

Abstract
Background and purpose:
Poly(ADP-ribose) polymerases (PARP)-1 and PARP-2 play complementary tasks in the maintenance of genomic integrity, but their role in cell death or survival processes is rather different. A recently described series of selective PARP-2 inhibitors (UPF-1035, UPF-1069) were used to study the role of PARP-1 and PARP-2 in post-ischaemic brain damage.
Experimental approach:
We evaluated post-ischaemic brain damage in two different in vitro models: rat organotypic hippocampal slices exposed to oxygen-glucose deprivation (OGD) for 20–30 min, a model characterized by apoptosis-like cell death and mouse mixed cortical cell cultures exposed to 60 min OGD, a model in which cells die with mostly necrosis-like features.
Key results:
In organotypic hippocampal slices, PARP-2 inhibition with UPF-1069 (0.01–1 µmol·L−1) caused a concentration-dependent exacerbation (up to 155%) of OGD-induced CA1 pyramidal cell death. Higher concentrations, acting on both PARP-1 and PARP-2, had no effect on OGD injury. In mouse mixed cortical cells exposed to OGD, on the contrary, UPF-1069 (1–10 µmol·L−1) significantly reduced post-ischaemic damage.
Conclusion and implications:
Selective PARP-2 inhibitors increased post-OGD cell death in a model characterized by loss of neurons through a caspase-dependent, apoptosis-like process (hippocampal slice cultures), but they reduced post-OGD damage and increased cell survival in a model characterized by a necrosis-like process (cortical neurons). UPF-1069 may be a valuable tool to explore the function of PARP-2 in biological systems and to examine the different roles of PARP isoenzymes in the mechanisms of cell death and survival.
Keywords: apoptosis, cardiac arrest, ischaemia, necrosis, neuroprotection, PARP-2, poly(ADP-ribose) polymerase, tankyrases, stroke
Introduction
Poly(ADP-ribose) polymerases (PARPs) are a relatively large family of enzymes that catalyse the transfer of ADP-ribose units from NAD+ to acceptor proteins. They are involved in key cellular functions including DNA repair, telomere integrity, gene expression, cell division, cell survival and cell death. Members of this family share a conserved catalytic domain but have different molecular structures and subcellular localization (Shall and de Murcia, 2000; Ame et al., 2004; Bürkle, 2005; Jagtap and Szabo, 2005; Koh et al., 2005; Schreiber et al., 2006; Hassa et al., 2006).
PARP enzymic activity was identified in the early 60s (Chambon et al., 1963), and for at least 30 years it was ascribed to a single enzyme (PARP-1), a 113 kDa protein that is still the most studied member of the family. In the last decade, genetic approaches allowed the identification of 18 putative PARP sequences in the human genome, and significant information is available for at least six different isoenzymes (Hassa et al., 2006). PARP-2 is a 62 kDa protein that was discovered in the late 90s as a result of studies on residual PARP activity in cells derived from PARP-1 knockout mice (Ame et al., 1999). Its catalytic domain shares 69% similarity with that of PARP-1, and both enzymes are localized in the nucleus and are activated by DNA strand breaks (Huber et al., 2004). Structural differences between PARP-1 and PARP-2 are mostly localized in the proximity of the PAR acceptor site, suggesting that the two enzyme isoforms may specifically interact with different substrate proteins (Oliver et al., 2004). Other members of the PARP family have been described and are thought to play important roles in cellular physiology (Schreiber et al., 2006). For example, the PARP family member tankyrase-1 (142 kDa) is present both in the Golgi apparatus and nucleus of most cell types. Using RNA interference (RNAi) technology it has been demonstrated that it is essential for the control of telomere cohesion and bipolar spindle formation during cell division (Chang et al., 2005).
In cellular extracts, PARP-1 has been shown to be responsible for approximately 80% while PARP-2 accounts for approximately 10–15% of the total, maximally stimulated, PARP activity (Oliver et al., 2004). Studies in PARP-1 and PARP-2 null animals revealed that the two enzymes may participate in similar biological process and share a number of common partner proteins for the repair of damaged DNA. Mice lacking both PARP-1 and PARP-2 do not survive, suggesting that the two enzymes may share complementary functions that are essential for the life of the animals (Schreiber et al., 2002). Recently, however, significant differences in the functional role of these enzymes have emerged (Yelamos et al., 2008). It is particularly interesting to note that when PARP-1−/− (Endres et al., 1997) or PARP-2−/− (Kofler et al., 2006) mice were exposed to focal ischaemia by means of middle cerebral artery occlusion (MCAO), their infarct volumes were significantly reduced as compared with those of wild-type controls, thus indicating that a reduction of PARP activity may be beneficial in reducing post-ischaemic brain injury after stroke (Moroni, 2008). On the other hand, when PARP-2-deficient mice were exposed to cardiac arrest followed by cardiopulmonary resuscitation (that leads to transient global forebrain ischaemia), the resulting selective death of hippocampal CA1 pyramidal cells was enhanced (Kofler et al., 2006), suggesting that activation of PARP-2 may facilitate the survival of hippocampal neurons while PARP-2 inhibition may increase post-ischaemic damage in selected neuronal populations.
We have previously observed that in murine mixed cortical cells exposed to oxygen-glucose deprivation (OGD), which resembles the type of cell death that occurs in models of focal ischaemia in vivo, cell death is mostly necrotic, whereas a caspase-dependent apoptotic-like form of neurodegeneration occurs in CA1 pyramidal cells of organotypic hippocampal slices exposed to OGD, an in vitro model of the hippocampal damage typical of transient global ischaemia in vivo (Moroni et al., 2001). PARP inhibitors interacting with both PARP-1 and PARP-2 significantly reduce brain injury following severe excitotoxicity, OGD in cortical cells and transient or permanent MCAO (Moroni, 2008). Conflicting results, however, have been obtained in global ischaemia models with PARP inhibitor treatment resulting in exacerbation (Nagayama et al., 2000), neuroprotection (Hamby et al., 2007) or no effects (Moroni et al., 2001; Meli et al., 2004).
