

Abstract
Aims
Hypercholesterolaemia and myeloperoxidase (MPO) overexpression are two well-recognized risk factors for ischaemic heart disease. Peroxisome proliferator-activated receptor-γ (PPARγ) agonists have recently been shown to reduce ischaemic heart injury in hypercholesterolaemic animals. However, whether PPARγ agonists may exert their cardioprotective effects by eliminating those risk factors that increase ischaemic injury remains unknown.
Methods and results
Male New Zealand rabbits were fed with a normal or a high-cholesterol diet for 8 weeks, treated with vehicle or rosiglitazone (RSG, 3 mg/kg/day for the last 5 weeks) and subjected to myocardial ischaemia/reperfusion (1 h/4 h). MPO expression, activity, and distribution, cardiac caspase-3 activity, and myocardial infarct size were determined. Diet-induced hypercholesterolaemia caused a significant increase in neutrophil MPO expression/activity (7.2-/5.4-fold). Hypercholesterolaemia also tripled MPO activity in ischaemic/reperfused hearts when compared with rabbits fed with a normal diet. Surprisingly, MPO immunostaining was not only observed in perivascular and extracellular spaces in ischaemic/reperfused hearts, but also in cardiomyocytes. This intracardiomyocyte MPO staining was further intensified by hypercholesterolaemia. There is a strong positive correlation between cardiac MPO activity and caspase-3 activity, and treatment with an MPO inhibitor significantly reduced post-ischaemic caspase-3 activation. Treatment with RSG markedly inhibited hypercholesterolaemia-induced leucocyte MPO overexpression and activation, reduced MPO activity in ischaemic/reperfused hearts, decreased caspase-3 activity, and reduced myocardial infarct size (P < 0.01).
Conclusion
Our results demonstrated that hypercholesterolaemia and MPO overexpression are causally related and that PPARγ agonists may have great therapeutic value in ischaemic heart disease patients with multiple complications such as hypercholesterolaemia and diabetes.
Keywords: Cholesterol homeostasis, Myocardial inflammation, Myocyte apoptosis and necrosis, Reperfusion injury
1. Introduction
It is well recognized that hypercholesterolaemia plays a role in not only initiating myocardial ischaemia (MI), but also in propagating ischaemic damage.1 Although enhanced reactive oxygen species production and resultant endothelial dysfunction, lipid accumulation and subsequent development of atherosclerotic plaques, and coronary spasm around existing atherosclerotic lesions have been shown to play causative roles in hypercholesterolaemia-initiated MI, the mechanisms responsible for enhanced cardiomyocyte injury following ischaemia/reperfusion in patients with hypercholesterolaemia remain poorly defined.
Emerging evidence obtained from animal studies as well as clinical observations suggests that myeloperoxidase (MPO), an enzyme that is primarily expressed in neutrophils, is a critical inflammatory mediator and an independent risk factor for coronary artery disease.2 MPO stimulates endothelial apoptosis,3 induces endothelial dysfunction,4 and increases plaque vulnerability,5 thus contributing to the development of MI. Moreover, clinical studies have demonstrated that MPO is a major risk factor for ischaemic heart disease and that the increased leucocyte MPO levels may not only contribute to the development of ischaemic heart disease, but also increase cardiomyocyte injury after ischaemia.6,7 Although previous studies have demonstrated that hypercholesterolaemia is associated with an increased inflammatory response,8 the specific contribution of MPO remains unclear. Moreover, although it is well recognized that MI/reperfusion initiates a typical inflammatory reaction in which neutrophils play a critical role,9 the role of MPO in neutrophil-mediated ischaemia/reperfusion injury remains largely unknown.
Peroxisome proliferator-activated receptor-γ (PPARγ), a member of the nuclear receptor superfamily of ligand-activated transcription factors, is a key regulator of adipogenesis and lipid metabolism.10 Besides their well-described insulin-sensitizing property, synthetic PPARγ agonists, such as rosiglitazone (RSG), have been recently shown to possess strong anti-inflammatory properties.11 In vitro treatment with a PPARγ agonist reduces intercellular adhesion molecule-1 (ICAM-1) expression in activated endothelial cells,12 inhibits production of proinflammatory cytokines (TNF-α, IL-6, and IL-1β) by activated monocytes,13 decreases transcription of monocyte chemoattractant protein,14 and significantly reduces monocyte/macrophage homing to atherosclerotic plaques.12 In a recent study, we have demonstrated that treatment with RSG in vivo significantly reduces MI/reperfusion injury in hypercholesterolaemic rabbits.15 However, whether the anti-inflammatory property of RSG that has been demonstrated in in vitro experiments may contribute to its cardioprotective effect remains to be determined. Moreover, a recent study reported that a PPARγ agonist markedly inhibits MPO expression in macrophages stimulated with granulocyte/macrophage colony-stimulating factor (GMCSF).16 Whether a PPARγ agonist may exert a similar effect on neutrophil MPO expression in a pathologically relevant in vivo model, such as hypercholesterolaemia, has never been previously studied.
