

Abstract
AIM: To evaluate the synergistic targeting and killing of human hepatocellular carcinoma (HCC) cells lacking p53 by the oncolytic autonomous parvovirus (PV) H-1 and chemotherapeutic agents and its dependence on functional promyelocytic leukemia protein (PML).
METHODS: The role of p53 and PML in regulating cytotoxicity and gene transfer mediated by wild-type (wt) PV H-1 were explored in two pairs of isogenic human hepatoma cell lines with different p53 status. Furthermore, H-1 PV infection was combined with cytostatic drug treatment.
RESULTS: While the HCC cells with different p53 status studied were all susceptible to H-1 PV-induced apoptosis, the cytotoxicity of H-1 PV was more pronounced in p53-negative than in p53-positive cells. Apoptosis rates in p53-negative cell lines treated by genotoxic drugs were further enhanced by a treatment with H-1 PV. In flow cytometric analyses, H-1 PV infection resulted in a reduction of the mitochondrial transmembrane potential. In addition, H-1 PV cells showed a significant increase in PML expression. Knocking down PML expression resulted in a striking reduction of the level of H-1 PV infected tumor cell death.
CONCLUSION: H-1 PV is a suitable agent to circumvent the resistance of p53-negative HCC cells to genotoxic agents, and it enhances the apoptotic process which is dependent on functional PML. Thus, H-1 PV and its oncolytic vector derivatives may be considered as therapeutic options for HCC, particularly for p53-negative tumors.
Keywords: Autonomous parvovirus, Apoptosis, p53, Promyelocytic leukemia protein, Human hepatocellular carcinoma, Hepatocytes
INTRODUCTION
Abrogation of function of tumor suppressors such as p53 or the promyelocytic leukemia protein (PML) are common events in human tumors and lead to more aggressive cancer phenotypes[1,2]. At early stages during the process of carcinogenesis, activated oncogenes sensitize primary cells towards the p53-dependent stress response, in which the nuclear phosphoprotein p53 serves as a genomic stabilizer, inhibitor of cell cycle progression and angiogenesis, and facilitator of apoptosis[3–5]. In order to overcome this endogenous defense mechanism, cancer cell variants are strongly selected for p53 mutations, with p53 gene alterations identified in approximately half of all human tumors[6–8]. Thus, loss of p53 function usually results in a more aggressive cancer phenotype and worse clinical outcome. Studies using in vitro cell cultures and in vivo animal models demonstrated that the deficiency of p53 correlates with enhanced tumorigenesis, tumor-induced angiogenesis, and an increased resistance towards intracellular stresses which is mainly due to abrogation of an effective apoptotic response to chemotherapy or radiation[9].
The tumor suppressor PML is predominantly localized in distinct nuclear domains that are termed PML-nuclear bodies (PML-NBs), and consist of multiprotein complexes implicated in apoptosis regulation, cellular senescence, and antiviral response[10,11]. PML expression results in potent growth-suppressive[12] and apoptosis-inducing effects[13,14], and PML-deficient mice and cells exhibit defects in multiple apoptosis pathways[15]. One major goal of therapeutic oncology is to identify ways to kill the tumor cells that became resistant to conventional treatments due to their lack of functional p53 or PML[16]. The rapid expansion of the field of gene transfer technologies led to the development of retroviral or adenoviral p53 expression vectors and their use to restore sensitivity to genotoxic agents or to directly induce apoptosis in preclinical tumor models[17–20].
Promising new approaches to tumor-directed therapy include oncolytic parvoviruses (PVs), which are of particular interest, since they are endowed with oncolytic properties and also increase the host immune response by priming effector immune cells against the tumors[21–24]. The autonomous PV of the rat (H-1 PV) and its close relatives, such as the minute virus of mice (MVM) and in addition the most commonly used herpes simplex virus and adenovirus are emerging as promising candidates because of a number of their properties[25].
Notably, these viruses preferentially replicate in and kill transformed and tumor-derived cells in culture[21,22,26,27]. In addition, recombinant PVs have recently been produced with the aim to increase the anti-tumor effect of the natural viruses[28]. In particular, PVs may be suitable to target and kill tumor cells and simultaneously deliver appropriate transgenes, e.g. genes coding for immuno-stimulatory factors[25]. As H-1 PV is seldom pathogenic to its natural adult hosts[29] and infects humans without any apparent consequences[21,28], the prospects for the clinical use of PVs are intriguing. In vivo, these viruses may combat tumor development or repress established tumors, what makes them promising tools in cancer therapy[25].
The factors controlling the sensitivity of target (in particular human) cells to PV-induced killing are still largely unknown. Cells transformed with oncogenes display both an enhanced capacity for accumulating the viral cyotoxic nonstructural (NS) protein[22] and a greater intrinsic responsiveness to NS1-mediated killing[30]. On the other hand, our previous investigation of the cytotoxicity of H-1 PV in hepatoma cell cultures failed to go into a requirement for functional p53. So far, the inactivation of p53 was only found in human leukemia cells and transformed rat fibroblasts and correlates with a greater susceptibility to H-1 PV-induced cell killing[31].
Thus, in order to better understand the role of the cell genetic background and effector pathways in the H-1 PV-induced cytotoxicity, we compared two isogenic pairs of p53-positive versus negative human tumor cell lines of hepatocellular carcinoma (HCC) origin. This system allowed us to assess the impact of p53 on the susceptibility of host cells to both H-1 PV gene expression and killing activity, and H-1 PV vector-reduced reporter gene transduction. To further understand the molecular mechanism underlying H-1 PV-induced cell killing, another tumor suppressor, the PML, was investigated for its influence on H-1 PV infection. We used RNA interference to knock down PML expression in the described cellular systems, and determined the consequences for the outcome of H-1 PV infection with regard to the host cell p53 status.
The present study shows that H-1 PV triggers an apoptotic type of death in human HCC cells, and that p53 is dispensable for this process. In contrast, PML, which is induced by H-1 PV infection, helps PV killing carcinoma cells, irrespective of their p53 status. Given the known dependence of apoptosis induction by radio-chemotherapeutic agents on the target cell p53 status[32], PVs appear to be suitable adjuvants to eliminate tumor cell populations resistant against these agents by means of combined treatments.
MATERIALS AND METHODS
Tumor cells
The Hep3B cells were derived from a HCC[33] and HepG2 cells from a human hepatoblastoma[32]. HepG2 cells were propagated in Dulbecco’s modified Eagle medium (DMEM; Life Technologies GmbH, Karlsruhe, Germany), and Hep3B in Eagle minimal essential medium (Eurobio GmbH, Raunheim, Germany). Both media were supplemented with 10% fetal calf serum (FCS), 5 mol/L glutamine, 100 μg/mL penicillin, and 5 mol/L Hepes[21]. The Hep3B4P line is a Hep3B derivate stably transfected with tamoxifen-regulated wt p53-estrogen receptor chimera (p53-mERtm-pBabepuro)[34]. p53-mERtm-pBpuro contains the BamHI fragment of human cDNA p53 cloned in frame with and N-terminally to a modified estrogen receptor containing a gly to arg mutation at residue 525[35]. This mutation renders the hormone binding domain insensitive to estradiol but responsive to the synthetic estrogen 4-OH-tamoxifen[35]. To induce p53 in the experiments, tamoxifen was added at a concentration of 750 nmol/L 1 d before H-1 PV infection.