We recently characterized a series of isoquinolinone derivatives with PARP-2 inhibitory properties: among these the most selective were UPF-1035 and UPF-1069 (Pellicciari et al., 2008). In the present study, we further characterized the PARP-2 selectivity of these compounds and ruled out their possible effects on tankyrase-1, another PARP family member. Moreover, we tested the effects of UPF-1035 and UPF-1069 in rat organotypic hippocampal slices and in mixed cortical cells exposed to OGD. Our results show that selective inhibition of PARP-2 with these agents enhances OGD injury in hippocampal slices while inhibition of both PARP-1 and PARP-2 (obtained either with higher concentrations of these agents or with other PARP inhibitors acting on both PARP-1 and PARP-2) had no effect on neuronal survival. In mixed cortical cells exposed to OGD, UPF-1069 reduced post-ischaemic damage at both low and high concentrations
Methods
PARP-1 and PARP-2 activity assays
PARP activity was evaluated by utilizing commercially available recombinant bovine PARP-1 and mouse PARP-2 (Alexis, Vinci, Italy), as previously described (Moroni et al., 2001; Pellicciari et al., 2008). Briefly, the enzymatic reaction was carried out in 100 µL of 50 mmol·L−1 Tris-HCl (pH 8.0) containing 5 mmol·L−1 MgCl2, 2 mmol·L−1 dithiothreitol, 10 µg sonicated calf thymus DNA, 0.2 µCi [adenine-2,8-3H]NAD and recombinant enzyme PARP-1 or PARP-2 (0.03 U per sample). Different concentrations of the putative inhibitors were added, and the mixture was incubated for 1 h at 37°C. The reaction was terminated by adding 1 mL of 10% trichloroacetic acid (w/v) and centrifuged. Pellets were then washed twice with 1 mL of H2O and resuspended in 1 mL of 0.1 mol·L−1 NaOH. The radioactivity incorporated from [adenine-2,8-3H]NAD into proteins was evaluated by liquid scintillation spectrometry.
Nuclear extracts for PARP activity assay were prepared from PARP-1−/− and PARP-1+/+ mouse fibroblasts as previously described (Fossati et al., 2006). Briefly, immortalized fibroblasts from PARP-1−/− and PARP-1+/+ mice were grown in DMEM supplemented with 2 mmol·L−1 glutamine, 10% bovine serum and antibiotics at 37°C. Nuclear extracts were prepared as follows: 10 cm confluent plates were scraped in 1 mL of homogenization buffer composed of 75 mmol·L−1 sucrose, 225 mmol·L−1 mannitol and 5 mmol·L−1 Tris, pH 7.4 plus 10 µL protease inhibitor cocktail (Sigma-Aldrich, Milan, Italy). The cell suspension was homogenized with a teflon pestle, and the mixture was centrifuged at 600×g for 5 min at 4°C. The crude nuclear pellet was washed and resuspended in 1 mL of PARP assay buffer (5 mmol·L−1 MgCl2, 2 mmol·L−1 DTT, 50 mmol·L−1 Tris, pH 8) containing 100 µmol·L−1 N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) to fully activate PARP activity. Samples containing 100 µL of the resuspended nuclear pellet were incubated for 60 min at 37°C in the presence of 35.5 nmol·L−13H-NAD. The reaction was stopped with 1 mL of 10% trichloroacetic acid (w/v), and the mixture was centrifuged at 12 000×g for 10 min at 4°C. The reaction was terminated by the addition of 1 mL of 10% trichloroacetic acid (w/v), and radioactivity of the suspension was measured by liquid scintillation spectrometry.
Evaluation of tankyrase-1 function
HeLa cells cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% heat-inactivated fetal calf serum were synchronized in mitosis by using 700 nmol·L−1 S-trityl-L-cysteine, fixed in paraformaldehyde 4% and processed for immunocytochemical evaluation using turbulent antibodies as described by Chang et al. (2005). In order to reduce the synthesis and function of tankyrase-1, cells were transfected with small interference RNA (siRNA) (control siRNA: 5-AATTCTCCGAACGTGTCACGT, tankyrase-1 siRNA: 5-AACAAUUCACCGUCGUCCUCU, Dharmacon, Lafayette, CO, USA) by using oligofectamine (Invitrogen, San Giuliano Milanese, Italy) as described by the manufacturer, and assayed 2 days post transfection. Imaging was performed by using a Nikon fluorescence microscope equipped with piezoelectric motorization and a CCD camera. Stacks of images were acquired through the depth of the section by sing Metamorph/Metafluor software (Molecular Devices, Downingtown, PA, USA) and deconvoluted by using Image Autodeblur software (MediaCybernetics, Bethesda, MD, USA). For each field, the number of mitosis and the ratio between abnormal and normal mitosis were evaluated. In each experiment, at least four microscopic fields were counted. The final values represent the mean of at least three independent experiments.
OGD in rat organotypic hippocampal slices
All animal care and the experimental procedures were formally approved by the ethical committee for animal care at the Department of Pharmacology of the University of Florence and were performed in compliance with the recommendations of the European Union (86/609/EEC). Organotypic hippocampal slice cultures were prepared as previously described (Pellegrini-Giampietro et al., 1999a,b). Briefly, the hippocampi were removed from the brains of 7–9-day-old Wistar rats, and transverse slices (420 µm) were prepared by using a McIlwain tissue chopper in a sterile environment. Isolated slices were first placed in ice-cold Hanks' balanced salt solution (HBSS), supplemented with 5 mg·mL−1 glucose and 1.5% Fungizone® (GIBCO-BRL, San Giuliano Milanese, Italy), then transferred to humidified semiporous membranes (30 mm Millicell-CM 0.4 mm tissue culture plate inserts, Millipore, Italy; four per membrane). These were placed in six-well tissue culture plates containing 1.2 mL culture medium containing 50% Eagle's minimal essential medium (MEM), 25% heat-inactivated horse serum, 25% HBSS, 5 mg·mL−1 glucose, 1 mmol·L−1 glutamine and 1.5% Fungizone®. Slices were maintained at 37°C, with 100% humidity and 95% air/5% CO2 atmosphere, and the medium was changed every 3 days. Experiments were carried out after 14 days in vitro.
Oxygen-glucose deprivation was induced as previously described in detail (Pellegrini-Giampietro et al., 1999a,b). Briefly, the slices were exposed to a serum-free medium saturated with 95% N2/5% CO2 at 37°C in a gassed incubator equipped with an oxygen controller (BioSpherix, New York, NY, USA). After 20 min, the cultures were transferred to oxygenated serum-free medium containing 5 mg·mL−1 glucose and returned to the incubator under normoxic conditions. Neuronal injury was evaluated 24 h later. Maximal damage was achieved in this system by exposing the slices to 10 mmol·L−1 glutamate for 24 h. Cell injury was assessed by using the fluorescent dye propidium iodide (PI), a highly polar compound that is normally excluded from cells with an intact membrane. When the membrane is damaged, PI can enter the cells and upon binding to exposed DNA becomes highly fluorescent. PI (5 µg·mL−1) was added to the medium at the end of the 24 h post-OGD recovery period. Thirty minutes later, fluorescence was viewed by using an inverted fluorescence microscope (Olympus IX-50; Solent Scientific, Segensworth, UK) equipped with a xenon-arc lamp, a low-power objective (4×) and a rhodamine filter. Images were digitized by using a video image obtained with a CCD camera (Diagnostic Instruments Inc., Sterling Heights, MI, USA) controlled by software (InCyt Im1TM; Intracellular Imaging Inc., Cincinnati, OH, USA) and subsequently analysed by using the Image-Pro Plus morphometric analysis software (Media Cybernetics, Silver Spring, MD, USA). In order to quantify cell death, the CA1 hippocampal subfield was identified and encompassed in a frame using the drawing function in the image software (ImageJ; NIH, Bethesda, MD, USA), and the optical density of PI fluorescence was recorded. There was a linear correlation between CA1 PI fluorescence and the number of injured CA1 pyramidal cells, as indicated by morphological criteria.
