

Abstract
Nickel-containing carbon monoxide dehydrogenases (CODHs) reversibly catalyze the oxidation of carbon monoxide to carbon dioxide and are of vital importance in the global carbon cycle. The unusual catalytic CODH C-cluster has been crystallographically characterized as either a NiFe4S4 or a NiFe4S5 metal center, the latter containing a fifth, additional sulfide that bridges Ni and a unique Fe site. To determine whether this bridging sulfide is catalytically relevant and to further explore the mechanism of the C-cluster, we obtained crystal structures of the 310 kDa bifunctional CODH/acetyl-CoA synthase complex from Moorella thermoacetica bound both with a substrate H2O/OH− molecule and with a cyanide inhibitor. X-ray diffraction data were collected from native crystals and from identical crystals soaked in a solution containing potassium cyanide. In both structures, the substrate H2O/OH− molecule exhibits binding to the unique Fe site of the C-cluster. We also observe cyanide binding in a bent conformation to Ni of the C-cluster, adjacent the substrate H2O/OH− molecule. Importantly, the bridging sulfide is not present in either structure. As these forms of the C-cluster represent the coordination environment immediately before the reaction takes place, our findings do not support a fifth, bridging sulfide playing a catalytic role in the enzyme mechanism. The crystal structures presented here, along with recent structures of CODHs from other organisms, have led us toward a unified mechanism for CO oxidation by the C-cluster, the catalytic center of an environmentally important enzyme.
Carbon monoxide dehydrogenases (CODHs) are key enzymes in the global carbon cycle and catalyze the reversible conversion of CO to CO2. In some anaerobic bacteria, including the phototroph Rhodospirillum rubrum and the thermophile Carboxydothermus hydrogenoformans, monofunctional Ni-containing CODHs allow these organisms to use CO as their sole carbon and energy source (1,2). CODH activity accounts for the removal of ∼108 tons of CO from the environment every year (3).
Acetogenic bacteria, including well-characterized Moorella thermoacetica, couple CODH-catalyzed CO2 reduction with acetyl-CoA synthesis in the bifunctional enzyme complex CODH/acetyl-CoA synthase (ACS) as part of the Wood−Ljungdahl carbon fixation pathway (4−7). Briefly, in the “eastern” branch of the pathway, one molecule of CO2 is reduced to a methyl group in a series of folate-dependent steps. The methyl group is then transferred from methyltetrahydrofolate to the corrinoid iron−sulfur protein (CFeSP) by methyl-H4folate:CFeSP methyltransferase (MeTr). In the “western” branch (Scheme 1), where Ni-containing CODH/ACS is the principal player, a second molecule of CO2 is reduced to a CO intermediate by the CODH active site C-cluster. CO then travels ∼70 Å through a remarkable tunnel within the enzyme to the ACS active site A-cluster (8−12), where it is combined with the CFeSP-derived methyl group and coenzyme A to form acetyl-CoA. Acetyl-CoA can encounter one of two cellular fates. It can be converted to biomass; alternatively, the high energy of the thioester bond can be harnessed first by phosphotransacetylase to produce acetyl phosphate and then by acetate kinase, which transfers the phosphate group to ADP, producing acetate and ATP. In this manner, it is estimated that globally anaerobic acetogens are responsible for roughly 10% of the total biological output of ∼1011 U.S. tons of acetate per year (13). Methanogenic archea also harbor a CODH/ACS variant, referred to as acetyl-CoA decarbonylase/synthase (ACDS), which operates in the reverse direction by degrading acetyl-CoA to the greenhouse gases methane and CO214,15.
Scheme 1. Reactions of CODH/ACS.

All CODHs characterized thus far are homodimeric, can catalyze both forward and reverse reactions, and contain five metalloclusters: two buried active site NiFeS C-clusters, two Fe4S4 B-clusters, and one Fe4S4 D-cluster located at the dimeric interface near the surface of the enzyme. The CODH component of methanogenic ACDS also harbors two E- and two F-clusters, which are both Fe4S4 centers. The B- and D-clusters are typical cubane Fe4S4 clusters and are involved in electron transfer. The unusual C-cluster is the site of CO2 reduction/CO oxidation, whose structure and catalytic mechanism have been the subject of considerable debate. Initial crystal structures of CODHs from R. rubrum (RrCODH) (16), C. hydrogenoformans (ChCODH) (17), and the bifunctional 310 kDa CODH/ACS from M. thermoacetica (MtCODH/ACS) (8,10) all show the same overall C-cluster geometry: Ni is part of a distorted cubane-like NiFe3S4 cluster, and an additional, unique Fe, often referred to as ferrous component II (FCII), is connected to a sulfide of the cubane (Figure 1). However, one major difference between these structures is apparent: the ChCODH structure contains a fifth sulfide that bridges the unique Fe and the Ni of the cubane (Figure 1a), while no such sulfide bridge is observed in the RrCODH and MtCODH/ACS structures (Figure 1b). As the bridging sulfide occupies the putative substrate binding sites, its presence or absence is of considerable catalytic significance.
Figure 1.

Representative structures of CODH C-clusters (a) with and (b) without the bridging sulfide. The C-cluster shown in (a) is from the structure of ChCODH (PDB ID 1SU8) and is shown in cyan cartoon with residue numbering following the ChCODH sequence. The bridging sulfide is circled in red. The C-cluster in (b) is from the structure of MtCODH/ACS solved previously (PDB ID 1MJG) and is shown in gray cartoon with residue numbering following the MtCODH/ACS sequence. C-clusters are depicted in ball-and-stick, while ligands are shown as sticks: Ni in green, Fe in orange, S in yellow, and N in blue. Ni and the unique Fe of the C-cluster are labeled Ni and Fe, respectively. All figures were made using PyMOL (60).
Since 2001, the catalytic relevance and mechanistic role of this bridging sulfide has been hotly contested. For ChCODH, the observation that the bridging sulfide is displaced and the enzyme becomes inactivated by long exposures to CO (>20 h) prompted speculation that MtCODH/ACS and RrCODH structures lack the bridging sulfide due to CO inhibition (18). However, the same CO treatment that led to inactivation of ChCODH did not affect the activity of MtCODH/ACS, even after >600 h of exposure (19). Furthermore, when sodium sulfide was added to active samples of RrCODH and MtCODH/ACS in an attempt to generate a sulfide bridge, these enzymes became inactivated (19). As the crystals of RrCODH and dissolved crystals of MtCODH/ACS are active (10,16), and their structures do not contain the sulfide bridge, we have suggested that sulfide does not play a catalytic role (reviewed in ref (20)). Instead, we have proposed that the sulfide-containing ChCODH structures may represent a precatalytic state and that the sulfide would be displaced upon substrate binding.
