

Abstract
Ribose-based nucleoside 5′-diphosphates and triphosphates and related nucleotides were compared in their potency at the P2Y receptors with the corresponding nucleoside 5′-phosphonate derivatives. Phosphonate derivatives of UTP and ATP activated the P2Y2 receptor but were inactive or weakly active at P2Y4 receptors. Uridine 5′-(diphospho)phosphonate was approximately as potent at the P2Y2 receptor as at the UDP-activated P2Y6 receptor. These results suggest that removal of the 5′-oxygen atom from nucleotide agonist derivatives reduces but does not prevent interaction with the P2Y2 receptor. Uridine 5′-(phospho)phosphonate as well as the 5′-methylphosphonate equivalent of UMP were inactive at the P2Y4 receptor and exhibited maximal effects at the P2Y2 receptor that were ≤50% of that of UTP suggesting novel action of these analogues.
The P2Y receptors are a family of eight G protein-coupled receptors (GPCRs) of class A that respond to diverse nucleotides.1 The human P2Y1, P2Y12, and P2Y13 receptors are activated preferentially by ADP, while the human P2Y11 receptor is activated preferentially by ATP. The human P2Y4, P2Y6, and P2Y14 subtypes respond exclusively to various uracil nucleotides, and the human P2Y2 receptor responds to both UTP and ATP. Depending on the subtype, the effector coupling of these receptors is typically through Gq or Gi proteins, to stimulate phospholipase or to inhibit of adenylate cyclase, respectively.
P2Y receptors are targets for therapeutic approaches, some of which are currently being explored at an early stage.2 Several clinical applications of P2Y receptor ligands are more advanced, such as P2Y12 receptor antagonists as antithrombotic agents3 and P2Y2 receptor agonists as drugs for cystic fibrosis and dry eye disease.4
To aid in the design of P2Y receptor ligands, we have reported rhodopsin-based molecular models of all known subtypes of P2Y receptors.5,6 Various amino acid residues have been proposed to coordinate the bound nucleotide ligands in P2Y1 receptors and other P2Y subtypes. The medicinal chemistry of P2Y receptors that are responsive to uracil nucleotides has recently been explored.7–11 The conformational constraint or replacement of the hydroxyl moieties of the ribose moiety have been introduced as a means for increasing the selectivity at P2Y1, P2Y2, or P2Y6 receptors.12,13
In the present study, we have explored the removal of the 5′-oxygen or its replacement by carbon moieties, resulting in 5′-phosphonate derivatives. The activity of various phosphonate derivatives was compared with the native ribosides at various P2Y receptor subtypes. Potency was best preserved at the P2Y2 receptor following this modification. However, the apparent efficacy of activation of the P2Y2 receptor by several phosphonate derivatives was variable and significantly less than 100%, suggesting a potential change in the mode of interaction with the receptor. The introduction of the phosphonate linkage is also intended to increase stability of the alpha-phosphate toward hydrolysis by nucleotidases, which is frequently a limitation in the pharmacological use of nucleotide derivatives in in vitro and in vivo experiments.
Various adenine and uracil nucleotide phosphonate derivatives were synthesized and evaluated in functional assays of different P2Y receptors (Table 1). The derivatives in which the 5′-oxygen was omitted, i.e., 1, 2, and 4–6, were prepared by known synthetic routes.14 The synthetic route to the elongated 5′-phosphonate derivatives 3 and 7 is shown in Scheme 1 following earlier reported strategies.15,19 5′, 6′-Vinyl phosphonate 9 was synthesized by oxidation of 2′, 3′-O-isopropylideneuridine (8) to the 5′-aldehyde intermediate, which was immediately reacted with freshly prepared [(diethoxyphosphinyl)methylidene] triphenylphosphorane.16 The E-configuration of the resulting alkene could be inferred from the large coupling constant (3J = 17.3 Hz). Catalytic hydrogenation of the resulting vinyl phosphonate ester 9 in the presence of palladium on carbon gave the corresponding saturated phosphonate ester 10. Simultaneous deprotection of the phosphonate diester and the acetonide was achieved upon treatment with TMSBr, yielding the corresponding 5′-phosphonate 7.
