

Abstract
Aims
Increased expression of several subtypes of prostaglandin E2 receptors (EP1–4) has recently been described in clinical and experimental myocardial ischaemia/reperfusion (I/R) injury. However, their pathophysiological significance in I/R remains obscure. Thus, we determined whether the activation of the prostanoid receptor, EP4, suppresses myocardial I/R injury.
Methods and results
To analyse the role of EP4, we administered an EP4 selective agonist (EP4RAG, 1 or 3 mg/kg) or vehicle to rats with myocardial I/R injury. After 7 days of reperfusion, I/R rats exhibited left ventricular (LV) dilatation and contractile dysfunction with myocyte hypertrophy and interstitial fibrosis. EP4RAG significantly reduced infarction area/ischaemic myocardium (72.4 ± 0.7 vs. 23.3 ± 0.6%; P < 0.05) and improved LV contraction and dilatation compared with that of the vehicle. EP4RAG also attenuated the recruitment of inflammatory cells, especially macrophages, and interstitial fibrosis in hearts. Monocyte chemoattractant protein (MCP)-1 and other cytokines were increased in both non-ischaemic (area not at risk, ANAR) and ischaemic (area at risk, AAR) myocardium; however, western blot analysis and RNase protection assay showed that EP4RAG suppressed these changes. Gelatin zymography revealed EP4RAG significantly reduced matrix metalloproteinase-2 and -9 activities in both ANAR and AAR. Chemoattractant assay demonstrated that EP4RAG suppressed the migration of cytokine-stimulated macrophages and decreased the level of MCP-1 production in the supernatant (587.3 ± 55.3 vs. 171.5 ± 47.5 pg/mL; P < 0.05).
Conclusion
The data suggest that the EP4 agonist is effective for attenuation of I/R injury by suppressing MCP-1 and the infiltration of inflammatory cells, especially macrophages.
Keywords: Prostaglandins, Inflammation, Reperfusion injury
1. Introduction
Ischaemia/reperfusion (I/R) is a common antecedent event that predisposes a patient to congestive heart failure. Loss of cardiac function following I/R occurs in the context of myocyte death and interstitial fibrosis and is referred to ventricular remodelling.1 Recent studies have demonstrated that inflammatory responses may cause myocardial damage and fibrosis, leading to progressive impairment of cardiac function.2 Specifically, macrophages are important mediators in I/R pathogenesis.3 Activated macrophages release various cytokines, such as tumour necrosis factor (TNF)-α, interleukin (IL)-1β, -6, and -12, interferon-γ, chemokines like monocyte chemoattractant protein (MCP)-1,4 matrix metalloproteinase (MMP),5 and transforming growth factors-β6 through an autocrine/paracrine mechanism. This is possibly representing a vicious cycle operative in the development of myocardial remodelling. Considering the harmful infiltration of macrophages, inhibition of the production of MCP-1 is very important, and to prevent the deposition of fibroblasts, reduction of the production of MMPs is also effective to attenuate left ventricular (LV) remodelling after I/R.
Prostaglandin E2 (PGE2) is produced during inflammatory responses and controls a variety of both innate and adaptive immunity through four different receptor subtypes (EP1–4) with various signalling cascades.7 However, the precise roles of each receptor have not yet been elucidated. The use of gene-targeted mice and a selective agonist/antagonist responsible for each receptor has gradually revealed that each receptor functions via a distinct signal cascade and plays a unique role in a variety of disease conditions, including rheumatoid arthritis8 and tumour growth-associated angiogenesis.9 It has recently been shown that PGE2 protects the reperfused myocardium from ischaemic injury via its receptor EP4, suggesting a particular role of this subtype for cardio protection.10 Nevertheless, it is unknown how PGE2 protects myocardium, and to what cells PGE2 acts on. Nowadays, the close relationships between I/R injury and inflammatory responses by macrophages are reported,3 and a previous study also revealed that PGE2 suppressed chemokine production in macrophages through EP4.11 We hypothesized that cardioprotective effect of PGE2 through EP4 would relate to suppression of infiltration and chemokine production in macrophages. Therefore, the purpose of this study was to determine whether EP4 selective agonist signalling can inhibit the infiltration of macrophages, production of MCP-1, deposition of fibroblasts through reduction of the production of MMPs, and can attenuate the progressive LV dysfunction in a rat I/R model using a selective agonist of EP4, EP4RAG.
2. Methods
2.1. Rat myocardial ischaemia/reperfusion injury models
Eight- to 10-week-old male Sprague–Dawley rats were used in this experiment. Rats were anaesthetized with 40 mg/kg sodium pentobarbital intraperitoneally immediately before operation. Rats were intubated orally with a polyethylene tube for artificial respiration (SN-480-7, Shinano, Tokyo, Japan). The left anterior descending coronary artery was visualized using a microscopy and ligated with 6-0 silk suture. Myocardial ischaemia was confirmed by epicardial cyanosis and wall asynergy. After 30 min of coronary artery occlusion, reperfusion was made by loosening the suture and verified visually. The chest wall and the skin were then closed with 3-0 silk suture.12 Animals used in this study were maintained in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (NIH Publication No. 85-23, revised 1996).
2.2. Treatment protocols
A novel EP4 selective agonist (EP4RAG), (2E)-17,18,19,20-tetranor-16-(3-biphenyl)-2,3,13,14-tetradehydro-PGE1 (Molecular Weight: 446.55) was kindly provided by Taisho Pharmaceutical Co., Ltd. The EP4RAG selectively bound to mouse EP4 receptor with a Ki value of 2.8 nM, compared with PGE2. The compound hardly bound to other mouse receptors EP1, EP2, and EP3 with a Ki value of >1000, 357.0, and 491.0, respectively. The serum pharmacokinetic parameters of EP4RAG were obtained after subcutaneous injection of EP4RAG (1 mg/kg, reconstituted in PBS) into male rats. Maximum plasma concentration (Cmax) was 414.41 ± 55.31 ng/mL, and half-life (t1/2) was 0.64 ± 0.30 h. Area under the concentration–time curve (AUCinf) was 272.26 ± 35.11 ng h/mL. The trough levels were <10 nM. Dosage regimen of EP4RAG in animal experiments was determined based on pharmacokinetic parameters.
The animals were assigned randomly to one of three treatment groups (n = 6 each), as follows: (a) subcutaneous injection of EP4RAG (3 mg/kg) 15 min before reperfusion and every 12 h to day 7; (b) subcutaneous injection of EP4RAG (1 mg/kg) 15 min before reperfusion and every 12 h to day 7; (c) subcutaneous injection of PBS 15 min before reperfusion and every 12 h to day 7. To clarify the dose-dependent responses of the in vivo normal rat hearts of the drug, we administered PBS, 1 or 3 mg/kg EP4RAG to the rats without I/R injury (n = 3 each).