To investigate where substrates bind in the well-characterized MtCODH/ACS, we have solved two liganded structures. First, to identify the binding site for a substrate water molecule on the C-cluster of MtCODH/ACS, we grew crystals in the complete absence of CO to prevent turnover. This lack of CO has also allowed us to explore the relationship between the bridging sulfide and CO exposure. The resulting native structure shows H2O/OH− bound to the unique iron of the C-cluster and no bridging sulfide, despite the absence of CO. Second, we wished to identify the binding site for substrate CO on the C-cluster of MtCODH/ACS; however, infrared spectroscopy has indicated that there does not exist a single, stable CO binding location that would be amenable to crystallographic observation (21). CO was also recently observed to exchange rapidly to form CO2 in ChCODH even in the absence of external electron acceptors (22). Therefore, to identify the CO binding site, we used cyanide, a known competitive inhibitor of monofunctional CODHs (23). This cyanide-liganded structure, also obtained in the absence of CO, shows cyanide bound to the Ni of the C-cluster. Despite the fact that MtCODH/ACS is the most thoroughly characterized CODH regarding substrate binding, no corroborating crystal structures with substrates or analogues bound have been captured until now.
As we were carrying out these crystallographic studies, a third set of ChCODH structures were reported which no longer contained the bridging sulfide and instead depicted CO2 bound to the C-cluster in its place (24). Until this point, no CODH structures had substrates bound to the C-cluster, and thus the mechanism of the C-cluster and the role of sulfide in catalysis remained elusive. Even more recently, a structure of the CODH component of methanogenic ACDS from Methanosarcina barkeri (MbCODH) with a putative CO molecule and H2O/OH− bound also did not contain the bridging sulfide (25). With these CODH structures and our new MtCODH/ACS structures presented here, we are now in a unique position to compare the C-clusters bound with different ligands, across the three types of CODHs: monofunctional CODH, bifunctional CODH/ACS, and CODH-containing methanogenic ACDS. This new crop of structures has led us toward a unified mechanistic view of the CODH C-cluster.
Methods
Protein Purification and Crystallization
M. thermoacetica (formerly Clostridium thermoaceticum strain ATCC 39073) was cultured on glucose in the absence of CO as described (26). Therefore, any CO present would only be due to CO produced as an intermediate during the growth of the organism. CO production in growth under glucose has been found to be small but not insignificant, with a maximum of 53 ppm of CO in the gas phase (27). CODH/ACS was purified as previously described (28,29) under strictly anaerobic conditions, with the final sample exhibiting specific activity for CO oxidation to indicate that no CO inactivation had occurred (366 units/mg). Crystals were grown anaerobically as described (10) in an anaerobic chamber (Coy Laboratories) in the absence of CO with 0.25 μL of a microcrystal solution to aid in crystal growth (9). Native crystals were flash-cooled in liquid nitrogen using 20% glycerol as a cryoprotectant. Cyanide-bound crystals were obtained by soaking native crystals in the precipitant solution supplemented with 100 μM KCN for 1 h at room temperature under anaerobic conditions before flash-cooling.
Data Collection and Refinement
X-ray diffraction data of native and cyanide crystals were collected at Stanford Synchrotron Radiation Laboratory (SSRL) beamline 11-1 and at Advanced Light Source (ALS) beamline 5.0.2, respectively. The asymmetric unit was composed of two 310 kDa α2β2 tetramers. Native and cyanide data were processed using MOSFLM/SCALA (30,31) and DENZO/SCALEPACK (32), respectively, with good quality diffraction data to 2.15 Å for both structures. The first published MtCODH/ACS structure (PDB ID 1MJG) was used for initial rigid body refinement, which resulted in R factors below 25%. To obtain test set reflections that were of higher resolution than the published MtCODH/ACS structure, 5% of the reflections from this region of the data were randomly generated. Subsequent rounds of refinement were carried out in REFMAC (33) from the CCP4 Program Suite (31) with model building in COOT (34), using noncrystallographic symmetry (NCS) restraints. Occupancy refinements of water and cyanide ligands to the C-cluster were carried out in PHENIX (35). The final model contains residues 2−674 (of 674) for CODH chains A−D and residues 2−729 (of 729) for ACS chains M−P. Ramachandran analysis of the final model was calculated in PROCHECK (36), and final data collection and refinement statistics are shown in Table 1.
Table 1. Data Collection and Refinement Statistics.
| native | cyanide bound | |
|---|---|---|
| Data Collection | ||
| space group | P1 | P1 |
| cell dimensions | ||
| a, b, c (Å) | 99.65, 136.87, 140.86 | 99.83, 136.77, 141.61 |
| α, β, γ (deg) | 101.26, 109.11, 104.08 | 101.23, 109.18, 103.87 |
| wavelength (Å) | 0.95369 | 1.0000 |
| resolution (Å) | 36.96−2.15 (2.21−2.15) | 50.0−2.15 (2.23−2.15) |
| Rsym (%) | 6.7 (40.6) | 5.1 (16.7) |
| mean I/σI | 12.2 (2.3) | 25.9 (6.8) |
| completeness (%) | 92.3 (90.4) | 96.3 (86.6) |
| redundancy | 2.1 | 3.5 |
| Refinement | ||
| resolution (Å) | 36.96−2.15 | 48.39−2.15 |
| Rwork/Rfree (%) | 18.6/24.2 | 17.2/22.1 |
| no. of atoms | ||
| protein | 43732 | 43619 |
| heteroatoms (Fe, S, Ni, Cu) | 124 | 124 |
| ligands | 16 | 24 |
| waters | 1413 | 2153 |
| other (ions and glycerol) | 28 | 28 |
| average B-factor, all atoms (Å2) | 36.9 | 35.6 |
| protein | 37.1 | 35.7 |
| C-clusters | 28.2 | 25.2 |
| C-cluster ligands | 31.7 | 32.1 |
| heteroatoms (Fe, S, Ni, Cu) | 31.1 | 28.4 |
| ligands | 56.5 | 55.1 |
| waters | 31.2 | 33.8 |
| rms deviations | ||
| bond lengths (Å) | 0.021 | 0.019 |
| bond angles (deg) | 1.871 | 1.650 |
| Ramachandran plot (%) | ||
| most favored | 88.1 | 89.8 |
| additionally allowed | 10.5 | 8.9 |
| generously allowed | 0.9 | 0.7 |
| disallowed | 0.6 | 0.5 |
Highest resolution shell is shown in parentheses.
