e-fold/−69 mV. The voltage dependence of the verapamil off-rate in low [K+]o (e-fold/−69 mV) was comparable with the voltage dependence of the verapamil off-rate in high [K+]o (e-fold/−79 mV) and high [Rb+]o (e-fold/−83 mV) respectively. From the verapamil off-rate constant (at −160 mV) of 3 ± 0.31 s−1 (mean ± SD, n= 3) in low [K+]o and 26 ± 6.7 s−1 (mean ± SD, n= 4) in high [K+]o we conclude that the voltage-dependent entry of extracellular K+ into the channel, as had been proposed by Armstrong (1971) for the relief of internal TEA+ block, could be a possible explanation for the voltage-dependent off-rate of verapamil in hKv1.3 channels.
Figure 6.
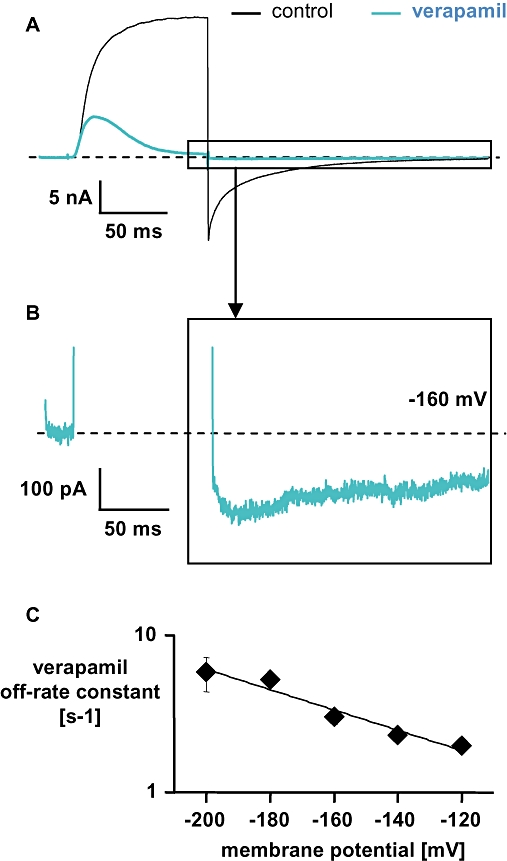
Current through the H399T mutant channel in the absence and presence of 100 µmol·L−1 verapamil in high [Na+]o solution (used as a low [K+]o condition). (A) Current was elicited in the whole-cell configuration from a holding potential of −120 mV with 100-ms depolarizing pulse to +60 mV followed by a 200 ms hyperpolarizing pulse to −160 mV. (B) Enlargement of the same current trace with 100 µmol·L−1 verapamil as shown in (A), to illustrate the hook. (C) Off-rate constants (mean ± SD, n= 3) for verapamil were obtained as described in the legend to Figure 4C and plotted against the applied membrane potential. The straight line represents a mono-exponential fit to the data with a slope of −0.015 mV−1 indicating a voltage dependence of the verapamil off-rate in [Na+]o of e-fold/−69 mV. Error bars are shown when they exceed the size of the symbols.
Influence of the duration of the depolarization on the tail current time course in H399T channels
Another explanation for the voltage dependence of the verapamil off-rate was that it was due to the gating of the channel (state dependence), i.e. that there is more than one open state. In an attempt to generate kinetic evidence for two open states of the hKv1.3 channel, we measured the deactivation time course after different prepulse durations, as one indication for a second open state would be the dependence of the deactivation time course on the duration of the depolarization. For this purpose we used the mutant channel H399T to exclude the influence of inactivation.
We elicited tail currents through H399T channels in high [K+]i and high [K+]o in the outside-out recording mode from a holding potential of −120 mV to a strong depolarization pulse to +100 mV with variable duration of the depolarization followed by hyperpolarized potentials of −100 mV or −160 mV respectively. After a 15-ms step to the depolarizing pulse at +100 mV the channels activate and undergo a transition from the closed to the open state (C → O). The following hyperpolarizing pulse to −100 mV deactivates the channels with a time constant of 44 ms (Figure 7A). After a 500-ms step to +100 mV the deactivation at −100 mV was slowed to a time constant of 67 ms (Figure 7A). Compared with the 15-ms step to +100 mV the deactivation was decreased by a factor of ∼1.5 after 500-ms depolarization. In several other cells we observed a similar effect on the deactivation time constant where after the 500-ms depolarizing step the factor was 2 ± 0.5 (mean ± SD, n= 3). Figure 7B demonstrates the slower tail current decay at −100 mV after a 500-ms depolarizing pulse compared with the 15-ms step to +100 mV more clearly indicating dependence between the duration of the depolarization and deactivation. The closing of the channel at the hyperpolarizing pulse −160 mV was faster than that at −100 mV. After 15 ms to +100 mV and the following hyperpolarizing pulse to −160 mV the channel closed with a deactivation time constant of 18 ms (Figure 7C). The deactivation time constant at −160 mV was 33 ms after a 500-ms depolarization (Figure 7C). Compared with the 15-ms step to +100 mV the closing of the channel was slowed by a factor of ∼1.8 after a 500-ms depolarizing pulse (Figure 7D) and, overall, the factor was 1.6 ± 0.2 (mean ± SD, n= 3) after a 500-ms step to +100 mV.