2.3. Echocardiographic measurements
Rats were anaesthetized mildly with sodium pentobarbital (40 mg/kg). Trans-thoracic echocardiography was performed with a commercially available ultrasound equipment (Nemio, Toshiba, Tokyo, Japan) before the operation, immediately, 1, 4, and 7 days after reperfusion. A 7 MHz annular array transducer was used. Hearts were imaged in the two-dimensional mode in short-axis views at the level of papillary muscle. M-mode views were used to measure the LV dimensions according to the American Society for Ecocardiography leading edge method.13 Left ventricular end-diastolic dimension (LVDd) and end-systolic dimension (LVDs), and fractional shortening (%FS = [(LVDd − LVDs)/LVDd] × 100) were calculated from the M-mode recordings.
2.4. Haemodynamic measurements
Blood pressure and heart rate of all rats were evaluated on days 0, 1, 4, and 7. Blood pressure (systolic, diastolic, and mean pressure) was measured in conscious rats by using a tail-cuff system (BP-98A, Softron Co., Tokyo, Japan).14 Before the study was initiated, rats were adapted to the apparatus for at least 5 days.
2.5. Measurement of area at risk and infarct size
At day 7 after reperfusion, the anaesthetized rats were intubated and thoracotomy was repeated (n = 6 in each group). Immediately after harvesting, heart weights were measured. The left anterior descending artery was religated tightly and Evans Blue dye (1 mL of 2.0% solution) was infused via an inferior vena cava to determine the non-ischaemic zone (area not at risk, ANAR). Hearts were then sliced transversely into four slices and incubated in 2.0% triphenyl tetrazolium chloride (TTC) (Sigma, Tokyo, Japan) for 15 min at 37°C to verify the viable and necrotic area in the ischaemic myocardium (area at risk, AAR) as described earlier.15 Each slice was weighed and photographed, and the areas of infarction, AAR, and left ventricle were evaluated using computer-assisted planimetry (Scion Image β4.0.2) by observers blinded to the treatment protocol. The volumes of each area were determined by the following process: volume of AREA = (A1 × Wt1) + (A2 × Wt2) + (A3 × Wt3) + (A4 × Wt4), where A is the percentage of each area by planimetry from subscripted numbers 1–4 indicating sections and Wt is the weight of the same numbered sections.12
2.6. Pathology
To perform myocardial pathology of reperfused myocardium, rat hearts were cut at the level of the papillary muscles and harvested in 10% formalin solution (n = 6 in each group). We obtained four transverse sections per heart for histopathological examination. Apex, mid-ventricular, and basal level slices were stained with HE and Mallory. The number of infiltrating cells in myocardium was counted as the sum of the cell counts on three fields at ×400 magnification in the HE staining.16 The area of myocardium and surrounding tissue affected by I/R (consisting of inflammatory cells, myocardial necrosis, and fibroblast) was determined with a computer-assisted analyser (Scion Image beta 4.0.2) in the Mallory staining. The area ratio (affected/LV as a percentage) was calculated as described previously.17 Values for three ventricular regions were averaged for each heart, and the mean percentage of affected area for each group was calculated. All data were analysed in a blind fashion by two independent investigators and averaged.
2.7. Immunohistochemistry
To perform immunohistochemistry of reperfused myocardium, rat hearts were cut at the level of the papillary muscles and frozen in OCT compound (Sakura Finetek, Tokyo, Japan). Each section (5 µm) was then incubated with anti-rat antibodies to CD68 (ED1, AbD serotec, Oxford, UK), CD4 (OX-35), CD8 (OX-8) (PharMingen, San Diego, CA), MCP-1 (R17), MMP-2 (H-76), or MMP-9 (H-129) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) (each at 1–10 µg/mL) for 8 h at 4°C and then with histofine simple stain rat (Nichirei, Tokyo, Japan). Finally, each section was reacted with AEC matrix solution for 5–20 min. Immunostained type- and class-matched non-immune PBS was used as the negative controls for each antibody. Intensity of expression was scored as follows: 0, no visible staining; 1, few cells with faint staining; 2, few cells with moderate staining; 3, some cells with moderate staining; 4, diffuse cells with moderate staining; or 5, diffuse and intense staining. Scores of two independent reviewers were averaged.18
2.8. Western blot analysis
Proteins were separated by SDS–PAGE, transferred to a nitrocellulose membrane, and incubated with monoclonal antibodies to MCP-1 (Santa Cruz Biotechnology) or to actin (MAB3128, Millipore, Billerica, MA, USA) at 4°C overnight. The membranes were incubated with a secondary antibody (Amersham Biosciences, Piscataway, NJ, USA) for 2 h and developed with ECL reagent (Amersham Biosciences). Enhanced chemiluminescence was detected with an LAS-1000 (Fujifilm, Tokyo, Japan).12
2.9. Zymography
Zymography was performed using myelin basic protein (MBP) polymerized in an SDS gel as the substrate. Cell supernatants were separated by SDS–PAGE, and following the renaturation of the proteins with 2.5% Triton X-100, the gels were incubated for 22 h in a buffer containing Tris–HCl (50 mmol/l), CaCl2 (5 mmol/L), and NaN3 (0.02%). Gels were then stained with Coomassie Blue and MBP degradation assessed densitometrically.18
2.10. Ribonuclease protection assay
The harvested hearts were homogenized in Trizol reagent (Life Technologies, Grand Island, NY, USA) and frozen at −80°C. Cytokine mRNA expression was measured by (ribonuclease protection assay) RPA using rCK1 template (Riboquany kit, PharMingen). Levels of mRNA expression were quantified and normalized to GAPDH mRNA using densitometry.19
2.11. THP-1 cell culture
The human monocytic cell line THP-1 was obtained from the American Type Culture Collection. THP-1 cells were maintained in RPMI-1640 supplemented with 10% foetal bovine serum (Sigma, St Louis, MO, USA), l-glutamine (200 mmol/L), non-essential amino acids, and 1 mmol /L sodium pyruvate. THP-1 cells (2 × 106 cells/10 mm dish) were differentiated by stimulation with PMA (50 ng/mL, final concentration) for 1 day to obtain a macrophage-like phenotype that closely resembles human monocyte-derived macrophages.20
2.12. Migration assay of macrophage
Monocyte chemotaxis was measured using a 6-well Micro Chemotaxis Chamber. First, THP-1 macrophages were cultured for 15 min in the absence (control) or presence of EP4RAG (10, 50, and 100 nM). After incubation, the medium with 1 × 104cells/mL (defined as conditioned medium) was transferred to the lower chamber of the Micro Chemotaxis Chamber. The lower and upper chambers were separated by a polycarbonate membrane with a pore size of 8 µm (Coster). An aliquot of THP-1 monocyte cell suspension (3.5 × 105 cells/mL) was added to the upper chamber, and IL-1β (10 ng/mL) was added, after that, the cells were allowed to transmigrate for 24 h. After transmigration, the surface of the membrane facing the THP-1 cell suspension was scraped and washed three times according to the manufacturer’s instructions. The migrated cells, on the side of the membrane facing the conditioned medium, were fixed and then counted. The number of cells was counted under a light microscope at a magnification of ×400. At least five fields in each well were counted. There were three repeats for each experimental condition. The extent of cell migration was expressed as the number of cells per field.21
2.13. Enzyme-linked immunosorbent assay of monocyte chemoattractant protein-1
The concentrations of MCP-1 protein in the upper supernatants were determined by ELISA (BioSource, Camarillo, CA, USA) according to the manufacturer’s instructions.12
2.14. Statistical analysis
Values are given as mean ± SD. Groups were compared with Scheffé’s ANOVA (Stat View, SAS Institute, Inc.). We used a two-way multiple ANOVA for statistical analyses of haemodynamics in Tables 1 and 2. We used Student’s t-test for comparisons between two groups in the second experiment. Differences were considered statistically significant at a value of P < 0.05.