Results
The Native C-Cluster
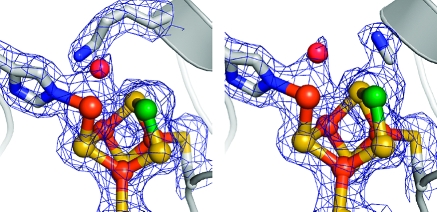
Following the initial CODH structure determinations, a primary point of contention regarding the geometry and mechanism of the C-cluster involves the presence or absence of a fifth sulfide bridging Ni and the unique Fe. The structures of RrCODH (16), MtCODH/ACS (8−10), and MbCODH (25) lack this bridging sulfide (Figure 1b); however, it is present in some structures of the ChCODH C-cluster (17,18) (Figure 1a). When explaining the absence of the bridging sulfide in the RrCODH and MtCODH/ACS structures, it was argued that these proteins could have been inhibited by the CO to which they were exposed, leading to an inactivated C-cluster (18). Here we present two structures of MtCODH/ACS obtained from proteins purified and crystallized in the absence of CO, one containing a native C-cluster (Figure 2a,b) and one containing a C-cluster bound by cyanide (Figure 2c,d). Both C-clusters have atomic positions consistent with those seen in all prior CODH structures, and neither contain density for the bridging sulfide (Figure 2e).
Figure 2.

Native C-cluster of MtCODH/ACS (a) before and (b) after H2O/OH− is modeled. H2O/OH− is bound 2.03 Å to the unique Fe and 2.60 Å to Lys587. Cyanide-bound C-cluster of MtCODH/ACS (c) before and (d) after H2O/OH− and cyanide are modeled. H2O/OH− is bound 2.05 Å to the unique Fe, and cyanide is bound 1.99 Å to Ni with an average Ni−C−N angle of 114°. For (a)−(d), 2Fo − Fc (1.5σ) and Fo − Fc (4.0σ) electron density are shown in blue and green mesh, respectively. Ni and the unique Fe of the C-cluster are labeled in (a) as Ni and Fe, respectively, with residues following MtCODH/ACS numbering. (e) C-cluster model of ChCODH with the bridging sulfide (PDB ID 1SU8), shown in cyan cartoon following ChCODH numbering, superimposed on the 2Fo − Fc density (blue mesh, 2.0σ) of the cyanide-bound C-cluster of MtCODH/ACS. (f) Residues surrounding the cyanide binding site, including Ile591 (MtCODH/ACS numbering) which hinders linear Ni−cyanide binding. Cyanide and Ile591 are shown in spheres with van der Waals radii and carbons in violet. All MtCODH/ACS chains are depicted in gray cartoon, C-clusters are shown in ball-and-stick, and ligands are shown as sticks: Ni in green, Fe in orange, S in yellow, and N in blue. For clarity, not all protein ligands to the cluster are shown.
In our native structure, a positive Fo − Fc electron density peak greater than 4σ is present, indicating a ligand bound to the unique Fe of the C-cluster (Figure 2a). As a monatomic ligand best fit the density, we placed a H2O/OH− molecule in this location. After refinement, no Fo − Fc difference density was present in this region, with H2O/OH− positioned 2.03 Å from the unique Fe and 2.60 Å to Lys587 (Figure 2b). All reported distances are derived from chain B of the final model, as this chain has the lowest B-factors of the four CODH chains present in the asymmetric unit. To confirm that this density does not represent a sulfide, we also modeled a sulfur atom into this site; after refinement, negative Fo − Fc difference density appeared near the sulfur, indicating that sulfur is an incorrect assignment (Supporting Information Figure S1).
The occupancies of the bound H2O/OH− ligands in all chains were then refined. Occupancies and B-factors of atoms are interrelated and thus cannot be refined simultaneously; when the occupancies for the H2O/OH− ligands were set to 1.0, as in the final model, their B-factors were generally somewhat higher than the average B-factor of the C-cluster atoms. Correspondingly, when B-factors of the H2O/OH− ligands were set to the average B-factor of their respective C-cluster atoms, occupancies generally fell to an average value of 0.9, denoting 90% occupancy of the oxo ligand. The results of these occupancy and B-factor refinements are shown in Supporting Information Table S1.
Previous structures of the C-cluster without substrates or the bridging sulfide indicate that an open coordination site on the unique Fe is present before substrate binding (8−10,16). When the C-cluster binds the H2O/OH− substrate as seen in our native structure, the coordination environment of the unique Fe converts from trigonal pyramidal to a distorted tetrahedral geometry. The positions of the cluster sulfide, Cys317, and His283 ligands to the unique Fe remain largely unchanged, as do the positions of the other C-cluster atoms.
The Cyanide-Bound C-Cluster
Our structure of cyanide-treated MtCODH/ACS crystals also shows density consistent with a H2O/OH− molecule (Figure 2c and Supporting Information Figure S2) bound in an analogous position as in the native structure, with a 2.05 Å Fe−oxo bond. However, we saw additional electron density for a larger ligand bound to Ni of the C-cluster, represented in both the 2Fo − Fc and the Fo − Fc electron density maps (Figure 2c). Because the only difference between the native and cyanide data sets was that, with the latter, crystals were soaked in a solution containing cyanide before data collection, we considered if the electron density represents a cyanide ligand by refining molecules of different sizes into this density. Assignment of this new site as a H2O/OH− molecule proved to be a poor fit to the density, as refinement moved the molecule to a distance of 2.47 Å from Ni, with some positive Fo − Fc density still remaining (Supporting Information Figure S2). Refinement was also carried out with a sulfur atom in this location; however, strong negative Fo − Fc density peaks were observed, signifying that sulfur was also an incorrect assignment (Supporting Information Figure S2). Fo − Fc density was eliminated only when a cyanide ligand was modeled into this region (Figure 2d).
Occupancies of the cyanide and H2O/OH− ligands were also refined. As with the native structure, when occupancies of the cyanide and H2O/OH− atoms were set to 1.0, their B-factors were somewhat higher than the average B-factor of the C-cluster. When B-factors of the cyanide and H2O/OH− atoms were set to the average B-factor of their respective C-cluster atoms, occupancies generally fell to an average value of 0.7, denoting 70% occupancy. The results of these occupancy and B-factor refinements are shown in Supporting Information Table S2.
Cyanide binds to Ni of the C-cluster in a bent conformation (Figure 2d), with a C−Ni distance of 1.99 Å, a C−H2O/OH− distance of 3.54 Å, and an N−H2O/OH− distance of 2.94 Å. The Ni−C−N bond angle is nonlinear and is not uniform across the four CODH subunits: chain A, 115°; chain B, 111°; chain C, 101°; and chain D, 130°; for an average of 114°. These discrepancies may be due to differences in the relative order and disorder of the four chains in the asymmetric unit, as chains C and D have higher B-factors, indicating more disorder. Three refinement programs, REFMAC (33), CNS (37), and PHENIX (35), consistently produced a bent Ni−CN coordination; when cyanide was fixed linearly to Ni and the cyanide carbon assigned as sp hybridized, refinement nonetheless routinely moved the cyanide ligand back into its favored bent conformation. Ile591 is found in close proximity to the cyanide nitrogen, presenting a potential steric clash that prevents linear binding of cyanide (Figure 2f).
Previous structures of the C-cluster without substrate ligands or the bridging sulfide indicated possible empty coordination sites on Ni of the C-cluster before substrate binding. In our cyanide-bound structure presented here, binding of the substrate mimic results in a distorted tetrahedral geometry of Ni. This geometry is in contrast to the structures of ChCODH that contain the bridging sulfide, which depicts square planar-like geometry. The positions of the other atoms and ligands to the C-cluster do not change significantly as a result of cyanide binding.