OGD in cortical cell cultures
Primary cultures of mixed cortical cells containing both neuronal and glial elements were prepared as previously described (Pellegrini-Giampietro et al., 1999a,b). Briefly, cerebral cortices were dissected from fetal mice at 14–15 days of gestation, minced by using medium stock [MS, composed of Eagle's MEM (with Earle's salts, glutamine- and NaHCO3-free) supplemented with 38 mmol·L−1 NaHCO3, 22 mmol·L−1 glucose, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin] and incubated for 10 min at 37°C in MS with 0.25% trypsin and 0.05% DNase. Enzymatic digestion was terminated by a second incubation (10 min at 37°C) in MS supplemented with 10% heat-inactivated horse serum and 10% fetal bovine serum, after which the cells were mechanically disrupted and counted. After brief centrifugation, cells were resuspended (approximately 4 × 105 cells·mL−1) and plated in 15 mm multi-well vessels on a layer of confluent astrocytes by using a plating medium of MS supplemented with 10% heat-inactivated horse serum, 10% fetal bovine serum and 2 mmol·L−1 glutamine. Cultures were kept in an incubator at 37°C, with 100% humidity and 95% air/5% CO2 atmosphere. After 4–5 days in vitro, non-neuronal cell division was halted by the application of 3 mmol·L−1 cytosine arabinoside for 24 h. Cultures were then shifted to a maintenance medium identical to the plating medium but lacking fetal bovine serum, which was then partially replaced twice a week. Experiments were done with mature cultures (14–15 days in vitro).
Oxygen-glucose deprivation was induced in mixed cortical cultures as previously described in detail (Pellegrini-Giampietro et al., 1999a,b). Briefly, the culture medium was replaced by thorough exchange with a glucose-free balanced salt solution (composition in mmol·L−1: 116 NaCl, 5.4 KCl, 0.8 MgSO4, 1 NaH2PO4, 26 NaHCO3, 1.8 CaCl2; with 10 mg·L−1 phenol red) previously saturated with 95% N2/5% CO2 and heated to 37°C. Multi-wells were then placed in an airtight incubation chamber equipped with inlet and outlet valves, and 95% N2/5% CO2 was blown through the chamber for 10 min to ensure maximal removal of oxygen. The chamber was then sealed and placed in the incubator at 37°C for 60 min. OGD was terminated by removing the cultures from the chamber, replacing the exposure solution with oxygenated MS and returning the multi-wells to the incubator under normoxic conditions. The extent of neuronal death was assessed 24 h later. In order to achieve maximal neuronal injury, the cultures were exposed for 24 h to 1 mmol·L−1 glutamate in MS at 37°C, with 100% humidity and 95% air/5% CO2 atmosphere. Cell damage was evaluated by measuring the amount of lactate dehydrogenase (LDH) released from injured cells into the extracellular fluid 24 h after exposure to OGD or glutamate. Background LDH release was determined in control cultures not exposed to OGD and was subtracted from all experimental values. The resulting value correlated linearly with the degree of cell loss estimated by observing the cultures under phase-contrast microscopy or under bright-field optics after 5 min incubation with 0.4% Trypan blue, which stains debris and nonviable cells.
Statistical procedures
Concentration–response curves of PARP inhibitors were analysed, and IC50 values (mean ± SEM) were calculated with the Prism software package (GraphPAD Software, Inc., San Diego, CA, USA). Statistical significance of differences between OGD-induced LDH release or PI fluorescence intensities was evaluated by performing analysis of variance (anova) followed by the Tukey's w-test for multiple comparisons. The P-value was calculated using a two-tailed test.
Materials
UPF-1035, UPF-1069, UPF-1066 and thieno[2,3-c]isoquinolin-5-one (TIQ-A) were synthesized in the Department of Chemistry and Drug Technology, University of Perugia, as previously described (Chiarugi et al., 2003; Pellicciari et al., 2008). Benzamide was purchased from Sigma-Aldrich (Milano, Italy). Tissue culture reagents were purchased from Gibco-BRL (San Giuliano Milanese, MI, Italy) and Sigma-Aldrich (Milano, Italy). Activity of LDH was quantified by using the Cytotoxicity Detection Kit (LDH) from Roche Diagnostics (Mannheim, Germany).
Results
Characterization of UPF-1069 as a PARP-2 subtype-specific inhibitor
UPF-1035 and UPF-1069 are compounds synthesized in the course of studies aimed at obtaining subtype-selective PARP antagonists (Pellicciari et al., 2008). Figure 1 shows their chemical structures, their concentration–response inhibitory curves against recombinant PARP-1 and PARP-2 activity and their IC50 values. The data for UPF-1066, another isoquinolinone derivative that was inactive in the PARP-1 and PARP-2 assays, are also reported.
Figure 1.
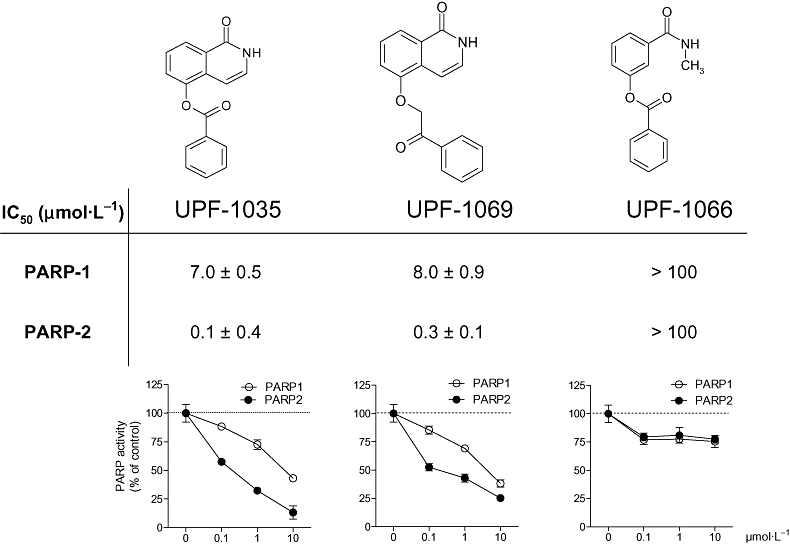
Molecular structures, IC50 values and inhibitory activity against poly(ADP-ribose) polymerase (PARP)-1 and PARP-2 of UPF-1035, UPF-1069 and UPF-1066. The compounds UPF-1035 and UPF-1069 were identified as numbers 54 and 55 respectively, in a previous report (Pellicciari et al., 2008). The structural analogue UPF-1066 was used as a negative control. Values in curves are expressed as percentage of control PARP activity and were obtained by incubating the inhibitors with 0.2 µCi [adenine-2,8-3H]NAD and 0.03 U recombinant bovine PARP-1 or mouse PARP-2 for 1 h at 37°C. Each point represents the mean ± SEM of at least three experiments performed in triplicate.