Figure 7.
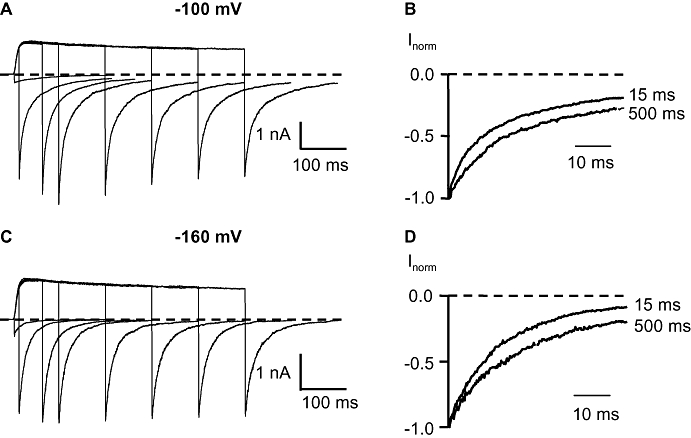
Influence of the duration of the depolarization on the tail current time course in H399T channels. (A) Tail currents at −100 mV after 5-, 15-, 65-, 100-, 200-, 300-, 400- and 500-ms depolarizing pulses to +100 mV in high internal and external K+ solution. (B) Normalized tail currents after 15- and 500-ms depolarizing pulse from (A). (C) Tail currents at −160 mV after 5-, 15-, 65-, 100-, 200-, 300-, 400- and 500-ms depolarizing pulses to +100 mV in high [K+]i and high [K+]o. (D) Normalized tail currents after 15- and 500-ms depolarizing pulse from (C).
Discussion
The aim of this study was to see whether internal and external cations could modulate verapamil affinity in the hKv1.3 channel. Intracellular K+ and Rb+ changed verapamil affinity although a part of this change was presumably due to a change in C-type inactivation. Extracellular K+ and Rb+ influenced the verapamil off-rate at hyperpolarizations. Although we cannot rule out that this effect is due to the voltage-dependent entry of the cations into the channel thereby pushing verapamil out of its binding site, we favour the idea that the voltage-dependent verapamil off-rate is due to the voltage-dependent closure of the channel. This idea would involve the existence of a second open state in the hKv1.3 which was confirmed by the kinetic properties of the deactivation time course after variable durations of the depolarized prepulse.
Role of intracellular K+ and Rb+ on the verapamil affinity
In the wild-type hKv1.3 channel the effect of verapamil was clearly influenced by the intracellular cation. The change in the verapamil affinity induced by the intracellular cations was presumably not caused by a change in C-type inactivation for the following reasons: (i) a faster as well as a slower transition to the C-type inactivated state resulted in a decrease of the verapamil affinity (Figure 2); and (ii) earlier measurements demonstrated that C-type inactivation under physiological conditions does affect the verapamil action since it was assumed that the verapamil affinity was identical for the open and the inactivated state of the channel (Röbe and Grissmer, 2000). In the wild-type channel the decrease of the peak current after the application of verapamil is influenced not only by the transition O → OB (Figure 1) but also by the transition OB → IB and the recovery from the inactivated state after 30 s. Therefore, changes in the occupancy of the inactivated state of the channel will have an influence on the accumulated peak current reduction in the presence of verapamil assuming that recovery from the inactivated state is not affected by a change in the intracellular cations. To avoid the complications with the influence of the C-type inactivation on verapamil affinity we used the mutant channel H399T where the transitions to IB and I can be neglected. Using this mutant channel we observed that the change in the verapamil affinity was smaller compared with the wild-type channel indicating that in the wild-type channel a part of the change in the verapamil affinity was presumably due to the inactivated state and another part due to the intracellular cations. The change in the verapamil affinity through a change in the intracellular cations could be caused by the cation binding sites in the selectivity filter (Zhou and MacKinnon, 2003). In intracellular and extracellular low K+ the structure at the selectivity filter region is changed which could result in a modified verapamil affinity. In intracellular and extracellular high Rb+ the protein structure is similar to the structure in high K+ except there are only three instead of four ion binding sites in the selectivity filter (Zhou and MacKinnon, 2003). This could lead to differences in the verapamil binding site compared with conditions in intracellular and extracellular high K+, which was experimentally observed since in the mutant channel the effect of verapamil changed in high [Rb+]i by a factor of ∼1.5 compared with high [K+]i. We therefore conclude that intracellular cations influence verapamil affinity in the wild-type hKv1.3 channel by (i) a modification of C-type inactivation; and (ii) the occupancy of ion binding sites in the selectivity filter. A similar observation was obtained in hKv1.5 channels with nifedipine by Lin et al. (2001). The effect of nifedipine was changed in high Rb+ by a factor of ∼2 compared with high K+ which is comparable to our data. Lin et al. (2001) concluded that the binding site of permeating ions within the channel pore could be close to the binding site of nifedipine and the cations might affect the binding of nifedipine.