Discussion
The CODH C-cluster is responsible for the catalysis of one of the most fundamental chemical reactions in biology: the reversible interconversion of CO and CO2. Despite the importance of this metallocluster, there has been significant disagreement regarding the structural elements required for activity. One point of contention has been whether the fifth (bridging) sulfide observed in structures of ChCODH (17,18), but not in other CODHs (8−10,16,25), is catalytically relevant in all CODH enzymes, represents a precatalytic state in all CODH enzymes, or has different roles in different CODH variants. The possibility that interspecies variations could explain the presence of the bridging sulfide in ChCODH and the lack of the sulfide in CODHs from other organisms is intriguing. However, given the CODH sequence homology, it is unlikely that C-clusters performing the same chemistry would employ different mechanisms with different substrate binding features, and there is a lack of biochemical or biophysical evidence supporting this explanation. The suggestion that a sulfide bridge could be important for maintaining the structural integrity of the C-cluster in a precatalytic state, while not being directly involved in catalysis, has precedence in the proposed mechanism of some [NiFe] hydrogenases. In this case, a sulfide ligand bridging Ni and Fe has been suggested to exist in a precatalytic, inactive state to protect metals in the active site prior to catalysis (38,39). In any case, the purpose of the fifth sulfide in the catalytic cycle of the C-cluster had been difficult to evaluate before structural data on substrate and analogue binding was acquired.
In our native structure of MtCODH/ACS, crystallized in the absence of CO, no bridging sulfide is observed. The C-cluster instead depicts a H2O/OH− molecule bound to the unique Fe. Electron density maps following refinement of this ligand as sulfur and not H2O/OH− indicated that assignment as sulfur does not fit the density (Supporting Information Figure S1). Our native structure thus represents the first time that the substrate H2O/OH− molecule has been observed in MtCODH/ACS, the best characterized CODH. Although it is difficult to unambiguously prove that a ligand is a H2O/OH− molecule using crystallography, our assignment is consistent with electron−nuclear double resonance (ENDOR) studies of MtCODH, which indicated that H2O/OH− binds to the unique Fe (40). Structural comparisons are also valuable in assigning H2O/OH− as the ligand; recent ChCODH and MbCODH structures also contain H2O/OH− bound in the same position on Fe (Figure 3), a site that is unavailable in structures that contain the bridging sulfide. H2O/OH− in ChCODH was modeled as the ligand partly based on spectroscopic work performed on MtCODH/ACS. Previous MtCODH/ACS structures (8−10), however, did not contain density for H2O/OH−. Of the three stable oxidation states of the C-cluster (Cox, Cred1, Cred2), it has been shown that both CO and H2O/OH− substrates bind only the Cred1 state, with oxidation of CO to CO2 further reducing the cluster to the Cred2 state, the only state that binds CO240−43. It is possible that in the previous MtCODH/ACS structures heterogeneity in the electronic states of the clusters prevented full occupancy of bound H2O/OH−, where some oxidation to the Cox state and some turnover to the Cred2 state due to CO exposure may have occurred. In the structures presented here, the purification and crystallization protocol appears to have maintained the Cred1 state for H2O/OH− binding at ∼90% or greater occupancy. The H2O/OH− substrate molecule is bound 2.03 Å from the unique Fe, compared to 1.94 and 2.14 Å in the ChCODH and MbCODH structures, respectively.
Figure 3.

(a) Superposition of C-clusters from cyanide-bound MtCODH/ACS (gray) and putative CO-bound MbCODH (teal), with MtCODH/ACS numbering. (b) Superposition of the same C-clusters as (a) and that of CO2-bound ChCODH (pink). C-clusters are depicted in ball-and-stick, and ligands are shown as sticks: Ni in green, Fe in orange, S in yellow, and N in blue. Ni and the unique Fe of the C-cluster are labeled as Ni and Fe, respectively. For clarity, not all protein ligands to the cluster are shown.
H2O/OH− is also 2.60 Å from a conserved Lys587, whose role in the reaction mechanism of the C-cluster is intriguing. In general, hydrogen atoms cannot be observed crystallographically due to the limits in resolution; therefore, we cannot assign with confidence the protonation state of H2O/OH− or Lys587. As Fe-bound H2O must be deprotonated before nucleophilic attack on CO, the proximity of Lys587 to H2O/OH− in our structure may lead one to propose that Lys587 acts as the general base. Due to pKa considerations, it would be atypical for lysine to accept protons from water; however, the proximity of both Lys587 and the unique Fe of the C-cluster could lower the pKa of H2O. It is also possible that Lys587 stabilizes the OH− form, serving to activate hydroxide attack on Ni-bound CO. Indeed, mutation of Lys587 has been shown to reduce the activity of MtCODH (44).
Because infrared spectroscopy has shown that MtCODH/ACS does not bind CO in a single, stable location (21), we have used cyanide, which is isosteric and isoelectronic to CO, to probe the CO substrate binding site. The literature is inconsistent regarding the location of the cyanide binding site on the C-cluster of CODH, with some spectroscopic studies suggesting cyanide binds to iron and others suggesting multiple, perhaps bridging, binding sites (22,23,40,41,45−48). However, recent extended X-ray absorption fine structure (EXAFS) data on ChCODH show cyanide as a competitive inhibitor of C-cluster activity, binding to the same location as CO (23). In our cyanide-bound MtCODH/ACS structure presented here, cyanide is bound to Ni at a distance of 1.99 Å. For ChCODH, EXAFS data on cyanide binding are also consistent with cyanide binding to Ni, with a Ni to C distance in the range of 1.81−1.84 Å (23). Therefore, while some of the spectroscopy on the cyanide-bound CODH is inconsistent with our structure, the recent EXAFS results on ChCODH are consistent.
