

Abstract
AIM: To explore the role of Bcl-xL and Myeloid cell leukaemia (Mcl)-1 for the apoptosis resistance of colorectal carcinoma (CRC) cells towards current treatment modalities.
METHODS: Bcl-xL and Mcl-1 mRNA and protein expression were analyzed in CRC cell lines as well as human CRC tissue by Western blot, quantitative PCR and immunohistochemistry. Bcl-xL and Mcl-1 protein expression was knocked down or increased in CRC cell lines by applying specific siRNAs or expression plasmids, respectively. After modulation of protein expression, CRC cells were treated with chemotherapeutic agents, an antagonistic epidermal growth factor receptor (EGFR1) antibody, an EGFR1 tyrosine kinase inhibitor, or with the death receptor ligand TRAIL. Apoptosis induction and cell viability were analyzed.
RESULTS: Here we show that in human CRC tissue and various CRC cell lines both Bcl-xL and Mcl-1 are expressed. Bcl-xL expression was higher in CRC tissue than in surrounding non-malignant tissue, both on protein and mRNA level. Mcl-1 mRNA expression was significantly lower in malignant tissues. However, protein expression was slightly higher. Viability rates of CRC cells were significantly decreased after knock down of Bcl-xL expression, and, to a lower extent, after knock down of Mcl-1 expression. Furthermore, cells with reduced Bcl-xL or Mcl-1 expression was more sensitive towards oxaliplatin- and irinotecan-induced apoptosis, and in the case of Bcl-xL also towards 5-FU-induced apoptosis. On the other hand, upregulation of Bcl-xL by transfection of an expression plasmid decreased chemotherapeutic drug-induced apoptosis. EGF treatment clearly induced Bcl-xL and Mcl-1 expression in CRC cells. Apoptosis induction upon EGFR1 blockage by cetuximab or PD168393 was increased by inhibiting Mcl-1 and Bcl-xL expression. More strikingly, CD95- and TRAIL-induced apoptosis was increased by Bcl-xL knock down.
CONCLUSION: Our data suggest that Bcl-xL and, to a lower extent, Mcl-1, are important anti-apoptotic factors in CRC. Specific downregulation of Bcl-xL is a promising approach to sensitize CRC cells towards chemotherapy and targeted therapy.
Keywords: Colorectal carcinoma, Bcl-xL, Myeloid cell leukaemia-1, Epidermal growth factor receptor 1, Apoptosis, 5-fluorouracil, Irinotecan, Oxaliplatin
INTRODUCTION
Colorectal carcinoma (CRC) is one of the most common malignancies in the Western world. In palliative care, novel treatment approaches including combination of chemotherapy and targeted therapies, such as epidermal growth factor receptor (EGFR) 1 blockage, have improved survival of cancer patients[1]. However, 5-year survival of patients with metastatic CRC remains < 5%. Current established systemic therapy options include 5-fluorouracil (5-FU), oxaliplatin, irinotecan, the EGFR1 antibody cetuximab and the vascular endothelial growth factor (VEGF)-A antibody bevacizumab.
Apoptosis is a genetically programmed process of controlled suicide, which is critical for multicellular organisms during development and for tissue homeostasis. However, in cancer, tumor cells acquire resistance to apoptosis. Thus, the ratio of apoptosis and cell division is altered, resulting in a net gain of malignant tissue. Additionally, defects in apoptosis signalling in cancer cells impair response to therapy and contribute to the limited efficacy of different therapy regimens in metastatic disease[2].
Stabilization of mitochondrial integrity is a key mechanism for the survival of a malignant cell and its resistance to therapy[3]. Mitochondrial integrity is regulated by pro- and anti-apoptotic members of the Bcl-2 family, such as Bcl-xL and Mcl-1 (Myeloid cell leukaemia-1, anti-apoptotic) and Bid, Bad and Bax (pro-apoptotic). Mcl-1 is essential for development, differentiation and survival in a variety of cell types[4,5]. It is involved in important interactions of Bcl-2 family members and thereby regulates mitochondrial activation[6]. Mcl-1 protein levels are elevated in various human tumors, such as hepatocellular carcinoma[7] and non-small cell lung cancer[8]. Importantly, it contributes to the resistance of cancer cells towards apoptosis induction[6,9]. Downregulation of Mcl-1 has been shown to sensitize cancer cells towards apoptosis induction, e.g. after treatment with the death receptor ligand TRAIL [tumor necrosis factor (TNF)-related apoptosis-inducing ligand][10,11]. In addition, Mcl-1 degradation is necessary for mitochondrial activation after genotoxic stress[12]. Like Mcl-1, Bcl-xL is known to promote cell survival by counteracting pro-apoptotic Bcl-2 family members, such as Bim, Bax and Bid. In cancer, overexpression of Bcl-xL is associated with tumor progression, poor prognosis and resistance to chemotherapy. In CRC, Bcl-xL expression is correlated with an advanced disease stage[13]. A role of Bcl-xL in cancer was first suggested when it was found that expression of Her-2/Neu in breast cancer cells increased Bcl-xL expression and rendered cells resistant to tamoxifen-induced apoptosis[14]. Ectopic expression of Bcl-xL in CRC blocks curcumin-induced apoptosis[15]. On the other hand, downregulation of Bcl-xL by antisense technique induces cell death, e.g. after treatment with chemotherapeutic drugs[16,17]. An important trigger for Bcl-xL expression in CRC is NF-κB[18]. CRC cells frequently harbor genetic aberrations that promote NF-κB-mediated induction of Bcl-xL.
The last decade has ushered in new advances for the treatment of patients with CRC. The older cytotoxic chemotherapy drug 5-FU underwent new formulation, and two new drugs, oxaliplatin and irinotecan, were investigated as adjunctive therapies. Finally, targeted therapies, including monoclonal antibodies against VEGF-A (bevacizumab) and EGFR1 (cetuximab), are now standard treatment for metastatic CRC. For patients with metastatic disease, the survival rate has doubled. Among others, a promising approach to overcome apoptosis resistance of CRC cells is the engagement of the death receptors belonging to the tumor necrosis factor receptor gene superfamily with the death ligand TRAIL (Apo2L)[19].
In our study, we investigated the role of the anti-apoptotic Bcl-2 family members Bcl-xL and Mcl-1 for the apoptosis sensitivity of CRC. Both Bcl-2 family proteins were specifically modulated in CRC cells by RNA interference and overexpression, respectively, and the impact on apoptosis sensitivity towards chemotherapy and targeted therapy including TRAIL and EGFR1 blockage was explored.
MATERIALS AND METHODS
Reagents and cell lines
SW480, HT29, Caco-2 (all isolated from primary tumor tissue) and SW620 (derived from lymph node metastasis), all human CRC cell lines (adenocarcinomas), were purchased from ATCC. Cell lines were cultured in RPMI 1640 (Invitrogen, Karlsruhe, Germany), supplemented with 10% fetal calf serum (FCS, Biochrom, Berlin, Germany), Pen/Strep (1%) (PAA Laboratories, Pasching, Austria), HEPES (1%) (Cambrex, Verviers, Belgium) and L-Glutamin (1%) (Cambrex). Cells were cultivated in reduced medium (FCS concentration decreased to 0.5%) in all experiments. Reagents were purchased from the following suppliers: chemotherapeutic agents from Sigma (Deisenhofen, Germany); TRAIL (with enhancer, applied in a concentration of 1 ng/mL) from Alexis Biochemicals (San Diego, CA, USA); PD168393 from Calbiochem (Schwalbach, Germany); Protein A (for co-treatment with anti-APO-1, in a concentration of 10 ng/mL) and EGF from Sigma. Cetuximab was supplied by Merck Pharma (Darmstadt, Germany). Anti-APO-1 was kindly provided by Peter H. Krammer (German Cancer Research Center).