Table 1. In vitro.
pharmacological data for various nucleotide 5′-phosphonate derivatives analogues and the corresponding native ribosides (X = OCH2) in the stimulation of PLC at recombinant human P2Y receptors expressed in 1321N1 astrocytoma cells.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| No | Name | R | n | B | X | Receptor | EC50, μMa | Reference comp. (X = OCH2) EC50, μM7–11 |
| 1 | adenosine 5′-(diphospho)-phosphonate | HO | 2 | A | CH2 | P2Y2 | 1.4 ± 0.6 | 0.085 ± 0.012 |
| P2Y4 | <50% at 10 μM | Antagonist12 | ||||||
|
| ||||||||
| 2 | uridine 5′-(diphospho)-phosphonate | HO | 2 | U | CH2 | P2Y2 | 8.6 ± 3.1 | 0.049 ± 0.012 |
| P2Y4 | NE | 0.073 ± 0.02 | ||||||
| P2Y6 | NE | >1010 | ||||||
|
| ||||||||
| 3 | uridine 5′-(diphospho)- vinylphosphonate | HO | 2 | U | trans -CH=CH | P2Y2 | 1.0 ± 0.4 | 0.049 ± 0.012 |
| P2Y4 | <50% at 10 μM | 0.09 ± 0.0121 | ||||||
| P2Y6 | NE | NE | ||||||
|
| ||||||||
| 4 | uridine 5′-phospho- phosphonate | HO | 1 | U | CH2 | P2Y2 | 3.4 ± 1.1 (partial ag) | NE |
| P2Y4 | NE | NE | ||||||
| P2Y6 | 1.9 ± 0.7 | 0.046 ± 0.020 | ||||||
|
| ||||||||
| 5 | di-(uridine- phosphonate) |
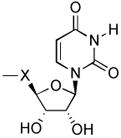
|
1 | U | CH2 | P2Y2 | <50% at 10 μM | NE8 |
| P2Y4 | NE | NE8 | ||||||
| P2Y6 | NE | 1.26 ± 0.17b | ||||||
|
| ||||||||
| 6 | uridine 5′-(glucose[1′]phos- pho)phosphonate |
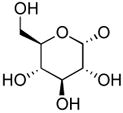
|
1 | U | CH2 | P2Y14 | NE | 0.301 ± 0.07 |
|
| ||||||||
| 7 | uridine 5 ′-methylene-phosphonate | HO | 0 | U | CH2 | P2Y2 | 1.6 ± 0.4 (partial ag) | NE |
| P2Y4 | NE | NE | ||||||
| P2Y6 | NE | NE | ||||||
Agonist potencies were calculated using a four-parameter logistic equation and the GraphPad software package (GraphPad, San Diego, CA). EC50 values (mean ± standard error) represent the concentration at which 50% of the maximal effect was achieved. A maximal effect (100% apparent efficacy) equivalent to that of the cognate agonist for the tested receptor was observed unless where indicated for two compounds (1 and 3) that exhibited <50% of maximal effect at the highest 10 μM concentration tested at the P2Y4 receptor and for two compounds (4 and 7) that produced apparently saturable activation that was much less than that observed with UTP at the P2Y2 receptor. The data for 4 and 7 are denoted as consistent with partial agonist (partial ag.) activity. N = 4 – 6 conc.-effect curves for each analogue.
Up2U. NE - no effect at 10 μM.
Scheme 1.