In our structure, cyanide is bound to Ni of the C-cluster in an analogous conformation as the proposed CO ligand modeled in the MbCODH structure (25). That ligand’s identification as CO was only inferred from the electron density, and the authors indicated that the ligand could also be a formyl group. If assignment as CO was correct, it is puzzling that turnover did not occur; the authors attribute this to the low pH of the crystallization conditions which may have disfavored the deprotonation necessary for activity. In contrast, we can more confidently report our ligand’s identity as cyanide, as the sole difference between our native and cyanide data was that, with the latter, crystals were soaked in a solution containing cyanide before data collection. As mentioned, no bridging sulfide is seen in this structure, and such a bridge would not be possible with CN/CO bound to Ni in this position. Interestingly, we observe cyanide bound to Ni in a bent conformation, with an average angle of 114°. A diatomic ligand seen in the MbCODH structure, modeled as CO, also exhibits a similar bent conformation (Figure 3a), with a Ni−C−O bond angle of 103°. In the MbCODH structure, when modeled CO was fixed in a linear fashion with respect to Ni, a steric clash was observed between CO and an isoleucine residue near the C-cluster. This isoleucine was seen in close proximity to the oxygen of CO, preventing linear binding of a diatomic molecule. Here, we similarly observe Ile591 impart a potential steric clash that cannot be overcome during refinement (Figure 2f). There is precedence for a bent cyanide metal ligand in several heme-containing metalloproteins, including catalases (49), peroxidases (50,51), sulfite reductase hemoprotein (52), hemoglobin and myoglobin (53), cytochrome cd1 nitrite reductase (54), and cytochrome P450 enzymes (55,56). A study of the non-heme mononuclear iron enzyme superoxide reductase also identifies bent CN geometry, where the Fe−C−N bond angle was observed as 123° and 133° for two different substrates (57). In all cases, bent geometry was attributed to steric constraints and hydrogen bonding upon adopting a bent conformation. In our structure, not only does Ile591 similarly impart a steric hindrance to linear binding, the bent conformation could also allow the cyanide ligand to form a hydrogen bond with the Fe-bound water molecule at 2.94 Å away or perhaps with His113, which is at a farther distance of 3.28 Å (Figure 2f).
Although cyanide binds Ni in a bent fashion in our structure, this does not require that CO in this bent arrangement represents the conformation needed for hydroxide attack on the CO carbon. Indeed, Gong et al. propose that before nucleophilic attack a change in CO binding occurs (25). Starting from the bent arrangement seen in their MbCODH structure, the authors propose that the CO carbon moves toward the H2O/OH− ligand, causing Ni to switch from the tetrahedral-like geometry observed in our structure to a more square planar geometry, while the CO oxygen does not move. Such a “carbon shift” causes CO to be bent in the opposite direction, allowing the carbon to be closer to the attacking hydroxide, without movement of the CO oxygen. This proposal stemmed from the authors’ observation that when compared with the ChCODH CO2-bound structure, discussed below, the position of the CO oxygen remains essentially constant after conversion to CO2, while the CO2 carbon was shifted, altering the Ni coordination geometry.
Further, Gong et al. propose that deprotonation of water to hydroxide may control the suggested “carbon shift” and thus the direction of CO bending. Here, CO is initially bent toward H2O, allowing for hydrogen bonding. Presumably, as H2O is not the catalytic substrate, CO cannot undergo the proposed “carbon shift”, as this would place CO and water, two nonreacting species, too close in proximity. Only after water is deprotonated to the active hydroxide species may the “carbon shift” take place, positioning the carbon closer to the hydroxide for chemistry to occur. Pointing to the fact that their CO-bound structure was obtained at pH 4.6, the authors suggest that their observed bent conformation may be attributed to the acidic conditions making deprotonation of water, and catalysis, more unfavorable and that bending in the opposite direction may be observed at neutral pH. Gong et al. thus suspect that their protein was inactive and did not turn over to form CO2 because of these acidic conditions.
Although our cyanide-bound structure presented was obtained from samples crystallized at pH 7.6, we nonetheless clearly observe the same bent conformation as was seen with the MbCODH structure in the presence of CO. Unlike CO, where a partial positive charge exists on carbon when bound to a metal ion, cyanide is not subject to attack by hydroxide and functions as an inhibitor, binding to the same site as CO without initiating catalysis. As such, had cyanide undergone the “carbon shift” to be bent in the opposite direction, the distance between the cyanide carbon and H2O/OH− would again be too close, approximately the length of a covalent bond, which is sterically unfavorable between two nonreacting species. Perhaps, because cyanide does not react with H2O/OH−, it cannot undergo the “carbon shift” due to either steric or electronic constraints and thus does not exhibit bending in the opposite direction.
A recent ChCODH crystal structure illustrates a CO2-bound C-cluster (24). Here, ChCODH crystals soaked in a solution containing sodium bicarbonate, buffered at pH 8.0, and with redox potential of −600 mV vs standard hydrogen electrode (SHE) yielded a C-cluster with electron density modeled as CO2 between Ni and the unique Fe. The bridging sulfide is not observed in this structure, as the modeled CO2 molecule now occupies the region where the ChCODH bridging sulfide was previously seen (17,18). The redox potential of −600 mV was attained using titanium(III) citrate with the intention of obtaining the Cred2 state for CO2 binding; however, the authors did not indicate why CO2 bound in the Cred2 state did not induce turnover to CO and water. In this structure, CO2 is modeled with the carbon bound 1.96 Å to Ni, one oxygen is bound 2.05 Å to the unique Fe, and the second oxygen is located 2.89 Å from His93 (His113 in MtCODH/ACS). In comparing our cyanide structure with the CO2-bound C-cluster, the locations of both Fe-bound oxygens are essentially the same, and the cyanide nitrogen is in the same position as the second CO2 oxygen. Similar to the MbCODH structure, the only significant difference is once again the location of the carbon, suggestive of the proposed “carbon shift”. To illustrate, a superposition of all three structures is shown in Figure 3b.
As the cyanide inhibitor is not subject to nucleophilic attack by hydroxide, we have captured a state of the enzyme immediately before the reaction or any “carbon shift” takes place, with both substrates lined up for catalysis. In this crucial stage, we observe no evidence of a bridging sulfide. The same scenario was observed with the putative CO-bound MbCODH. As the recent CO2-bound ChCODH structure also does not contain the bridging sulfide, and as it represents the cluster environment immediately after hydroxide attack on CO, this structure further provides strong support of our original position that a bridging sulfide must not be catalytically relevant. Our structures of MtCODH/ACS together with other recent CODH structures (24,25) and biochemical data (19) should put to rest the proposition that a bridging sulfide is catalytically relevant and make possible a unified mechanistic view of C-cluster chemistry.
In our proposed reaction mechanism for CO oxidation, shown in Figure 4, a water molecule is bound to the unique Fe of the C-cluster in the Cred1 redox state, as observed in the native structure of MtCODH/ACS shown here. This state is referred to in Figure 4 as state I. Then, CO is proposed to bind Ni in a bent conformation (state II), as seen in the cyanide-bound crystal structure described here, with the carbon too distant from the Fe-bound water to undergo reaction. In the next step, deprotonation of water to the reactive hydroxide species may promote a “carbon shift” that would place the carbon closer to the Fe-bound hydroxide, in a position that would promote the reaction between CO and OH− (state III). Following nucleophilic attack of hydroxide onto Ni-bound CO, the Ni−COOH intermediate (state IV) must also be deprotonated to give a Ni−COO− species (state V). CO2 is then released and the C-cluster becomes reduced to the Cred2 state (state VI). Finally, two electrons are transferred to the B-cluster, the D-cluster, and ultimately an electron acceptor protein, ferredoxin, hydrogenase, or pyruvate:ferredoxin oxidoreductase (PFOR) (58,59). This electron transfer reaction reoxidizes the C-cluster to the Cred1 state (state I), and the catalytic cycle continues.