Tissue samples
CRC tissue samples as well as non-neoplastic colorectal tissues were obtained from patients undergoing elective surgery for colorectal cancer at the University of Mainz. Analysis of CRC samples was approved by the local ethics committee. The morphological classification of the carcinomas was conducted according to WHO specifications. Tissues samples were used for immunhistochemical staining as well as for mRNA extraction.
Immunohistochemical staining
Paraffin-embedded tissue sections (which all included carcinoma as well as normal epithelial cells in one section) were subjected to immunostaining, using a biotin/streptavidin-peroxidase technique (Vector Laboratories Inc., Burlingame, CA). They were deparaffinized in xylene and dehydrated in ethanol, and dried in a steamer with 10 mmol Na-citrate buffer (pH 6.0). Endogenous peroxidase activity was blocked by incubating the slices for 5 min with 3.0% hydrogen peroxide at room temperature, followed by washing in TPBS (0.5% TWEEN in PBS). The sections were then incubated for 30 min at room temperature with TNB (1% BSA, 0.5% protein-blocking reagent in TBS) prior to an overnight incubation at 4°C with polyclonal rabbit Bcl-xS/L (clone S-18, Santa Cruz Biotechnology Inc., Santa Cruz, California) or polyclonal rabbit Mcl-1 (S-19, Santa Cruz). Both were diluted 1:160 in TNB. Bound antibody was detected using biotinylated anti-rabbit IgG secondary antibody (Vector) and streptavidin-peroxidase complex (Vector), followed by incubating with diaminobenzidine as substrate. Sections were counterstained with Mayer’s haematoxylin. As negative controls, sections were incubated in the presence of nonimmunized rabbit IgG as first antibody.
Viability test
Cell viability was determined by a colorimetric 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. CRC cell lines were seeded onto 12-well plates. On day 1 after seeding, cells were treated as indicated. 100 μL MTT (5 mg/mL) was added to each well. After 4 h incubation at 37°C the supernatant was discarded and cells were washed with PBS. For cell lysis 0.5 mL 1-propanol was added for another 20 min. After transfer of 100 μL of each sample to a flat-bottomed 96-well microtiter plate; the optical density was determined at 550 nm. The viability of “1” was defined as the absorbance obtained from mock transfected cells or untreated cells, respectively.
Detection of apoptosis
CRC cell lines were seeded onto 12-well plates. On day 1 after seeding, cells were treated as indicated. Cells were then collected, washed, and resuspended in lysis buffer containing 0.1% (w/v) sodium citrate, 0.1% (v/v) Triton X-100 and 50 μg/mL propidium iodide (Sigma). After overnight incubation at 4°C, nuclei from apoptotic cells were quantified by flow cytometry according to the method of Nicoletti et al[20].
Cell lysis and Western blotting
Cell lysis and Western blotting were performed as described before[7]. Immunodetection was performed using the indicated primary antibodies: anti-Mcl-1 (S19) (Santa Cruz Biotechnology, Heidelberg, Germany), anti-Bcl-xL (Labvision/NeoMarkers, Warm Springs Blvd. Fremont, Canada), and mouse anti-α-Tubulin clone B-5-1-2 (Sigma).
RNAi and transfection
For small interfering RNA (siRNA)-mediated knock down of Mcl-1 and Bcl-xL, the following siRNA sequences were applied (MWG Biotech, Ebersberg, Germany): Mcl-1, 5'-aaguaucacagacguucucTT-3’ (sense) and 5’-gagaacgucugugauacuuTT-3’ (antisense). Bcl-xL, 5’-gcu ugggauaaagaugcaaTT-3’ (sense) and 5’-uugcaucuuuau cccaagcAG-3’ (antisense). As a non-silencing control, siRNA specific for green fluorescent protein (GFP) was used: 5’-ggcuacguccaggagcgcaccTT-3’ (sense) and 5’-ggu gcgcuccuggacguagccTT-3’ (antisense), where capitals represent deoxyribonucleotides and lower case letters represent ribonucleotides. SW480 cells were transiently transfected with Lipofectamin RNAiMAX (Invitrogen, Karlsruhe, Germany) according to the manufacturer's protocol and analyzed 24 h after transfection. For Mcl-1 and Bcl-xL expression, we applied specific expression vectors (pEF4Mcl-1 or pcDNA3Bcl-xL) or the corresponding empty vectors (pEF4empty or pcDNA3empty, respectively), all kindly provided by Peter H. Krammer, German Cancer Research Center (Heidelberg, Germany). SW480 cells were transfected with plasmids using Transfectin (Biorad, München, Germany) according to the manufacturer’s protocol.
Real-time quantitative polymerase chain reaction (RT-QPCR)
To analyze RNAi efficiency, total RNA from CRC cells was extracted using RNeasy Mini Kit (Qiagen) 24 h after transfection of siRNA. One μg of total RNA was reverse transcribed using an oligo-dT primer with the Omniscript RT kit (Qiagen) and afterwards analyzed for specific mRNA expression by RT-QPCR using the QuantiTect SYBR Green PCR Kit (Qiagen) and the following primers: Actin forward: 5’-GGACTTCGAGCAAGAGAT GG-3’, Actin reverse: 5’-AGCACTGTGTTGGCGTAC AG-3’, Mcl-1 forward: 5’-TAAGGACAAAACGGGACT GG-3’, Mcl-1 reverse: 5’-ACCAGCTCCTACTCCAGC AA-3’. Bcl-xL forward: 5’-GTAAACTGGGGTCGC ATTGT-3’, Bcl-xL reverse: 5’-TGCTGCATTGTTCCC ATAGA-3’. The relative increase in reporter fluorescent dye emission was monitored. The level of Mcl-1 or Bcl-xL (gene of interest, GOI) mRNA, respectively, relative to actin, was calculated using the formula: Relative GOI mRNA expression = 2 [Ct (GOIcontrol) - Ct (GOItreated) + Ct (Actintreated) - Ct (Actincontrol)], where Ct is defined as the number of the cycle in which emission exceeds an arbitrarily defined threshold. For evaluation of Bcl-xL and Mcl-1 mRNA expression in tumor as well as non-neoplastic colon tissues, RPII instead of actin was measured as housekeeping gene: RPII forward: 5’-GCACCACGTCCAATGACAT-3’, RPII reverse: 5’-GTGCGGCTGCTTCCATAA-3’.
Statistical analysis
All results are expressed as mean ± SD. Data were analyzed by Student’s t-test (paired, two sided). P < 0.05 was considered significant.
RESULTS
Expression of the anti-apoptotic Bcl-2 family members Bcl-xL and Mcl-1 in CRC
Apoptosis resistance is a well-known phenomenon which counteracts chemotherapeutic drug-induced cell death of CRC cells. Anti-apoptotic Bcl-2 family members such as Bcl-xL and Mcl-1 contribute to the apoptosis resistance in different tumor entities. First, we analyzed expression of Bcl-xL and Mcl-1 in various CRC cell lines. All cell lines tested showed a profound expression of Mcl-1 on protein level (Figure 1A). Bcl-xL expression was rather low in HT29 and Caco-2 and high in SW480 cells (Figure 1A).
Figure 1.