HepG2 303 is a HepG2 derivative stably transfected with a dominant-negative p53 mutant (dn-p53) kindly provided by A. Levine (ΔV143A) as described by Schuler et al[36]. Since the dn-p53 transfection plasmid contained the puromycin resistance gene, HepG2 303 cells were selected with puromycin (0.5 μg/mL) for 4 wk (4 consecutive days each week).
Virus infection
For infection, H-1 PV was produced in NB-E cells and purified over cesium chloride gradients as described earlier. Wild-type (wt) H-1 PV titration by plaque assays was performed according to published methods[26]. The multiplicity of infection (MOI) is given by the number of plaque-forming units (pfu) inoculated per cell. For experimental infections, exponentially growing cell cultures were incubated for 1 h with H-1 PV at indicated MOIs. Cells were cultured for up to 8 d post infection (p.i.).
Cell treatments
For combined treatment with H-1 PV and chemotherapeutics, cells were first infected with H-1 PV (MOI = 20 pfu/cell) in complete medium. One hour after infection, the chemotherapeutic agents Irinotecan (100 μg/mL), 5-Fluorouracil (5-FU) (5 μg/mL), or Cisplatin (0.25 μg/mL) were added, and cells were further incubated for 3 d at 37°C. Apoptosis rates were then quantitatively determined by FACS. Herein, the treated cells were harvested via trypisinization, washed with PBS, stained with propidium iodide and annexin V, and apoptosis levels were assessed, using the FACScan flow cytometry with CellQuest software (Becton Dickinson, San Jose, CA). Anticancer agents were purchased from Pfizer (Irinotecan), Hexal AG (5-FU), and Gry Pharma GmbH (Cisplatin).
Analysis of virus protein expression
Cultures were infected with H-1 PV at a MOI of 20 pfu/cell. After washing with PBS, cells were lysed in RIPA buffer (10 mol/L Tris-HCl, 150 mol/L NaCl, 1 mol/L EDTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, 5% SDS) containing protease inhibitors. Protein concentrations were determined using the Bio-Rad protein assay (Bio-Rad, Munich, Germany). Total proteins (50 μg) were diluted into an equal volume, subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane (Amersham Pharmacia Biotech, Freiburg, Germany). Non-specific binding sites were blocked by incubating the membrane for 2 h in PBS containing 10% low-fat milk powder and 0.2% Tween-20 (Sigma, Deisenhofen, Germany). The blot was further incubated with the rabbit polyclonal antibody SP8 directed against carboxy-terminal peptides of NS1[37], then with an anti-rabbit peroxidase-conjugated antibody, and processed for enhanced chemoluminescence detection (Amersham Pharmacia Biotech, Freiburg, Germany).
Characterisation of the p53 and PML status of human tumor cells
Cultures grown for 48 h were processed for Western blotting as described above for viral proteins, p53 was detected using the monoclonal DO-7 antibody[38]. Actin was used as an internal loading control.
RNA interference
To knock down PML expression by RNA interference, the targeting oligonucleotide 5’-GAGCTCAAG TGCGACATCA-3’ (PML sense) was inserted into the pSUPER vector. This target region is present in all PML isoforms and was verified by BLAST searches to confirm specificity. For control experiments, empty pSUPER vectors were used.
Measurement of PV-induced cell lysis
Hep3B and HepG2 cells were infected with H-1 PV at a MOI of 20 pfu/cell and further grown for 1 to 3 d. Cell permeabilization was then measured by using a standard toxicity assay (Toxilight, Cambrex Bio Science, Rockland Inc., USA) assessing the concentration of cellular adenylate kinase (AK) in culture supernatants according to the manufacturer’s recommendations[39].
FACScan analysis of apoptosis
For quantification of the percentage of apoptotic cells in H-1 PV-infected cultures (MOI = 20 pfu/cell), adherent cells were dissociated with 0.25% trypsin and collected, together with cells floating in the medium, by centrifugation at 800 g. Cells were washed twice with PBS and stained with propidium iodide and annexin V (Becton Dickinson, Heidelberg, Germany). Fluorescence was measured with a minimum of 10 000 events per sample in a FACScan according to the manufacturer’s instructions (Becton Dickinson). Data analysis was performed using the software Cell Quest (Becton Dickinson)[21].
Analysis of mitochondrial membrane potential
To measure the mitochondrial transmembrane potential, the cationic lipophilic fluorochrome JC-1 (5, 5, 6, 6-tetrachloro-1, 1, 3, 3-tetraethylbenzimidazolyl-carbocyanine iodide) (Molecular Probes, Inc., Eugene, OR) was used. JC-1 exists as a monomer in solution, emitting green fluorescence. In a reaction driven by the mitochondrial transmembrane potential, JC-1 can adopt a dimeric configuration and emit red fluorescence[40,41]. Mock- or H-1 PV-infected cultures (5 × 104 cells/mL) were incubated with JC-1 (5 μg/mL) for 20 min at room temperature in the dark, washed once in PBS and immediately analyzed by flow cytometry (FACScan, Becton Dickinson, Heidelberg, Germany) using Cellquest software. The red fluorescence of JC-1 indicates intact mitochondria, whereas green fluorescence shows monomeric JC-1 that remained unprocessed due to breakdown of the mitochondrial membrane potential[42]. After gating out small sized (i.e., non-cellular) debris, 10 000 events were collected for each analysis. The emitted green fluorescence signals were used as a measure for the loss of mitochondrial membrane potential[43].
Statistical analysis
Protein and (real time) gene expression values were analyzed for differences, using the one-sided Student's t-test. A P-value lower than 0.05 was considered as statistically significant.
RESULTS
Characterization of the p53 status in the hepatoma cell line pairs
In order to validate the system chosen to analyze the role of p53 and its effector pathways in H-1 PV-induced cellular cytotoxicity, we first confirmed the differential activity and expression of p53 in the two pairs of isogenic p53-positive and p53-negative HCC cell lines. The cell line HepG2 expresses functional wt p53 while Hep3B is a p53-null (p53-/-) cell line[44]. Hep3B4P cells transfected with a tamoxifen-regulated p53-estrogen receptor chimera[34] were cultured with different concentrations of 4-OH-tamoxifen to induce p53. Upon transfection of a p53-transactivated luciferase construct (pConluc/pgupluc), 4-OH-tamoxifen induced dose-dependent the p53 activity, up to 25 times at 750 nmol/L of 4-OH-tamoxifen (data not shown). At this concentration, the induction of p53 expression was determined to have no detectable effects on the growth of non-infected cells in accordance to previous reports[38,45]. The cell lines studied differ in expression of p53 as expected (Figure 1A). HepG2 and HepG2 303 show p53 bands in both cases, because HepG2 303 is a HepG2 derivative stably transfected with a dominant-negative p53 mutant (dn-p53).
Figure 1.