Figure 4.

Proposed mechanism of the C-cluster, consistent with current structural and biochemical knowledge that supports the lack of a bridging sulfide. Our structures presented here represent states I and II of this mechanism; the putative CO-bound MbCODH structure represents state II; and the CO2-bound ChCODH structure represents state IV or V, which differ only in the protonation state of the intermediate and would thus be difficult to distinguish using X-ray crystallography. States I and VI are structurally identical and differ only in the redox state of the cluster. “B” refers to a basic residue involved in deprotonation. The identity of the base has not been resolved, and candidates include His113 and Lys587.
Two deprotonations are carried out during the course of the reaction, but identifying the catalytic base(s) responsible is difficult. For both deprotonations, Lys587 is most optimally positioned, but as discussed, pKa considerations may limit the lysine to playing a role in stabilizing the negatively charged hydroxide and the Ni-bound carboxylate intermediate. If this is the case, His113 could instead be the basic residue (Figure 2f), as we have previously suggested (16), and the recent CO2-bound ChCODH shows the water-derived oxygen located 3.93 Å away from the corresponding histidine, His93. Further, a base may not be necessary for the first deprotonation, as the pKa of the water molecule may be dramatically lowered in its distinctive environment, bound to the unique Fe of the C-cluster.
Protons generated by the reaction must be delivered from the C-cluster to the exterior of the protein to access the bulk solvent, and the first CODH structures revealed a channel that emanates from Lys587, contains water molecules, and is lined with several conserved histidine residues. At that time, we had suggested that this channel could serve as a proton transfer network (16). Mutational studies have since shown that several of the residues that make up the channel are important for activity (44). The structures presented here contain substrates that now mark the site of catalysis and support the involvement of these residues in proton transfer. Figure 5 shows the positions of cyanide and H2O/OH− relative to the conserved residues that make up the proton transfer network, Lys587, His113, His116, His119, His122, and Asn284, which is probably not directly involved in proton transfer but contributes hydrogen bonds that may be structurally important.
Figure 5.

Proposed proton transfer network. MtCODH/ACS is depicted in gray cartoon, residues involved in the proton transfer network are shown as slate sticks, and the C-cluster atoms with cyanide and H2O/OH− ligands are shown in ball-and-stick: Ni in green, Fe in orange, S in yellow, O in red, and N in blue.
Conclusion
The structures presented here of native and cyanide-bound C-clusters of the well-characterized MtCODH/ACS have confirmed our initial proposal that a sulfide bridge does not play a catalytic role in CO oxidation and CO2 reduction in M. thermoacetica. Recent crystal structures of ChCODH have further indicated that a bridging sulfide is also not catalytic in C. hydrogenoformans. Whether the bridging sulfide plays a physiological role in a precatalytic state remains to be determined, but the catalytic C-cluster should be described as a NiFe4S4 and not a NiFe4S5 cluster. In this way, our structures along with other recent CODH structures have dispelled previous claims that the sulfide bridge participates in catalysis.
With the first structure of the H2O/OH−-bound C-cluster of MtCODH/ACS, we have also finally linked spectroscopic work that was carried out using MtCODH/ACS that pointed to Fe-bound H2O/OH− with crystallographic evidence. Our data also provide the first three-dimensional structural view of cyanide bound to Ni of the C-cluster. After years of considerable debate, these structures contribute toward a unified view of the chemistry performed by the CODH C-cluster, as for the first time there is consistency between structures of ligand-bound CODHs, with crystallographic snapshots of H2O/OH−-, CO-, CN-, and CO2-bound C-clusters providing a portrait of CODH reactivity.
Due to the clear importance of CODHs in the global carbon cycle and in light of the current crisis of global climate change, the details surrounding CO2/CO interconversion in nature have been eagerly anticipated. Our structures of the CODH C-cluster and the mechanistic insights they offer may have broader applications in the conception and development of model systems that can be used for the removal of environmental CO2 and CO.
Acknowledgments
Portions of this research were carried out at the Stanford Synchrotron Radiation Laboratory (SSRL), a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The SSRL Structural Molecular Biology Program is supported by the Department of Energy, Office of Biological and Environmental Research, and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract DE-AC02-05CH11231.
Supporting Information Available
Refinement trials after modeling atoms of varying sizes as ligands to the native (Figure S1) and cyanide (Figure S2) C-clusters and occupancy refinements for native (Table S1) and cyanide (Table S2) C-cluster ligands. This material is available free of charge via the Internet at http://pubs.acs.org.
This work is supported by NIH Grants GM69857 (C.L.D.) and GM39451 (S.W.R.) and the MIT Energy Initiative.
The atomic coordinates and structure factors (PDB accession numbers 3I01 and 3I04) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics (http://www.pdb.org).
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Svetlichny V. A.; Sokolova T. G.; Gerhardt M.; Ringpfeil M.; Kostrikina N. A.; Zavarzin G. A. (1991) Carboxydothermus hydrogenoformans gen. nov., sp. nov., a CO-utilizing thermophilic anaerobic bacterium from hydrothermal environments of Kunashir Island. Syst. Appl. Microbiol. 14, 254–260. [Google Scholar]
- Uffen R. L. (1976) Anaerobic growth of a Rhodopseudomonas species in the dark with carbon monoxide as sole carbon and energy substrate. Proc. Natl. Acad. Sci. U.S.A. 73, 3298–3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomew G. W.; Alexander M. (1979) Microbial metabolism of carbon monoxide in culture and in soil. Appl. Environ. Microbiol. 37, 932–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl P. A. (2002) The Ni-containing carbon monoxide dehydrogenase family: Light at the end of the tunnel?. Biochemistry 41, 2097–2105. [DOI] [PubMed] [Google Scholar]
