

Fibroblast growth factor-23 (FGF23)
Maintenance of physiologic phosphate balance is important for essential cellular functions [1]. Dysregulation of the phosphate balance in the form of hypophosphataemia can lead to the development of myopathy, cardiac dysfunction, haematological abnormalities and bone mineralization defects [1]. In contrast, hyperphosphataemia can cause vascular and soft tissue calcification [2,3]. Studies have convincingly demonstrated that FGF23 is a master regulator of systemic phosphate homeostasis [4–9].
FGF23 is a 30 kDa protein that is proteolytically processed to generate smaller N-terminal (∼18 kDa) and C-terminal (∼12 kDa) fragments. The N-terminal fragment of FGF23 contains the FGF receptor-binding domain, while the C-terminal fragment is proposed to be necessary for interaction with Klotho (a type 1 membrane protein with homology to ß-glucosidase), which is believed to be a cofactor in FGF23–FGF receptor interactions [10]. FGF23 is a circulating phosphaturic factor that controls systemic phosphate homeostasis by regulating renal inorganic phosphate reabsorption [5]. The expression of members of the sodium phosphate co-transporter family (Na/Pi-2a and Na/Pi-2c) that mediate phosphate uptake in proximal tubular epithelial cells can be suppressed by FGF23 [11]. By suppressing Na/Pi co-transporter activity, FGF23 can reduce renal phosphate reabsorption, thereby increasing urinary phosphate excretion. The in vivo phosphaturic effect of FGF23 is convincingly demonstrated in animal studies. For instance, transgenic mice overexpressing human or mouse FGF23/Fgf23 have severe urinary phosphate wasting due to the suppression of renal Na/Pi co-transporter activity [12–14]. Inactivation of Fgf23 function in mice resulted in increased serum accumulation of phosphate and ectopic soft tissue calcification [2,3]. Genetically restoring the systemic actions of human FGF23 in Fgf23 knockout mice reversed this hyperphosphataemia to hypophosphataemia and prevented ectopic calcification [15].
The pathologic significance of FGF23 is also demonstrated in various human diseases; for example, activating and inactivating mutations in the human FGF23 gene are associated with autosomal dominant hypophosphataemic rickets [16] and hyperphosphataemic familial tumoral calcinosis, respectively [17]. The clinical features of tumoral calcinosis were also noted in a homozygous loss-of-function mutation in the Klotho gene, an important molecule necessary for FGF23 function. In a 13-year-old patient, a mutation in the Klotho gene was found to be associated with ectopic calcification, despite significantly elevated serum levels of FGF23 [18]. The lack of Klotho function in this patient could attenuate the ability of FGF23 to exert its phosphate-lowering effects, which would, in turn, explain the severe hyperphosphataemia and ectopic calcifications seen in the patient [18].
Experimental studies have convincingly demonstrated that Klotho is essential for the FGF23-mediated systemic regulation of phosphate homeostasis. A major breakthrough in how FGF23 exerts its bioactivities has been achieved by the recent demonstration of strikingly similar physical and biochemical phenotypes of Fgf23 and klotho knockout mice [19–21]. Such extreme similarity in the phenotypes of both Fgf23 and klotho knockout mice suggests a functional relationship between these molecules. These observations have led to the identification of Klotho as a cofactor for FGF23 and its receptor interactions and subsequent signalling [22]. Human studies have found that the expression of Klotho mRNA and protein is significantly reduced in chronic kidney disease (CKD) patients [23]. Recent clinical and experimental research works have focused on understanding the role of FGF23 in the progression of CKD patients [24].