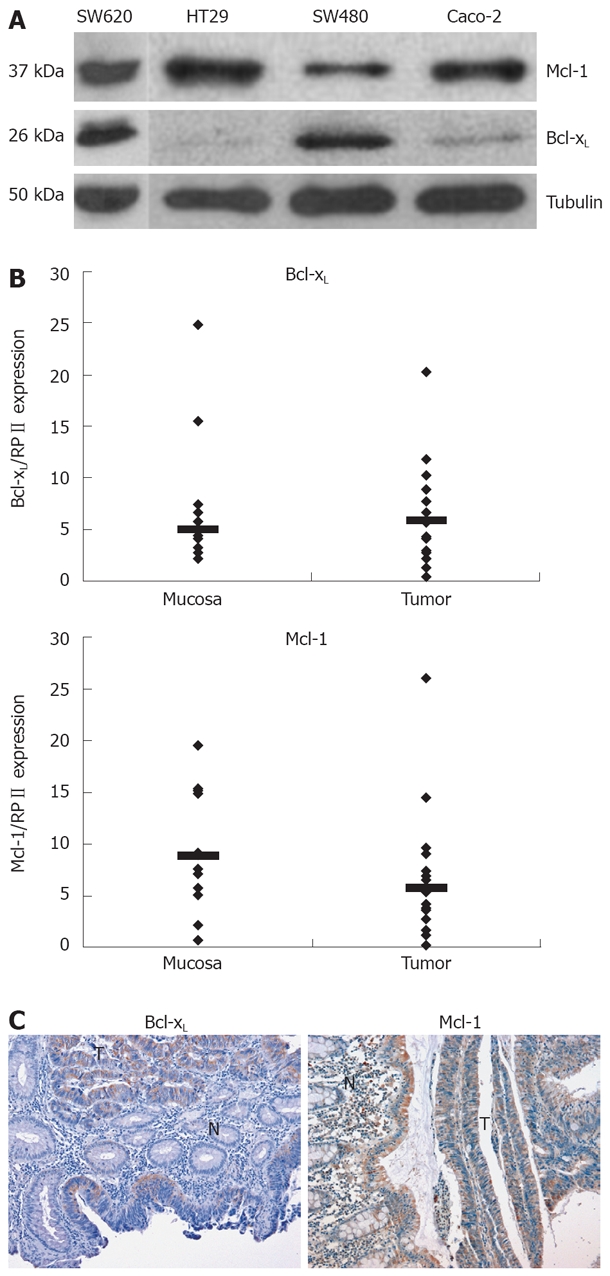
Mcl-1 and Bcl-xL expression in CRC. A: The CRC cell lines, HT29, SW620, SW480, and Caco-2, were analyzed for the basal expression of the Bcl-2 family members Bcl-xL and Mcl-1. Whole cell lysates were prepared, separated, and immunoblotted with antibodies against Bcl-xL, Mcl-1 and α-tubulin; B: CRC tissues and normal colorectal tissues were tested for mRNA expression (n = 9 patients). mRNA expression levels of Bcl-xL, Mcl-1 and RPII were measured in all tissue samples by quantitative real-time PCR. mRNA expression levels of Bcl-xL or Mcl-1 were normalized to RPIIin each sample. Each PCR reaction was run in triplicates. Median is added; C: Immunohistochemical analysis of human CRC tissues was performed as described in the Methods section. All sections included carcinoma as well as normal epithelial tissues to directly compare Bcl-xL as well as Mcl-1 expression in neoplastic and non-malignant tissues. Representative analysis of immunoperoxidase detection of Bcl-xL and Mcl-1 in paraffin embedded carcinoma tissue (T) and adjacent non-tumor tissue (N) is presented.
Next, we analyzed expression of Bcl-xL and Mcl-1 mRNA in human CRC tissues by quantitative PCR. Bcl-xL levels were higher in CRC tissues compared to non-malignant, adjacent tissue (Median of relative expression: 1.2, n = 9, P < 0.2, not significant, Figure 1B). Six of 9 patients showed a higher Bcl-xL expression, 2 patients showed a lower expression, and in 1 patient, expression was virtually equal. Mcl-1 mRNA expression was significantly lower in carcinoma tissue compared to non-malignant tissue (Median of relative expression: 0.41, n = 9, P < 0.01). In addition, we performed immunohistochemical analysis of Bcl-xL and Mcl-1 in CRC. In all tissues tested (n = 6), expression of Bcl-xL was profoundly higher in carcinoma cells compared to surrounding epithelial cells (Figure 1C). Furthermore, Mcl-1 expression was also (slightly) higher compared to surrounding epithelial cells in all probes tested (n = 4).
Sensitivity of Bcl-xL and Mcl-1 expressing CRC cells towards chemotherapeutic drug- induced apoptosis and EGFR1 inhibition
Subsequently, we tested the sensitivity of CRC cell lines towards chemotherapeutic drug-induced apoptosis. We treated SW480 cells with different agents frequently applied for the treatment of patients with CRC: the chemotherapeutic agents irinotecan, oxaliplatin and 5-FU (Figure 2). After 48 h, oxaliplatin (10 μg/mL) and irinotecan (40 μg/mL) induced apoptosis in more than 50% of the cells. 5-FU (10 μg/mL) induced apoptosis in nearly 45% of CRC cells. Treatment with the antagonistic EGFR1 antibody cetuximab induced apoptosis in 18% of cells after 48 h (compared to 13% of apoptosis in control cells, P = 0.07, Figure 2). Apoptosis induction in cells treated with the EGFR1 tyrosine kinase inhibitor PD168393 was not significant (16% vs 13%, P = 0.15).
Figure 2.
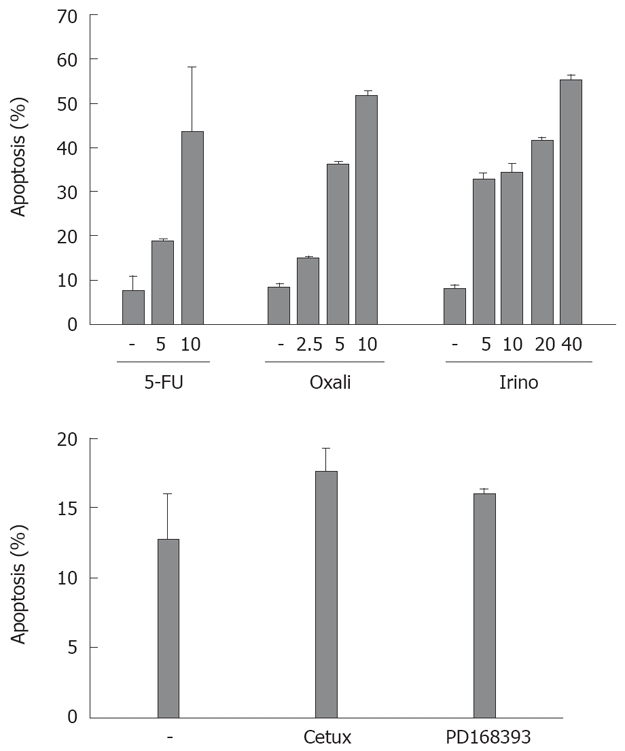
Drug-induced apoptosis in CRC cells. SW480 cells were treated with the chemotherapeutic agents 5-FU, oxalipatin (“oxali”) and irinotecan (“irino”) for 48 h (concentrations as indicated, upper panel). In addition, cells were treated with the EGFR1 antibody cetuximab (“cetux”, 20 μg/mL) or the EGFR1 tyrosine kinase inhibitor PD168393 (7 μmol/L, lower panel) in FCS reduced medium. Cells were then harvested and analyzed for apoptosis induction by flow cytometry. Assays were performed in triplicates. Values are mean ± SD.
Modulation of Bcl-xL and Mcl-1 expression and its impact on chemotherapeutic drug-induced apoptosis
In order to analyze the functional contribution of Bcl-xL and Mcl-1 on apoptosis sensitivity of CRC cells, we specifically modulated their expression. Transfection of specific siRNA sequences effectively knocked down expression of Bcl-xL and Mcl-1 mRNA and protein in SW480 cells (Figures 3A and 4A). For example, 24 h after transfecting 20 nmol/L of specific siRNAs, mRNA expressions were reduced by 75% or 74%, respectively. For 10, 20 and 40 nmol/L of siRNA concentration, an efficient knock down of Bcl-xL and Mcl-1 was observed after 24 h (data not shown). As could be detected in Western blot assays, Bcl-xL and Mcl-1 protein expression were also drastically reduced 24 h after siRNA transfection. On the other hand, transfection of expression plasmids for Bcl-xL increased protein expression (Figure 3C). Next, we tested the effect of specific knock down of Mcl-1 vs Bcl-xL on apoptosis sensitivity of SW480 cells. Knock down of Bcl-xL significantly enhanced apoptosis induction in untreated cells compared to control transfected cells (21% to 38%, P < 0.05, Figure 3B). These results were confirmed in viability assays: Viability was decreased by 33% (Figure 3B). In contrast, Mcl-1 knock down only moderately enhanced spontaneous apoptosis rates (20% to 25%, P < 0.05, Figure 4B). Correspondingly, viability was not decreased in CRC cells after Mcl-1 knock down (Figure 4B).