- Müller V., Imkamp F., Rauwolf A., Küsel K., and Drake H. L. (2004) Molecular and Cellular Biology of Acetogenic Bacteria, in Strict and Facultative Anaerobes: Medical and Environmental Aspects (Nakano M. M., and Zuber P. A., Eds.) Horizon Bioscience, Wymondham, U.K., pp 251−281. [Google Scholar]
- Ragsdale S. W. (1997) The eastern and western branches of the Wood/Ljungdahl pathway: How the east and west were won. Biofactors 6, 3–11. [DOI] [PubMed] [Google Scholar]
- Ragsdale S. W.; Pierce E. (2008) Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim. Biophys. Acta 1784, 1873–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnault C.; Volbeda A.; Kim E. J.; Legrand P.; Vernede X.; Lindahl P. A.; Fontecilla-Camps J. C. (2003) Ni-Zn-[Fe4-S4] and Ni-Ni-[Fe4-S4] clusters in closed and open subunits of acetyl-CoA synthase/carbon monoxide dehydrogenase. Nat. Struct. Biol. 10, 271–279. [DOI] [PubMed] [Google Scholar]
- Doukov T. I.; Blasiak L. C.; Seravalli J.; Ragsdale S. W.; Drennan C. L. (2008) Xenon in and at the end of the tunnel of bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase. Biochemistry 47, 3474–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doukov T. I.; Iverson T. M.; Seravalli J.; Ragsdale S. W.; Drennan C. L. (2002) A Ni-Fe-Cu center in a bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase. Science 298, 567–572. [DOI] [PubMed] [Google Scholar]
- Maynard E. L.; Lindahl P. A. (1999) Evidence of a molecular tunnel connecting the active sites for CO2 reduction and acetyl-CoA synthesis in acetyl-CoA synthase from Clostridium thermoaceticum. J. Am. Chem. Soc. 121, 9221–9222. [Google Scholar]
- Seravalli J.; Ragsdale S. W. (2000) Channeling of carbon monoxide during anaerobic carbon dioxide formation. Biochemistry 39, 1274–1277. [DOI] [PubMed] [Google Scholar]
- Drake H. L., Daniel S. L., Matthies C., and Küsel K. (1994) Acetogenesis, acetogenic bacteria, and the acetyl-CoA pathway: past and current perspectives, in Acetogenesis (Drake H. L., Ed.) pp 3−60, Chapman and Hall, New York. [Google Scholar]
- Grahame D. A. (1991) Catalysis of acetyl-CoA cleavage and tetrahydrosarcinapterin methylation by a carbon monoxide dehydrogenase-corrinoid enzyme complex. J. Biol. Chem. 266, 22227–22233. [PubMed] [Google Scholar]
- Terlesky T. C.; Nelson M. J. K.; Ferry J. G. (1986) Isolation of an enzyme complex with carbon monoxide dehydrogenase activity containing corrinoid and nickel from acetate-grown Methanosarcina thermophila. J. Bacteriol. 168, 1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drennan C. L.; Heo J.; Sintchak M. D.; Schreiter E.; Ludden P. W. (2001) Life on carbon monoxide: X-ray structure of Rhodospirillum rubrum Ni-Fe-S carbon monoxide dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 98, 11973–11978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbek H.; Svetlitchnyi V.; Gremer L.; Huber R.; Meyer O. (2001) Crystal structure of a carbon monoxide dehydrogenase reveals a [Ni-4Fe-5S] cluster. Science 293, 1281–1285. [DOI] [PubMed] [Google Scholar]
- Dobbek H.; Svetlitchnyi V.; Liss J.; Meyer O. (2004) Carbon monoxide induced decomposition of the active site [Ni-4Fe-5S] cluster of CO dehydrogenase. J. Am. Chem. Soc. 126, 5382–5387. [DOI] [PubMed] [Google Scholar]
- Feng J.; Lindahl P. A. (2004) Effect of sodium sulfide on Ni-containing carbon monoxide dehydrogenases. J. Am. Chem. Soc. 126, 9094–9100. [DOI] [PubMed] [Google Scholar]
- Drennan C. L.; Peters J. W. (2003) Surprising cofactors in metalloenzymes. Curr. Opin. Struct. Biol. 13, 220–226. [DOI] [PubMed] [Google Scholar]
- Chen J.; Huang S.; Seravalli J.; Gutzma H. J.; Swartz D. J.; Ragsdale S. W.; Bagley K. A. (2003) Infrared studies of carbon monoxide binding to carbon monoxide dehydrogenase/acetyl-CoA synthase from Moorella thermoacetica. Biochemistry 42, 14822–14830. [DOI] [PubMed] [Google Scholar]
- Seravalli J.; Ragsdale S. W. (2008) 13C NMR characterization of an exchange reaction between CO and CO2 catalyzed by carbon monoxide dehydrogenase. Biochemistry 47, 6770–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S.-W.; Korbas K.; Klepsch M.; Meyer-Klaucke W.; Meyer O.; Svetlitchnyi V. (2007) Interaction of potassium cyanide with the [Ni-4Fe-5S] active site cluster of CO dehydrogenase from Carboxydothermus hydrogenoformans. J. Biol. Chem. 282, 10639–10646. [DOI] [PubMed] [Google Scholar]
- Jeoung J.-H.; Dobbek H. (2007) Carbon dioxide activation at the Ni,Fe-cluster of anaerobic carbon monoxide dehydrogease. Science 318, 1461–1464. [DOI] [PubMed] [Google Scholar]
- Gong W.; Hao B.; Wei Z.; Ferguson D. J. J.; Tallant T.; Krzycki J. A.; Chan M. K. (2008) Structure of the α2ε2 Ni-dependent CO dehydrogenase component of the Methanosarcina barkeri acetyl-CoA decarbonylase/synthase complex. Proc. Natl. Acad. Sci. U.S.A. 105, 9558–9563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungdahl L. G.; Andreesen J. R. (1978) Formate dehydrogenase, a selenium-tungsten enzyme from Clostridium thermoaceticum. Methods Enzymol. 53, 360–372. [DOI] [PubMed] [Google Scholar]
- Diekert G.; Hansch M.; Conrad R. (1984) Acetate synthesis from 2 CO2 in acetogenic bacteria: Is carbon monoxide an intermediate?. Arch. Microbiol. 138, 224–228. [Google Scholar]
- Andreesen J. R.; Schaupp A.; Neurauter C.; Brown A.; Ljungdahl L. G. (1973) Fermentation of glucose, fructose, and xylose by Clostridium thermoaceticum: Effect of metals on growth yield, enzymes, and the synthesis of acetate from CO2. J. Bacteriol. 114, 743–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale S. W.; Ljungdahl L. G.; DerVartanian D. V. (1983) Isolation of carbon monoxide dehydrogenase from Acetobacterium woodii and comparison of its properties with those of the Clostridium thermoaceticum enzyme. J. Bacteriol. 155, 1224–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie A. G. W. (1992) Recent changes to the MOSFLM package for processing film and image plate data, Joint CCPE + ESF-EAMCB Newsletter on Protein Crystallography No. 26. [Google Scholar]
- Collaborative Computational Project, Number 4 (1994) The CCP4 suite: Programs for protein crystallography, Acta Crystallogr. D50, 760−763. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Grosse-Kunstleve R. W.; Hung L.-W.; Ioerger T. R.; McCoy A. J.; Moriarty N. W.; Read R. J.; Sacchettini J. C.; Sauter N. K.; Terwilliger T. C. (2002) PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr. D58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; MacArthur M. W.; Thornton J. M. (1993) PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. [Google Scholar]