FGF23 and CKD
The main functions of the kidney are to maintain water, electrolyte and mineral ion balance and to eliminate metabolic waste. Most CKD patients progress to irreversible renal failure without therapeutic intervention [25–27], where this affects water, electrolyte and mineral ion balances. Studies have shown that the circulating levels of FGF23 are significantly increased in CKD patients, partly due to decreased renal clearance of FGF23 [28]. The other cause of increased levels of FGF23 may be a compensatory phenomenon to the hyperphosphataemia seen in CKD, given that human studies have shown that a dietary phosphorus load can increase serum FGF23 levels [29,30]. Calcitriol therapy in patients with CKD may also contribute to increased serum levels of FGF23 [31]. There is an inverse in vivo correlation between FGF23 and 1,25-dihydroxyvitamin D. FGF23 can reduce serum levels of 1,25-dihydroxyvitamin D by suppressing the expressions of a key converting enzyme, 1-alpha hydroxylase [32]; conversely, elevated serum 1,25-dihydroxyvitamin D can induce an increase in the serum level of FGF23 [33]. Mice bearing FGF23-expressing Chinese hamster ovary cells have been shown to exhibit suppressed 1-alpha hydroxylase synthesis in the kidney [34]. In contrast, mice lacking Fgf23, klotho or both genes have higher renal expression of 1-alpha hydroxylase with a concomitant increase in serum levels of 1,25-dihydroxyvitamin D [21]. In a separate study, Saito and colleagues suggested that both serum phosphorus and 1,25-dihydroxyvitamin D could regulate circulating FGF23 levels independently of each other [33].
A deficiency of 1,25-dihydroxyvitamin D and excessive FGF23 are suggested to be associated with increased mortality in CKD patients, while a deficiency of Fgf23 and excessive 1,25-dihydroxyvitamin D are associated with increased mortality in experimental animals [19,20,35,36]. Such an inverse relationship of FGF23 and 1,25-dihydroxyvitamin D in CKD patients and experimental animals is typically associated with a common pathology, hyperphosphataemia, which is likely to be one of the important determinants of mortality in these cases, irrespective of other associated biochemical changes. It is worth mentioning that serum phosphate levels in chronic renal diseases can be influenced by numerous factors such as dietary intake, use of phosphate lowering drugs, abnormal skeletal conditions, etc. Therefore, serum phosphate could be misleading for risk assessment, particularly when the level is within the normal range. Recent studies have suggested that under normophosphataemic conditions, FGF23 may be a better biomarker for risk assessment [37]. The pathologic importance and prognostic significance of increased serum levels of FGF23 in patients with CKD and its influence on associated biochemical changes, however, need additional studies.
Another aspect that requires further study is whether elevated serum FGF23 levels can influence increased serum parathyroid hormone (PTH) levels in patients with CKD. Phosphate retention and subsequent hyperphosphataemia, together with decreased production and reduced circulating levels of 1,25-dihydroxyvitamin D, are the major biochemical changes detected in patients with CKD. PTH normally guarantees maintenance of phosphate balance, not only by promoting phosphate excretion but also by reducing urinary calcium excretion and stimulating renal production of active vitamin D metabolites. However, despite increased serum levels of PTH in patients with CKD, PTH fails to reduce serum phosphate towards normal values in the long term. This results in the development of secondary hyperparathyroidism [38]. Interestingly, elevated FGF23 levels have been suggested to be an important predictor of future secondary hyperparathyroidism in patients undergoing dialysis treatment [39].
As mentioned above, despite the demonstration of elevated serum levels of FGF23 in patients with CKD, the exact pathological significance of the increase is not clear. Whether the risk of FGF23 toxicity is independent of other known risk factors of CKD patients (e.g. race, diabetes, hypertension and advanced age) needs additional carefully designed clinical studies. FGF23 has been proposed to be an important biomarker of mortality in patients with early renal diseases, particularly in patients where the serum FGF23 level increased before the development of hyperphosphataemia [37]. In a similar line of work, a recent study suggested that FGF23 toxicity might contribute to renal death [36].
Elevated serum FGF23 and mortality
Abnormal mineral balance is an early complication in patients with CKD, where the altered calcium/phosphate balance is believed to be a risk factor for both cardiovascular complications [40] and renal dysfunction [41]. Such mineral ion imbalance favours the progression of CKD. Studies have convincingly demonstrated that disturbed calcium–phosphate metabolism affects cardiovascular morbidity and mortality in patients with CKD, particularly in those with end-stage disease [42]. Abnormal function of vitamin D metabolites, PTH and FGF23 is actively involved in impaired calcium–phosphate metabolism in patients with CKD.