The voltage-dependent recovery from verapamil block
From measurements reported by DeCoursey (1995) we knew that the block of Kv1.3 by verapamil was state-dependent and the rate of recovery from verapamil block (verapamil off-rate) was described as voltage-dependent. We wanted to find out whether the voltage-dependent verapamil off-rate was due to the positively charged verapamil sensing the membrane electric field leaving its binding site because of the negative voltage (‘voltage-dependence’) or whether it was due to the closing of the channel (‘state-dependence’). The similarities in the voltage dependence of channel closure (e-fold/−35 mV in mKv1.3; e-fold/−40 mV in rKv1.3) and the voltage dependence of verapamil off-rate (e-fold/−36 mV in mKv1.3; e-fold/−43 mV in rKv1.3) reported by Röbe and Grissmer (2000) as well as by DeCoursey (1995) and our own results suggested to us that the voltage dependence of the verapamil off-rate may be caused by the voltage dependence of channel closure. The reasons for the steeper voltage dependence of both deactivation and verapamil off-rate reported by Röbe and Grissmer (2000) and DeCoursey (1995) compared with our results could be due to a species difference (mouse/rat vs. human) and/or due to the measuring technique (whole cell vs. outside-out). One way to experimentally distinguish the voltage dependence from the state dependence of recovery from verapamil block would be to create conditions where the channels stay open longer at potentials more negative than −60 mV, compared with control conditions. If recovery from verapamil under these conditions is identical to those under control conditions then this recovery would be mainly voltage-dependent. Alternatively, if recovery from verapamil under these conditions is slower compared with those under control conditions then the recovery process would be mainly state-dependent. For this purpose we created conditions using high [Rb+]o in the extracellular solution in order to slow down channel closing compared with control conditions (high [K+]o). This procedure resulted in a prolongation of the open state of the channel by a factor of ∼5, comparable to earlier reports (Cahalan et al., 1985). As recovery from verapamil block under those conditions was also slowed down compared with those under control conditions we initially concluded that recovery from verapamil block could be affected by the voltage-dependent closing (deactivation) of the channel and not by the voltage-dependent entry of K+ and Rb+ respectively. However, our observations under physiological conditions in low [K+]o suggested to us that the voltage-dependent entry of K+ into the channel might displace verapamil from its blocking site as the verapamil off-rate constant at the hyperpolarized potential of −160 mV was almost 10-fold slower in low [K+]o= 4.5 mmol·L−1, compared with high [K+]o, close to the difference in K+ concentration in those solutions. From this result, it also becomes clear that verapamil action is optimal under real physiological conditions (low [K+]o+ high [K+]i+ membrane potential more positive than −80 mV) since hardly any K+ will be able under these conditions to displace verapamil from its binding site.
Although the voltage-dependent entry of K+ into the channel might be able to explain our results, we cannot exclude the possibility that the voltage-dependent recovery from verapamil block was due to the voltage-dependent closure of the channel. If this, however, were the case we would have to postulate the existence of a second open state of the channel with a lower verapamil affinity. Therefore, we examined the kinetic properties of channel closure in an attempt to create kinetic indications for another open state of the hKv1.3 channel.