- Brünger A. T.; Adams P. D.; Clore M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J.-S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. (1998) Crystallography & NMR system: A new software suite for marcromolecular structure determination. Acta Crystallogr. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- Higuchi Y.; Yagi T. (1999) Liberation of hydrogen sulfide during the catalytic action of Desulfovibrio hydrogenase under the atmosphere of hydrogen. Biochem. Biophys. Res. Commun. 255, 295–299. [DOI] [PubMed] [Google Scholar]
- Higuchi Y.; Ogata H.; Miki N.; Yosuoka N.; Yagi T. (1999) Removal of the bridging ligand atom at the Ni-Fe active site of [NiFe] hydrogenase upon reduction with H2, as revealed by X-ray structure analysis at 1.4 Å resolution. Structure 7, 549–556. [DOI] [PubMed] [Google Scholar]
- DeRose V. J.; Telser J.; Andersen M. E.; Lindahl P. A.; Hoffman B. M. (1998) A multinuclear ENDOR study of the C-cluster in CO dehydrogenase from Clostridium thermoaceticum: Evidence for HxO and histidine coordination to the [Fe4S4] center. J. Am. Chem. Soc. 120, 8767–8776. [Google Scholar]
- Andersen M. E.; Lindahl P. A. (1994) Organization of clusters and internal electron pathways in CO dehydrogenase from Clostridium thermoaceticum: Relevance to the mechanism of catalysis and cyanide inhibition. Biochemistry 33, 8702–8711. [DOI] [PubMed] [Google Scholar]
- Andersen M. E.; Lindahl P. A. (1996) Spectroscopic states of the CO oxidation/CO2 reduction active site of carbon monoxide dehydrogenase and mechanistic implications. Biochemistry 35, 8371–8390. [DOI] [PubMed] [Google Scholar]
- Seravalli J.; Kumar M.; Lu W.-P.; Ragsdale S. W. (1997) Mechanism of carbon monoxide oxidation by the carbon monoxide dehydrogenase/acetyl-CoA synthase from Clostridium thermoaceticum: Kinetic characterization of the intermediates. Biochemistry 36, 11241–11251. [DOI] [PubMed] [Google Scholar]
- Kim E. J.; Feng J.; Bramlett M. R.; Lindahl P. A. (2004) Evidence for a proton transfer network and a required persulfide-bond-forming cysteine residue in Ni-containing carbon monoxide dehydrogenases. Biochemistry 43, 5728–5734. [DOI] [PubMed] [Google Scholar]
- Ensign S. A.; Bonam D.; Ludden P. W. (1989) Nickel is required for the transfer of electrons from carbon monoxide to the iron-sulfur center(s) of carbon monoxide dehydrogenase from Rhodospirillum rubrum. Biochemistry 28, 4968–4973. [DOI] [PubMed] [Google Scholar]
- Ensign S. A.; Hyman M. R.; Ludden P. W. (1989) Nickel-specific, slow-binding inhibition of carbon monoxide dehydrogenase from Rhodospirillum rubrum by cyanide. Biochemistry. 28, 4973–4979. [DOI] [PubMed] [Google Scholar]
- Andersen M. E.; DeRose V. J.; Hoffman B. M.; Lindahl P. A. (1993) Identification of a cyanide binding site in CO dehydrogenase from Clostridium thermoaceticum using EPR and ENDOR spectroscopies. J. Am. Chem. Soc. 115, 12204–12205. [Google Scholar]
- Hu Z.; Spangler H. J.; Andersen M. E.; Xia J.; Ludden P. W.; Lindahl P. A.; Münck E. (1996) Nature of the C-cluster in Ni-containing carbon monoxide dehydrogenases. J. Am. Chem. Soc. 118, 830–845. [Google Scholar]
- Al-Mustafa J.; Sykora M.; Kincaid J. R. (1995) Resonance Raman investigation of cyanide ligated beef liver and Aspergillus niger catalases. J. Biol. Chem. 270, 10449–10460. [DOI] [PubMed] [Google Scholar]
- Al-Mustafa J.; Kincaid J. R. (1994) Resonance Raman study of cyanide-ligated horseradish peroxidase. Detection of two binding geometries and direct evidence for the “push-pull” effect. Biochemistry 33, 2191–2197. [DOI] [PubMed] [Google Scholar]
- Blair-Johnson M.; Fiedler T.; Fenna R. (2001) Human myeloperoxidase: Structure of a cyanide complex and its interaction with bromide and thiocyanate substrates at 1.9 Å resolution. Biochemistry 40, 13990–13997. [DOI] [PubMed] [Google Scholar]
- Han S.; Madden J. F.; Siegel L. M.; Spiro T. G. (1989) Resonance Raman studies of Escherichia coli sulfite reductase hemoprotein. 3. Bound ligand vibrational modes. Biochemistry 28, 5477–5485. [DOI] [PubMed] [Google Scholar]
- Bolognesi M.; Rosana C.; Losso R.; Borassi A.; Rizzi M.; Wittenberg J. B.; Boffi A.; Ascenzi P. (1999) Cyanide binding to Lucina pectinata hemoglobin I and to sperm whale myoglobin: An X-ray crystallographic study. Biophys. J. 77, 1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafferji A.; Allen J. W. A.; Ferguson S. J.; Fülöp V. (2000) X-ray crystallographic study of cyanide binding provides insights into the structure-function relationship for cytochrome cd1 nitrite reductase from Paracoccus pantotrophus. J. Biol. Chem. 275, 25089–25094. [DOI] [PubMed] [Google Scholar]
- Simianu M. C.; Kincaid J. R. (1995) Resonance Raman spectroscopic detection of both linear and bent Fe-CN fragments for the cyanide adducts of cytochrome P-450 camphor and its substrate-bound forms. Relevance to the “charge relay” mechanism. J. Am. Chem. Soc. 117, 4628–4636. [Google Scholar]
- Deng T.; Macdonald I. D. G.; Simianu M. C.; Sykora M.; Kincaid J. R.; Sligar S. G. (2001) Hydrogen-bonding interactions in the active sites of cytochrome P450cam and its site-directed mutants. J. Am. Chem. Soc. 123, 269–278. [DOI] [PubMed] [Google Scholar]
- Clay M. D.; Yang T.-C.; Jenney F. E. J.; Kung I. Y.; Cosper C. A.; Krishnan R.; Kurtz D. M. J.; Adams M. W. W.; Hoffman B. M.; Johnson M. K. (2006) Geometric and electronic structures of cyanide adducts of the non-heme iron active site of superoxide reductases: Vibrational and ENDOR studies. Biochemistry 45, 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale S. W.; Clark J. E.; Ljungdahl L. G.; Lundie L. L.; Drake H. L. (1983) Properties of purified carbon monoxide dehydrogenase from Clostridium thermoaceticum, a nickel, iron-sulfur protein. J. Biol. Chem. 258, 2364–2369. [PubMed] [Google Scholar]
- Menon S.; Ragsdale S. W. (1996) Unleashing hydrogenase activity in carbon monoxide dehydrogenase/acetyl-CoA synthase and pyruvate:ferredoxin oxidoreductase. Biochemistry 35, 15814–15821. [DOI] [PubMed] [Google Scholar]
- DeLano W. L. (2002) The PyMOL molecular graphics system, DeLano Scientific, Palo Alto, CA: (http://www.pymol.org).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.