Recently, Gutierrez et al. [36] showed that an increased serum level of FGF23 was associated with increased mortality in a case-control study of 400 incident haemodialysis patients. The investigators further found that the predictive value of high serum FGF23 levels was conserved at different degrees of hyperphosphataemia, except for the highest quartile (>5.5 mg/dL), which itself was associated with a 20% increase in adjusted risk of death [36]. Whether the increase in mortality was due to cardiovascular events requires further evaluation. Although there is a consensus regarding the phosphaturic effects of FGF23 in man [5], it is not yet clear whether FGF23 also directly affects cardiovascular function.
Available animal models may shed light on the issue of a possible direct effect of FGF23 on cardiac and/or vascular morphology and function. Genetic ablation of klotho in mice (klotho−/−) resulted in increased serum accumulation of Fgf23, as high as 2000-fold over controls [22], with a shortened lifespan of around 15 weeks and sudden death. Studies have linked the early sudden death of klotho−/− mice to sinoatrial node dysfunction [43]. Whether high serum Fgf23 levels in klotho−/− mice contribute per second to cardiac dysfunction and sudden death remains to be seen. Molecular insight into the possible direct actions of FGF23/Klotho at the cardiac and/or vascular level is needed to explain the pathological events leading to increased mortality in patients undergoing haemodialysis in association with often dramatically high serum levels of FGF23. It is worth mentioning that, in the kidneys of patients with CKD, the expression of Klotho mRNA and protein is significantly reduced [23].
A detailed molecular, functional and comparative analysis of klotho−/− mice with high serum FGF23 levels relative to klotho−/− mice with normal or low serum levels may help us determine the toxic effects of high serum Fgf23 on various organs and subsequent mortality in klotho−/− mice. Of relevance, recently generated klotho−/−/Fgf23−/− double knockout mice will be helpful in such a comparative analysis [21]. The initial results with these mice, however, did not indicate a significant survival advantage of the double knockout mice over klotho−/− single knockout mice [21]. Since klotho−/− mice have extremely high serum Fgf23 level, any potential toxic effects of Fgf23 in klotho−/− mice should disappear in klotho−/−/Fgf23−/− double knockout mice. However, there were no significant biochemical and morphological differences between klotho−/− mice and klotho−/−/Fgf23−/− double knockout mice [21], ruling out any potential Fgf23 toxicity in these genetically altered mice. It is worth mentioning that both klotho−/−/Fgf23−/− and klotho−/− mice develop severe hyperphosphataemia [21]. This is likely to influence increased mortality in these mutant mice, emphasizing that hyperphosphataemia is an important determinant of mortality in these experimental animals, irrespective of serum Fgf23 levels. Despite ruling out the potential adverse effects of high serum Fgf23 levels in the survival of klotho−/− mice, the human relevance of the in vivo experimental observations will require additional studies.
Concluding remarks
How elevated serum levels of FGF23 might exert toxic effects in CKD patients, whose tissue expression of Klotho is low, is an unresolved question [23]. One possible explanation may be that, at higher concentrations, FGF23 might exert non-specific effects without Klotho (Figure 1) via low affinity binding to its receptors [22]. The potential mechanisms and organs affected by non-specific effects of FGF23 in patients with CKD, however, require additional carefully designed studies. In view of the fact that there are close molecular interactions between 1,25-dihydroxyvitamin D, PTH, phosphate and FGF23, the current approach of reducing both serum phosphate and PTH and correcting vitamin D insufficiency/deficiency in patients with CKD may be more efficient than any one of these approaches alone, possibly by fine-tuning the activity of FGF23 [44]. Such a therapeutic approach might also reduce any potential toxic effects of high serum levels of FGF23 in CKD patients.
Fig. 1.