Therefore, the aims of the present study were (i) to investigate whether a diet-induced hypercholesterolaemia may enhance MPO expression; if so, (ii) to determine whether increased MPO activity may contribute to enhanced MI/reperfusion injury in hypercholesterolaemic animals; and (iii) to determine whether treatment with RSG may downregulate MPO expression and thus reduce post-ischaemic myocardial injury.
2. Methods
2.1. Animals and dietary protocol
Adult male New Zealand white rabbits weighing 2.0–2.5 kg were provided with food and water ad libitum. Blood was drawn from the central ear artery of each rabbit, and baseline plasma lipids, leucocyte count, and leucocyte MPO content (Western) and activity were determined. On Day 0 of the experiment, rabbits were assigned to either a normal or a high-cholesterol (HC, normal rabbit diet supplemented with 1% cholesterol) diet for 8 weeks. Three weeks after being fed a normal or an HC diet, the rabbits’ blood was redrawn and plasma lipids were determined once again. Rabbits were then randomized to receive either vehicle or RSG (3 mg/kg/day, oral gavage) during the remaining 5 weeks.15 All rabbit diets were prepared by and purchased from Zeigler Bros., Inc. (Gardners, PA, USA). The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and was approved by the Thomas Jefferson University Committee on Animal care.
2.2. Experimental preparation
At the end of 8 weeks of the experimental period, rabbits were anaesthetized with sodium pentobarbital (30 mg/kg, iv) and ventilated with a Harvard small animal respirator. After midline thoracotomy, a 4-0 silk ligature was placed around the major marginal branch of the left circumflex coronary 10–12 mm from its origin and MI was initiated by complete ligation of the marginal coronary artery. After 60 min of ischaemia, the ligature was untied and the ischaemic myocardium was reperfused (R) for 4 h. At the end of reperfusion, the heart was quickly excised and ischaemic-reperfused tissue (area-at-risk, AAR) was isolated for apoptotic, biochemical assays, and western blots as described below. Sham MI and reperfusion rabbits were subjected to the same surgical procedures performed on MI/R rabbits, except that the coronary artery was left untied.
2.3. Measurement of myeloperoxidase content and activity
Immediately after animals were anaesthetized and before any surgery procedure was performed, peripheral blood (2 mL) was drawn from the central ear artery and mixed with 200 IU of heparin sodium. Neutrophils were isolated and lysed as described previously7 and neutrophil MPO content was measured by western blotting analysis and ELISA according to procedures recommended by the manufacturer (Assay Design Inc.). Leucocyte and cardiac MPO activity was determined as described in our previous study.17 MPO activity was calculated as IU/g protein and results were expressed as fold change over normal diet control.
2.4. Measurement of caspase-3 activity in cardiac tissue
Myocardial caspase-3 activity was determined using colorimetric assay kits (Chemicon International, Inc.) according to the manufacturer’s instructions. Results were expressed as fold change over sham control.
2.5. Assessment of area-at-risk and infarct size
At the end of the 4 h reperfusion period, the ligature around the coronary artery was re-occluded through the previous ligation. Ischaemic reperfused area (AAR) was identified by negative Evans Blue dye staining and infarcted area was identified by negative triphenyltetrazolium chloride (TTC) staining. Evan’s Blue stained area (area-not-at-risk, ANAR), TTC stained area (ischaemic but viable tissue), and TTC-negative stained area (infarcted myocardium) were digitally measured using an IPLab 3.6. Myocardial infarct size was expressed as a percentage of infarct area over total AAR.
2.6. Immunohistochemical detection of myeloperoxidase and intercellular adhesion molecule-1 expression in the ischaemic/reperfused heart
Optimal cutting temperature compound-embedded tissues were cut into 6 µm thick and stained with primary antibody (anti-MPO monoclonal antibody, Abcam; anti-ICAM-1 monoclonal antibody, BD Biosciences) and biotinylated secondary antibody. MPO was detected with alkaline phosphatase kit (VECTASTAIN® ABC-AP kit with Vector Red as a substrate, Vector Laboratories) and ICAM-1 expression was detected with horseradish-peroxidase kit (VECTASTAIN® ABC kit, Vector Laboratories). Negative control slides were prepared by omitting the primary antibody and were developed with 3,3 diaminobenzidine for 5 min.