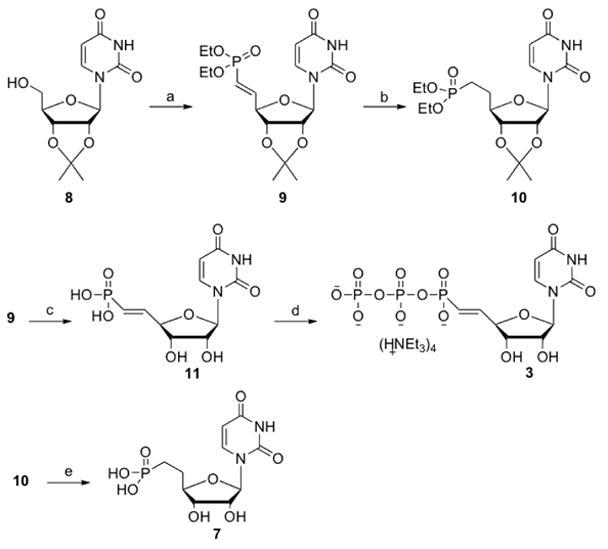
Reagents and conditions: (a) (i) IBX, CH3CN; (ii) (EtO)2P(O)C=PPh3, DMSO (44%); (b) H2, Pd/C, MeOH (84%); (c) TMSBr, CH2Cl2 (62%); (d) Bu3N, CDI, DMF; (Bu3NH)2H2P2O7; TEAB 1M (22%); (e) TMSBr, CH2Cl2 (65%).
To prepare the (diphospho)phosphonate derivative 3, vinyl phosphonate 11, obtained by hydrolysis of both the phosphonate esters and the acetonide upon treatment with TMSBr,15 was activated with CDI in DMF, followed by the addition of bis(tri-n-butylammonium)pyrophosphate.
For biological assays, the human P2Y1, P2Y2, P2Y4, and P2Y6 receptors, which are coupled to activation of phospholipase C (PLC) through Gq, were stably expressed in 1321N1 human astrocytoma cells, which lack a native response to nucleotides.17,18 Activity at the human P2Y14 receptor was followed by quantification of phosphoinositide hydrolysis in COS-7 cells coexpressing a phospholipase C-activating chimeric G protein, Gαq/i, which is activated by Gi-coupled receptors.20 Concentration effect curves were generated for each of the phosphonate derivatives at the relevant receptor(s), and the EC50 values were compared to the potency of the corresponding nucleotide in the native riboside series (Table 1). Representative data for the uridine 5′-(diphospho)phosphonate 2 and the vinylicdiphosphophosphonate 3 at the P2Y2 receptor are illustrated in Fig. 1.
Figure 1.
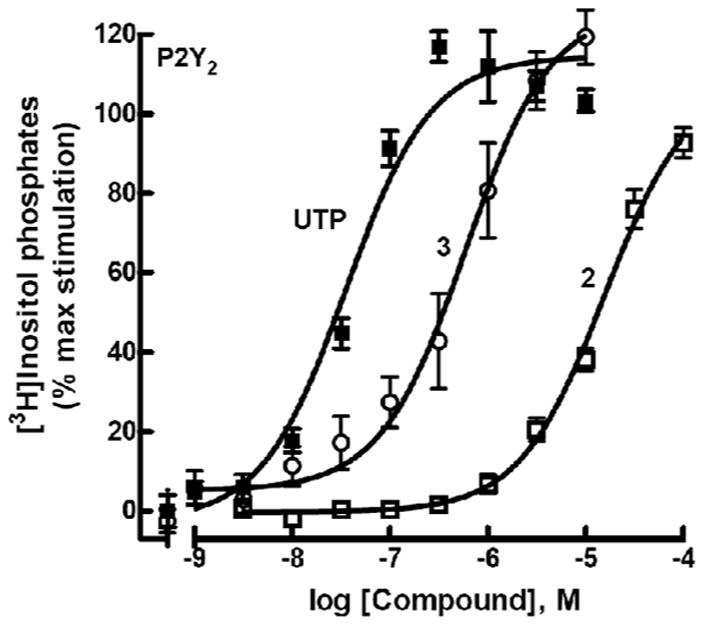
Activity of agonists 2 and 3 at P2Y2 receptors as indicated by activation of PLC in stably transfected astrocytoma cells. The effect of UTP corresponds to 100%.