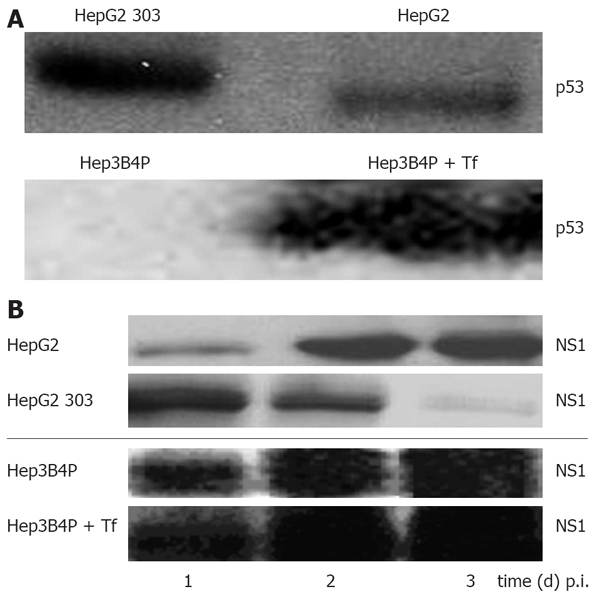
A: Characterisation of the p53 status and analysis of parvoviral proteins in different human tumor cells by Western blot. Cells were cultured for 2 d and lysed with RIPA buffer, and 50 µg of total protein was subjected to SDS-PAGE. For p53 protein detection, blots were incubated with the monoclonal DO-7 antibody; B: Production of parvoviral proteins in H-1 PV-infected p53 different tumor cells. Hep3B4P and HepG2 cells were H-1 PV-infected (MOI = 20 pfu/cell) and grown for 1 to 3 d. After lysis with RIPA buffer, 50 μg of total proteins were equally diluted and separated on SDS-PAGE. For parvoviral protein detection, blots were incubated with the NS1-specific antibody[37].
Production of parvoviral non-structural proteins in virus-infected p53 different tumor cells
In order to assess the effect of p53 on H-1 PV replication, expression of the replicative cytotoxic NS1 protein was analyzed in infected tumor cell line pairs. As illustrated in Figure 1B, all cultures were proficient in NS1 accumulation within a few days p.i., as detected by Western blot analysis. It was noteworthy; however the p53 expression somehow impaired the capacity for NS1 production, as apparent from the delayed appearance (HepG2 system) or reduced steady-state level (Hep3B4P system) of NS1 in the p53-positive cells. As previously reported[21], NS1 expression levels were in accordance with the respective amounts of viral DNA intermediates as well as luciferase activities of parvoviral vectors.
H-1 PV toxicity for human tumor cell pairs differing in their p53 status
We previously observed that a p53-negative hepatoma cell line was lightly susceptible to H-1 PV-induced cytotoxicity[21]. This prompted us to compare the p53-deficient cells with their p53-positive counterparts in terms of their relative sensitivities to H-1 PV-induced killing. The levels of cytotoxicity in H-1 PV-inoculated Hep3B4P and HepG2 cultures were first monitored by measuring cell permeabilization through the release of adenylate kinase (AK) in the medium for up to 3 d p.i. (Figure 2A). Compared to their mock-treated controls, increased cell death was observed for all hepatoma cell cultures.
Figure 2.
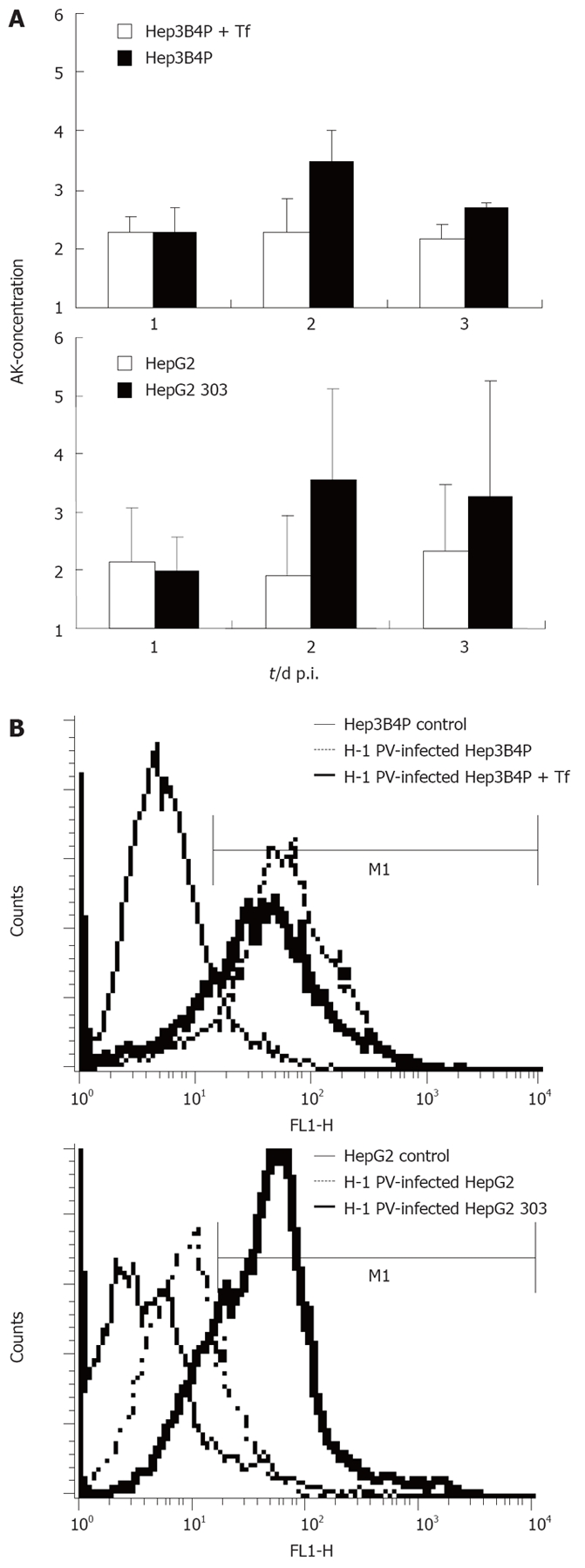
A: Cytotoxicity and induction of apoptosis in H-1 PV-infected p53 different tumor cell line pairs. The early damage of tumor cells upon H-1 PV infection was measured with a standardized toxicity test via supernatant adenylate kinase (AK) concentration; B: Induction of apoptosis in H-1 PV-infected tumor cells is shown in histograms for annexin V (FL1-H) of Hep3B4P and HepG2 cells. Data are given as mean values of triplicates.
In keeping with their above-mentioned greater efficiency in cytotoxic NS1 production, the p53-negative cell cultures moved to be significantly more sensitive to the toxic effect of H-1 PV than their p53-positive derivative. Indeed, supernatants from HepG2 303 cultures contained about twice the amounts of AK than those from the HepG2 parent after H-1 PV infection. Likewise, tamoxifen induction of p53 in the Hep3B4P cells correlated with an increase in their resistance to H-1 PV cytotoxicity.
This difference was confirmed by FACS analysis quantifying the expression of annexin V, a known marker of apoptosis[30] (Figure 2B). In agreement with the above viability assay, H-1 PV infection induced annexin V in all hepatoma cells tested, yet to a higher level in p53-negative compared to p53-positive lines. Thus, p53 status correlated with sensitivity of hepatoma cells to the induction of H-1 PV-mediated apoptosis.
H-1 PV infection enhances depolarization of the inner mitochondrial membrane
The depolarization of the inner mitochondrial membrane has been associated with apoptosis in tumor cells exposed to different cytostatic agents or viruses[46]. This prompted us to investigate the effect of H-1 PV infection on this parameter in hepatoma cells. To this end, the profiles of JC-1 fluorescence were compared between mock- and H-1 PV-infected Hep3B4P cells. As illustrated in Figure 3, H-1 PV infection correlated with a striking increase in the fraction of hepatoma cells displaying depolarized mitochondrial membranes. In keeping with above data, this change occurred as soon as 1 d p.i. (Figure 3A) in p53-negative (Hep3B4P) in contrast to positive (Hep3B4P + Tf) cells. The p53-positive cells show an increase of the depolarization of membran potential not before 2 d p.i. (Figure 3B).