2.7. Western blotting
Leucocytes were isolated as described above and lysed with lysis buffer. Leucocyte proteins were separated by electrophoresis on SDS–PAGE and transferred to nitrocellulose membranes. After being blocked with 5% milk, the immunoblots were probed with anti-MPO antibody (1:1000, Abcam) overnight at 4°C. Nitrocellulose membranes were then incubated with HRP-conjugated anti-mouse IgG antibody (1:2000, Cell Signaling) for 1 h and the blot was developed with a Supersignal chemiluminescence detection kit (Pierce). The immunoblotting was visualized with a Kodak Image Station 400 and the blot densities were analysed with Kodak 1D software. Results were expressed as fold change over normal diet control.
2.8. Statistical analysis
All values in the text, table, and figures are presented as means ± SEM of n independent experiments. All data (except western blot density) were subjected to ANOVA followed by Bonferroni correction for post hoc t test. Western blot densities were analysed with the Kruskal–Wallis test followed by Dunn’s post test. Probabilities of 0.05 or less were considered to be statistically significant.
3. Results
3.1. High-cholesterol diet markedly increased plasma cholesterol and LDL levels
Eight weeks of an HC diet resulted in a dramatic increase in plasma cholesterol (2549 ± 311 vs. 44.8 ± 3.9 mg/dL, P < 0.01) and LDL levels (1648 ± 158 vs. 17.4 ± 2.5 mg/dL, P < 0.01). There were no significant differences between groups in plasma lipid profiles before the onset of the study (time 0), after 3 weeks of an HC diet (before RSG treatment), and at the end of 8 weeks of cholesterol-feeding (after 5 weeks of treatment with vehicle or RSG) (Table 1). These results indicate that RSG failed to improve plasma lipid profiles in this diet-induced hypercholesterolaemic model, and therefore, any cardiovascular effects observed could not be attributed to this mechanism. In addition, 8 weeks of an HC diet did not result in a significant change in total leucocyte count (Table 1), and treatment with RSG had no effect on total leucocyte count.
Table 1.
Lipid profiles and leucocyte counts in vehicle-treated (n = 11) and rosiglitazone-treated (n = 12) groups
| CHOL (mg/dL) |
LDL (mg/dL) |
Leucocyte counts (mL) |
||||
|---|---|---|---|---|---|---|
| Vehicle | RSG | Vehicle | RSG | Vehicle | RSG | |
| 0 | 44.8 ± 3.9 | 32.4 ± 3.9 | 17.4 ± 2.5 | 16.4 ± 2.3 | 7284 ± 118 | 7354 ± 169 |
| 3 weeks | 1318 ± 166 | 1231 ± 214 | 948 ± 115 | 879 ± 140 | 7415 ± 154 | 7198 ± 154 |
| 8 weeks | 2549 ± 311 | 2485 ± 235 | 1648 ± 158 | 1598 ± 147 | 7370 ± 139 | 7299 ± 168 |
3.2. Hypercholesterolaemia significantly increased leucocyte myeloperoxidase level and activity
Hypercholesterolaemia and increased leucocyte MPO levels are two well-recognized risk factors for ischaemic heart disease. Although previous studies have demonstrated that patients with hypercholesterolaemia also have an increased inflammatory response, whether hypercholesterolaemia may cause increased expression of MPO has never been previously studied. As illustrated in Figure 1, 8 weeks of an HC diet resulted in a marked increase in leucocyte MPO level as determined by western blotting (Figure 1A) and ELISA (Figure 1B). To further determine whether this increased MPO expression may result in a concomitant increase in MPO activity, leucocytes were isolated from time-matched control rabbits or hypercholesterolaemic rabbits and MPO activity was determined. As summarized in Figure 1C, a 6.3-fold increase in MPO activity was observed in leucocytes isolated from hypercholesterolaemic rabbits compared with leucocytes isolated from control animals. These results demonstrated for the first time that hypercholesterolaemia increases leucocyte MPO content and activity.
Figure 1.