Table 1.
Time-dependent changes of FS and mean blood pressure
| TX group | Fractional shortening (%) |
Mean blood pressure (mmHg) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Day 0 before operation | Day 0 after operation | Day 1 | Day 4 | Day 7 | Day 0 | Day 1 | Day 4 | Day 7 | |
| Vehicle (3 mg/kg) | 57.5 ± 1.1 | 29.5 ± 1.8 | 32.5 ± 1.1 | 26.5 ± 0.8 | 23.8 ± 1.4 | 84.6 ± 3.6 | 74.6 ± 1.3 | 73.3 ± 1.7 | 67.0 ± 3.3 |
| EP4RAG (1 mg/kg) | 57.8 ± 1.3 | 26.8 ± 1.1 | 33.0 ± 0.6 | 35.2 ± 2.0 | 39.8 ± 0.5* | 88.5 ± 2.5 | 73.0 ± 1.5 | 74.5 ± 3.0* | 80.6 ± 2.1* |
| EP4RAG (3 mg/kg) | 57.6 ± 1.0 | 26.8 ± 1.4 | 37.8 ± 1.0 | 42.5 ± 1.6* | 48.5 ± 2.3* | 86.8 ± 1.8 | 77.3 ± 2.9* | 82.2 ± 2.0*,** | 87.8 ± 2.1*,** |
Mean ± SEM pathological values were compared among the groups using two-way ANOVA and Bonferroni post-tests.
*P < 0.05 compared with rat given vehicle.
**P < 0.05 compared with rat given EP4RAG (1 mg/kg).
Table 2.
Histological findings of the myocardium in native, I/R + vehicle and I/R + EP4RAG rats 7 days after operation
| TX group | Myocardial infiltration (counts) | Myocardial fibrosis (%) | Immunohistochemsity (0–5) |
|||||
|---|---|---|---|---|---|---|---|---|
| ED1 | CD4 | CD8 | MCP-1 | MMP-2 | MMP-9 | |||
| Vehicle (3 mg/kg) | 644 ± 71 | 26 ± 4.3 | 2.8 ± 0.7 | 3.1 ± 0.9 | 2.8 ± 1.2 | 3.0 ± 0.9 | 2.6 ± 0.6 | 2.5 ± 0.8 |
| EP4RAG (3 mg/kg) | 79 ± 15* | 6.9 ± 2.4 | 1.3 ± 0.5* | 1.4 ± 0.5* | 0.6 ± 0.4* | 1.2 ± 0.5* | 1.1 ± 0.6* | 0.7 ± 0.5* |
| Native | O ± O* | O ± O* | O ± O* | O ± O* | O ± O* | O ± O* | O ± O* | O ± O* |
Mean ± SEM pathological values were compared among the groups using Scheffe’s ANOVA.
*P < 0.05 compared with rat given vehicle.
3. Results
3.1. EP4RAG prevented myocardial dysfunction after infarction
M-mode echocardiogram of a normal rat heart showed good LV contraction without any impaired wall movement. The vehicle-treated rat heart on day 7 after reperfusion showed that the wall motion in antero-lateral region of LV was decreased remarkably. However, EP4RAG (3 mg/kg) improved the regional wall motion in rats 7 days after reperfusion. Serial changes in %FS are presented in Table 1. There was no difference in %FS at baseline and at immediately after I/R between the EP4RAG- and the vehicle-treated groups. However, 7 days after reperfusion, %FS of the EP4RAG-treated groups was dose-dependently improved compared with the vehicle-treated groups (Table 1; see Supplementary material online, Data 1). EP4RAG (3 mg) did not affect the %FS in the animals without I/R injury (58 ± 1% before the injection of EP4RAG, and 57 ± 1% 1 day after the injection of EP4RAG).
3.2. Blood pressure and heart rate
There was no significant difference in heart rate among the three groups, but mean blood pressure of the EP4RAG-treated groups were dose-dependently improved compared with the vehicle-treated groups on 7 days after reperfusion (Table 1). EP4RAG (3 mg) did not statistically affect mean blood pressure (91.6 ± 3 mmHg before the injection of EP4RAG, and 74.1 ± 3 mmHg 1 day after the injection of EP4RAG) and heart rates (445 ± 3 b.p.m. before the injection of EP4RAG, and 438 ± 9 b.p.m. 1 day after the injection of EP4RAG) in the animals without I/R injury.
3.3. EP4RAG reduced ischaemia/reperfusion-induced myocardial damage
AAR/LV ratios were not significantly different between the EP4RAG- and vehicle-treated groups [EP4RAG (3 mg/kg) 49.0 ± 0.9%; EP4RAG (1 mg/kg) 51.0 ± 0.8%; vehicle 49.8 ± 1.1%]. However, infarct/AAR was significantly different between the EP4RAG- and vehicle-treated groups on day 7 [EP4RAG (3 mg/kg) 23.3 ± 0.6%; EP4RAG (1 mg/kg) 38.0 ± 1.6%; vehicle 72.4 ± 0.7%; P < 0.05]. EP4RAG also significantly decreased infarct/LV compared with the vehicle-treated groups [EP4RAG (3 mg/kg) 11.4 ± 0.3%; EP4RAG (1 mg/kg) 19.3 ± 1.0%; vehicle 36.1 ± 0.8%; P < 0.05] (Figure 1).
Figure 1.