Figure 3.

Analysis of mitochondrial membrane potential. Kinetics of reduction of the mitochondrial membrane potential. Hepatoma cells were left untreated (control) or infected (H-1 PV) for the indicated periods of time and analyzed by flow cytometry using the fluorochrome JC-1. The percentage of cells with decreased mitochondrial membrane potential is shown. After one (A) and two (B) days incubation, the percentage of cells with decreased mitochondrial membrane potential were determined by flow cytometry.
Interplay of the tumor suppressor PML and H-1 PV infection
With the intention to identify factors influencing the sensitivity of hepatoma cells for H-1 PV, we determined whether the expression of the tumor suppressor protein PML was affected by H-1 PV-inoculation, and conversely, whether it had an impact on the outcome of infection. As shown in Figure 4, H-1 PV infection caused a strong increase in PML expression both on the protein (Figure 4A) and on the RNA (Figure 4B) level. To assess a possible role of PML in the control of H-1 PV-induced apoptosis in hepatoma cells, PML expression was reduced by RNA interference through expression of a short hairpin (sh) RNA (pSUPER-PML) which specifically targets PML. In contrast to the control pSUPER vector, the shPML construct strongly inhibited expression of endogenous PML in transfected HepG2 cells, irrespective of their p53 status (Figure 4B). Interestingly, knocking down of PML expression was found to correlate with a marked reduction of the efficiency of H-1 PV in inducing apoptosis in both HepG2 cells and their p53-negative HepG2 303 derivatives (Figure 4C). This result reveals PML to be centrally involved in regulation of apoptosis of hepatoma cells upon H-1 PV infection.
Figure 4.

H-1 PV-induced apoptosis is mediated by PML. HepG2 (p53+) and HepG2 303 (p53-) cells were transfected with pSUPER or pSUPER-PML as indicated, and infected with H-1 PV for 48 h. Cells were harvested and subjected to Western blot (A) or PCR analysis (B). Hepatoma cells were treated as described, harvested, and subjected to cytotoxicity assay. Apoptosis was determined as described in Material and Methods (C).
Treatment with chemotherapeutic agents combined with H-1 PV infection
The genetic drift of cancer cells leads to the appearance of variants resisting conventional genotoxic anticancer treatments. This prompted us to test whether hepatoma cells escaping chemotherapy may still be killed by H-1 PV, i.e. whether the combination of chemotherapeutics with H-1 PV meant an advantage. In a first step, this possibility was explored in vitro by determining whether H-1 PV infection enhanced the fraction of apoptotic hepatoma cells in cultures treated with cisplatin (Cis), irinotecan (Iri), or 5-FU. As illustrated in Figure 5B, in p53-deficient HepG2 303 cultures, H-1 PV was found to cooperate with all three agents in enhancing the overall fraction of treated cells undergoing apoptosis. Interestingly, the beneficial effect of the combined treatment or either of its individual components was not (cisplatin, irinotecan) or hardly (5-FU) significant in the p53-positive parental line HepG2 (Figure 5A). Therefore, p53 appeared to impair the cooperation of H-1 PV with genotoxic agents, possibly due to the fact that the chemotherapeutic agents enhanced the above-mentioned negative impact of p53 on the parvoviral life cycle. It is noteworthy that the p53-deficient HepG2 303 cells were less (irinotecan, 5-FU) or even more (cisplatin) sensitive to the chemotherapeutic tested, compared with the p53-positive HepG2 parent (Figure 5). This was surprising, given the usually lower susceptibility of p53-negative cells to the induction of apoptosis by genotoxic agents, but is not without precedent as p53 can be functionally replaced by related products in drug-induced killing of some tumor cells[47].
Figure 5.
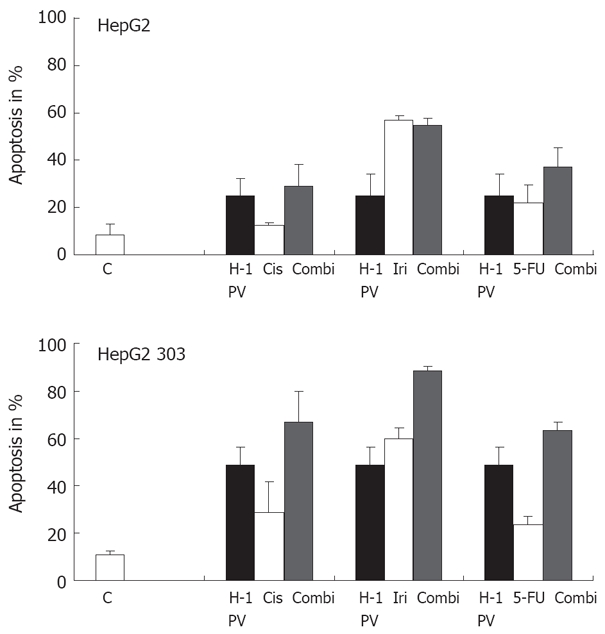
Treatment of p53 different tumor cells with chemotherapeutic agents. The p53 different HepG2 cells were treated with various chemotherapeutic agents alone or combined with H-1 PV infection (MOI = 20 pfu/cell). H-1 PV- or mock-infected cells were seeded into 6-well plates, and 1 h after infection cultures were treated with chemotherapeutic agents as indicated. Apoptosis was measured on day 3 by FACScan analysis. Data given represent mean values of triplicates.
DISCUSSION
The development of gene transfer techniques for tumor suppressor protein negative cancers is a rapidly expanding field: For HCC, different viruses have been assessed to specifically target p53-negative tumors or to transfer the wt p53 gene to reconstitute apoptotic pathways in tumor cells[17–20,44,48–51]. However, several of the viral delivery systems are limited by their immunogenic or pathogenic side effects[52,53]. We have shown in this paper that oncolytic PV H-1 may be effective in the treatment of HCC. Even more, these oncolytic viruses directly targeting p53-negative carcinomas may be attractive alternative vectors as well as ideal tools for combination with classical chemotherapeutic agents. As the rat PV H-1 is seldom pathogenic to its natural adult hosts and infects humans without any apparent clinical consequences[28,29], we considered the PV H-1 for tumor cell-targeted therapy, in particular in p53-negative tumors. In addition, immune reaction to PVs, such as AAV or H-1 PV might not induce any severe side effects[21–24].