When tested against PARP activity in nuclear extracts obtained either from wild-type or from PARP-1−/− fibroblasts, the non-specific inhibitors benzamide (100 µmol·L−1) and the PARP-1 and PARP-2 inhibitor TIQ-A (at 10 µmol·L−1) were able to reduce enzymatic activity to a similar extent (approximately by 75%) both in wild-type and PARP-1-deficient cells (Figure 2). Conversely, 10 µmol·L−1 UPF-1035 (not shown) and UPF-1069 (Figure 2) decreased by approximately 80% the residual PARP activity of PARP-1-deficient fibroblasts, but the enzymic activity in wild-type fibroblasts was only slightly inhibited (15%).
Figure 2.
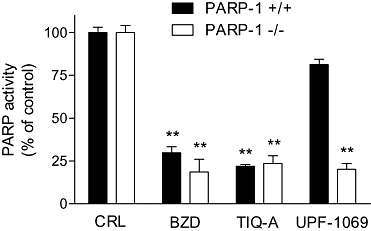
Effects of various PARP inhibitors on PARP activity in nuclear extracts from PARP-1+/+ and PARP-1−/− mouse fibroblasts. Values are expressed as percentage of control PARP activity and were obtained by incubating benzamide (BZD, 100 µmol·L−1), TIQ-A (10 µmol·L−1) and UPF-1069 (1 µmol·L−1) with nuclear extracts in the presence of 100 µmol·L−1 MNNG and 35.5 nmol·L−1 of added [adenine-2,8-3H]NAD for 1 h at 37°C. Benzamide and TIQ-A-reduced PARP activity both in wild-type and in PARP-1−/− extracts, whereas UPF-1069 reduced PARP activity only in PARP-1−/− extracts. Each bar represents the mean ± SEM of at least three experiments performed in triplicate. **P < 0.01 versus respective control. CRL, control; MNNG, N-methyl-N′-nitro-N-nitrosoguanidine; PARP, poly(ADP-ribose) polymerase; TIQ-A, thieno[2,3-c]isoquinolin-5-one.
In order to rule out a possible interaction between the selective PARP-2 inhibitors and tankyrase-1 (another PARP family member), we assessed the telomere function of this enzyme in HeLa cells by counting the number of abnormal spindles and mitosis during cell division. It has been shown that a reduction of tankyrase-1 activity by RNAi leads to the formation of multipolar spindles and to abnormal mitosis (Chang et al., 2005). Figure 3 shows that, whereas siRNA against tankyrase-1, or the non-specific PARP inhibitor benzamide promoted the appearance of a significant number of multipolar spindles, UPF-1069, and UPF-1035, even when used at concentrations (100 µmol·L−1) that were much larger than those required to inhibit PARP-2, did not affect spindle formation. In a similar manner, the PARP-1 and PARP-2 inhibitor TIQ-A (0.1–100 µmol·L−1), which belongs to a different chemical class, did not increase the percentage of multipolar spindles (Figure 3).
Figure 3.
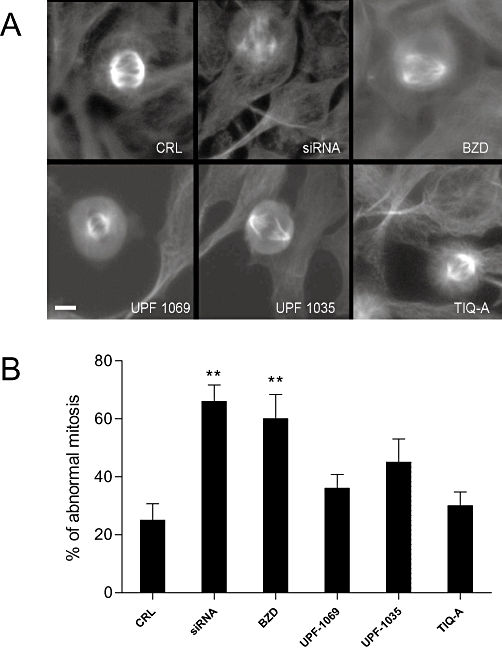
Effects of PARP inhibitors on the telomeric function of tankyrase-1 in HeLa cells. (A) Morphological appearance of mitotic spindles in HeLa cells treated with tankyrase-1 siRNA, benzamide (BZD, 100 µmol·L−1), UPF-1069 (100 µmol·L−1), UPF-1035 (100 µmol·L−1) and TIQ-A (100 µmol·L−1). Tankyrase-1 siRNA and benzamide induce the appearance of a significant number of abnormal, multipolar spindles. (B) Quantitative analysis of the extent of abnormal mitosis in HeLa cells exposed to tankyrase-1 siRNA or to PARP inhibitors at 100 µmol·L−1. Values are expressed as percentage of abnormal mitosis (calculated as the ratio between abnormal and normal mitosis) and are the mean ± SEM from at least four microscopic fields from three independent experiments. **P < 0.01 versus control; Scale bar: 5 µm. CRL, control; PARP, poly(ADP-ribose) polymerase; siRNA, small interference RNA; TIQ-A, thieno[2,3-c]isoquinolin-5-one.
UPF-1069 exacerbates OGD damage in organotypic hippocampal slices
As repeatedly described (Pellegrini-Giampietro et al., 1999a,b; Moroni et al., 2001), exposure of organotypic hippocampal slices to 30 min OGD produces, 24 h later, an increase in PI fluorescence in the CA1 region that is approximately 75–80% of the maximal fluorescence intensity obtained by exposing the slices to 10 mmol·L−1 glutamate for 24 h (100% neuronal damage). Exposure to OGD for a shorter (20 min) period causes a less extensive death of CA1 pyramidal cells (approximately 40–60% of the maximal damage), a strategy that can be used to detect both reduction and exacerbation of OGD injury by experimental drugs (Pellegrini-Giampietro et al., 1999a; Werner et al., 2007). Using both morphological and biochemical approaches, we have previously demonstrated that, after this injury, most of CA1 pyramids undergo a caspase-dependent, apoptotic-like neuronal death (Moroni et al., 2001) and that the CA1 damage in this model reproduces the injury pattern found in transient global forebrain ischaemia in vivo (Kirino, 1982; Pulsinelli et al., 1982).