Figure 3.

Modulation of Bcl-xL expression alters chemotherapeutic drug-induced apoptosis. A: SW480 cells were transfected with siRNA specific for Bcl-xL or transfected with siRNA specific for GFP as control (20 nmol/L). After the indicated time post transfection, total RNA was extracted and analyzed for Bcl-xL expression by quantitative real-time PCR (left panel). Relative expression was calculated as described in the materials and methods section. In addition, after the indicated time post transfection, cells were lyzed and analyzed for Bcl-xL expression by Western blot (right panel). α-Tubulin expression was used to control equal loading; B: SW480 cells were transfected with siRNA specific for Bcl-xL or transfected with siRNA specific for GFP as control. 24 h post transfection, cells were treated with 5-FU (10 μg/mL), oxaliplatin (16 μg/mL) and irinotecan (60 μg/mL) for further 24 h. Cells were then harvested and analyzed for apoptosis induction (left panel). In addition, cell viability was measured by MTT assay and is shown relative to mock treated controls (right panel); C: SW480 cells were transfected with pcDNA3 Bcl-xL or pcDNA3 empty vector as control. 24 h post transfection, cells were analyzed for Bcl-xL expression using Western blotting (left panel). In addition, 24 h post transfection, cells were treated with 5-FU (15 μg/mL), oxaliplatin (10 μg/mL) and irinotecan (40 μg/mL) for further 24 h. Cells were then harvested and analyzed for apoptosis induction by flow cytometry. (B) and (C) assays were performed in triplicates. Values are means ± SD, aP < 0.05.
Figure 4.

Modulation of Mcl-1 expression alters chemotherapeutic drug-induced apoptosis. A: SW480 cells were transfected with siRNA specific for Mcl-1, or transfected with siRNA specific for GFP as control (20 nmol/L). After the indicated time post transfection, total RNA was extracted and analyzed for Mcl-1 expression by quantitative real-time PCR (left panel). Relative expression was calculated as described in the materials and methods section. In addition, after the indicated time post transfection, cells were lyzed and analyzed for Mcl-1 expression by Western blot (right panel). α-Tubulin expression was used to control equal loading; B: SW480 cells were transfected with siRNA specific for Mcl-1 or transfected with control siRNA. 24 h post transfection cells were treated with 5-FU (10 μg/mL), oxaliplatin (16 μg/mL) and irinotecan (60 μg/mL) for further 24 h. Apoptosis induction was measured by flow cytometry (left panel) and MTT assay (right panel). Values are mean ± SD, aP < 0.05.
Subsequently, we treated CRC cells with the chemotherapeutic agents 5-FU, oxaliplatin and irinotecan. Knock down of Bcl-xL in combination with chemotherapy resulted in an increase of apoptosis induction (5-FU: 34% (chemotherapy alone) to 53% (chemotherapy plus Bcl-xL knock down); oxaliplatin: 35% to 47% and irinotecan: 58% to 72%, P < 0.05, Figure 3B). Silencing of Mcl-1 expression led to a slight enhancement of irinotecan-induced apoptosis (33% to 35%, not significant, Figure 4B) and to a moderate, but significant increase of oxaliplatin-induced apoptosis (27% to 33%, P < 0.05). Surprisingly, 5-FU-induced apoptosis was decreased in cells with lower Mcl-1 expression (32% to 28%, not significant, Figure 4B). Transfection of the Bcl-xL expression plasmid enhanced viability of CRC cells: spontaneous apoptosis rates were 20% instead of 36% in control transfected cells (P < 0.05, Figure 3C). Moreover, 5-FU (33% vs 46%, P < 0.05), and irinotecan (26% vs 34%, P < 0.05)-induced apoptosis, but not oxaliplatin-induced apoptosis (29% vs 30%, n.s.), was reduced by transfecting Bcl-xL (Figure 3C). After transfection of Mcl-1 expression plasmids, no significant impact on chemotherapeutic-drug-induced apoptosis was observed (data not shown).
Modulation of Bcl-xL and Mcl-1 expression and its impact on EGFR1 blockage
We next analyzed apoptosis induction in CRC cells after targeted therapy approaches. Inhibition of EGFR1 signalling has already entered clinical routine in the treatment of patients with CRC. Cetuximab, a monoclonal antibody against EGFR1, demonstrates anti-tumor efficacy both as a single agent and in combination with irinotecan- and oxaliplatin-based chemotherapy. EGF is known to contribute to an apoptosis resistant phenotype of carcinoma cells. First, we tested the influence of EGF treatment on Bcl-xL and Mcl-1 expression in vitro. EGF treatment of CRC cells induced expression of Bcl-xL and Mcl-1 after 1.5 and 2.5 h, respectively (Figure 5A). We next analyzed the role of Mcl-1 and Bcl-xL for the resistance towards targeted therapy. First, we treated CRC cells with cetuximab. Treatment with cetuximab alone (100 μg/mL) did not induce apoptosis SW480 cells after 24 h (Figure 5B). However, in cells with reduced Bcl-xL, a significant increase in apoptosis induction after treatment with cetuximab (44% vs 37%, P < 0.05, Figure 5B) or the EGFR1 tyrosine kinase inhibitor PD168393 (0.7 μmol/L; 41% vs 29%, P < 0.01, Figure 5B) was observed. Moreover, knock down of Mcl-1 also resulted in a moderate, but significant sensitization towards cetuximab (28% vs 23%; Figure 5B, P < 0.01) and PD168393 (32% vs 24%, Figure 5B, P < 0.05).
Figure 5.

Bcl-xL and Mcl-1 knock down enhances apoptosis induction after EGFR1 inhibition. A: SW480 and HT29 cells were treated with EGF (100 ng/mL) for the time indicated. Whole cell lysates were prepared, separated, and immunoblotted with antibodies against Bcl-xL, Mcl-1 and α-tubulin; B: SW480 cells were transfected with siRNA specific for Bcl-xL (upper panel), or Mcl-1 (lower panel), respectively, or transfected with siRNA specific for GFP as control. 24 h post transfection cells were treated with cetuximab (100 μg/mL) or PD168393 (0.7 μmol/L) for further 24 h. Cells were then harvested and analyzed for apoptosis induction by flow cytometry. Assays were performed in triplicates and are representative for at least two independent experiments. Values are means ± SD, aP < 0.05.
Modulation of Bcl-xL and Mcl-1 expression and its impact on TRAIL- and CD95-mediated apoptosis
The death receptor ligand TRAIL is a promising anti-cancer agent (for recent review[21]) and already has been tested in clinical studies in CRC patients. Thus, we analyzed the impact of Bcl-xL and Mcl-1 modulation on TRAIL-induced apoptosis of CRC cells. TRAIL efficiently induced apoptosis in SW480 cells (22% vs 3% apoptosis in control cells, P < 0.05, Figure 6A). Importantly, Bcl-xL downregulation enhanced TRAIL-induced apoptosis more than two-fold (46% vs 22%, P < 0.001, Figure 6A). To further evaluate the sensitizing effect of Bcl-xL knock down for TRAIL efficacy, we included another CRC cell line, SW620, in our study. After Bcl-xL knock down, TRAIL-induced apoptosis was increased from 16% to 27% (P < 0.05, Figure 6A). Mcl-1 knock down sensitized SW480 cells to TRAIL (17% vs 11%, P < 0.05, Figure 6B). Upregulation of Bcl-xL by transfection of expression plasmids decreased TRAIL-induced cell death (P < 0.05, Figure 6C). Subsequently, we tested the effect of Bcl-xL and Mcl-1 knock down on CD95-induced apoptosis of CRC cells (Figure 6D). SW480 cells were highly susceptible to CD95 stimulation by the agonistic CD95 antibody anti-APO-1 (65% vs 3% after 24 h of anti-APO-1 treatment, Figure 6D). Bcl-xL knock down further increased anti-APO-1 induced apoptosis from 65% to 80% (P < 0.001). Mcl-1 knock down did not further enhance CD95-triggered apoptosis of CRC cells (Figure 6D).