Thus, we first characterized the susceptibility to H-1 PV infection and cell killing of pairs of human tumor cells which differ in their wt p53 levels. In concordance with earlier data[21], H-1 PV-induced killing of tested tumor cells was dependent on MOI and correlated with NS1 expression levels. Similarly, amenability to gene transfer after recombinant PV infection was higher in p53 lacking cells[21]. Despite both isogenic HepG2 cells were susceptible to H-1 PV-induced apoptosis, cell death was more pronounced in HepG2 303. As well in other human and rodent cell systems studied so far, susceptibility to H-1 PV-induced cell killing correlated with the capacity of the host cells to sustain both, parvoviral DNA amplification and NS1 protein expression[20,28,54–56]. With this regard, earlier data revealed cellular processes underlying the PV-induced tumor cell killing. Further on p53 displayed a key role in the G1/S checkpoint in response to DNA damage[57] as a regulator of cell cycle progression and a mediator of apoptosis in many cell lines[58]. Thus, p53 could prevent cell progression to S-Phase. According to this some S-phase factors such as p53 have been involved in the regulation of PV DNA replication[31,35]. Indeed, the rare H-1 PV-resistant variant clones named KS cells, isolated from the H-1 PV-susceptible human p53-negative erythroleukemia cell line K562, differed from the parental wt p53-positive cells by a reduced oncogenic potential in immunocompromised mice. Similarly, rat fibroblasts overexpressing mutant p53 protein were more sensitive to H-1 PV infection than parental cells[31].
Our data further demonstrate that H-1 PV induces significantly expression of PML on the RNA and protein level and, thus, increases the susceptibility to cell death in H-1 PV-infected tumor cells. The significance of this effect was clearly demonstrated by the knock down of PML in H-1 PV-infected cells with the consequence of impaired apoptosis upon H-1 PV infection. Recently, we showed that the hepatotropic hepatitis C virus (HCV) was able to impair apoptosis in hepatoma cells by inhibition of p53 function via interaction with PML[59]. Polypeptides from other viruses were also shown to interact with PML, and to disable its biological function in apoptosis regulation, growth suppression and cellular senescence. For example, adenoviral E1A protein abrogates oncogenic Ras- and PML-IV-induced cellular senescence by overriding PML function[60].
Furthermore PODs/PML bodies have been associated with transcription, cell growth, and antiviral responses[61,62]. DNA and RNA viruses also frequently target PODs, presumably to facilitate the early stages of transcription and replication[63–67]. PODs can also be targeted for reorganization following viral infection[66]. For example, adenovirus protein E4-ORF3 localizes to PODs/PML bodies, thereby causing a physical restructuring of the bodies from spherical to extended fibril-like structures termed nuclear tracks[66]. Additionally, many viruses induce interferon expression, what increases the size and number of PODs[62,68]. Although PV infection did not induce an interferon response, a dramatic relocalization of PODs has been seen late in MVM infection. However, Cziepluch et al[69] suggested that H-1 virus does not target known nuclear bodies for DNA replication but rather induced the formation of a novel structure in the nucleus of infected cells. Within that study, PML-expression was not directly investigated.
In addition, PVs were recently reported to replicate in association with distinct nuclear bodies[69], which appear to lately merge with PML and PML-NBs[70]. PML may interact with viral products and participate in the regulation of virus replication. PML proteins may thus modulate viral cytopathic effects, in keeping with the above-mentioned involvement of PML in growth inhibition and death processes. As PV H-1 infection enhances PML expression and function, this might be a central molecular mechanism for an effective treatment of HCC.
We furthermore examined the effects of various chemotherapeutic agents in combination with H-1 PV infection on growth inhibition using isogenic cancer cells with different p53 status. H-1 PV infection enhanced the cytotoxicity of chemotherapeutic agents in the treatment of two HCC. In p53-deficient HepG2 303 cultures, H-1 PV was found to cooperate with all three agents in enhancing the overall fraction of treated cells undergoing apoptosis. The treatment with Irinotecan, 5-Fluorouracil and Cisplatin combined with H-1 PV infection more strongly inhibited the growth of the p53-negative HepG2 303 cells than treatment with chemotherapeutics alone. Therefore H-1 PV infection enhances cytostatic drug therapy in p53-negative tumors. Furthermore, irrespective of the p53 status, H-1 PV was able to induce programmed cell death in these human tumor cells, as it was also shown for other PVs[71]. Chemotherapeutic treatment alone did not induce such a high apoptosis rate compared to combined treatment with H-1 PV. Comparable data have also been published for other oncolytic viruses[72–75]. Thus, our data show a beneficial interaction between chemotherapy and oncolytic viral therapy and suggest that H-1 PV infection may enhance the effectiveness of chemotherapeutic agents in the treatment of HCC.
In summary, our results strongly suggest that p53-impaired tumors-which have a poor prognosis-may be particularly suitable to PV H-1-induced therapy[39,43,45]. Though p53 deficiency in tumors may induce resistance to chemotherapeutic agents, this will not affect the tumor cell susceptibility to H-1 PV-induced oncolytic infections[51]. As recombinant H-1 PV had a high capacity to transduce transgenes in these cells, the therapeutic potential of H-1 PV-based recombinant vectors carrying suicide genes or cytokines should be further assessed in p53-negative tumors. The PV H-1 may then also overcome other tumor resistance mechanisms against autocrine and paracrine apoptotic triggers developed in these tumor entities[50,51]. We conclude that our strategy using H-1 PV infection in combination with chemotherapeutic treatment can enhance the cytotoxic effect of anti-cancer agents. Furthermore, H-1 PV induced the expression of PML, thus increased the susceptibility to cell death in H-1 PV-infected tumor cells. So PML may operate as a positive element which controls in a direct or indirect way the susceptibility of hepatoma cells to apoptosis-activity of H-1 PV.
COMMENTS
Background
Oncolytic parvoviruses (PVs) are endowed with oncolytic properties and also increase the host immune response against the tumor by priming effector cells.
Research frontiers
Authors evaluated the synergistic targeting and killing of human hepatocellular carcinoma (HCC) cells lacking p53 by PV H-1 and chemotherapeutic agents.
Innovations and breakthroughs
Analysing the regulating the cell killing pathways and gene transfer mediated by PV H-1 in pairs of human hepatocellular cell lines with different p53 status, H-1 PV is quite a suitable agent to circumvent the resistance of p53-negative HCC to genotoxic agents, and enhances the apoptotic process which is dependent on functional PML.
Applications
Especially for p53-negative human tumors authors consider PV H-1 as therapeutic option for human HCC.
Peer review
This manuscript described that H-1 PV is a novel agent for treating p53 negative HCC via the induction of PML and apoptosis. This study is a well designed, well exerted, and well written manuscript.
Acknowledgments
The authors wish to thank the lab assistants Petra Schaefer and Sandra Weyer for excellent technical assistance. The manuscript is based on at least in part of the data from the PhD thesis of Maike Sieben and of the MD thesis of Vera Heinrichs.