Second open state observed by tail currents
Our experiments shown in Figure 7 indicated a second open state of the hKv1.3 channel: increasing the duration of the depolarizing pulse slowed down the deactivation time course at the hyperpolarizing potential. Therefore, we conclude due to the dependency between the deactivation time course and the duration of the depolarized prepulse that the wild-type hKv1.3 channel undergoes at least two different conformational changes before finally closing (Figure 8). The closure of the channel itself could influence the verapamil off-rate with or without a possible voltage-dependent K+ competition mechanism, as proposed above. The first conformational change would push verapamil out of its binding site but would still allow ions to pass and the second conformational change would finally close the channel. Therefore, our results also suggest a second open state of the channel that might not be blocked by verapamil. In that case the first open state OI has a low affinity for verapamil, whereas the second open state OII has a high affinity. We propose that Rb+ stabilizes the second open state and therefore impede the transition from OII to OI and finally to the deactivated, closed state. This might also explain why the deactivation in high [Rb+]o is decreased by a factor of ∼5 compared with the control conditions.
Figure 8.
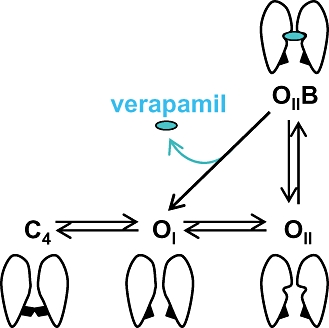
Simplified scheme of hKv1.3 transitions by de- and hyperpolarizations and cartoons showing the possible conformational changes of the channel. C4 is the last closed state, OI and OII are the open and conducting states and OIIB is the open-blocked state. The inactivated state of the channel is not included.
Comparison with other channels
In the hKv1.5 channel, another voltage-gated K+ channel, two open states (Rich and Snyders, 1998) were postulated as the closing of the channel (deactivation) due to hyperpolarization was slower when the depolarizing pulse was longer. In our experiments with hKv1.3 channels, using variable pulse conditions similar to those used by Rich and Snyders (1998), we also observed that increasing the duration of the depolarizing pulse resulted in a slower deactivation time course compared with short depolarizations. Other K+ channels with two or more open states have already been proposed for the hyperpolarization activated, cyclic nucleotide-gated HCN channels (Männikköet al., 2005; Elinder et al., 2006) and MaxiK channels (Benzinger et al., 2006), also mainly for kinetic reasons similar to our measurements.
In homomeric Kv7.1 (KCNQ1) channels, Pusch et al. (2001) observed at least two open states. The authors measured Na+ block in these channels and found that after short depolarizing pulses (50 ms) the block with Na+ was smaller compared with after long depolarizing pulses (200 ms) where the currents through the homomeric Kv7.1 channel was fully blocked. The authors concluded that in the homomeric Kv7.1 channel, the Na+ block provided evidence for the existence of at least two open states with different Na+ affinity. The first open state had a low Na+ affinity whereas the second open state had a high affinity to Na+. In the absence of Na+, the open states showed a similar conductance, that is, the transition between the open states, produced no changes in the current. The verapamil affinity in hKv1.3 channels might be similar to the Na+ affinity in homomeric Kv7.1 channel. We suggest that the first open state in hKv1.3 shows a low verapamil affinity whereas the second open state has a high affinity.
In conclusion, our results indicate a change in the verapamil affinity caused by intracellular cations. Part of this change in verapamil affinity was influenced by the modified C-type inactivated state induced by the intracellular cations and the other part by the occupation by K+ and Rb+ of the ion binding sites in the selectivity filter. The voltage-dependent entry of extracellular cations could displace verapamil from its blocking site inducing a voltage-dependent verapamil off-rate. Although we could not exclude this mechanism to explain the voltage-dependent off-rate of verapamil at hyperpolarizations, we favour the idea that the voltage-dependent verapamil off-rate might be caused by the voltage-dependent closure of the channel. As a consequence of this hypothesis, a second open state of the hKv1.3 channel has to be postulated with a low verapamil affinity in one open state and a high affinity in the other open state and we present kinetic evidence for such a state through the dependency of the deactivation time course on the duration of the depolarization.
Acknowledgments
The authors would like to thank Ms Katharina Ruff for her excellent technical assistance. This work was supported by grants from the 4SC AG (Martinsried, Germany), the Land Baden-Württemberg (1423/74) and the Deutsche Forschungsgemeinschaft (Gr848/14-1).