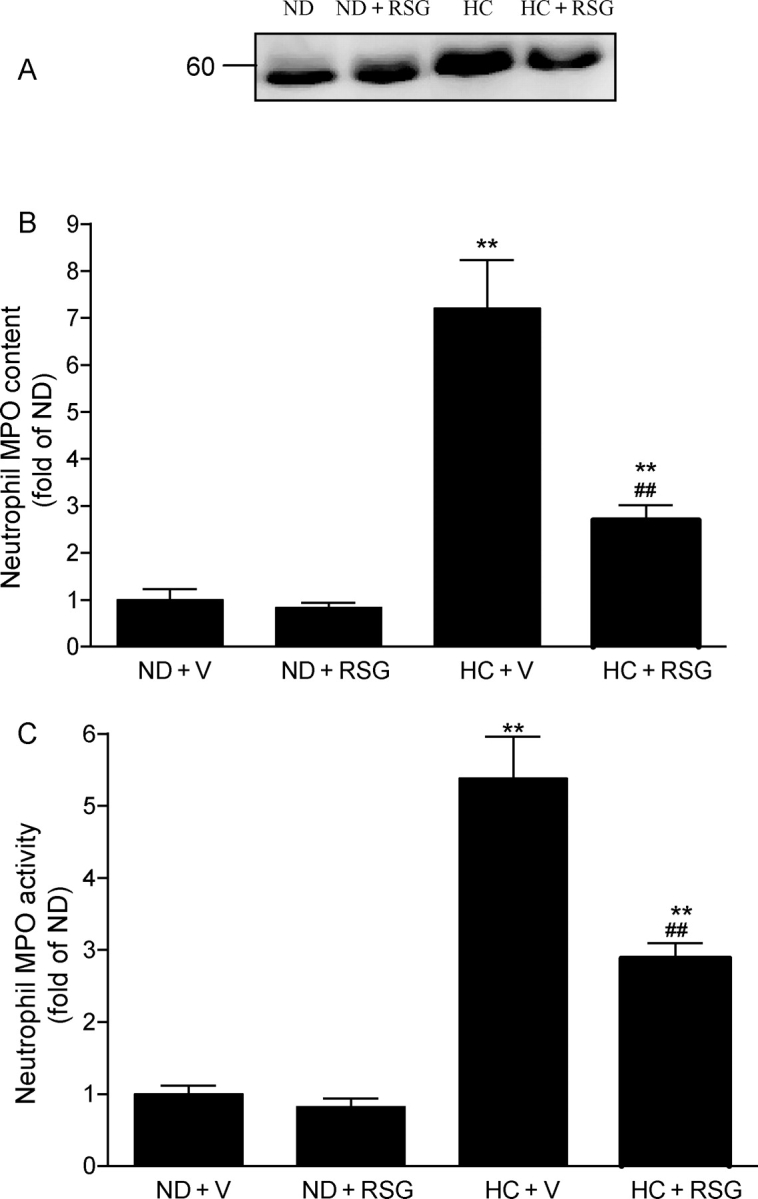
Hypercholesterolaemia increases neutrophil myeloperoxidase (MPO) expression (A, typical western blots from 3 to 5 rabbits/group), MPO content (B, ELISA results from 8 to 10 rabbits/group), and MPO activity (C, spectrophotometry measurement from 10 to 14 rabbits/group) and their blockade by RSG. Results were expressed as fold change over rabbits fed with a normal diet. ND, normal diet; HC diet, high-cholesterol diet; V, vehicle; RSG, rosiglitazone. The same abbreviations were used for all figures. **P < 0.01 vs. ND + V. ##P < 0.01 vs. HC + V.
3.3. Treatment with rosiglitazone markedly inhibited hypercholesterolaemia-induced myeloperoxidase upregulation
A recent in vitro experiment demonstrated that PPARγ agonists differentially regulate MPO expression in macrophages stimulated by GMCSF or macrophage colony-stimulating factor (MCSF).16 To determine whether PPARγ agonists may also regulate leucocyte MPO content in vivo, the effect of RSG treatment on leucocyte MPO content and MPO activity was determined in rabbits fed with a normal or an HC diet. As illustrated in Figure 1, administration of RSG had no significant effect on MPO content and activity in rabbits fed with a normal diet. However, the same treatment resulted in a greater than 60% reduction in hypercholesterolaemia-induced leucocyte MPO upregulation and MPO activity. These results indicate that although RSG has no significant effect on leucocyte MPO content under basal conditions, it markedly blocked hypercholesterolaemia-induced MPO overexpression.
3.4. Treatment with rosiglitazone attenuated the ischaemia/reperfusion-induced increase in cardiac myeloperoxidase activity in both normal and high-cholesterol diet groups
Having demonstrated that administration of RSG significantly reduced hypercholesterolaemia-induced leucocyte MPO upregulation, we next determined whether this treatment may also reduce MPO activity in ischaemic/reperfused cardiac tissue, thus contributing to its previously reported cardiac protective effects. Consistent with results from other investigators as well as results from our laboratory, ischaemia/reperfusion markedly increased MPO activity in ischaemic/reperfused cardiac tissue. MPO activity in ischaemic/reperfused cardiac tissue was further increased in hypercholesterolaemic rabbits (Figure 2A). This hypercholesterolaemia-induced additional increase in MPO activity was completely abolished when hypercholesterolaemic rabbits were treated with RSG for 5 weeks (Figure 2A). Interestingly, although administration of RSG had no significant effect on leucocyte MPO content in rabbits fed with a normal diet, this treatment significantly reduced cardiac MPO activity in normal diet-fed rabbits subjected to ischaemia/reperfusion (Figure 2A), suggesting that RSG may inhibit the ischaemia/reperfusion-initiated inflammatory response by other mechanisms in addition to inhibiting leucocyte MPO expression.