Supported by Grants from the German Cancer Aid (Deutsche Krebshilfe) No. 10-2183/102322 and local university research grants, MAIFOR program, No. 9728053 and 9728275
Peer reviewer: Dr. Toru Ikegami, Department of Surgery and Science, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582, Japan
S- Editor Li DL L- Editor Mihm S E- Editor Ma WH
References
- 1.Gurrieri C, Nafa K, Merghoub T, Bernardi R, Capodieci P, Biondi A, Nimer S, Douer D, Cordon-Cardo C, Gallagher R, et al. Mutations of the PML tumor suppressor gene in acute promyelocytic leukemia. Blood. 2004;103:2358–2362. doi: 10.1182/blood-2003-07-2200. [DOI] [PubMed] [Google Scholar]
- 2.Alves VA, Nita ME, Carrilho FJ, Ono-Nita SK, Wakamatsu A, Lehrbach DM, de Carvalho MF, de Mello ES, Gayotto LC, da Silva LC. p53 immunostaining pattern in Brazilian patients with hepatocellular carcinoma. Rev Inst Med Trop Sao Paulo. 2004;46:25–31. doi: 10.1590/s0036-46652004000100005. [DOI] [PubMed] [Google Scholar]
- 3.Clarke AR, Purdie CA, Harrison DJ, Morris RG, Bird CC, Hooper ML, Wyllie AH. Thymocyte apoptosis induced by p53-dependent and independent pathways. Nature. 1993;362:849–852. doi: 10.1038/362849a0. [DOI] [PubMed] [Google Scholar]
- 4.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 5.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 6.Baker SJ, Fearon ER, Nigro JM, Hamilton SR, Preisinger AC, Jessup JM, vanTuinen P, Ledbetter DH, Barker DF, Nakamura Y, et al. Chromosome 17 deletions and p53 gene mutations in colorectal carcinomas. Science. 1989;244:217–221. doi: 10.1126/science.2649981. [DOI] [PubMed] [Google Scholar]
- 7.Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hostetter R, Cleary K, Bigner SH, Davidson N, Baylin S, Devilee P. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342:705–708. doi: 10.1038/342705a0. [DOI] [PubMed] [Google Scholar]
- 8.Chang F, Syrjanen S, Syrjanen K. Implications of the p53 tumor-suppressor gene in clinical oncology. J Clin Oncol. 1995;13:1009–1022. doi: 10.1200/JCO.1995.13.4.1009. [DOI] [PubMed] [Google Scholar]
- 9.Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74:957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 10.Sternsdorf T, Jensen K, Will H. Evidence for covalent modification of the nuclear dot-associated proteins PML and Sp100 by PIC1/SUMO-1. J Cell Biol. 1997;139:1621–1634. doi: 10.1083/jcb.139.7.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell. 2002;108:165–170. doi: 10.1016/s0092-8674(02)00626-8. [DOI] [PubMed] [Google Scholar]
- 12.Le XF, Yang P, Chang KS. Analysis of the growth and transformation suppressor domains of promyelocytic leukemia gene, PML. J Biol Chem. 1996;271:130–135. doi: 10.1074/jbc.271.1.130. [DOI] [PubMed] [Google Scholar]
- 13.Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, Pandolfi PP, Will H, Schneider C, Del Sal G. Regulation of p53 activity in nuclear bodies by a specific PML isoform. EMBO J. 2000;19:6185–6195. doi: 10.1093/emboj/19.22.6185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W, Pandolfi PP. The function of PML in p53-dependent apoptosis. Nat Cell Biol. 2000;2:730–736. doi: 10.1038/35036365. [DOI] [PubMed] [Google Scholar]
- 15.Wang Z, Seliger B, Mike N, Momburg F, Knuth A, Ferrone S. Molecular analysis of the HLA-A2 antigen loss by melanoma cells SK-MEL-29.1.22 and SK-MEL-29.1.29. Cancer Res. 1998;58:2149–2157. [PubMed] [Google Scholar]
- 16.El-Deiry WS. Insights into cancer therapeutic design based on p53 and TRAIL receptor signaling. Cell Death Differ. 2001;8:1066–1075. doi: 10.1038/sj.cdd.4400943. [DOI] [PubMed] [Google Scholar]
- 17.Fujiwara T, Cai DW, Georges RN, Mukhopadhyay T, Grimm EA, Roth JA. Therapeutic effect of a retroviral wild-type p53 expression vector in an orthotopic lung cancer model. J Natl Cancer Inst. 1994;86:1458–1462. doi: 10.1093/jnci/86.19.1458. [DOI] [PubMed] [Google Scholar]
- 18.Wills KN, Maneval DC, Menzel P, Harris MP, Sutjipto S, Vaillancourt MT, Huang WM, Johnson DE, Anderson SC, Wen SF. Development and characterization of recombinant adenoviruses encoding human p53 for gene therapy of cancer. Hum Gene Ther. 1994;5:1079–1088. doi: 10.1089/hum.1994.5.9-1079. [DOI] [PubMed] [Google Scholar]
- 19.Sandig V, Brand K, Herwig S, Lukas J, Bartek J, Strauss M. Adenovirally transferred p16INK4/CDKN2 and p53 genes cooperate to induce apoptotic tumor cell death. Nat Med. 1997;3:313–319. doi: 10.1038/nm0397-313. [DOI] [PubMed] [Google Scholar]
- 20.Kirn D, Martuza RL, Zwiebel J. Replication-selective virotherapy for cancer: Biological principles, risk management and future directions. Nat Med. 2001;7:781–787. doi: 10.1038/89901. [DOI] [PubMed] [Google Scholar]
- 21.Moehler M, Blechacz B, Weiskopf N, Zeidler M, Stremmel W, Rommelaere J, Galle PR, Cornelis JJ. Effective infection, apoptotic cell killing and gene transfer of human hepatoma cells but not primary hepatocytes by parvovirus H1 and derived vectors. Cancer Gene Ther. 2001;8:158–167. doi: 10.1038/sj.cgt.7700288. [DOI] [PubMed] [Google Scholar]
- 22.Rommelaere J, Cornelis JJ. Autonomous Parvoviruses[A] In: Hernáiz Driever P, Rabkin SD., editors. Replication-Competent Viruses for Cancer Therapy. Monographs in Virology. Basel: Karger; 2001. pp. 100–129. [Google Scholar]
- 23.Moehler M, Zeidler M, Schede J, Rommelaere J, Galle PR, Cornelis JJ, Heike M. Oncolytic parvovirus H1 induces release of heat-shock protein HSP72 in susceptible human tumor cells but may not affect primary immune cells. Cancer Gene Ther. 2003;10:477–480. doi: 10.1038/sj.cgt.7700591. [DOI] [PubMed] [Google Scholar]
- 24.Moehler MH, Zeidler M, Wilsberg V, Cornelis JJ, Woelfel T, Rommelaere J, Galle PR, Heike M. Parvovirus H-1-induced tumor cell death enhances human immune response in vitro via increased phagocytosis, maturation, and cross-presentation by dendritic cells. Hum Gene Ther. 2005;16:996–1005. doi: 10.1089/hum.2005.16.996. [DOI] [PubMed] [Google Scholar]
- 25.Cornelis JJ, Salome N, Dinsart C, Rommelaere J. Vectors based on autonomous parvoviruses: novel tools to treat cancer? J Gene Med. 2004;6 Suppl 1:S193–S202. doi: 10.1002/jgm.502. [DOI] [PubMed] [Google Scholar]
- 26.Chen YQ, de Foresta F, Hertoghs J, Avalosse BL, Cornelis JJ, Rommelaere J. Selective killing of simian virus 40-transformed human fibroblasts by parvovirus H-1. Cancer Res. 1986;46:3574–3579. [PubMed] [Google Scholar]