Glossary
Abbreviations:
- GFP
green fluorescent protein
- Kv channels
voltage-gated potassium channels
- MaxiK channels
Ca2+-activated K+ channels with large conductance
Conflict of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edn. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong CM. Interaction of tetraethylammonium ion derivates with the potassium channels of giant axons. J Gen Physiol. 1971;58:413–437. doi: 10.1085/jgp.58.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzinger GR, Xia X-M, Lingle CJ. Direct observation of a preinactivated, open state in BK channels with β2 subunits. J Gen Physiol. 2006;127:119–131. doi: 10.1085/jgp.200509425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan MD, Chandy KG, DeCoursey TE, Gupta S. A voltage-gated potassium channel in human T lymphocytes. J Physiol. 1985;358:197–237. doi: 10.1113/jphysiol.1985.sp015548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE. State-dependent inactivation of K+ currents in rat type II alveolar epithelial cells. J Gen Physiol. 1990;95:617–646. doi: 10.1085/jgp.95.4.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE. Mechanism of K+ channel block by verapamil and related compounds in rat alveolar epithelial cells. J Gen Physiol. 1995;106:745–779. doi: 10.1085/jgp.106.4.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE, Chandy KG, Gupta S, Cahalan MD. Voltage-gated K+ channels in human T lymphocytes: a role in mitogenesis? Nature. 1984;307:465–468. doi: 10.1038/307465a0. [DOI] [PubMed] [Google Scholar]
- Dreker T, Grissmer S. Investigation of the phenylalkylamine binding site in hKv1.3 (H399T), a mutant with a reduced C-type inactivated state. Mol Pharmacol. 2005;68:966–973. doi: 10.1124/mol.105.012401. [DOI] [PubMed] [Google Scholar]
- Elinder F, Männikkö R, Pandey S, Larsson HP. Mode shifts in the voltage gating of the mouse and human HCN2 and HCN4 channels. J Physiol. 2006;575:417–431. doi: 10.1113/jphysiol.2006.110437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Calvo M, Leonard RJ, Novick J, Stevens SP, Schmalhofer W, Kaczorowski GJ, et al. Purification, characterization, and biosynthesis of margatoxin, a component of Centruroides margartatus venom that selectively inhibits voltage-dependent potassium channels. J Biol Chem. 1993;268:18866–18874. [PubMed] [Google Scholar]
- Grissmer S, Dethlefs B, Wasmuth JJ, Goldin AL, Gutman GA, Cahalan MD, et al. Expression and chromosomal localization of a lymphocyte K+ channel gene. Proc Natl Acad Sci USA. 1990;87:9411–9415. doi: 10.1073/pnas.87.23.9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Kalman K, Pennington MW, Lanigan MD, Nguyen A, Rauer H, Mahnir V, et al. ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. J Biol Chem. 1998;273:32697–32707. doi: 10.1074/jbc.273.49.32697. [DOI] [PubMed] [Google Scholar]
- Lin S, Wang Z, Fedida D. Influence of permeating ions on Kv1.5 channel block by nifedipine. Am J Physiol Heart Circ Physiol. 2001;280:H1160–H1172. doi: 10.1152/ajpheart.2001.280.3.H1160. [DOI] [PubMed] [Google Scholar]
- López-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Männikkö R, Pandey S, Larsson HP, Elinder F. Hysteresis in the voltage dependence of HCN channels: conversion between two modes affects pacemaker properties. J Gen Physiol. 2005;125:305–326. doi: 10.1085/jgp.200409130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Ferrera L, Friedrich T. Two open states and rate-limiting gating steps revealed by intracellular Na+ block of human KCNQ1 and KCNQ1/KCNE1 K+ channels. J Physiol. 2001;533(1):135–144. doi: 10.1111/j.1469-7793.2001.0135b.x. Pt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauer H, Grissmer S. Evidence for an internal phenylalkylamine action on the voltage-gated potassium channel Kv1.3. Mol Pharmacol. 1996;50:1625–1634. [PubMed] [Google Scholar]
- Rauer H, Grissmer S. The effect of deep pore mutations on the action of phenylalkylamines on the Kv1.3 potassium channel. Br J Pharmacol. 1999;127:1065–1074. doi: 10.1038/sj.bjp.0702599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich TC, Snyders DJ. Evidence for multiple open and inactivated states of the hKv1.5 delayed rectifier. Biophys J. 1998;75:183–195. doi: 10.1016/S0006-3495(98)77505-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röbe RJ, Grissmer S. Block of the lymphocyte K+ channel mKv1.3 by the phenylalkylamine verapamil: kinetic aspects of block and disruption of accumulation of block by a single point mutation. Br J Pharmacol. 2000;131:1275–1284. doi: 10.1038/sj.bjp.0703723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, MacKinnon R. The occupancy of ions in the K+ selectivity filter: charge balance and coupling of ion binding to a protein conformational change underlie high conduction rates. J Mol Biol. 2003;333:965–975. doi: 10.1016/j.jmb.2003.09.022. [DOI] [PubMed] [Google Scholar]
- Zhou Y, MacKinnon R. Ion binding affinity in the cavity of the KcsA potassium channel. Biochemistry. 2004;43:4978–4982. doi: 10.1021/bi049876z. [DOI] [PubMed] [Google Scholar]