Figure 2.
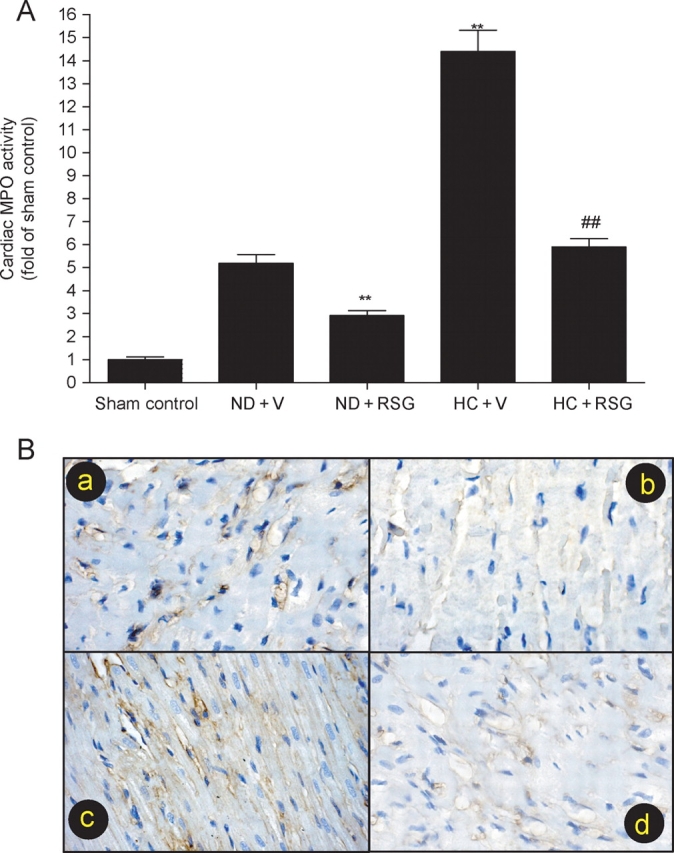
(A) Cardiac myeloperoxidase (MPO) activity in ischaemic/reperfused area expressed as fold change over rabbits fed with a normal diet subjected to sham myocardial ischaemia/reperfusion. **P < 0.01 vs. ND + V. ##P < 0.01 vs. HC + V. n = 10–14/group. (B) Typical photographs for intercellular adhesion molecule-1 immunostaining in area-at-risk (ischaemia/reperfused). (a) normal diet treated with vehicle; (b) normal diet treated with rosiglitazone (RSG); (c) high-cholesterol (HC) diet treated with vehicle; (d) HC diet treated with RSG.
Previous studies have demonstrated that PPARγ agonists inhibit ICAM-1 expression in cultured endothelial cells. To determine whether in vivo administration of RSG may also inhibit ICAM-1 expression in ischaemic/reperfused hearts and thus inhibit leucocyte accumulation, an additional experiment was performed. As illustrated in Figure 2B, ischaemia/reperfusion caused a significant upregulation of ICAM-1 expression in rabbits fed with a normal diet [Figure 2B(a)], which was almost completely abolished by RSG treatment [Figure 2B(b)]. Ischaemia/reperfusion-induced ICAM-1 expression was further increased in hypercholesterolaemic rabbits [Figure 2B(c)], which was also significantly reduced when rabbits were treated with RSG [Figure 2B(d)]. Using a histology expression score system originally described by Weyrich et al.,18 per cent ICAM-1 positive staining coronary microvessels were calculated. Treatment with RSG significantly reduced ICAM-1-positive microvessels in the normal diet group (from 49 ± 5.4% to 9.1 ± 1.8%, P < 0.01) as well as in the HC diet group (from 78.4 ± 6.9% to 17.6 ± 2.5%, P < 0.01). Taken together, these results strongly suggest that RSG may exert its anti-inflammatory effect by multiple mechanisms, including inhibiting ischaemia/reperfusion-induced adhesion molecular expression and blocking hypercholesterolaemia-induced MPO overexpression.