Figure 6.

TRAIL- and CD95-induced apoptosis after modulation of Mcl-1 and Bcl-xL expression. A: SW480 or SW620 cells were transfected with siRNA specific for Bcl-xL or GFP as control. 24 h post transfection cells were treated with TRAIL (0.1 μg/mL) for further 24 h. Cells were then harvested and analyzed for apoptosis induction by flow cytometry; B: SW480 cells were transfected with siRNA specific for Mcl-1 or GFP as control. 24 h post transfection cells were treated with TRAIL (0.01 μg/mL) for further 24 h. Cells were then harvested and analyzed for apoptosis induction by flow cytometry; C: SW480 cells were transfected with pcDNA3 Bcl-xL or pcDNA3 empty vector as control. 24 h post transfection, cells were treated with TRAIL (0.1 μg/mL) for further 12 h. Cells were then harvested and analyzed for apoptosis induction by flow cytometry; D: SW480 cells were transfected with siRNA specific for Mcl-1 (left panel) or Bcl-xL (right panel) or with control siRNA. 24 h post transfection cells were treated with anti-APO-1 (0.1 μg/mL) for further 24 h. Cells were then harvested and analyzed for apoptosis induction by flow cytometry. All assays were performed in triplicates and are representative for at least three independent experiments. Values are mean ± SD, aP < 0.05.
DISCUSSION
In the present study we demonstrate an important role of the anti-apoptotic Bcl-2 family members Bcl-xL and, to a lower extent, Mcl-1, for the apoptosis sensitivity of CRC cells. Bcl-xL expression is considerably enhanced in CRC tissue compared to adjacent non-malignant tissue. After knock down of Bcl-xL by RNA interference, CRC cells prove to be more sensitive towards chemotherapy, EGFR1 blockage, CD95 triggering and treatment with the death receptor ligand TRAIL. The sensitizing effect of Mcl-1 knock down is comparatively moderate. Our data suggest, that Bcl-2 family members, such as Bcl-xL, are promising targets to improve treatment of patients with CRC. Since strategies to inhibit Bcl-xL activity have already been applied in preclinical studies, our data are of particular interest[22].
In the past two decades, substantial progress has been made in the treatment of colon cancer, the second most common cancer in western countries. However, therapy resistance of CRC remains a common clinical problem, so that recurrence and metastasis of CRC remain major obstacles in oncology. Thus, new strategies to overcome resistance to current treatment options are needed.
Numerous defects in apoptosis signalling have been described in CRC. These defects appear to be involved in colorectal tumorigenesis, by facilitating tumor cell progression[23]. In addition, defects in apoptosis signalling represent principle mechanisms through which cancer cells are enabled to survive therapy, since chemotherapy and irradiation induce cell death mainly by apoptosis induction[24]. These defects include, among others, stabilization of mitochondria, inactivation of death receptor signalling and overexpression of EGFR1[2].
Anti-apoptotic proteins of the Bcl-2 family, such as Bcl-2, Bcl-xL and Mcl-1 critically regulate mitochondrial integrity. Increased expression of anti-apoptotic Bcl-2 proteins counteracts chemotherapeutic drug-induced apoptosis in cancer cells. In our study, human CRC tissues revealed enhanced Bcl-xL expression both on mRNA and protein levels. In line with our study, previous studies also observed enhanced Bcl-xL expression in CRC tissues[13,25]. In contrary, Northern blot as well as immunohistochemical analysis failed to detect Bcl-2 expression in CRC[25]. The reason for increased Bcl-xL expression in CRC remains elusive. However, a consistent finding is that oncogenic tyrosine kinases induce expression of Bcl-xL and enhance protein stability. Among these tyrosine kinases is EGFR1, which has been described to upregulate Bcl-xL in other tumor models[26]. In line with this observation, we show a significant upregulation of Bcl-xL in CRC cells upon treatment with EGF in this study. Since overexpression of EGFR1 is a frequent finding in CRC cells, EGFR1 signalling may represent a major cause for the induction of Bcl-xL expression in CRC. Another mechanism which triggers Bcl-xL activity in tumor cells is suppression of deamidation[27]. In a recent immunohistochemical evaluation of human CRC tissues, hypoxia-inducible factor-1 has been discussed to induce expression of Bcl-xL[28].
We also explored expression of the anti-apoptotic protein Mcl-1 in CRC. In other cancer entities, e.g. in hepatocellular carcinoma, a significant correlation of Bcl-xL and Mcl-1 with apoptosis resistance was observed[29]. In our study, Mcl-1 mRNA expression was significantly lower in CRC tissue compared to non-neoplastic cells. In contrast, no profound difference was observed on protein level in immunohistochemistry. Previous studies, however, observed decreased Mcl-1 expression relative to normal mucosa and adenomas, also in immunohistochemistry[13]. At the same time, decrease in Mcl-1 expression has been discussed as a later event in the progression of colorectal tumors, since adenomas show no decreased Mcl-1 expression[13]. The significant difference in Mcl-1 expression we observed on mRNA level, is in line with these previously published data. Another important aspect about Mcl-1 expression is the staining pattern: in contrast to normal mucosa cells, Mcl-1 has been described to be diffusely expressed in tumour samples in previous studies[30]. Such a diffuse expression could also be detected in our study.
Next, we confirmed that Mcl-1 has a relatively short half-life in CRC cells. Inhibition of translational events in different CRC cell lines resulted in a much more rapid decrease in Mcl-1 compared to Bcl-xL expression. This is in line with the well-known fact that Mcl-1 provides short-term cell viability protection against cell death during critical transitions in the cell fate[6]. The rapid decrease of Mcl-1 expression can be explained mainly by efficient proteasomal degradation. In addition, we could show that Mcl-1 expression is induced upon treatment with EGF. This might be mediated at least in part by activation of the MEK/ERK-pathway. In lung cancer, it has been shown that EGF enhanced Mcl-1 protein level in an ERK-dependent manner[8]. In our study, EGF also induced expression of Bcl-xL and ERK might also be involved in this context. Chemotherapeutic agents such as oxaliplatin have been shown to decrease anti-apoptotic proteins like Bcl-xL in SW480 cells[31]. This effect at least in part explains the apoptosis-inducing capacity of oxaliplatin. However, other chemotherapeutic agents such as 5-FU and paclitaxel did not downregulate Bcl-xL in CRC cells in previous studies[32].
Our results demonstrate that a specific knock down of Bcl-xL and, to a lower extent, knock down of Mcl-1 by RNAi sensitize CRC cells to chemotherapeutic drugs frequently applied in CRC therapy (5-FU, oxaliplatin and irinotecan). Notably, Bcl-xL knock down alone already significantly induces apoptosis in untreated cells. Thus, Bcl-xL expression is important for the survival of CRC cells. Our results extend studies which have already shown that knock down of Bcl-xL effectively blocks proliferation of CRC cells[33]. On the other hand, overexpression of Bcl-xL counteracts chemotherapeutic drug-induced apoptosis. In previous studies, overexpression of Bcl-xL has been reported to enhance resistance to various chemotherapeutic agents in leukaemia cells[34]. However, in a previous study, resistance of CRC cells was not observed in Bcl-xL overexpressing cells treated with 5-FU or TRAIL[33] In our study, Bcl-xL overexpression only reduced apoptosis rates in CRC cells treated with 5-FU, irinotecan and oxaliplatin.