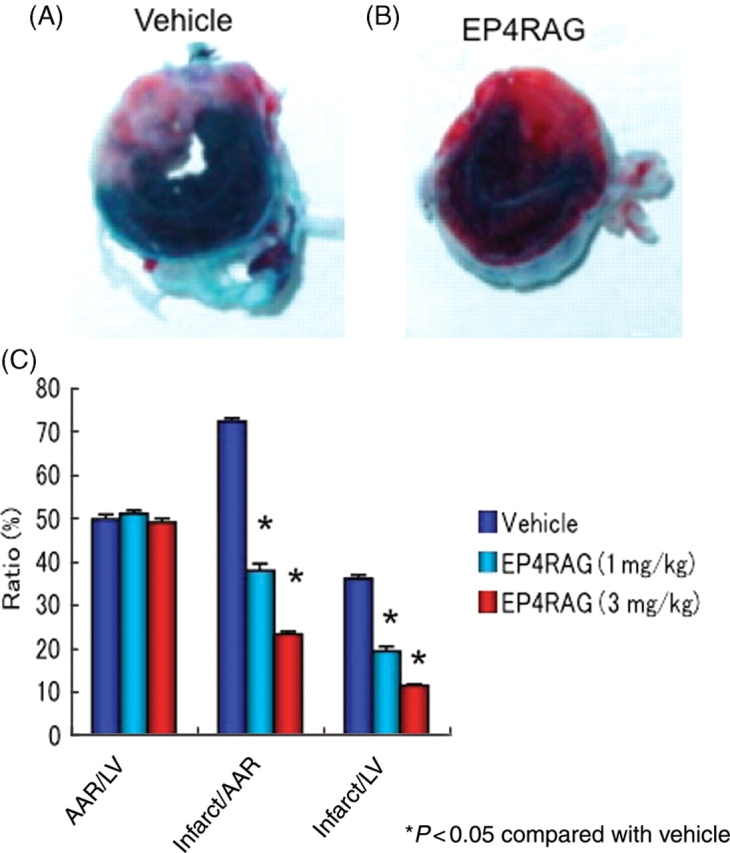
Evans Blue and TTC staining. Hearts were stained by Evans Blue and TTC to confirm the infarct area, the AAR and the ANAR after 30 min of ischaemia and 7 days of reperfusion. EP4RAG (3 mg/kg) treatment (B) significantly decreased infarct/AAR and infarct/LV compared with the vehicle-treated group (A). Statistical analyses of infarct size and AAR in hearts from the vehicle- and EP4RAG-treated rats (C). *P < 0.05 compared with rat given vehicle.
3.4. Myocardial pathology
Consistent with the echocardiographic data, EP4RAG-treated I/R rats had significantly smaller LV diameters compared with those of control. There was a statistical difference in heart weight-to-body weight ratios at day 7 after reperfusion between PBS-treated control (4.9 ± 1.2%) and 3 mg/kg EP4RAG-treated (3.9 ± 0.2%, P < 0.05) samples. Photomicrographs of LV sections showed an increased myocyte cross-sectional area in I/R, while it was significantly attenuated by EP4RAG. The number of infiltrating cells was increased in both AAR and ANAR in I/R, which was inhibited by EP4RAG. Although interstitial fibrosis was increased in I/R, EP4RAG also suppressed this change (Figure 2; Table 2). Statistically, there were comparable results between 1 and 3 mg/kg EP4RAG-treated groups. However, there was a difference between the results of control and 1 mg/kg EP4RAG.
Figure 2.
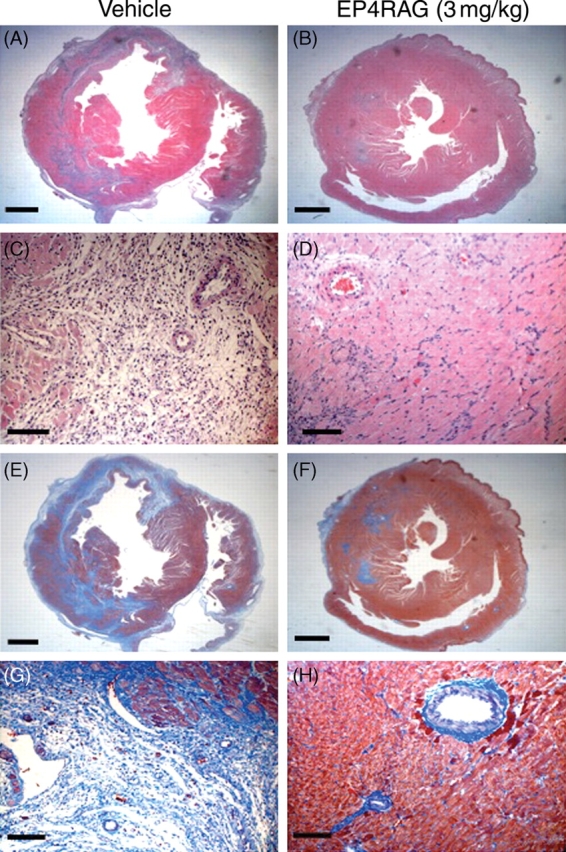
HE and Masson-trichrome staining. Low-power (A, B, E, and F) and high-power (C, D, G, and H) photomicrographs of HE-stained (A through D) and Masson-trichrome-stained (E through H) LV cross-sections obtained from I/R (A, C, E, and G), I/R + EP4RAG (3 mg/kg) (B, D, F, and H) rats 7 days after operation. EP4RAG-treated I/R rats had significantly smaller LV diameters compared with those of vehicle. EP4RAG attenuated infiltration of inflammatory cells and LV fibrosis (D and H) compared with vehicle (C and G). Scale bars: 2 mm (A, B, E, and F) and 100 µm (C, D, G, and H).
3.5. Myocardial immunohistochemistry
The immunoreactive staining for EP4 receptor was barely detectable in the LV obtained from native rats. In contrast, in post-I/R animals on day 7, the expression of EP4 receptor was clearly increased in AAR in LV (Supplementary material online, Data 2). In the vehicle-treated I/R rats, the expression of MCP-1 and severe myocardial macrophage infiltration was observed at day 7. However, EP4RAG significantly attenuated the expression of MCP-1 and infiltration of macrophages. T lymphocytes (CD4 and CD8), to which MCP-1 can be also chemotactic, were detected in the I/R rats, which were also reduced by EP4RAG. MMP-2 and -9 were expressed in both LV of EP4RAG- and vehicle-treated groups on day 7, but the expression was much stronger in vehicle-treated groups (Figure 3; Table 2). Although there were comparable results between 1 and 3 mg/kg EP4RAG-treated groups, there was a difference between control and 1 mg/kg EP4RAG.
Figure 3.