3.5. Myeloperoxidase distribution in ischaemic/reperfused cardiac tissue
Although many investigators (including us) have previously reported that MPO activity was significantly increased in ischaemic/reperfused cardiac tissue, MPO activity was used as a quantitative marker for neutrophil accumulation. MPO distribution (e.g. intravascular vs. extravascular, extracellular vs. intracellular) in ischaemic/reperfused tissue has not been previously determined. To our surprise, although only scattered MPO immunostaining was observed in vascular tissue in non-ischaemic areas (Figure 3A, ANAR), intensive MPO immunostaining was observed not only in vascular tissue, but also in cardiomyocytes in ischaemic/reperfused areas (Figure 3A, AAR; Figure 3B). This result indicates that MPO was not only released from neutrophils infiltrating the ischaemic/reperfused heart, but also entered (either actively or passively) ischaemic/reperfused cardiomyocytes, where they may cause cardiomyocyte injury by multiple mechanisms.
Figure 3.
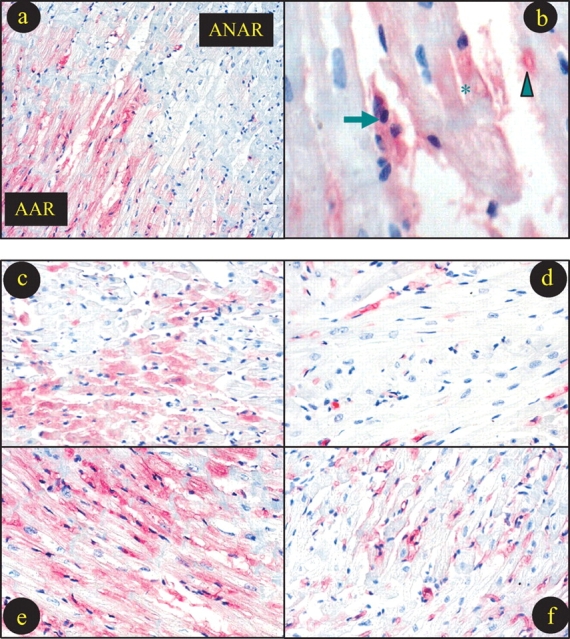
Typical photographs for myeloperoxidase (MPO) immunostaining. (a) Sample from a hypercholesterolaemic rabbit treated with vehicle showing that intensive MPO staining presents in area-at-risk (AAR), but not in area-not-at-risk. (b) High-power magnification photograph showing MPO staining in leucocytes (arrow), microvessel (arrowhead) and cardiomyocyte (star). Remaining photographs (c–f) were taken from AAR with different treatments. (b) Normal diet treated with vehicle; (c) normal diet treated with rosiglitazone (RSG); (d) high-cholesterol diet treated with vehicle; (e) high-cholesterol diet treated with RSG.
Consistent with our results described above that hypercholesterolaemia further enhanced MPO activity in ischaemic/reperfused cardiac tissue, intensity of MPO staining was also increased in hypercholesterolaemic rabbits (Figure 3E) when compared with normal diet-fed rabbits (Figure 3C). Moreover, treatment with RSG virtually abolished MPO immunostaining in normal diet-fed rabbits (Figure 3D) and significantly reduced MPO immunostaining in hypercholesterolaemic rabbits (Figure 3F).
3.6. Treatment with rosiglitazone reduced apoptosis and decreased infarct size after ischaemia
The results described above clearly demonstrated that RSG significantly attenuated hypercholesterolaemia-induced leucocyte MPO upregulation and blocked MPO infiltration into ischaemic/reperfused cardiomyocytes. To determine whether this anti-MPO property of RSG may contribute to its cardioprotective effect, several additional observations were made. Consistent with our previous results, treatment with RSG significantly attenuated myocardial apoptosis (as determined by caspase-3 activation) in both normal diet-fed rabbits and HC diet-fed rabbits (Figure 4A). In addition, our current study demonstrated that treatment with RSG also significantly reduced myocardial infarct size (Figure 4B).
Figure 4.