As PARP-2 deletion significantly increased neuronal cell loss in the hippocampal CA1 field in a model of 10 min cardiac arrest (Kofler et al., 2006), we tested the effects of UPF-1069 on organotypic hippocampal slices exposed to 20 min OGD both at concentrations (0.1–1 µmol·L−1) selectively acting on PARP-2 and at a higher concentration (10 µmol·L−1) that reduces both PARP-1 and PARP-2 activity (Pellicciari et al., 2008). Figure 4 shows that UPF-1069 at 0.1–1 µmol·L−1 significantly enhanced CA1 hippocampal damage. A qualitatively similar effect was present when the slices were exposed to 30 min OGD (data not shown). When UPF-1069 was tested at 10 µmol·L−1, there was no exacerbation of damage (Figure 4). In accordance with previous observations (Moroni et al., 2001), another PARP inhibitor (TIQ-A), acting both on PARP-1 or PARP-2, tested in a wide range of concentrations (0.01–100 µmol·L−1), neither decreased nor exacerbated OGD injury.
Figure 4.
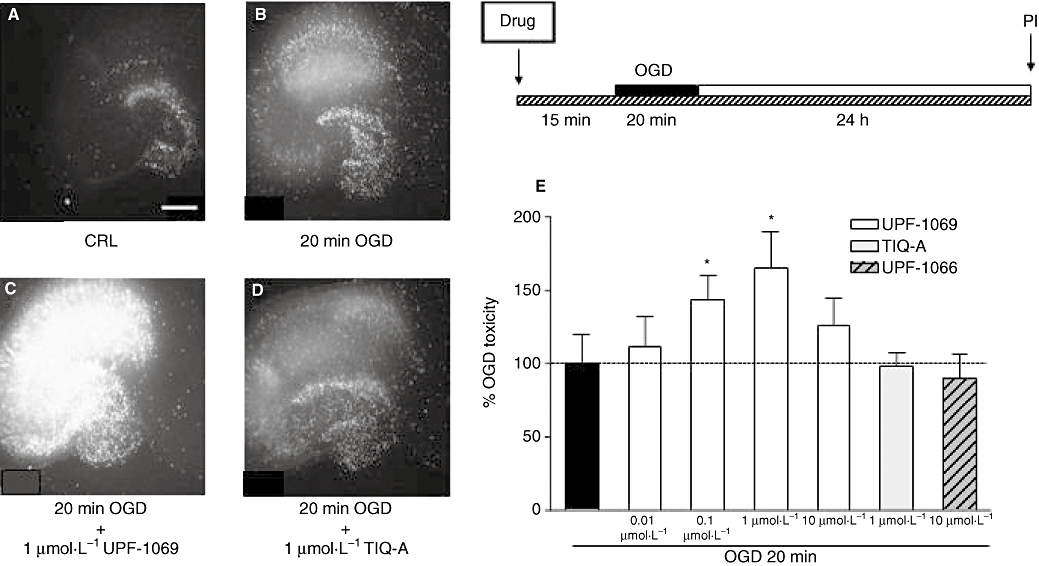
Effects of PARP inhibitors in organotypic rat hippocampal slices exposed to OGD. Slices in culture were exposed to 20 min OGD and 24 h later incubated with PI for fluorescence detection of optical density in the CA1 region. PARP inhibitors were added to the incubation medium 15 min prior to OGD and maintained during OGD and the subsequent 24 h recovery period. (A) Hippocampal slice under normoxic control conditions displaying background PI fluorescence. (B) Hippocampal slice exposed to 20 min OGD, displaying intense PI labelling in the CA1 sub-region. (C) Hippocampal slice exposed to 20 min OGD and treated with 1 µmol·L−1 UPF-1069, displaying exacerbation of CA1 and CA3 damage. (D) Hippocampal slice exposed to 20 min OGD and treated with 1 µmol·L−1 TIQ-A, displaying no effect on OGD injury. (E) Quantitative analysis of the effects of UPF-1069, TIQ-A and UPF-1066 in organotypic hippocampal slices exposed to 20 min OGD. CA1 damage was significantly increased when slices were treated with 0.01–1 µmol·L−1 UPF-1069 but not with 10 µmol·L−1 UPF-1069, 1 µmol·L−1 TIQ-A or 10 µmol·L−1 UPF-1066. Data are expressed as percentage of OGD toxicity and represent the mean ± SEM of at least three experiments performed in triplicate. *P < 0.05 versus 20 min OGD; Scale bar: 2 mm. CRL, control; OGD, oxygen-glucose deprivation; PARP, poly(ADP-ribose) polymerase; PI, propidium iodide; TIQ-A, thieno[2,3-c]isoquinolin-5-one.
Interestingly, UPF-1066, another isoquinolinone derivative that was inactive in the PARP-1 and PARP-2 assays (Figure 1) had no effect on the OGD-induced CA1 injury (Figure 4).
UPF-1069 attenuates OGD damage in mixed cortical cell cultures
Mixed cortical cells exposed to 60 min OGD undergo extensive neuronal damage that can be qualitatively evaluated with phase-contrast microscopy or other morphological approaches and quantitatively measured by monitoring the release of LDH from the damaged cells into the bathing medium (Pellegrini-Giampietro et al., 1999a,b). We have shown that most of cortical neurons exposed to OGD display electron microscopic and biochemical features of necrotic-like cell death and that PARP inhibitors drastically reduce neuronal loss (Moroni et al., 2001). When a number of PARP inhibitors with different molecular structures were tested, a direct correlation was obtained between their potency (EC50 values) in reducing OGD injury and their IC50 values in inhibiting PARP-1 activity (Chiarugi et al., 2003). Figure 5 shows that UPF-1069 displayed a significant neuroprotective activity both at a concentration (1 µmol·L−1) selectively acting on PARP-2 and at a concentration (10 µmol·L−1) that inhibits both PARP-1 and PARP-2 activities (Pellicciari et al., 2008). A similar protective effect was observed with the PARP-1 and PARP-2 inhibitor TIQ-A (Figure 5).
Figure 5.