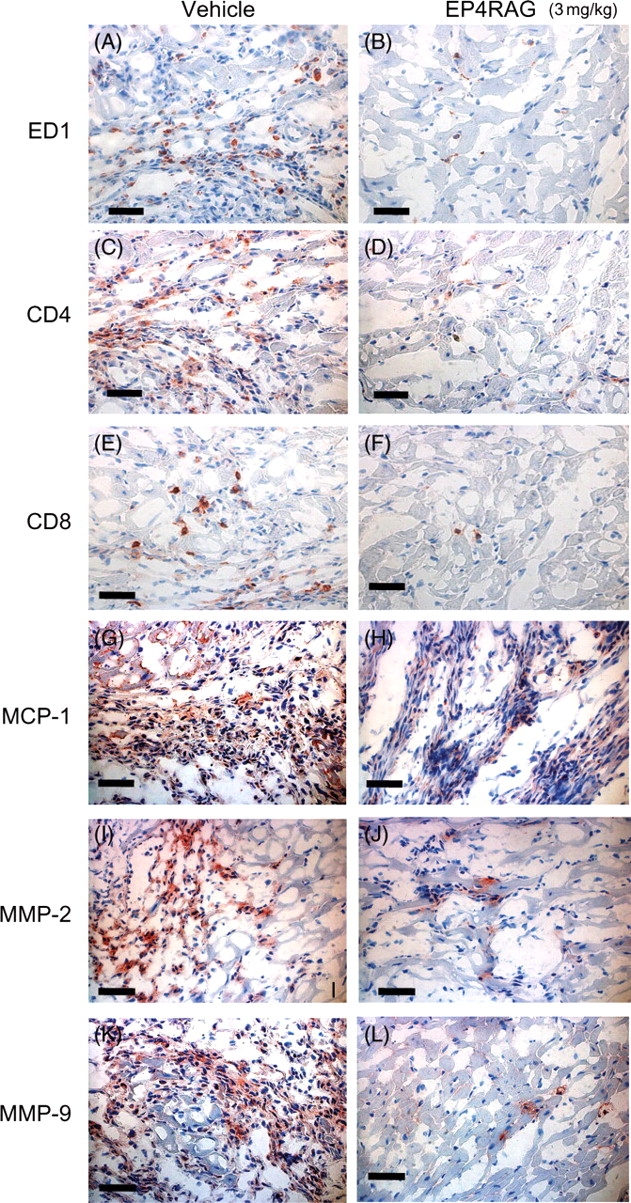
Inflammatory cell infiltration. Representative immunohistochemical findings of myocardium of the I/R rats at day 7. Left panels show myocardium from vehicle-treated rats at day 7; right panels are from those treated with EP4RAG (3 mg/kg) at day 7. ED1 (A and B), CD4 (C and D), CD8 (E and F), MCP-1 (G and H), MMP-2 (I and J), and MMP-9 (K and L) staining. Increased numbers of ED1-, CD4-, and CD8-positive cells were observed in vehicle-treated rats, while EP4RAG suppressed the cell numbers significantly. MCP-1, MMP-2, and -9 were strongly expressed on the infiltrating cells in vehicle-treated rats, but these changes were attenuated in EP4RAG-treated rats. Scale bars: 50 µm.
3.6. Myocardial expression of cytokines, chemokines and matrix metalloproteinases
MCP-1 levels were significantly higher in I/R than those in native animals. However, EP4RAG attenuated the increase in MCP-1 expression in both AAR and ANAR (Figure 4A and B). EP4RAG also attenuated inflammatory cytokines, including TNF-α, IL-1β, and IL-6, induced by I/R (Figure 4C and D). On day 7 after I/R, LV gelatinolytic activity of pro- and active-MMP-2 and -9 appeared in both EP4RAG- and vehicle-treated groups, but EP4RAG-treated groups showed significantly weaker activity not only in AAR but also in ANAR (Figure 5). Although there were comparable results between 1 and 3 mg/kg EP4RAG-treated groups, there was difference between control and 1 mg/kg EP4RAG.
Figure 4.
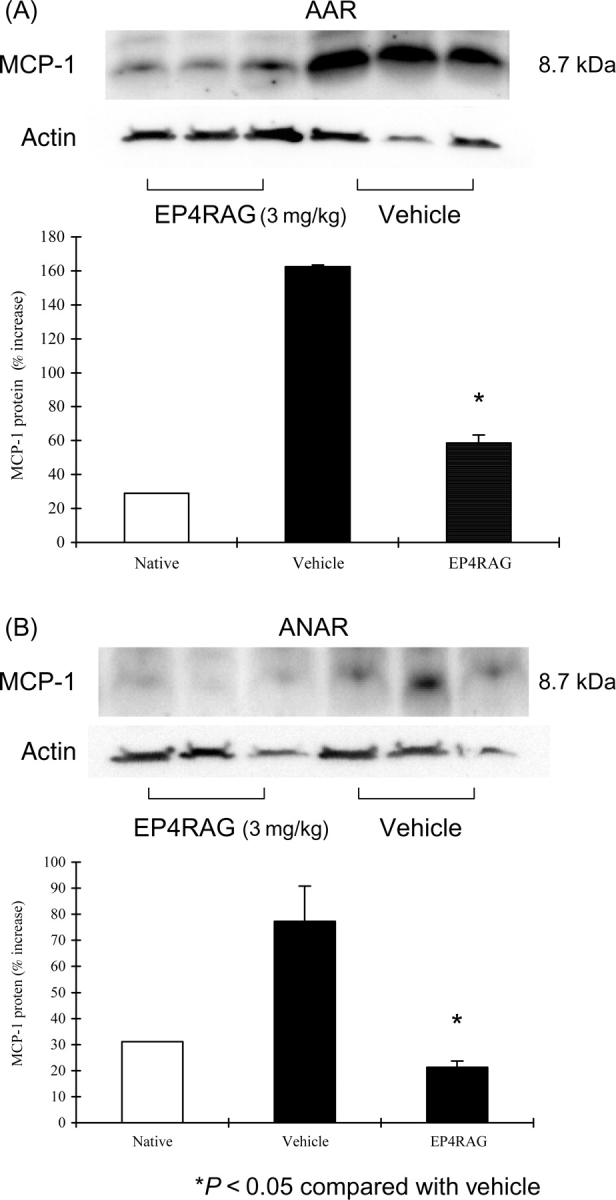

Western blot. Representative western blot and its densitometric analysis of myocardial MCP-1 and actin protein levels from native, I/R + vehicle, and I/R + EP4RAG (3 mg/kg) rats 7 days after operation in AAR (A) and ANAR (B). Representative RPA results using RNA from I/R + vehicle and I/R + EP4RAG (3 mg/kg) rats 7 days after operation in AAR and ANAR (C). Quantitative results of IL-1β, IL-6, and TNF-α mRNA are presented (D). Levels of MCP-1 protein and cytokine mRNA for IL-β, -6, and TNF-α in both AAR and ANAR were markedly suppressed by EP4RAG compared with vehicle. *P < 0.05 compared with rat given vehicle.
Figure 5.