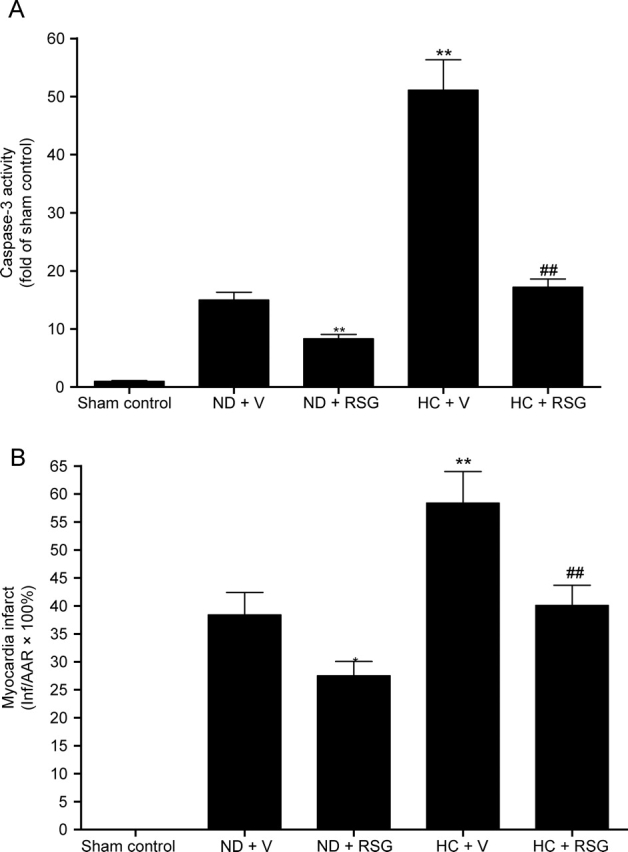
(A) Cardiac caspase-3 activity in ischaemic/reperfused area expressed as fold change over rabbits fed with a normal diet subjected to sham myocardial ischaemia/reperfusion. (B) Myocardial infarct size expressed as percent of area-at-risk. *P < 0.05; **P < 0.01 vs. ND + V; ##P < 0.01 vs. HC + V. n = 10–14/group.
To determine the contribution of MPO to post-ischaemic myocardial injury, the correlation between cardiac MPO activity and caspase activation in 48 animals (normal and HC diet) subjected to ischaemia/reperfusion (treated with vehicle and RSG) was analysed. As illustrated in Figure 5, a strong positive correlation (P < 0.01) between cardiac MPO activity and caspase-3 activity was observed. This result suggests that MPO may be a significant contributor to post-ischaemic myocardial injury.
Figure 5.

Correlation between myeloperoxidase (MPO) [area-at-risk (AAR), expressed as fold change over rabbits fed with a normal diet subjected to sham myocardial ischaemia/reperfusion) and caspase-3 activity (AAR, expressed as fold change over rabbits fed with a normal diet subjected to sham myocardial ischaemia/reperfusion). A strong positive correlation was found between MPO and caspase-3 activity (n = 48, P < 0.01).
3.7. Acute treatment of rabbits with an myeloperoxidase inhibitor significantly reduced cardiac myeloperoxidase activity and decreased cardiac caspase-3 activity
The results described in Figure 5 demonstrated that there is a strong positive correlation between cardiac MPO activity and caspase-3 activation. However, whether increased MPO activity is a cause of ischaemia/reperfusion-induced caspase-3 activation or is an inflammatory response caused by increased apoptotic cell death remains unknown. To establish a causative link between MPO infiltration and caspase-3 activation, an additional experiment was performed. Sixteen additional rabbits were subjected to MI and reperfusion as described above. Ten minutes before ischaemia, rabbits were treated with vehicle (10% DMSO in pH 6.5 PBS, 1 mL/kg) or 4-aminobenzoic acid hydrazide (ABAH, a specific MPO inhibitor19,20 that has been shown to reduce MPO-dependent apoptosis in HL-60 cells,21,22 6.6 µmol/kg, ip). In a recent study, this compound has been administered to the rabbit at a comparable dose for 8 weeks to inhibit MPO activity and no toxic effect is reported.23 As expected, treatment with ABAH almost completely abolished MPO activity in ischaemia/reperfused cardiac tissue. Most interestingly, this treatment also significantly reduced cardiac caspase-3 activity (Figure 6). Since direct addition of ABAH did not interfere with caspase-3 assay (data not shown), this result provides direct evidence that MPO is a significant contributor to post-ischaemic cardiomyocyte apoptosis. Therefore, those therapeutic interventions, such as RSG, that reduce MPO expression and/or MPO infiltration may exert their cardioprotective effects, at least in part, by their anti-MPO activity.
Figure 6.