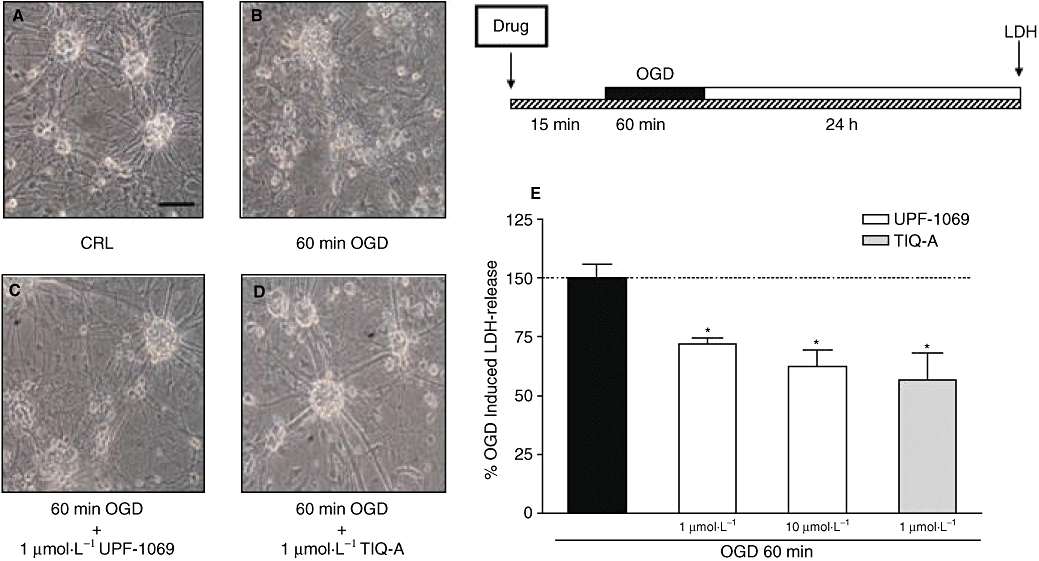
Effects of PARP inhibitors in murine mixed cortical cell cultures exposed to OGD. Cultures were exposed to 60 min OGD and 24 h later neuronal injury was assessed by phase-contrast microscopy or by measuring the release of LDH into the medium. PARP inhibitors were added to the incubation medium 15 min prior to OGD and maintained during OGD and the subsequent 24 h recovery period. (A) Cortical cultures under normoxic control conditions, displaying intact morphology. (B) Cortical cultures exposed to 60 min OGD, displaying extensive death of neuronal bodies that are replaced by cellular debris. (C,D) Cortical cultures exposed to 60 min OGD and treated with 1 µmol·L−1 UPF-1069 or 1 µmol·L−1 TIQ-A, displaying a reduction of OGD injury. (E) Quantitative analysis of the effects of UPF-1069 and TIQ-A in mixed cortical cells exposed to 60 min OGD. Neuronal death was significantly reduced when cultures were treated with 1–10 µmol·L−1 UPF-1069 and 1 µmol·L−1 TIQ-A. Data are expressed as percentage of OGD-induced LDH release represent the mean ± SEM of at least three experiments performed in triplicate. *P < 0.05 versus 60 min OGD. Scale bar: 50 µm. CRL, control; LDH, lactate dehydrogenase; OGD, oxygen-glucose deprivation; PARP, poly(ADP-ribose) polymerase; TIQ-A, thieno[2,3-c]isoquinolin-5-one.
Discussion
Our results show that UPF-1069 and its structural analogue UPF-1035 are potent and selective PARP-2 inhibitors able to reduce PAR formation both in recombinant enzyme preparations and in nuclear extracts from PARP-1−/− fibroblasts. Other PARP-2 inhibitors have been described (Perkins et al., 2001; Iwashita et al., 2005), but to our knowledge, UPF-1035 and UPF-1069 are the most selective available to date.
We show here that UPF-1069 increased CA1 pyramidal cell loss in organotypic hippocampal slices exposed to 20 or 30 min OGD. The CA1 hippocampal cell injury observed in OGD-exposed slices is morphologically and biochemically similar to that occurring in the same region in rodents (gerbils, mice and rats) exposed to transient global forebrain ischaemia (Kirino, 1982; Pulsinelli et al., 1982) or in humans after transient cardiac arrest (Petito et al., 1987): it is selective for CA1 pyramidal cells and, at least in rodents, has the electron microscopic features of an apoptosis-like process and is significantly reduced by caspase inhibitors (Moroni et al., 2001). Because UPF-1069 exacerbates this type of cell loss and, in a similar manner, PARP-2−/− mice exposed to short periods of cardiac arrest have increased CA1 pyramidal cell damage (Kofler et al., 2006) it seems that PARP-2 activation facilitates CA1 pyramidal cell survival. When hippocampal slices were exposed to longer OGD periods (45–60 min), cell loss and histological damage was not limited to the principal cells of the CA1 region (the model we decided to study here) but every hippocampal region and cell type, including most glial cells, were also damaged (DE Pellegrini-Giampietro et al., unpubl. obs.). We have not studied PARP inhibitors under those conditions, but experiments in vivo (global forebrain ischaemia of 20–30 min) suggest that PARP inhibition reduces the hippocampal damage mostly because of a decreased inflammatory cell infiltration (Hamby et al., 2007).
Interestingly, when both PARP-1 and PARP-2 were inhibited either with agents lacking subtype specificity for the two isoenzymes (such TIQ-A) or with larger concentrations (10 µmol·L−1) of UPF-1069, CA1 injury after OGD was neither enhanced nor reduced. Indeed, the inhibition of PARP-1 in addition to a persisting PARP-2 inhibition is the most likely explanation for the bell-shaped concentration–response curve observed in Figure 4 for UPF-1069.
Our results showing that PARP-2 inhibition may lead to exacerbation of an apoptosis-like cell death process is not surprising, as it has been previously shown that PARP-2 is a survival factor in other cell types such as CD4+CD8+ thymocytes (Yelamos et al., 2006). PARP-2−/− mice have a significantly reduced number of these cells possibly because of a constitutively increased expression of Noxa, a pro-apoptotic factor belonging to the Bcl2 family leading to an increased rate of thymocyte elimination (Yelamos et al., 2008). It is reasonable to propose that changes in gene expression similar to those described in PARP-2−/− thymocytes also occur in hippocampal pyramidal cells after UPF-1069 treatment.
Recently, the role of PARP-1 and PARP-2 in the expression of apoptosis-related genes has been thoroughly studied in HeLa cells exposed to DNA damaging agents. It has been clearly shown that PARP-1 inhibition may reduce the expression of a number of pro-apoptotic genes, such as Bcl10, c-Rel and tumour necrosis factor-related apoptosis-inducing ligand receptor-1 and 2 (Cohausz and Althaus, 2009). These findings may help to explain why, when PARP-1 was inhibited, the toxic effects of UPF-1069 were reduced and why, under some circumstances, PARP inhibitors may even improve the hippocampal damage in rodents exposed to global forebrain ischaemia (Hamby et al., 2007).