Full agonist activity was observed with 1, 2, and 3 at the P2Y2 receptor, although the potencies were reduced by 16–176-fold relative to ATP or UTP. Although UTP is also a potent agonist at the human P2Y4 receptor, neither uridine 5′-(diphospho)phosphonate 2, which is the simple phosphonate equivalent of UTP, nor the vinyl phosphonate derivative 3 exhibited activity at this receptor. Compounds 2 and 3 were inactive at the P2Y6 receptor. Uridine 5′-phosphophosphonate 4, which is the simple phosphonate equivalent of UDP, was a full agonist at the P2Y6 receptor exhibiting a potency approximately 40-fold less than UDP. In contrast, uridine 5′-(glucose-[1′]phospho)-phosphonate 6 exhibited no activity at the UDP-glucose activated P2Y14 receptor.
A surprising result from our tests of activity was that uridine 5′-phosphophosphonate 4 was essentially as potent for activation of the P2Y2 receptor as for activation of the P2Y6 receptor and had no activity at the P2Y4 receptor. However, the maximal effect observed with 4 for activation of the P2Y2 receptor was only ~50% of that observed with UTP (Fig. 2A). Although UMP has no effect, uridine 5′-methylene-phosphonate 7 also was a relatively potent (EC50 = 1.6 ± 0.4 μM) agonist at the P2Y2 receptor. Similar to 4, the maximal effect observed at the P2Y2 receptor with 7 was <50% of that observed with UTP (Fig. 2B). Analogue 7 had no effect on the P2Y4 and P2Y6 receptors.
Figure 2.
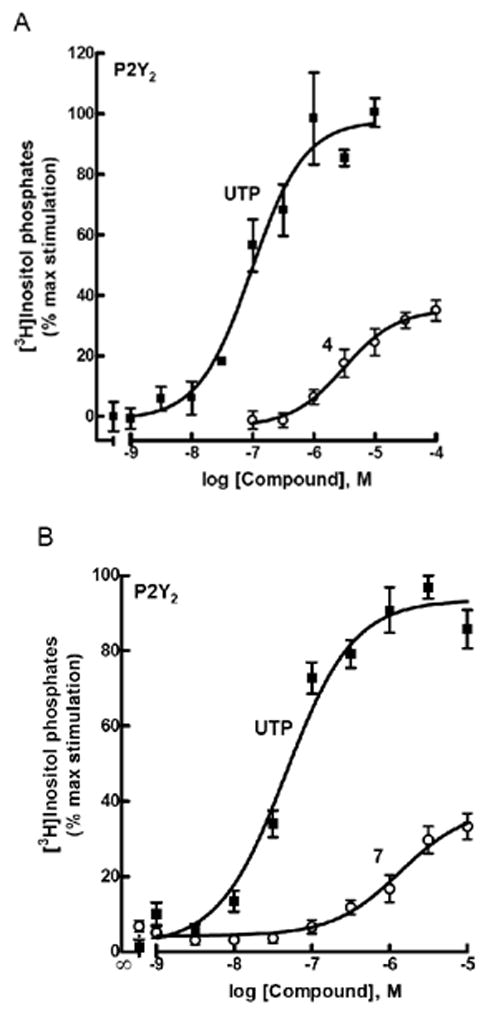
Partial agonist activity of 4 (A) and 7 (B) at P2Y2 receptors as indicated by activation of PLC in stably transfected astrocytoma cells. The effect of UTP corresponds to 100%.
Preliminary experiments examining the capacity of high concentrations of compounds 4 and 7 to antagonize activation of the P2Y2 receptor by UTP failed to provide convincing evidence that these molecules interact with the orthosteric binding site of the receptor. Therefore, these novel analogues potentially activate the P2Y2 receptor through an allosteric mechanism.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute of Diabetes & Digestive & Kidney Diseases and by an NIH grant GM38213.