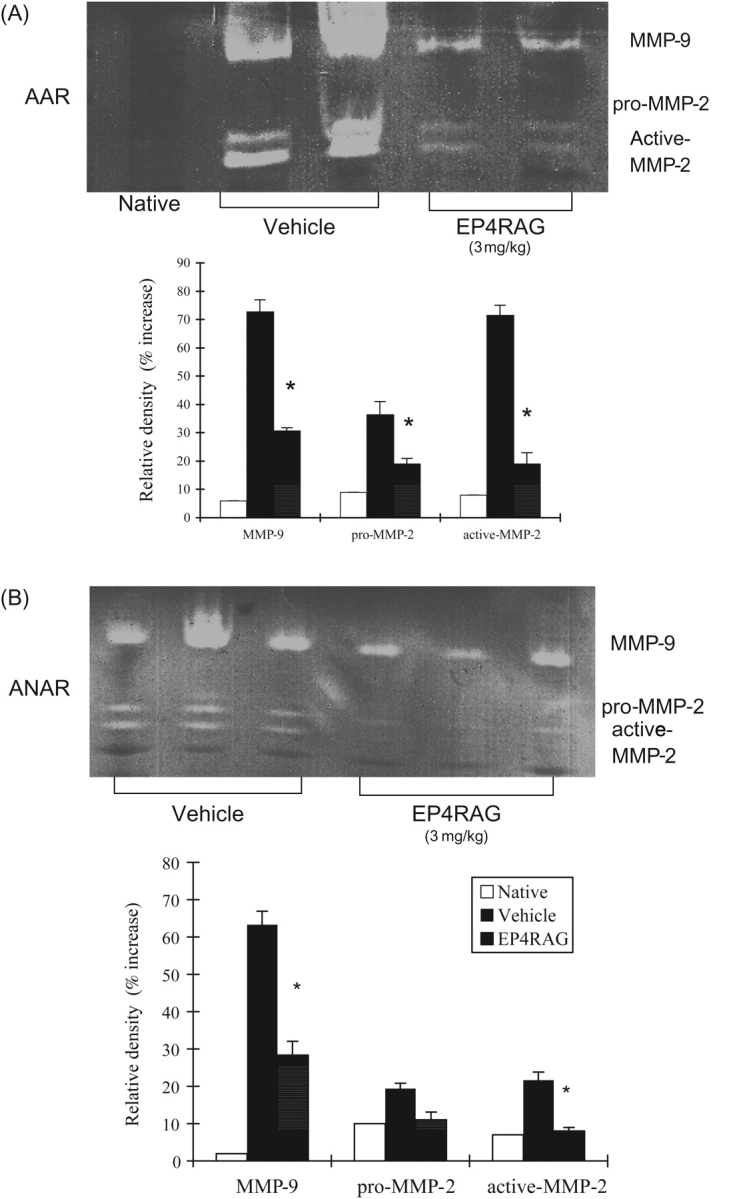
Zymography. Gelatin zymography of heart from native, I/R + vehicle and I/R + EP4RAG (3 mg/kg) rats 7 days after operation in AAR (A) and ANAR (B). Relative intensity of MMP-2 (pro- and active forms) and -9 are shown. In accordance with the LV fibrosis, vehicle-treated rats showed the high activity of MMP-2 and -9, but EP4RAG suppressed these changes. *P < 0.05 compared with rat given vehicle.
3.7. Induction of monocyte chemoattractant protein-1 and migration of THP-1 in vitro
IL-1β (10 ng/mL, 24 h) exposure caused an increase in MCP-1 protein levels in THP-1 monocytes, but this increase was dose-dependently inhibited by EP4RAG. MCP-1-mediated chemotaxis was also significantly attenuated by EP4RAG (Figure 6).
Figure 6.

MCP-1 production. Efficacy of EP4RAG in migration and MCP-1 production of THP-1 macrophage. Representative photographs of lower chamber in condition of 1 × 104 THP-1 macrophages/mL (IL-1β, 10 ng/mL, 24 h) in the absence (A) or presence (B) of EP4RAG (100 nM). (C) The number of migrated THP-1 macrophages was determined by counting four high-power microscope fields (×400). (D) Production of MCP-1 in the supernatant was estimated by ELISA. IL-1β (10 ng/mL, 24 h) exposure caused a increase in MCP-1 protein levels in THP-1 monocytes and many THP-1 cells migrated into the lower chamber, but these changes were dose-dependently inhibited by EP4RAG. *P < 0.05 compared with cells not treated with EP4RAG.
4. Discussion
This is the first report that provides the evidence of increased myocardial expression of EP4 in the inflammatory area and the beneficial effects of EP4 selective agonist on the development of myocardial infarction after I/R injury by suppressing MCP-1 and the infiltration of macrophages. These effects were associated with a decrease in interstitial fibrosis as well as macrophage infiltration and myocardial TNF-α and IL-1β gene expression. Our observations suggest that EP4 agonist strategy may be of therapeutic benefit against the evolution of post-I/R failure.
Prostaglandins mediate cardioprotective effect in various in vitro and animal models of I/R-associated myocardial injury.22–24 Earlier work suggested that the anti-ischaemic action of prostaglandins is mediated, at least in part, by receptors specific for E-type prostaglandins.25 Contrarily, recent work demonstrated that targeted disruption of the gene for microsomal prostaglandin E synthase-1 depressed PGE2 production and retarded atherogenesis, supporting that pro- and anti-inflammatory pathways in cardiovascular diseases involves PGE2.26 However, it is unclear that how PGE2 protects myocardium, and to what cells and receptors PGE2 acts on. Prostaglandins exert their effects via G-protein-coupled receptors. Therefore, prostaglandin-mediated cardioprotection involves one of the EP receptor subtypes, namely EP1, EP2, EP3, and EP4. Each receptor has their unique signalling pathways.27 EP1 couples to Gq and increases intracellular Ca2+. EP2 and EP4 couple to Gs and stimulate adenylate cyclase with subsequent increase in intracellular cAMP. A previous study showed that targeted disruption of the gene for EP4 decreased PGE2-mediated inhibition of TNF-α, IL-12,28 in LPS-activated mouse macrophage. PGE2 suppressed the production of MCP-111 in LPS-activated human macrophages via its receptor EP4, and we also suggested that the structure of EP4 itself was associated with the anti-inflammatory effect in human atheroma.29 These researches support the primary role of the EP4 receptor in the anti-inflammatory pathway in macrophages. In accordance with these facts, we showed that this EP4 agonist suppressed the migration of macrophages stimulated by IL-1β and decreased the production of MCP-1 protein in vitro. This fact is very important, because increased production of MCP-1 and subsequent monocyte infiltration are observed at the reperfusion phase of myocardial ischaemia, and sustained MCP-1 expression can lead to sustained cytokine expression and inflammatory responses that lead to inadvertent myocardial damage.30
MCP-1 is known as a chemotactic factor, and several studies have reported the role of MCP-1 in human coronary disease.31,32 It has been found that administration of antibody against MCP-1 significantly decreases the reperfusion injury in a rat model.33 In reperfused myocardium, monocytes and macrophages are recruited by MCP-1 and function along with polymorphonuclear neutrophils as the main sources of inflammatory cytokines.3 Furthermore, it has been reported that MCP-1 enhances the expression of MMPs in fibroblasts5 and macrophages34,35 and that IL-1β and MCP-1 work together to enhance their own production.36 Taken together, MCP-1 plays essential and multiple roles in the recruitment of inflammatory cells, which promote myocardial I/R injury and subsequent LV remodelling. Therefore, the suppression of MCP-1 expression by EP4 agonist is an effective way to prevent harmful inflammatory cell accumulation in reperfused myocardium. In addition, recent studies have revealed that MCP-1 may exert other biological activities than recruiting monocytes/macrophages, because the expression of MCP-1 does not always correlate with the extent of their infiltration.7 Therefore, the inhibition of MCP-1 by EP4 agonist shown in this study might not be attributable solely to its chemoattractant properties on inflammatory cells. In fact, EP4RAG reduced the expression of TNF-α in the post-I/R hearts in both AAR and ANAR. TNF-α has profound and wide-ranging effects that can initiate a cascade of myocyte hypertrophy and apoptosis. Therefore, a proposed mechanism of EP4RAG for reverse LV remodelling is related to the attenuation of this cytokine after I/R.