Bcl-xL exerts anti-apoptotic effects in cancer cells mainly by its interaction with pro-apoptotic Bcl-2 family members, e.g. Bax and Bak. Activation of Bax and Bak commit the cell to apoptosis by permeabilizing the outer mitochondrial membrane. However, interaction with Bcl-2 proteins such as Bcl-xL and Mcl-1 ablates pro-survival functions of Bax and Bak[35]. Bax expression is not reduced in CRC tissues[25]. Thus, Bcl-xL silencing might induce apoptosis via release of Bax and concomitant mitochondrial permeabilization. However, more studies are required to fully understand the roles of the Bcl-2 proteins and how they cooperate to regulate CRC cell survival.
The apoptosis-sensitizing effect of Mcl-1 modulation was less pronounced (e.g. for oxaliplatin and irinotecan-induced apoptosis) or not significant (e.g. for 5-FU-induced apoptosis), respectively. The reason might be the relatively low expression of Mcl-1 in CRC cells, which may at least in part be compensated by a higher Bcl-xL expression. Nevertheless, Mcl-1 knock down has sensitizing effects in our study. This effect may also be explained by the fact that Mcl-1, like Bcl-xL, guards Bax and Bak and thereby prevents them to activate mitochondria.
Notably, Bcl-xL knock down by siRNA also enhances death receptor-mediated apoptosis in CRC cells. CD95-mediated apoptosis was increased by Bcl-xL knock down in SW480 cells. However, this effect is supposed to be less relevant for the in vivo situation, since no CD95 expression has been detected in CRC tissues in previous studies[30]. Remarkably, Bcl-xL knock down considerably enhanced tumor necrosis factor alpha (TNF-alpha)-related apoptosis-inducing ligand (TRAIL)-induced apoptosis of SW480 as well as SW620 cells. TRAIL is a member of the TNF family, which has been reported to induce apoptosis in various tumor cells, but not in normal cells, thus representing a promising anticancer agent[36]. Agonistic TRAIL receptor antibodies have already entered clinical trials[37]. Recently, we have shown that treatment with TRAIL alone or in combination with chemotherapeutic drugs (with the exception of cisplatin) is not toxic for human hepatocytes[38]. In our study, treatment of CRC cells with TRAIL resulted in relatively low apoptosis rates. This is in line with previous studies on CRC cells, e.g. on SW620 cells[39] or HT29 cells[40]. The reason for restricted apoptosis induction is that TRAIL signalling also involves activation of anti-apoptotic pathways including PI3K/Akt, NF-κB and MEK/ERK[41]. NF-κB activation, for example, induces anti-apoptotic proteins such as Mcl-1 in HT29 cells, contributing to TRAIL resistance[39]. Bcl-xL has also been shown to be upregulated by NF-κB activity[42].
Several approaches have been exploited to sensitize cancer cells towards TRAIL. An important strategy, also pursued in this study, is downregulation of anti-apoptotic Bcl-2 proteins. In the present study, Bcl-xL knock down by siRNA efficiently sensitized CRC cells towards TRAIL-induced apoptosis. In contrary, Mcl-1 knock down only slightly sensitized CRC cells to TRAIL. These data correspond to previous studies of our group and others on hepatocellular carcinoma, where Mcl-1 knock down did not sensitize towards TRAIL[9]. However, in cholangiocellular carcinoma, where high Mcl-1 expression is frequently found, Mcl-1 knock down renders cancer cells susceptible to TRAIL[11]. Since TRAIL-induced apoptosis in cancer cells is hampered by NF-κB activation, inhibition of NF-κB is likely to augment TRAIL-induced death of CRC cells. Approaches to block NF-κB are, among others, peptidomimetic compounds that disrupt the IKK complex or multikinase inhibitors, such as sorafenib[43].
The epidermal growth factor receptor (EGFR1) is a receptor tyrosine kinase of the ErbB family that is abnormally activated in many epithelial tumors, such as CRC. EGFR1 is involved in survival signalling, cell migration, metastasis formation, angiogenesis, and reduced responses to chemotherapy. Clinical and survival benefits with anti-EGFR1 agents have been demonstrated in tumor patients (for review[44]). Monoclonal antibodies to EGFR1 are among promising novel targeted therapies being explored in CRC. One such agent that inhibits EGFR1 signalling by interfering with ligand-binding is cetuximab. Cetuximab is a human-mouse chimeric therapeutic monoclonal antibody that competitively binds to the extracellular domain of EGFR1. EGFR1 tyrosine kinase inhibitors, such as PD168393, also block EGFR1 signalling. In this study, knock down of Mcl-1 slightly sensitized CRC cells towards cetuximab and PD168393. Moreover, knock down of Bcl-xL sensitized CRC cells towards cetuximab as well as PD168393. These findings suggest that combining EGFR1 blockage with agents that directly destabilize or disable Bcl-xL and Mcl-1 will have therapeutic benefits.
The development of siRNA technology has made it possible to suppress the function of specific molecules and helps to develop new treatment strategies for cancer[45]. Our study suggests that Bcl-xL and Mcl-1 are suitable targets to sensitize CRC cells to death. The delivery of siRNA in vivo including specific uptake in tumor cells remains a challenging issue[46]. Many approaches use plasmid and viral vectors for transcription of short-hairpin RNAs, both in vitro and in vivo. However, human trials are still on the way to optimize delivery techniques. Another approach to specifically knock down Bcl-xL expression is by antisense oligonucleotides. Bispecific antisense oligonucleotides inhibiting both Bcl-2 and Bcl-xL may be useful to induce apoptosis of tumor cells[47]. Other promising strategies to downregulate Bcl-xL or Mcl-1 are application of small-molecule inhibitors. ABT-737 is an example of one of the first small-molecule inhibitors of Bcl-2/Bcl-xL shown to be efficacious in vivo, causing complete regression in small-cell lung carcinoma tumour xenografts in mice[22]. TW-37 has recently been described to simultaneously inhibit Bcl-2, Bcl-xL and Mcl-1 in lymphoma cells by targeting the BH3-binding groove of these Bcl-2 proteins[48]. Apart from direct suppression of Bcl-xL by siRNA or small-molecule inhibitors, suppression of oncogenic tyrosine kinases, such as Src kinases, which trigger Bcl-xL expression[26], is another promising approach to induce killing of CRC cells.
In conclusion, our findings clearly implicate the anti-apoptotic activity of Bcl-2 family members, such as Bcl-xL and, to a lower extent, Mcl-1, as important components of the treatment resistance of CRC cells. Efficacy of chemotherapy, EGFR1 blockage and treatment with TRAIL, might be substantially improved by co-suppression of the anti-apoptotic protein Bcl-xL.
COMMENTS
Background
Colorectal carcinoma (CRC) is a very common malignancy with an increasing incidence in recent decades. Defects in apoptosis signalling contribute to the resistance of CRC cells towards different treatment regimens. Thus, one of the main goals for oncologic treatment of patients suffering from CRC is to overcome resistance of tumor cells towards apoptosis.
Research frontiers
Decreased sensitivity of mitochondria towards apoptosis stimuli, such as chemotherapy, is a key mechanism for apoptosis resistance of CRC cells. Mitochondrial activation is determined by the interaction of pro- and anti-apoptotic Bcl-2 family proteins, such as Bcl-xL and Mcl-1. In CRC, anti-apoptotic Bcl-2 family proteins are highly expressed, thus contributing to apoptosis resistance.