Footnotes
Supporting material: Procedure for biological assays, the synthetic route for compounds 5 and 6, and detailed procedures for compounds 3 - 7 are provided.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Fumagalli M, King BF, Gachet C, Jacobson KA, Weisman GA. Pharmacol Rev. 2006;58:281. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jacobson KA, Ivanov AA, de Castro S, Harden TK, Ko H. Purinergic Signalling. 2009;5:75. doi: 10.1007/s11302-008-9106-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gachet C. Thromb Haemost. 2008;99:466. doi: 10.1160/TH07-11-0673. [DOI] [PubMed] [Google Scholar]
- 4.Nichols KK, Yerxa B, Kellerman DJ. Expert Opin Investig Drugs. 2004;13:47. doi: 10.1517/13543784.13.1.47. [DOI] [PubMed] [Google Scholar]
- 5.Ivanov AA, Costanzi S, Jacobson KA. J Comput Aided Mol Des. 2006;20:417. doi: 10.1007/s10822-006-9054-2. [DOI] [PubMed] [Google Scholar]
- 6.Moro S, Jacobson KA. Curr Pharmaceut Design. 2002;8:99. doi: 10.2174/1381612023392892. [DOI] [PubMed] [Google Scholar]
- 7.Brunschweiger A, Müller CE. Curr Med Chem. 2006;13:289. doi: 10.2174/092986706775476052. [DOI] [PubMed] [Google Scholar]
- 8.Shaver SR, Rideout JL, Pendergast W, Douglas JG, Brown EG, Boyer JL, Patel RI, Redick CC, Jones AC, Picher M, Yerxa BR. Purinergic Signaling. 2005;1:183. doi: 10.1007/s11302-005-0648-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Besada P, Shin DH, Costanzi S, Ko HJ, Mathé C, Gagneron J, Gosselin G, Maddileti S, Harden TK, Jacobson KA. J Med Chem. 2006;49:5532. doi: 10.1021/jm060485n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El-Tayeb A, Qi A, Müller CE. J Med Chem. 2006;49:7076. doi: 10.1021/jm060848j. [DOI] [PubMed] [Google Scholar]
- 11.Ivanov AA, Ko H, Cosyn L, Maddileti S, Besada P, Fricks I, Costanzi S, Harden TK, Van Calenbergh S, Jacobson KA. J Med Chem. 2007;50:1166. doi: 10.1021/jm060903o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim HS, Ravi RG, Marquez VE, Maddileti S, Wihlborg AK, Erlinge D, Malmsjö M, Boyer JL, Harden TK, Jacobson KA. J Med Chem. 2002;45:208. doi: 10.1021/jm010369e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Costanzi S, Joshi BV, Maddileti S, Mamedova L, Gonzalez-Moa MJ, Marquez VE, Harden TK, Jacobson KA. J Med Chem. 2005;48:8108. doi: 10.1021/jm050911p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raju N, Smee DF, Robins RK, Vaghefi MM. J Med Chem. 1989;32:1307. doi: 10.1021/jm00126a027. [DOI] [PubMed] [Google Scholar]
- 15.Jung K-Y, Hohl RJ, Wiemer AJ, Wiemer D. Bioorg Med Chem. 2008;8:2501. doi: 10.1016/s0968-0896(00)00183-8. [DOI] [PubMed] [Google Scholar]
- 16.Xu Y, Flavin MT, Xu ZQ. J Org Chem. 1996;61:7697. doi: 10.1021/jo9608275. [DOI] [PubMed] [Google Scholar]
- 17.Nicholas RA, Lazarowski ER, Watt WC, Li Q, Harden TK. Mol Pharmacol. 1996;50:224. [PubMed] [Google Scholar]
- 18.Bourdon DM, Wing MR, Edwards EB, Sondek J, Harden TK. Methods in Enzymology. 2006;406:489. doi: 10.1016/S0076-6879(06)06037-X. [DOI] [PubMed] [Google Scholar]
- 19.Jones GH, Moffatt JG. J Am Chem Soc. 1968;90:5337. doi: 10.1021/ja01021a086. [DOI] [PubMed] [Google Scholar]
- 20.Fricks I, Maddiletti S, Carter R, Lazarowski ER, Nicholas RA, Jacobson KA, Harden TK. J Pharm Exp Therap. 2008;325:588. doi: 10.1124/jpet.108.136309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ko H, Carter RL, Cosyn L, Petrelli R, de Castro S, Besada P, Zhou Y, Cappellacci L, Franchetti P, Grifantini M, Van Calenbergh S, Harden TK, Jacobson KA. Bioorg Med Chem. 2008;16:6319. doi: 10.1016/j.bmc.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.