MCP-1 can also directly stimulate collagen production via upregulation of IL-1β expression in cardiac fibroblasts. Therefore, the inhibition of interstitial fibrosis in I/R by EP4RAG could be attributable to the attenuation of IL-1β. However, we cannot exclude the possibility that the longer-term inhibition of MCP-1 signalling may enhance LV remodelling and even cause cardiac rupture. Although we did not experience cardiac rupture in our series of rat experiment, we apparently need additional studies to clarify this crucial issue.
Adding to the fact that EP4RAG suppressed the expression of MCP-1 and infiltration of inflammatory cells including macrophages, it also attenuated the expression and activities of MMP-2 and -9 in both AAR and ANAR. Sustained expression of MMP-2 and -9 leads to the myocardial fibrosis, LV enlargement, and the resultant LV failure,37 so this effect of EP4RAG is noteworthy. Cytokines including TNF-α and IL-1β evoke a secondary MCP-1 production from other types of cells, including cardiac myocytes and fibroblasts, and establish a positive loop, amplifying and sustaining the proinflammatory response. Thus, there is an intimate link between MCP-1 and cytokines, and MMPs in the hearts through an autocrine/paracrine mechanism, possibly representing a vicious cycle implicated in myocardial failure. Therefore, an inhibition of MCP-1 signalling by EP4RAG has a great potential for breaking the vicious feedback loop of inflammation and may attenuate the development of myocardial remodelling. Considering these cardioprotective effects of PGE2 via its receptor EP4, EP4 agonist may be useful for the treatment of cardiac dysfunction after myocardial infarction.
This study has a limitation. We administered EP4RAG 15 min before reperfusion and every 12 h for 7 days. Because clinical pre-treatment of patients prior to the onset of ischaemia may be difficult to achieve, further treatment protocols are needed.
In conclusion, the results of the present study demonstrate a robust cardioprotective effect against myocardial reperfusion injury by pre-treatment and continued treatment with the selective EP4 agonist. This cardioprotective effect is based on the suppression of macrophage infiltration and macrophage-derived MCP-1. It makes great sense that activation of EP4 results in reduction of necrotic area as well as in preservation of cardiac physiological function. Although further investigations are required to evaluate the anti-leucocyte therapy, EP4RAG could potentially become a part of strategy to prevent the I/R injury.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
This study was supported by grants from the Japan Cardiovascular Research Foundation, a Grant-in-Aid from the Japanese Ministry of Education, Science and Culture, a Grant-in-Aid from the Japan Society for the Promotion of Science, and the Takeda Science Foundation. The work on EP4 was initiated under a grant from the US National Heart Lung and Blood Institute HL34636 and from the Foundation Leducq to Dr Peter Libby, Brigham and Women’s Hospital and the Harvard Medical School.
Acknowledgements
We thank Libby for his encouragement and mentorship. We thank Ms Noriko Tamura and Ms Yasuko Matsuda for their excellent assistance.
Conflict of interest: T.T. and M.O. are employees of Taisho Pharmaceutical Co., Ltd. K.T. worked for Taisho Pharmaceutical Co., Ltd, until 2006. All other authors have no conflict of interest.
References
- 1.Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. 1990;81:1161–1172. doi: 10.1161/01.cir.81.4.1161. [DOI] [PubMed] [Google Scholar]
- 2.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 3.Herskowitz A, Choi S, Ansari AA, Wesselingh S. Cytokine mRNA expression in postischemic/reperfused myocardium. Am J Pathol. 1995;146:419–428. [PMC free article] [PubMed] [Google Scholar]
- 4.Nanki T, Nagasaka K, Hayashida K, Saita Y, Miyasaka N. Chemokines regulate IL-6 and IL-8 production by fibroblast-like synoviocytes from patients with rheumatoid arthritis. J Immunol. 2001;167:5381–5385. doi: 10.4049/jimmunol.167.9.5381. [DOI] [PubMed] [Google Scholar]
- 5.Yamamoto T, Eckes B, Mauch C, Hartmann K, Krieg T. Monocyte chemoattractant protein-1 enhances gene expression and synthesis of matrix metalloproteinase-1 in human fibroblasts by an autocrine IL-1 alpha loop. J Immunol. 2000;164:6174–6179. doi: 10.4049/jimmunol.164.12.6174. [DOI] [PubMed] [Google Scholar]
- 6.Gharaee-Kermani M, Denholm EM, Phan SH. Costimulation of fibroblast collagen and transforming growth factor beta1 gene expression by monocyte chemoattractant protein-1 via specific receptors. J Biol Chem. 1996;271:17779–17784. doi: 10.1074/jbc.271.30.17779. [DOI] [PubMed] [Google Scholar]
- 7.Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- 8.McCoy JM, Wicks JR, Audoly LP. The role of prostaglandin E2 receptors in the pathogenesis of rheumatoid arthritis. J Clin Invest. 2002;110:651–658. doi: 10.1172/JCI15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amano H, Hayashi I, Endo H, Kitasato H, Yamashita S, Maruyama T, et al. Host prostaglandin E(2)-EP3 signaling regulates tumor-associated angiogenesis and tumor growth. J Exp Med. 2003;197:221–232. doi: 10.1084/jem.20021408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao CY, Yuhki K, Hara A, Fujino T, Kuriyama S, Yamada T, et al. Prostaglandin E2 protects the heart from ischemia-reperfusion injury via its receptor subtype EP4. Circulation. 2004;109:2462–2468. doi: 10.1161/01.CIR.0000128046.54681.97. [DOI] [PubMed] [Google Scholar]
- 11.Takayama K, Garcia-Cardena G, Sukhova GK, Comander J, Gimbrone MA, Jr, Libby P. Prostaglandin E2 suppresses chemokine production in human macrophages through the EP4 receptor. J Biol Chem. 2002;277:44147–44154. doi: 10.1074/jbc.M204810200. [DOI] [PubMed] [Google Scholar]
- 12.Onai Y, Suzuki J, Kakuta T, Maejima Y, Haraguchi G, Fukasawa H, et al. Inhibition of IkappaB phosphorylation in cardiomyocytes attenuates myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;63:51–59. doi: 10.1016/j.cardiores.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Litwin SE, Katz SE, Morgan JP, Douglas PS. Serial echocardiographic assessment of left ventricular geometry and function after large myocardial infarction in the rat. Circulation. 1994;89:345–354. doi: 10.1161/01.cir.89.1.345. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki J, Ogawa M, Futamatsu H, Kosuge H, Tanaka H, Isobe M. A cyclooxygenase-2 inhibitor alters Th1/Th2 cytokine balance and suppresses autoimmune myocarditis in rats. J Mol Cell Cardiol. 2006;40:688–695. doi: 10.1016/j.yjmcc.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 15.Vivaldi MT, Kloner RA, Schoen FJ. Triphenyltetrazolium staining of irreversible ischemic injury following coronary artery occlusion in rats. Am J Pathol. 1985;121:522–530. [PMC free article] [PubMed] [Google Scholar]