Innovations and breakthroughs
In previous articles, the interaction of anti- and pro-apoptotic members of the Bcl-2 protein family and their role for the apoptosis sensitivity of carcinoma cells has been extensively studied. It has been shown that anti-apoptotic Bcl-2 family members are capable of blocking pro-apoptotic members of the family. Approaches to block the activity of anti-apoptotic Bcl-2 proteins, e.g. by RNA interference, have been evaluated and proven to be likely effective for the treatment of cancer patients.
Applications
In this article, authors show an important role of Bcl-xL and Mcl-1 for the apoptosis resistance of CRC cells. Thus, downregulation of these anti-apoptotic proteins is a promising approach for the treatment of patients with CRC. Here authors show that the use of RNA interference can effectively downregulate Bcl-xL and Mcl-1 expression in CRC cells. After downregulation, CRC cells are sensitized to chemotherapy and target therapy approaches. Other ways to downregulate these proteins is application of so called “BH3-only mimetics”. These drugs can interact with Bcl-xL and Mcl-1 and thereby induce the release of pro-apoptotic Bcl-2 proteins. “BH3-only mimetics” have already entered clinical trials in cancer patients.
Terminology
Apoptosis is also depicted as programmed cell death. It is characterized by typical morphological alterations, e.g. the condensation of chromatin in the nucleus. Bcl-2 proteins are a large family of proteins, which can be sub-divided in anti-apoptotic members, multidomain pro-apoptotic members and BH3-only pro-apoptotic members. Bcl-xL and Mcl-1 are both anti-apoptotic members of the Bcl-2 family. Receptors for epidermal growth factor (EGF) contribute to the growth of cancer cells. Therapeutic approaches in patients with CRC target this receptor (e.g. antibodies binding to the EGF receptor as well as small molecules which inhibit the kinase domain of the receptor) and have been proven to be effective anti-cancer reagents in clinical studies.
Peer review
This study shows that Mcl-1 and Bcl-xL are important anti-apoptotic factors in CRC. Downregulation of Bcl-xL is proven to be a promising approach to sensitize CRC towards chemotherapy and targeted therapy. Thus, a translational idea for the treatment of CRC is provided. This is a well written paper and the results are important.
Supported by A Research Grant of Merck Pharma GmbH, Darmstadt, Germany, to the University Clinic of Mainz
Peer reviewers: Shu Zheng, Professor, Scientific Director of Cancer Institute, Zhejiang University, Secondary Affiliated Hospital, Zhejiang University, 88# Jiefang Road, Hangzhou 310009, Zhejiang Province, China; Dr. John M Carethers, GI Section, 111D, VA San Diego Healthcare System, 3350 La Jolla Village Drive, San Diego CA 92161, United States; Wei Tang, MD, EngD, Assistant Professor, H-B-P Surgery Division, Artificial Organ and Transplantation Division, Department of surgery, Graduate School of Medicine, the University of Tokyo, Tokyo 113-8655, Japan
S- Editor Li DL L- Editor Li M E- Editor Ma WH
References
- 1.Majer M, Akerley W, Kuwada SK. Oncologists' current opinion on the treatment of colon carcinoma. Anticancer Agents Med Chem. 2007;7:492–503. doi: 10.2174/187152007781668742. [DOI] [PubMed] [Google Scholar]
- 2.Schulze-Bergkamen H, Krammer PH. Apoptosis in cancer--implications for therapy. Semin Oncol. 2004;31:90–119. doi: 10.1053/j.seminoncol.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 3.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 4.Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci USA. 1993;90:3516–3520. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Dev. 2000;14:23–27. [PMC free article] [PubMed] [Google Scholar]
- 6.Craig RW. MCL1 provides a window on the role of the BCL2 family in cell proliferation, differentiation and tumorigenesis. Leukemia. 2002;16:444–454. doi: 10.1038/sj.leu.2402416. [DOI] [PubMed] [Google Scholar]
- 7.Fleischer B, Schulze-Bergkamen H, Schuchmann M, Weber A, Biesterfeld S, Muller M, Krammer PH, Galle PR. Mcl-1 is an anti-apoptotic factor for human hepatocellular carcinoma. Int J Oncol. 2006;28:25–32. [PubMed] [Google Scholar]
- 8.Song L, Coppola D, Livingston S, Cress D, Haura EB. Mcl-1 regulates survival and sensitivity to diverse apoptotic stimuli in human non-small cell lung cancer cells. Cancer Biol Ther. 2005;4:267–276. doi: 10.4161/cbt.4.3.1496. [DOI] [PubMed] [Google Scholar]
- 9.Schulze-Bergkamen H, Fleischer B, Schuchmann M, Weber A, Weinmann A, Krammer PH, Galle PR. Suppression of Mcl-1 via RNA interference sensitizes human hepatocellular carcinoma cells towards apoptosis induction. BMC Cancer. 2006;6:232. doi: 10.1186/1471-2407-6-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han J, Goldstein LA, Gastman BR, Rabinowich H. Interrelated roles for Mcl-1 and BIM in regulation of TRAIL-mediated mitochondrial apoptosis. J Biol Chem. 2006;281:10153–10163. doi: 10.1074/jbc.M510349200. [DOI] [PubMed] [Google Scholar]
- 11.Taniai M, Grambihler A, Higuchi H, Werneburg N, Bronk SF, Farrugia DJ, Kaufmann SH, Gores GJ. Mcl-1 mediates tumor necrosis factor-related apoptosis-inducing ligand resistance in human cholangiocarcinoma cells. Cancer Res. 2004;64:3517–3524. doi: 10.1158/0008-5472.CAN-03-2770. [DOI] [PubMed] [Google Scholar]
- 12.Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev. 2003;17:1475–1486. doi: 10.1101/gad.1093903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krajewska M, Moss SF, Krajewski S, Song K, Holt PR, Reed JC. Elevated expression of Bcl-X and reduced Bak in primary colorectal adenocarcinomas. Cancer Res. 1996;56:2422–2427. [PubMed] [Google Scholar]
- 14.Kumar R, Mandal M, Lipton A, Harvey H, Thompson CB. Overexpression of HER2 modulates bcl-2, bcl-XL, and tamoxifen-induced apoptosis in human MCF-7 breast cancer cells. Clin Cancer Res. 1996;2:1215–1219. [PubMed] [Google Scholar]
- 15.Rashmi R, Kumar S, Karunagaran D. Ectopic expression of Bcl-XL or Ku70 protects human colon cancer cells (SW480) against curcumin-induced apoptosis while their down-regulation potentiates it. Carcinogenesis. 2004;25:1867–1877. doi: 10.1093/carcin/bgh213. [DOI] [PubMed] [Google Scholar]
- 16.Zhang L, Yu J, Park BH, Kinzler KW, Vogelstein B. Role of BAX in the apoptotic response to anticancer agents. Science. 2000;290:989–992. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]
- 17.Zangemeister-Wittke U, Schenker T, Luedke GH, Stahel RA. Synergistic cytotoxicity of bcl-2 antisense oligodeoxynucleotides and etoposide, doxorubicin and cisplatin on small-cell lung cancer cell lines. Br J Cancer. 1998;78:1035–1042. doi: 10.1038/bjc.1998.624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen C, Edelstein LC, Gelinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L) Mol Cell Biol. 2000;20:2687–2695. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–162. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- 21.Merino D, Lalaoui N, Morizot A, Solary E, Micheau O. TRAIL in cancer therapy: present and future challenges. Expert Opin Ther Targets. 2007;11:1299–1314. doi: 10.1517/14728222.11.10.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stauffer SR. Small molecule inhibition of the Bcl-X(L)-BH3 protein-protein interaction: proof-of-concept of an in vivo chemopotentiator ABT-737. Curr Top Med Chem. 2007;7:961–965. doi: 10.2174/156802607780906843. [DOI] [PubMed] [Google Scholar]