In mixed cortical cells exposed to OGD, selective PARP-2 inhibition appears to be sufficient to reduce the extent of ischaemia-induced neuronal death. Because mixed cortical cell cultures exposed to OGD may be considered a suitable model to study the molecular events occurring in injured cells after MCAO (Moroni et al., 2001; Meli et al., 2002), these data can be considered in good agreement with previous findings obtained in PARP-2−/− mice in vivo showing that these animals have a reduced brain infarct after middle cerebral occlusion (Kofler et al., 2006). It is reasonable to assume that the molecular mechanisms leading to cell death are different in the two ischaemic in vitro models we used and that OGD injury in the various cell populations present in brain tissue is mediated by a wide variety of processes.
In the past, post-ischaemic neuronal injury was thought to be mainly caused by the release of glutamate and the ensuing cascade involving stimulation of ionotropic and metabotropic glutamate receptors, increase in intracellular free Ca2+, activation of neuronal NO synthase and formation of NO or other highly reactive free radicals that produce DNA strand breaks, activation of PARP, depletion of NAD and ATP, and eventually energy deprivation resulting in necrotic cell death (Zhang et al., 1994; Szabò and Dawson, 1998; Virag and Szabo, 2002). A similar cascade may occur also in other cell types such as glia, pericytes or endothelia. In these cells, however, besides the possible PARP activation-induced energy failure it may be important to consider that PARP activation also leads to an increased expression of inflammatory agents, which are locally released and amplify the inflammatory reaction that constantly accompany tissue ischaemia (Barone and Feuerstein, 1999; Faraco et al., 2007). All these events appear to be attenuated by PARP-1 inhibitors (Moroni, 2008).
Although PARP-2 has a lower activity and has been much less studied, its inhibition appears to be enough to either increase or reduce post-ischaemic damage. The contribution of PARP-2 to the depletion of cellular NAD and ATP should, however, be marginal, because most of PARP activity under sub-maximally stimulated conditions has been ascribed to PARP-1 (Oliver et al., 2004). It is now emerging, however, that different mechanisms are operative in the decision processes of survival or death in cells exposed to an ischaemic insult and that the death pathways (apoptosis-like, necrosis-like or even necroptosis) activated under various ischaemic conditions are probably different not only in the different cell populations present in slice cultures (neurons, glia, microglia, endothelial cells), but also within the same subpopulation of neurons.
In conclusion, our findings indicate that the inhibition of a single PARP family enzyme (PARP-2) may exacerbate OGD toxicity in hippocampal CA1 pyramids and reduce it in cortical neurons. The exacerbation appears to be specific because at the concentration we used, UPF-1069 does not interact with other PARP family member enzymes (including tankyrase-1) and because a structural analogue (UPF-1066) lacking inhibitory activity on PARPs had no effect on OGD injury. Finally, the data with our PARP inhibitors are qualitatively similar to those obtained with gene-targeting approaches in PARP-1−/− or PARP-2−/− mice (Endres et al., 1997; Kofler et al., 2006).
In conclusion, selective inhibition of PARP-2 enzyme exacerbates OGD injury in the hippocampus. Inhibition of both PARP-1 and PARP-2 activity significantly decreases post-ischaemic neuronal damage in mixed cortical cells, an in vitro stroke model.
Acknowledgments
This work was supported by grants from the University of Florence, the University of Perugia, the Italian Ministry of Instruction, University and Research MIUR (PRIN 2006), the Ente Cassa di Risparmio di Firenze and Wyeth Pharmaceuticals.
Glossary
Abbreviations:
- LDH
lactate dehydrogenase
- MCAO
middle cerebral artery occlusion
- MNNG
N-methyl-N′-nitro-N-nitrosoguanidine
- OGD
oxygen-glucose deprivation
- PARP
poly(ADP-ribose) polymerase
- PI
propidium iodide
- RNAi
RNA interference
- siRNA
small interference RNA
- TIQ-A
thieno[2,3-c]isoquinolin-5-one
Conflict of interest
The authors have no conflict of interest.
References
- Ame JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, et al. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem. 1999;274:17860–17868. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- Barone FC, Feuerstein GZ. Inflammatory mediators and stroke: new opportunities for novel therapeutics. J Cereb Blood Flow Metab. 1999;19:819–834. doi: 10.1097/00004647-199908000-00001. [DOI] [PubMed] [Google Scholar]
- Bürkle A. Poly(ADP-ribose). The most elaborate metabolite of NAD+ FEBS J. 2005;272:4576–4589. doi: 10.1111/j.1742-4658.2005.04864.x. [DOI] [PubMed] [Google Scholar]
- Chambon P, Weill JD, Mandel P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun. 1963;11:39–43. doi: 10.1016/0006-291x(63)90024-x. [DOI] [PubMed] [Google Scholar]
- Chang P, Coughlin M, Mitchison TJ. Tankyrase-1 polymerization of poly(ADP-ribose) is required for spindle structure and function. Nat Cell Biol. 2005;7:1133–1139. doi: 10.1038/ncb1322. [DOI] [PubMed] [Google Scholar]
- Chiarugi A, Meli E, Calvani M, Picca R, Baronti R, Camaioni E, et al. Novel isoquinolinone-derived inhibitors of poly(ADP-ribose) polymerase-1: pharmacological characterization and neuroprotective effects in an in vitro model of cerebral ischemia. J Pharmacol Exp Ther. 2003;305:943–949. doi: 10.1124/jpet.103.048934. [DOI] [PubMed] [Google Scholar]
- Cohausz O, Althaus FR. Role of PARP-1 and PARP-2 in the expression of apoptosis-regulating genes in HeLa cells. Cell Biol Toxicol. 2009 doi: 10.1007/s10565-008-9092-8. in press. [DOI] [PubMed] [Google Scholar]
- Endres M, Wang Z-Q, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly(ADP-ribose) polymerase. J Cereb Blood Flow Metab. 1997;17:1143–1151. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- Faraco G, Fossati S, Bianchi ME, Patrone M, Pedrazzi M, Sparatore B, et al. High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J Neurochem. 2007;103:590–603. doi: 10.1111/j.1471-4159.2007.04788.x. [DOI] [PubMed] [Google Scholar]
- Fossati S, Formentini L, Wang ZQ, Moroni F, Chiarugi A. Poly(ADP-ribosyl)ation regulates heat shock factor-1 activity and the heat shock response in murine fibroblasts. Biochem Cell Biol. 2006;84:703–712. doi: 10.1139/o06-083. [DOI] [PubMed] [Google Scholar]