- 16.Yamashiro S, Takeya M, Kuratsu J, Ushio Y, Takahashi K, Yoshimura T. Intradermal injection of monocyte chemoattractant protein-1 induces emigration and differentiation of blood monocytes in rat skin. Int Arch Allergy Immunol. 1998;115:15–23. doi: 10.1159/000023825. [DOI] [PubMed] [Google Scholar]
- 17.Suzuki J, Ogawa M, Sagesaka YM, Isobe M. Tea catechins attenuate ventricular remodeling and graft arterial diseases in murine cardiac allografts. Cardiovasc Res. 2006;69:272–279. doi: 10.1016/j.cardiores.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 18.Cheung PY, Sawicki G, Wozniak M, Wang W, Radomski MW, Schulz R. Matrix metalloproteinase-2 contributes to ischemia-reperfusion injury in the heart. Circulation. 2000;101:1833–1839. doi: 10.1161/01.cir.101.15.1833. [DOI] [PubMed] [Google Scholar]
- 19.Futamatsu H, Suzuki J, Kosuge H, Yokoseki O, Kamada M, Ito H, et al. Attenuation of experimental autoimmune myocarditis by blocking activated T cells through inducible costimulatory molecule pathway. Cardiovasc Res. 2003;59:95–104. doi: 10.1016/s0008-6363(03)00334-1. [DOI] [PubMed] [Google Scholar]
- 20.Gianturco SH, Ramprasad MP, Lin AH, Song R, Bradley WA. Cellular binding site and membrane binding proteins for triglyceride-rich lipoproteins in human monocyte-macrophages and THP-1 monocytic cells. J Lipid Res. 1994;35:1674–1687. [PubMed] [Google Scholar]
- 21.Zhu P, Ding J, Zhou J, Dong WJ, Fan CM, Chen ZN. Expression of CD147 on monocytes/macrophages in rheumatoid arthritis: its potential role in monocyte accumulation and matrix metalloproteinase production. Arthritis Res Ther. 2005;7:R1023–R1033. doi: 10.1186/ar1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jugdutt BI, Hutchins GM, Bulkley BH, Becker LC. Dissimilar effects of prostacyclin, prostaglandin E1, and prostaglandin E2 on myocardial infarct size after coronary occlusion in conscious dogs. Circ Res. 1981;49:685–700. doi: 10.1161/01.res.49.3.685. [DOI] [PubMed] [Google Scholar]
- 23.Woditsch I, Schror K. Prostacyclin rather than endogenous nitric oxide is a tissue protective factor in myocardial ischemia. Am J Physiol. 1992;263:H1390–H1396. doi: 10.1152/ajpheart.1992.263.5.H1390. [DOI] [PubMed] [Google Scholar]
- 24.Schror K, Woditsch I. Endogenous prostacyclin preserves myocardial function and endothelium-derived nitric oxide formation in myocardial ischemia. Agents Actions Suppl. 1992;37:312–319. doi: 10.1007/978-3-0348-7262-1_43. [DOI] [PubMed] [Google Scholar]
- 25.Hohlfeld T, Meyer-Kirchrath J, Vogel YC, Schror K. Reduction of infarct size by selective stimulation of prostaglandin EP(3)receptors in the reperfused ischemic pig heart. J Mol Cell Cardiol. 2000;32:285–296. doi: 10.1006/jmcc.1999.1072. [DOI] [PubMed] [Google Scholar]
- 26.Wang M, Zukas AM, Hui Y, Ricciotti E, Pure E, FitzGerald GA. Deletion of microsomal prostaglandin E synthase-1 augments prostacyclin and retards atherogenesis. Proc Natl Acad Sci USA. 2006;103:14507–14512. doi: 10.1073/pnas.0606586103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Narumiya S, Sugimoto Y, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 28.Nataraj C, Thomas DW, Tilley SL, Nguyen MT, Mannon R, Koller BH, et al. Receptors for prostaglandin E(2) that regulate cellular immune responses in the mouse. J Clin Invest. 2001;108:1229–1235. doi: 10.1172/JCI13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takayama K, Sukhova GK, Chin MT, Libby P. A novel prostaglandin E receptor 4-associated protein participates in antiinflammatory signaling. Circ Res. 2006;98:499–504. doi: 10.1161/01.RES.0000204451.88147.96. [DOI] [PubMed] [Google Scholar]
- 30.Hayashidani S, Tsutsui H, Shiomi T, Ikeuchi M, Matsusaka H, Suematsu N, et al. Anti-monocyte chemoattractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2003;108:2134–2140. doi: 10.1161/01.CIR.0000092890.29552.22. [DOI] [PubMed] [Google Scholar]
- 31.de Lemos JA, Morrow DA, Sabatine MS, Murphy SA, Gibson CM, Antman EM, et al. Association between plasma levels of monocyte chemoattractant protein-1 and long-term clinical outcomes in patients with acute coronary syndromes. Circulation. 2003;107:690–695. doi: 10.1161/01.cir.0000049742.68848.99. [DOI] [PubMed] [Google Scholar]
- 32.Matsumori A, Furukawa Y, Hashimoto T, Yoshida A, Ono K, Shioi T, et al. Plasma levels of the monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 are elevated in patients with acute myocardial infarction. J Mol Cell Cardiol. 1997;29:419–423. doi: 10.1006/jmcc.1996.0285. [DOI] [PubMed] [Google Scholar]
- 33.Ono K, Matsumori A, Furukawa Y, Igata H, Shioi T, Matsushima K, et al. Prevention of myocardial reperfusion injury in rats by an antibody against monocyte chemotactic and activating factor/monocyte chemoattractant protein-1. Lab Invest. 1999;79:195–203. [PubMed] [Google Scholar]
- 34.Stuve O, Chabot S, Jung SS, Williams G, Yong VW. Chemokine-enhanced migration of human peripheral blood mononuclear cells is antagonized by interferon beta-1b through an effect on matrix metalloproteinase-9. J Neuroimmunol. 1997;80:38–46. doi: 10.1016/s0165-5728(97)00134-3. [DOI] [PubMed] [Google Scholar]
- 35.Okuma T, Terasaki Y, Kaikita K, Kobayashi H, Kuziel WA, Kawasuji M, et al. C-C chemokine receptor 2 (CCR2) deficiency improves bleomycin-induced pulmonary fibrosis by attenuation of both macrophage infiltration and production of macrophage-derived matrix metalloproteinases. J Pathol. 2004;204:594–604. doi: 10.1002/path.1667. [DOI] [PubMed] [Google Scholar]
- 36.Damas JK, Aukrust P, Ueland T, Odegaard A, Eiken HG, Gullestad L, et al. Monocyte chemoattractant protein-1 enhances and interleukin-10 suppresses the production of inflammatory cytokines in adult rat cardiomyocytes. Basic Res Cardiol. 2001;96:345–352. doi: 10.1007/s003950170042. [DOI] [PubMed] [Google Scholar]
- 37.Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. doi: 10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.