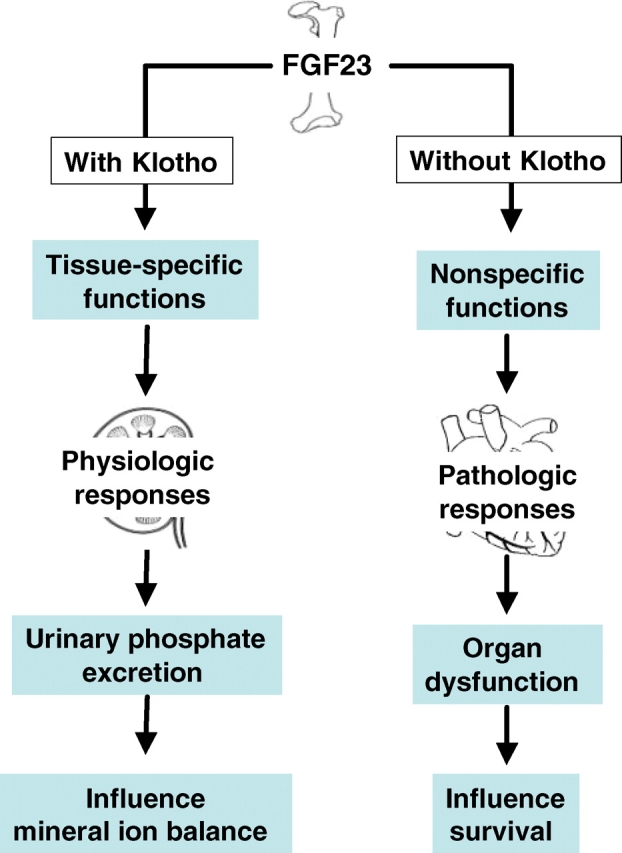
Simplified schematic outline of how FGF23 may exert physiological and pathological responses. Under physiological conditions, FGF23-mediated systemic regulation of phosphate homeostasis is a Klotho-dependent process. Under pathological conditions, however, an excessive amount of FGF23 may bind its receptors with low affinity without Klotho to exert non-specific effects that might influence organ functions.
Finally, despite a number of studies proposing differential roles for FGF23 in CKD patients, these studies have generated as many questions as they have answered. Whether a compensatory increased level of FGF23 in CKD patients is a protective response or a harmful response remains an unsettled issue, and any clinical interventions that manipulate FGF23 therapeutically require thoughtful consideration.
Acknowledgments
Part of the author's research work is supported by the grant (R01-DK077276) provided by NIH (NIDDK), and the institutional support from Harvard School of Dental Medicine, Boston, MA, USA.
Conflict of interest statement. None declared.
References
- 1.Gaasbeek A, Meinders AE. Hypophosphatemia: an update on its etiology and treatment. Am J Med. 2005;118:1094–1101. doi: 10.1016/j.amjmed.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 2.Razzaque MS, St-Arnaud R, Taguchi T, et al. FGF-23, vitamin D and calcification: the unholy triad. Nephrol Dial Transplant. 2005;20:2032–2035. doi: 10.1093/ndt/gfh991. [DOI] [PubMed] [Google Scholar]
- 3.Memon F, El-Abbadi M, Nakatani T, et al. Does Fgf23-klotho activity influence vascular and soft tissue calcification through regulating mineral ion metabolism? Kidney Int. 2008;74:566–570. doi: 10.1038/ki.2008.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berndt T, Kumar R. Phosphatonins and the regulation of phosphate homeostasis. Annu Rev Physiol. 2007;69:341–359. doi: 10.1146/annurev.physiol.69.040705.141729. [DOI] [PubMed] [Google Scholar]
- 5.Razzaque MS, Lanske B. The emerging role of the fibroblast growth factor-23-klotho axis in renal regulation of phosphate homeostasis. J Endocrinol. 2007;194:1–10. doi: 10.1677/JOE-07-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu X, White KE. FGF23 and disorders of phosphate homeostasis. Cytokine Growth Factor Rev. 2005;16:221–232. doi: 10.1016/j.cytogfr.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Yamashita T. Structural and biochemical properties of fibroblast growth factor 23. Ther Apher Dial. 2005;9:313–318. doi: 10.1111/j.1744-9987.2005.00288.x. [DOI] [PubMed] [Google Scholar]
- 8.Imel EA, Econs MJ. Fibroblast growth factor 23: roles in health and disease. J Am Soc Nephrol. 2005;16:2565–2575. doi: 10.1681/ASN.2005050573. [DOI] [PubMed] [Google Scholar]
- 9.Liu S, Gupta A, Quarles LD. Emerging role of fibroblast growth factor 23 in a bone-kidney axis regulating systemic phosphate homeostasis and extracellular matrix mineralization. Curr Opin Nephrol Hypertens. 2007;16:329–335. doi: 10.1097/MNH.0b013e3281ca6ffd. [DOI] [PubMed] [Google Scholar]
- 10.Goetz R, Beenken A, Ibrahimi OA, et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol. 2007;27:3417–3428. doi: 10.1128/MCB.02249-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miyamoto K, Ito M, Tatsumi S, et al. New aspect of renal phosphate reabsorption: the type IIc sodium-dependent phosphate transporter. Am J Nephrol. 2007;27:503–515. doi: 10.1159/000107069. [DOI] [PubMed] [Google Scholar]