- Hamby AM, Suh SW, Kauppinen TM, Swanson RA. Use of a poly(ADP-ribose) polymerase inhibitor to suppress inflammation and neuronal death after cerebral ischemia-reperfusion. Stroke. 2007;38:632–636. doi: 10.1161/01.STR.0000250742.61241.79. [DOI] [PubMed] [Google Scholar]
- Hassa PO, Haenni SS, Elser M, Hottiger M. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber A, Bai P, de Murcia JM, de Murcia G. PARP-1, PARP-2 and ATM in the DNA damage response: functional synergy in mouse development. DNA Repair. 2004;3:1103–1108. doi: 10.1016/j.dnarep.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Iwashita A, Hattori K, Yamamoto H, Ishida J, Kido Y, Kamijo K, et al. Discovery of quinazolinone and quinoxaline derivatives as potent and selective poly(ADP-ribose) polymerase-1/2 inhibitors. FEBS Lett. 2005;579:1389–1393. doi: 10.1016/j.febslet.2005.01.036. [DOI] [PubMed] [Google Scholar]
- Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4:421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- Kofler J, Otsuka T, Zhang ZZ, Noppens R, Grafe MR, Koh DW, et al. Differential effect of PARP-2 deletion on brain injury after focal and global cerebral ischemia. J Cereb Blood Flow Metab. 2006;26:135–141. doi: 10.1038/sj.jcbfm.9600173. [DOI] [PubMed] [Google Scholar]
- Koh DW, Dawson TM, Dawson VL. Poly(ADP-ribosyl)ation regulation of life and death in the nervous system. Cell Mol Life Sci. 2005;62:760–768. doi: 10.1007/s00018-004-4508-y. [DOI] [PubMed] [Google Scholar]
- Meli E, Picca R, Attucci S, Cozzi A, Peruginelli F, Moroni F, et al. Activation of mGlu1 but not mGlu5 metabotropic glutamate receptors contributes to postischemic neuronal injury in vitro and in vivo. Pharmacol Biochem Behav. 2002;73:439–446. doi: 10.1016/s0091-3057(02)00834-1. [DOI] [PubMed] [Google Scholar]
- Meli E, Pangallo M, Picca R, Baronti R, Moroni F, Pellegrini-Giampietro DE. Differential role of poly(ADP-ribose) polymerase-1 in apoptotic and necrotic neuronal death induced by mild or intense NMDA exposure in vitro. Mol Cell Neurosci. 2004;25:172–180. doi: 10.1016/j.mcn.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Moroni F. Poly(ADP-ribose)polymerase 1 (PARP-1) and postischemic brain damage. Curr Opin Pharmacol. 2008;8:96–103. doi: 10.1016/j.coph.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Moroni F, Meli E, Peruginelli F, Chiarugi A, Cozzi A, Picca R, et al. Poly(ADP-ribose) polymerase inhibitors attenuate necrotic but not apoptotic neuronal death in experimental models of cerebral ischemia. Cell Death Differell. 2001;8:921–932. doi: 10.1038/sj.cdd.4400884. [DOI] [PubMed] [Google Scholar]
- Nagayama T, Simon RP, Chen D, Henshall DC, Pei W, Stetler RA, et al. Activation of poly(ADP-ribose) polymerase in the rat hippocampus may contribute to cellular recovery following sublethal transient global ischemia. J Neurochem. 2000;74:1636–1645. doi: 10.1046/j.1471-4159.2000.0741636.x. [DOI] [PubMed] [Google Scholar]
- Oliver AW, Ame JC, Roe SM, Good V, de Murcia G, Pearl LH. Crystal structure of the catalytic fragment of murine poly(ADP-ribose) polymerase-2. Nucleic Acids Res. 2004;32:456–464. doi: 10.1093/nar/gkh215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Cozzi A, Peruginelli F, Leonardi P, Meli E, Pellicciari R, et al. 1-Aminoindan-1,5-dicarboxylic acid and (S)-(+)-2-(3′-carboxybicyclo[1.1.1]pentyl)-glycine, two mGlu1 receptor-preferring antagonists, reduce neuronal death in in vitro and in vivo models of cerebral ischemia. Eur J Neurosci. 1999a;11:3637–3647. doi: 10.1046/j.1460-9568.1999.00786.x. [DOI] [PubMed] [Google Scholar]
- Pellegrini-Giampietro DE, Peruginelli F, Meli E, Cozzi A, Albani Torregrossa S, Pellicciari R, et al. Protection with metabotropic glutamate 1 receptor antagonists in models of ischemic neuronal death: time-course and mechanisms. Neuropharmacology. 1999b;38:1607–1619. doi: 10.1016/s0028-3908(99)00097-0. [DOI] [PubMed] [Google Scholar]
- Pellicciari R, Camaioni E, Costantino G, Formentini L, Sabbatini P, Venturoni F, et al. On the way to selective PARP-2 inhibitors. Design, synthesis, and preliminary evaluation of a series of isoquinolinone derivatives. ChemMedChem. 2008;3:914–923. doi: 10.1002/cmdc.200800010. [DOI] [PubMed] [Google Scholar]
- Perkins E, Sun D, Nguyen A, Tulac S, Francesco M, Tavana H, et al. Novel inhibitors of poly(ADP-ribose) polymerase/PARP1 and PARP2 identified using a cell-based screen in yeast. Cancer Res. 2001;61:4175–4183. [PubMed] [Google Scholar]
- Petito CK, Feldmann E, Pulsinelli WA, Plum F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology. 1987;37:1281–1286. doi: 10.1212/wnl.37.8.1281. [DOI] [PubMed] [Google Scholar]
- Pulsinelli WA, Brierley JB, Plum F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann Neurol. 1982;11:491–498. doi: 10.1002/ana.410110509. [DOI] [PubMed] [Google Scholar]
- Schreiber V, Ame JC, Dolle P, Schultz I, Rinaldi B, Fraulob V, et al. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem. 2002;277:23028–23036. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- Shall S, de Murcia G. Poly(ADP-ribose) polymerase-1: what have we learned from the deficient mouse model? Mutat Res. 2000;460:1–15. doi: 10.1016/s0921-8777(00)00016-1. [DOI] [PubMed] [Google Scholar]
- Szabò C, Dawson VL. Role of poly(ADP-ribose) synthetase in inflammation and ischemia-reperfusion. Trends Pharmacol Sci. 1998;19:287–297. doi: 10.1016/s0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- Virag L, Szabo C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- Werner CG, Scartabelli T, Pancani T, Landucci E, Moroni F, Pellegrini-Giampietro DE. Differential role of mGlu1 and mGlu5 receptors in rat hippocampal slice models of ischemic tolerance. Eur J Neurosci. 2007;25:3597–3604. doi: 10.1111/j.1460-9568.2007.05614.x. [DOI] [PubMed] [Google Scholar]
- Yelamos J, Monreal Y, Saenz L, Aguado E, Schreiber V, Mota R, et al. PARP-2 deficiency affects the survival of CD4+CD8+ double-positive thymocytes. EMBO J. 2006;25:4350–4360. doi: 10.1038/sj.emboj.7601301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelamos J, Schreiber V, Dantzer F. Toward specific functions of poly(ADP-ribose) polymerase-2. Trends Mol Med. 2008;14:169–178. doi: 10.1016/j.molmed.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poli(ADP-ribose)synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]