- 27.Cornelis JJ, Becquart P, Duponchel N, Salome N, Avalosse BL, Namba M, Rommelaere J. Transformation of human fibroblasts by ionizing radiation, a chemical carcinogen, or simian virus 40 correlates with an increase in susceptibility to the autonomous parvoviruses H-1 virus and minute virus of mice. J Virol. 1988;62:1679–1686. doi: 10.1128/jvi.62.5.1679-1686.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rommelaere J, Cornelis JJ. Antineoplastic activity of parvoviruses. J Virol Methods. 1991;33:233–251. doi: 10.1016/0166-0934(91)90024-t. [DOI] [PubMed] [Google Scholar]
- 29.Jacoby RO, Ball-Goodrich LJ, Besselsen DG, McKisic MD, Riley LK, Smith AL. Rodent parvovirus infections. Lab Anim Sci. 1996;46:370–380. [PubMed] [Google Scholar]
- 30.Mousset S, Ouadrhiri Y, Caillet-Fauquet P, Rommelaere J. The cytotoxicity of the autonomous parvovirus minute virus of mice nonstructural proteins in FR3T3 rat cells depends on oncogene expression. J Virol. 1994;68:6446–6453. doi: 10.1128/jvi.68.10.6446-6453.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Telerman A, Tuynder M, Dupressoir T, Robaye B, Sigaux F, Shaulian E, Oren M, Rommelaere J, Amson R. A model for tumor suppression using H-1 parvovirus. Proc Natl Acad Sci USA. 1993;90:8702–8706. doi: 10.1073/pnas.90.18.8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muller M, Strand S, Hug H, Heinemann EM, Walczak H, Hofmann WJ, Stremmel W, Krammer PH, Galle PR. Drug-induced apoptosis in hepatoma cells is mediated by the CD95 (APO-1/Fas) receptor/ligand system and involves activation of wild-type p53. J Clin Invest. 1997;99:403–413. doi: 10.1172/JCI119174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ponchel F, Puisieux A, Tabone E, Michot JP, Froschl G, Morel AP, Frebourg T, Fontaniere B, Oberhammer F, Ozturk M. Hepatocarcinoma-specific mutant p53-249ser induces mitotic activity but has no effect on transforming growth factor beta 1-mediated apoptosis. Cancer Res. 1994;54:2064–2068. [PubMed] [Google Scholar]
- 34.Friedman SL, Shaulian E, Littlewood T, Resnitzky D, Oren M. Resistance to p53-mediated growth arrest and apoptosis in Hep 3B hepatoma cells. Oncogene. 1997;15:63–70. doi: 10.1038/sj.onc.1201149. [DOI] [PubMed] [Google Scholar]
- 35.Vater CA, Bartle LM, Dionne CA, Littlewood TD, Goldmacher VS. Induction of apoptosis by tamoxifen-activation of a p53-estrogen receptor fusion protein expressed in E1A and T24 H-ras transformed p53-/- mouse embryo fibroblasts. Oncogene. 1996;13:739–748. [PubMed] [Google Scholar]
- 36.Schuler M, Maurer U, Goldstein JC, Breitenbucher F, Hoffarth S, Waterhouse NJ, Green DR. p53 triggers apoptosis in oncogene-expressing fibroblasts by the induction of Noxa and mitochondrial Bax translocation. Cell Death Differ. 2003;10:451–460. doi: 10.1038/sj.cdd.4401180. [DOI] [PubMed] [Google Scholar]
- 37.Faisst S, Faisst SR, Dupressoir T, Plaza S, Pujol A, Jauniaux JC, Rhode SL, Rommelaere J. Isolation of a fully infectious variant of parvovirus H-1 supplanting the standard strain in human cells. J Virol. 1995;69:4538–4543. doi: 10.1128/jvi.69.7.4538-4543.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galmarini CM, Falette N, Tabone E, Levrat C, Britten R, Voorzanger-Rousselot N, Roesch-Gateau O, Vanier-Viornery A, Puisieux A, Dumontet C. Inactivation of wild-type p53 by a dominant negative mutant renders MCF-7 cells resistant to tubulin-binding agent cytotoxicity. Br J Cancer. 2001;85:902–908. doi: 10.1054/bjoc.2001.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olsson T, Gulliksson H, Palmeborn M, Bergstrom K, Thore A. Leakage of adenylate kinase from stored blood cells. J Appl Biochem. 1983;5:437–445. [PubMed] [Google Scholar]
- 40.Lawrence JW, Darkin-Rattray S, Xie F, Neims AH, Rowe TC. 4-Quinolones cause a selective loss of mitochondrial DNA from mouse L1210 leukemia cells. J Cell Biochem. 1993;51:165–174. doi: 10.1002/jcb.240510208. [DOI] [PubMed] [Google Scholar]
- 41.Loeffler M, Kroemer G. The mitochondrion in cell death control: certainties and incognita. Exp Cell Res. 2000;256:19–26. doi: 10.1006/excr.2000.4833. [DOI] [PubMed] [Google Scholar]
- 42.Reers M, Smith TW, Chen LB. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry. 1991;30:4480–4486. doi: 10.1021/bi00232a015. [DOI] [PubMed] [Google Scholar]
- 43.Scaffidi C, Schmitz I, Zha J, Korsmeyer SJ, Krammer PH, Peter ME. Differential modulation of apoptosis sensitivity in CD95 type I and type II cells. J Biol Chem. 1999;274:22532–22538. doi: 10.1074/jbc.274.32.22532. [DOI] [PubMed] [Google Scholar]
- 44.Vollmer CM, Ribas A, Butterfield LH, Dissette VB, Andrews KJ, Eilber FC, Montejo LD, Chen AY, Hu B, Glaspy JA, et al. p53 selective and nonselective replication of an E1B-deleted adenovirus in hepatocellular carcinoma. Cancer Res. 1999;59:4369–4374. [PubMed] [Google Scholar]
- 45.Ran Z, Rayet B, Rommelaere J, Faisst S. Parvovirus H-1-induced cell death: influence of intracellular NAD consumption on the regulation of necrosis and apoptosis. Virus Res. 1999;65:161–174. doi: 10.1016/s0168-1702(99)00115-x. [DOI] [PubMed] [Google Scholar]
- 46.Duverger V, Sartorius U, Klein-Bauernschmitt P, Krammer PH, Schlehofer JR. Enhancement of cisplatin-induced apoptosis by infection with adeno-associated virus type 2. Int J Cancer. 2002;97:706–712. doi: 10.1002/ijc.10077. [DOI] [PubMed] [Google Scholar]
- 47.Vayssade M, Haddada H, Faridoni-Laurens L, Tourpin S, Valent A, Benard J, Ahomadegbe JC. P73 functionally replaces p53 in Adriamycin-treated, p53-deficient breast cancer cells. Int J Cancer. 2005;116:860–869. doi: 10.1002/ijc.21033. [DOI] [PubMed] [Google Scholar]
- 48.Anderson SC, Johnson DE, Harris MP, Engler H, Hancock W, Huang WM, Wills KN, Gregory RJ, Sutjipto S, Wen SF, et al. p53 gene therapy in a rat model of hepatocellular carcinoma: intra-arterial delivery of a recombinant adenovirus. Clin Cancer Res. 1998;4:1649–1659. [PubMed] [Google Scholar]
- 49.Bookstein R, Demers W, Gregory R, Maneval D, Park J, Wills K. p53 gene therapy in vivo of herpatocellular and liver metastatic colorectal cancer. Semin Oncol. 1996;23:66–77. [PubMed] [Google Scholar]
- 50.Borresen-Dale AL. TP53 and breast cancer. Hum Mutat. 2003;21:292–300. doi: 10.1002/humu.10174. [DOI] [PubMed] [Google Scholar]
- 51.Picksley SM, Spicer JF, Barnes DM, Lane DP. The p53-MDM2 interaction in a cancer-prone family, and the identification of a novel therapeutic target. Acta Oncol. 1996;35:429–434. doi: 10.3109/02841869609109917. [DOI] [PubMed] [Google Scholar]