- 12.Bai X, Miao D, Li J, et al. Transgenic mice overexpressing human fibroblast growth factor 23(R176Q) delineate a putative role for parathyroid hormone in renal phosphate wasting disorders. Endocrinology. 2004;145:5269–5279. doi: 10.1210/en.2004-0233. [DOI] [PubMed] [Google Scholar]
- 13.Larsson T, Marsell R, Schipani E, et al. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha 1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology. 2004;145:3087–3094. doi: 10.1210/en.2003-1768. [DOI] [PubMed] [Google Scholar]
- 14.Shimada T, Urakawa I, Yamazaki Y, et al. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem Biophys Res Commun. 2004;314:409–414. doi: 10.1016/j.bbrc.2003.12.102. [DOI] [PubMed] [Google Scholar]
- 15.DeLuca S, Sitara D, Kang K, et al. Amelioration of the premature aging-like features of Fgf-23 knockout mice by genetically restoring the systemic actions of FGF-23. J Pathol. 2008;216:345–355. doi: 10.1002/path.2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. The ADHR Consortium. Nat Genet. 2000;26:345–348. doi: 10.1038/81664. [DOI] [PubMed] [Google Scholar]
- 17.Benet-Pages A, Orlik P, Strom TM, et al. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum Mol Genet. 2005;14:385–390. doi: 10.1093/hmg/ddi034. [DOI] [PubMed] [Google Scholar]
- 18.Ichikawa S, Imel EA, Kreiter ML, et al. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J Clin Invest. 2007;117:2684–2691. doi: 10.1172/JCI31330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Razzaque MS, Lanske B. Hypervitaminosis D and premature aging: lessons learned from Fgf23 and Klotho mutant mice. Trends Mol Med. 2006;12:298–305. doi: 10.1016/j.molmed.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 20.Razzaque MS, Sitara D, Taguchi T, et al. Premature aging-like phenotype in fibroblast growth factor 23 null mice is a vitamin D-mediated process. Faseb J. 2006;20:720–722. doi: 10.1096/fj.05-5432fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakatani T, Bara S, Ohnishi M, et al. In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (Fgf23)-mediated regulation of systemic phosphate homeostasis. Faseb J. doi: 10.1096/fj.08-114397. doi:10.1096. /fj.08–114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urakawa I, Yamazaki Y, Shimada T, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006;444:770–774. doi: 10.1038/nature05315. [DOI] [PubMed] [Google Scholar]
- 23.Koh N, Fujimori T, Nishiguchi S, et al. Severely reduced production of klotho in human chronic renal failure kidney. Biochem Biophys Res Commun. 2001;280:1015–1020. doi: 10.1006/bbrc.2000.4226. [DOI] [PubMed] [Google Scholar]
- 24.Larsson T, Nisbeth U, Ljunggren O, et al. Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int. 2003;64:2272–2279. doi: 10.1046/j.1523-1755.2003.00328.x. [DOI] [PubMed] [Google Scholar]
- 25.Razzaque MS, Taguchi T. Cellular and molecular events leading to renal tubulointerstitial fibrosis. Med Electron Microsc. 2002;35:68–80. doi: 10.1007/s007950200009. [DOI] [PubMed] [Google Scholar]
- 26.Razzaque MS. Does renal ageing affect survival? Ageing Res Rev. 2007;6:211–222. doi: 10.1016/j.arr.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 27.Taguchi T, Razzaque MS. The collagen-specific molecular chaperone HSP47: is there a role in fibrosis? Trends Mol Med. 2007;13:45–53. doi: 10.1016/j.molmed.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 28.Imanishi Y, Inaba M, Nakatsuka K, et al. FGF-23 in patients with end-stage renal disease on hemodialysis. Kidney Int. 2004;65:1943–1946. doi: 10.1111/j.1523-1755.2004.00604.x. [DOI] [PubMed] [Google Scholar]