- 23.Sinicrope FA, Ruan SB, Cleary KR, Stephens LC, Lee JJ, Levin B. bcl-2 and p53 oncoprotein expression during colorectal tumorigenesis. Cancer Res. 1995;55:237–241. [PubMed] [Google Scholar]
- 24.Fisher DE. Apoptosis in cancer therapy: crossing the threshold. Cell. 1994;78:539–542. doi: 10.1016/0092-8674(94)90518-5. [DOI] [PubMed] [Google Scholar]
- 25.Maurer CA, Friess H, Buhler SS, Wahl BR, Graber H, Zimmermann A, Buchler MW. Apoptosis inhibiting factor Bcl-xL might be the crucial member of the Bcl-2 gene family in colorectal cancer. Dig Dis Sci. 1998;43:2641–2648. doi: 10.1023/a:1026695025990. [DOI] [PubMed] [Google Scholar]
- 26.Karni R, Jove R, Levitzki A. Inhibition of pp60c-Src reduces Bcl-XL expression and reverses the transformed phenotype of cells overexpressing EGF and HER-2 receptors. Oncogene. 1999;18:4654–4662. doi: 10.1038/sj.onc.1202835. [DOI] [PubMed] [Google Scholar]
- 27.Zhao R, Yang FT, Alexander DR. An oncogenic tyrosine kinase inhibits DNA repair and DNA-damage-induced Bcl-xL deamidation in T cell transformation. Cancer Cell. 2004;5:37–49. doi: 10.1016/s1535-6108(03)00333-7. [DOI] [PubMed] [Google Scholar]
- 28.Wincewicz A, Sulkowska M, Koda M, Sulkowski S. Cumulative expression of HIF-1-alpha, Bax, Bcl-xL and P53 in human colorectal cancer. Pathology. 2007;39:334–338. doi: 10.1080/00313020701329765. [DOI] [PubMed] [Google Scholar]
- 29.Sieghart W, Losert D, Strommer S, Cejka D, Schmid K, Rasoul-Rockenschaub S, Bodingbauer M, Crevenna R, Monia BP, Peck-Radosavljevic M, et al. Mcl-1 overexpression in hepatocellular carcinoma: a potential target for antisense therapy. J Hepatol. 2006;44:151–157. doi: 10.1016/j.jhep.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 30.Backus HH, Van Groeningen CJ, Vos W, Dukers DF, Bloemena E, Wouters D, Pinedo HM, Peters GJ. Differential expression of cell cycle and apoptosis related proteins in colorectal mucosa, primary colon tumours, and liver metastases. J Clin Pathol. 2002;55:206–211. doi: 10.1136/jcp.55.3.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fujie Y, Yamamoto H, Ngan CY, Takagi A, Hayashi T, Suzuki R, Ezumi K, Takemasa I, Ikeda M, Sekimoto M, et al. Oxaliplatin, a potent inhibitor of survivin, enhances paclitaxel-induced apoptosis and mitotic catastrophe in colon cancer cells. Jpn J Clin Oncol. 2005;35:453–463. doi: 10.1093/jjco/hyi130. [DOI] [PubMed] [Google Scholar]
- 32.Wu S, Zhu H, Gu J, Zhang L, Teraishi F, Davis JJ, Jacob DA, Fang B. Induction of apoptosis and down-regulation of Bcl-XL in cancer cells by a novel small molecule, 2[[3-(2,3-dichlorophenoxy)propyl]amino]ethanol. Cancer Res. 2004;64:1110–1113. doi: 10.1158/0008-5472.can-03-2790. [DOI] [PubMed] [Google Scholar]
- 33.Zhu H, Guo W, Zhang L, Davis JJ, Teraishi F, Wu S, Cao X, Daniel J, Smythe WR, Fang B. Bcl-XL small interfering RNA suppresses the proliferation of 5-fluorouracil-resistant human colon cancer cells. Mol Cancer Ther. 2005;4:451–456. doi: 10.1158/1535-7163.MCT-04-0162. [DOI] [PubMed] [Google Scholar]
- 34.Schmitt E, Cimoli G, Steyaert A, Bertrand R. Bcl-xL modulates apoptosis induced by anticancer drugs and delays DEVDase and DNA fragmentation-promoting activities. Exp Cell Res. 1998;240:107–121. doi: 10.1006/excr.1998.4003. [DOI] [PubMed] [Google Scholar]
- 35.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med. 1999;5:157–163. doi: 10.1038/5517. [DOI] [PubMed] [Google Scholar]
- 37.Marini P, Denzinger S, Schiller D, Kauder S, Welz S, Humphreys R, Daniel PT, Jendrossek V, Budach W, Belka C. Combined treatment of colorectal tumours with agonistic TRAIL receptor antibodies HGS-ETR1 and HGS-ETR2 and radiotherapy: enhanced effects in vitro and dose-dependent growth delay in vivo. Oncogene. 2006;25:5145–5154. doi: 10.1038/sj.onc.1209516. [DOI] [PubMed] [Google Scholar]
- 38.Ganten TM, Koschny R, Sykora J, Schulze-Bergkamen H, Buchler P, Haas TL, Schader MB, Untergasser A, Stremmel W, Walczak H. Preclinical differentiation between apparently safe and potentially hepatotoxic applications of TRAIL either alone or in combination with chemotherapeutic drugs. Clin Cancer Res. 2006;12:2640–2646. doi: 10.1158/1078-0432.CCR-05-2635. [DOI] [PubMed] [Google Scholar]
- 39.Vaculova A, Hofmanova J, Soucek K, Kozubik A. Different modulation of TRAIL-induced apoptosis by inhibition of pro-survival pathways in TRAIL-sensitive and TRAIL-resistant colon cancer cells. FEBS Lett. 2006;580:6565–6569. doi: 10.1016/j.febslet.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 40.Tillman DM, Izeradjene K, Szucs KS, Douglas L, Houghton JA. Rottlerin sensitizes colon carcinoma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis via uncoupling of the mitochondria independent of protein kinase C. Cancer Res. 2003;63:5118–5125. [PubMed] [Google Scholar]
- 41.Falschlehner C, Emmerich CH, Gerlach B, Walczak H. TRAIL signalling: decisions between life and death. Int J Biochem Cell Biol. 2007;39:1462–1475. doi: 10.1016/j.biocel.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 42.Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der Eb AJ, Vogelstein B. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327:293–297. doi: 10.1038/327293a0. [DOI] [PubMed] [Google Scholar]
- 43.Ricci MS, Kim SH, Ogi K, Plastaras JP, Ling J, Wang W, Jin Z, Liu YY, Dicker DT, Chiao PJ, et al. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer Cell. 2007;12:66–80. doi: 10.1016/j.ccr.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 44.Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol. 2006;33:369–385. doi: 10.1053/j.seminoncol.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 45.Hannon GJ. RNA interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 46.Wall NR, Shi Y. Small RNA: can RNA interference be exploited for therapy? Lancet. 2003;362:1401–1403. doi: 10.1016/S0140-6736(03)14637-5. [DOI] [PubMed] [Google Scholar]
- 47.Zangemeister-Wittke U, Leech SH, Olie RA, Simoes-Wust AP, Gautschi O, Luedke GH, Natt F, Haner R, Martin P, Hall J, et al. A novel bispecific antisense oligonucleotide inhibiting both bcl-2 and bcl-xL expression efficiently induces apoptosis in tumor cells. Clin Cancer Res. 2000;6:2547–2555. [PubMed] [Google Scholar]
- 48.Mohammad RM, Goustin AS, Aboukameel A, Chen B, Banerjee S, Wang G, Nikolovska-Coleska Z, Wang S, Al-Katib A. Preclinical studies of TW-37, a new nonpeptidic small-molecule inhibitor of Bcl-2, in diffuse large cell lymphoma xenograft model reveal drug action on both Bcl-2 and Mcl-1. Clin Cancer Res. 2007;13:2226–2235. doi: 10.1158/1078-0432.CCR-06-1574. [DOI] [PubMed] [Google Scholar]