- 52.Alt M, Caselmann WH. Liver-directed gene therapy: molecular tools and current preclinical and clinical studies. J Hepatol. 1995;23:746–758. doi: 10.1016/0168-8278(95)80044-1. [DOI] [PubMed] [Google Scholar]
- 53.Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, Ng L, Nye JA, Sampson-Johannes A, Fattaey A, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274:373–376. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- 54.Dupressoir T, Vanacker JM, Cornelis JJ, Duponchel N, Rommelaere J. Inhibition by parvovirus H-1 of the formation of tumors in nude mice and colonies in vitro by transformed human mammary epithelial cells. Cancer Res. 1989;49:3203–3208. [PubMed] [Google Scholar]
- 55.Ries SJ, Brandts CH, Chung AS, Biederer CH, Hann BC, Lipner EM, McCormick F, Korn WM. Loss of p14ARF in tumor cells facilitates replication of the adenovirus mutant dl1520 (ONYX-015) Nat Med. 2000;6:1128–1133. doi: 10.1038/80466. [DOI] [PubMed] [Google Scholar]
- 56.St George JA. Gene therapy progress and prospects: adenoviral vectors. Gene Ther. 2003;10:1135–1141. doi: 10.1038/sj.gt.3302071. [DOI] [PubMed] [Google Scholar]
- 57.Ciciarello M, Mangiacasale R, Casenghi M, Zaira Limongi M, D’Angelo M, Soddu S, Lavia P, Cundari E. p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J Biol Chem. 2001;276:19205–19213. doi: 10.1074/jbc.M009528200. [DOI] [PubMed] [Google Scholar]
- 58.Cui Q, Yu JH, Wu JN, Tashiro S, Onodera S, Minami M, Ikejima T. P53-mediated cell cycle arrest and apoptosis through a caspase-3- independent, but caspase-9-dependent pathway in oridonin-treated MCF-7 human breast cancer cells. Acta Pharmacol Sin. 2007;28:1057–1066. doi: 10.1111/j.1745-7254.2007.00588.x. [DOI] [PubMed] [Google Scholar]
- 59.Herzer K, Weyer S, Krammer PH, Galle PR, Hofmann TG. Hepatitis C virus core protein inhibits tumor suppressor protein promyelocytic leukemia function in human hepatoma cells. Cancer Res. 2005;65:10830–10837. doi: 10.1158/0008-5472.CAN-05-0880. [DOI] [PubMed] [Google Scholar]
- 60.Ferbeyre G, de Stanchina E, Lin AW, Querido E, McCurrach ME, Hannon GJ, Lowe SW. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol. 2002;22:3497–3508. doi: 10.1128/MCB.22.10.3497-3508.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doucas V. The promyelocytic (PML) nuclear compartment and transcription control. Biochem Pharmacol. 2000;60:1197–1201. doi: 10.1016/s0006-2952(00)00413-5. [DOI] [PubMed] [Google Scholar]
- 62.Lavau C, Marchio A, Fagioli M, Jansen J, Falini B, Lebon P, Grosveld F, Pandolfi PP, Pelicci PG, Dejean A. The acute promyelocytic leukaemia-associated PML gene is induced by interferon. Oncogene. 1995;11:871–876. [PubMed] [Google Scholar]
- 63.Ahn JH, Hayward GS. Disruption of PML-associated nuclear bodies by IE1 correlates with efficient early stages of viral gene expression and DNA replication in human cytomegalovirus infection. Virology. 2000;274:39–55. doi: 10.1006/viro.2000.0448. [DOI] [PubMed] [Google Scholar]
- 64.Day PM, Roden RB, Lowy DR, Schiller JT. The papillo-mavirus minor capsid protein, L2, induces localization of the major capsid protein, L1, and the viral transcription/replication protein, E2, to PML oncogenic domains. J Virol. 1998;72:142–150. doi: 10.1128/jvi.72.1.142-150.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Doucas V, Ishov AM, Romo A, Juguilon H, Weitzman MD, Evans RM, Maul GG. Adenovirus replication is coupled with the dynamic properties of the PML nuclear structure. Genes Dev. 1996;10:196–207. doi: 10.1101/gad.10.2.196. [DOI] [PubMed] [Google Scholar]
- 66.Everett RD, Maul GG. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 1994;13:5062–5069. doi: 10.1002/j.1460-2075.1994.tb06835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu FY, Ahn JH, Alcendor DJ, Jang WJ, Xiao J, Hayward SD, Hayward GS. Origin-independent assembly of Kaposi’s sarcoma-associated herpesvirus DNA replication compartments in transient cotransfection assays and association with the ORF-K8 protein and cellular PML. J Virol. 2001;75:1487–1506. doi: 10.1128/JVI.75.3.1487-1506.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fabunmi RP, Wigley WC, Thomas PJ, DeMartino GN. Interferon gamma regulates accumulation of the proteasome activator PA28 and immunoproteasomes at nuclear PML bodies. J Cell Sci. 2001;114:29–36. doi: 10.1242/jcs.114.1.29. [DOI] [PubMed] [Google Scholar]
- 69.Cziepluch C, Lampel S, Grewenig A, Grund C, Lichter P, Rommelaere J. H-1 parvovirus-associated replication bodies: a distinct virus-induced nuclear structure. J Virol. 2000;74:4807–4815. doi: 10.1128/jvi.74.10.4807-4815.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Young PJ, Jensen KT, Burger LR, Pintel DJ, Lorson CL. Minute virus of mice NS1 interacts with the SMN protein, and they colocalize in novel nuclear bodies induced by parvovirus infection. J Virol. 2002;76:3892–3904. doi: 10.1128/JVI.76.8.3892-3904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Poole BD, Karetnyi YV, Naides SJ. Parvovirus B19-induced apoptosis of hepatocytes. J Virol. 2004;78:7775–7783. doi: 10.1128/JVI.78.14.7775-7783.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eisenberg DP, Adusumilli PS, Hendershott KJ, Yu Z, Mullerad M, Chan MK, Chou TC, Fong Y. 5-fluorouracil and gemcitabine potentiate the efficacy of oncolytic herpes viral gene therapy in the treatment of pancreatic cancer. J Gastrointest Surg. 2005;9:1068–1077; discussion 1077-1079. doi: 10.1016/j.gassur.2005.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mullerad M, Bochner BH, Adusumilli PS, Bhargava A, Kikuchi E, Hui-Ni C, Kattan MW, Chou TC, Fong Y. Herpes simplex virus based gene therapy enhances the efficacy of mitomycin C for the treatment of human bladder transitional cell carcinoma. J Urol. 2005;174:741–746. doi: 10.1097/01.ju.0000164730.38431.5c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Raykov Z, Grekova S, Galabov AS, Balboni G, Koch U, Aprahamian M, Rommelaere J. Combined oncolytic and vaccination activities of parvovirus H-1 in a metastatic tumor model. Oncol Rep. 2007;17:1493–1499. [PubMed] [Google Scholar]
- 75.Toyoizumi T, Mick R, Abbas AE, Kang EH, Kaiser LR, Molnar-Kimber KL. Combined therapy with chemotherapeutic agents and herpes simplex virus type 1 ICP34.5 mutant (HSV-1716) in human non-small cell lung cancer. Hum Gene Ther. 1999;10:3013–3029. doi: 10.1089/10430349950016410. [DOI] [PubMed] [Google Scholar]