- 29.Antoniucci DM, Yamashita T, Portale AA. Dietary phosphorus regulates serum fibroblast growth factor-23 concentrations in healthy men. J Clin Endocrinol Metab. 2006;91:3144–3149. doi: 10.1210/jc.2006-0021. [DOI] [PubMed] [Google Scholar]
- 30.Ferrari SL, Bonjour JP, Rizzoli R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J Clin Endocrinol Metab. 2005;90:1519–1524. doi: 10.1210/jc.2004-1039. [DOI] [PubMed] [Google Scholar]
- 31.Nishi H, Nii-Kono T, Nakanishi S, et al. Intravenous calcitriol therapy increases serum concentrations of fibroblast growth factor-23 in dialysis patients with secondary hyperparathyroidism. Nephron Clin Pract. 2005;101:c94–c99. doi: 10.1159/000086347. [DOI] [PubMed] [Google Scholar]
- 32.Shimada T, Hasegawa H, Yamazaki Y, et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res. 2004;19:429–435. doi: 10.1359/JBMR.0301264. [DOI] [PubMed] [Google Scholar]
- 33.Saito H, Maeda A, Ohtomo S, et al. Circulating FGF-23 is regulated by 1-alpha, 25-dihydroxyvitamin D3 and phosphorus in vivo. J Biol Chem. 2005;280:2543–2549. doi: 10.1074/jbc.M408903200. [DOI] [PubMed] [Google Scholar]
- 34.Shimada T, Mizutani S, Muto T, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci USA. 2001;98:6500–6505. doi: 10.1073/pnas.101545198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fukagawa M, Nii-Kono T, Kazama JJ. Role of fibroblast growth factor 23 in health and in chronic kidney disease. Curr Opin Nephrol Hypertens. 2005;14:325–329. doi: 10.1097/01.mnh.0000172717.49476.80. [DOI] [PubMed] [Google Scholar]
- 36.Gutierrez OM, Mannstadt M, Isakova T, et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med. 2008;359:584–592. doi: 10.1056/NEJMoa0706130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gutierrez O, Isakova T, Rhee E, et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J Am Soc Nephrol. 2005;16:2205–2215. doi: 10.1681/ASN.2005010052. [DOI] [PubMed] [Google Scholar]
- 38.Felsenfeld AJ, Rodriguez M, Aguilera-Tejero E. Dynamics of parathyroid hormone secretion in health and secondary hyperparathyroidism. Clin J Am Soc Nephrol. 2007;2:1283–1305. doi: 10.2215/CJN.01520407. [DOI] [PubMed] [Google Scholar]
- 39.Nakanishi S, Kazama JJ, Nii-Kono T, et al. Serum fibroblast growth factor-23 levels predict the future refractory hyperparathyroidism in dialysis patients. Kidney Int. 2005;67:1171–1178. doi: 10.1111/j.1523-1755.2005.00184.x. [DOI] [PubMed] [Google Scholar]
- 40.Go AS, Chertow GM, Fan D, et al. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–1305. doi: 10.1056/NEJMoa041031. [DOI] [PubMed] [Google Scholar]
- 41.Schwarz S, Trivedi BK, Kalantar-Zadeh K, et al. Association of disorders in mineral metabolism with progression of chronic kidney disease. Clin J Am Soc Nephrol. 2006;1:825–831. doi: 10.2215/CJN.02101205. [DOI] [PubMed] [Google Scholar]
- 42.Block GA, Klassen PS, Lazarus JM, et al. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J Am Soc Nephrol. 2004;15:2208–2218. doi: 10.1097/01.ASN.0000133041.27682.A2. [DOI] [PubMed] [Google Scholar]
- 43.Takeshita K, Fujimori T, Kurotaki Y, et al. Sinoatrial node dysfunction and early unexpected death of mice with a defect of klotho gene expression. Circulation. 2004;109:1776–1782. doi: 10.1161/01.CIR.0000124224.48962.32. [DOI] [PubMed] [Google Scholar]
- 44.Razzaque MS. Can fibroblast growth factor 23 fine-tune therapies for diseases of abnormal mineral ion metabolism? Nat Clin Pract Endocrinol Metab. 2007;3:788–789. doi: 10.1038/ncpendmet0667. [DOI] [PubMed] [Google Scholar]