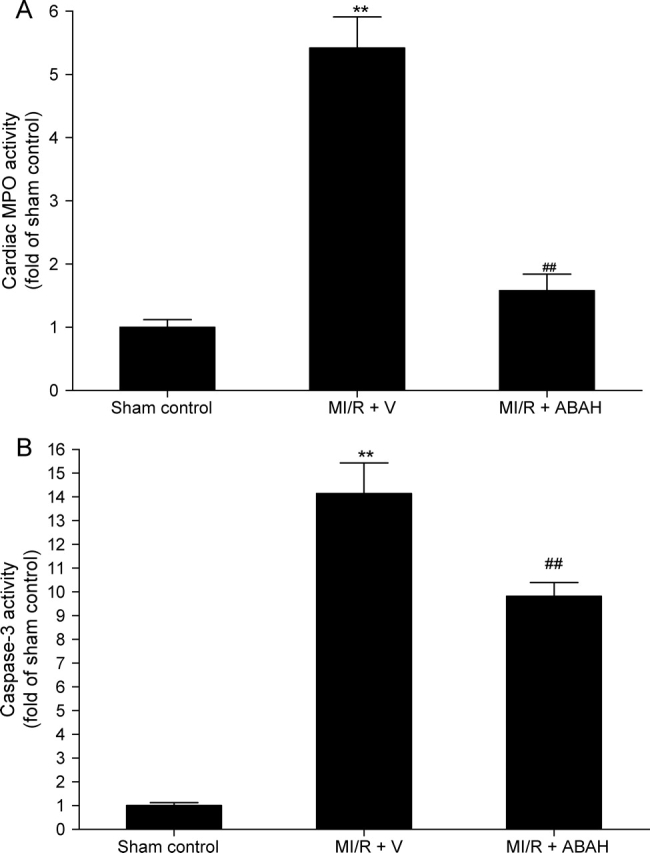
Effect of acute treatment with ABAH, a selective myeloperoxidase (MPO) inhibitor, on cardiac MPO and caspase-3 activity in ischaemic/reperfused area. ABAH, 4-aminobenzoic acid hydrazide. **P < 0.01 vs. ND + V; ##P < 0.01 vs. HC + V. n = 8/group.
4. Discussion
Three novel findings have been uncovered from the current study. First, we have demonstrated for the first time that hypercholesterolaemia markedly increases leucocyte MPO content, suggesting that hypercholesterolaemia may exert its adverse effect on ischaemia/reperfusion injury by enhancing the MPO-mediated inflammatory response. Secondly, we have provided the first evidence that in the ischaemic/reperfused heart, MPO is not only present in the perivascular area and extracellular spaces, but is also present within cardiomyocytes. Treatment with an MPO inhibitor significantly reduced caspase-3 activation induced by ischaemia/reperfusion, suggesting that MPO may cause cardiomyocyte injury by producing toxic molecules within the cardiomyocytes. Thirdly, we have demonstrated that treatment with RSG markedly inhibited hypercholesterolaemia-induced MPO upregulation in neutrophils and reduced MPO activity in ischaemic/reperfused cardiac tissue. Although general anti-inflammatory properties of PPARγ agonists have been recently reported, the specific effect of a PPARγ agonist on MPO expression has never been previously studied in a pathologically relevant in vivo model.
4.1. Increased myeloperoxidase content may contribute to enhanced myocardial ischaemia/reperfusion injury in hypercholesterolaemia
Hypercholesterolaemia has long been recognized as a major risk factor for coronary artery disease and acute myocardial infarction.1 However, the majority of previously published studies were focused on the effect of hypercholesterolaemia on endothelial function, vascular injury, and development of atherosclerosis.24,25 Several previous studies have demonstrated that hypercholesterolaemia markedly increases MI/reperfusion injury as evidenced by increased apoptotic and necrotic cell death, and enlarged infarct size.26,27 Although it is certain that hypercholesterolaemia-induced vascular injury plays a critical role in the development of MI, the increased cardiomyocyte injury after ischaemia/reperfusion in hypercholesterolaemic animals cannot be completely attributed to hypercholesterolaemia-induced endothelial dysfunction and vascular injury.
In the present study, we have demonstrated that 8 weeks of an HC diet markedly increased leucocyte MPO content and enhanced leucocyte MPO activity. Although previous experimental and clinical studies have demonstrated that hypercholesterolaemia is associated with increased inflammatory responses such as enhanced endothelial adhesion molecule expression and increased plasma cytokine levels,8 the current study is the first to demonstrate that hypercholesterolaemia increases leucocyte MPO content. Since little is known at the present time about the factors that regulate MPO expression in vivo,28 it remains unclear how hypercholesterolaemia results in MPO upregulation. Answering this critical question will require a substantial number of additional experiments that are currently under way in our laboratory. Nevertheless, our present experiments have demonstrated that hypercholesterolaemia and increased MPO content, two of the most recognized risk factors for ischaemic heart disease, are causally related. Since recent studies have demonstrated that increased MPO expression is a direct contributor to myocardial injury, current experimental results strongly suggest that hypercholesterolaemia may enhance cardiomyocyte susceptibility to ischaemia/reperfusion injury partially by this novel MPO upregulation mechanism.
MPO is a highly-expressed haemoprotein in neutrophils (∼5% of neutrophil protein) that plays a major role in a variety of inflammatory responses.28 Substantial evidence exists that neutrophils play a critical role in myocardial reperfusion injury, and many therapeutic interventions that block neutrophil accumulation have been shown reduced post-ischaemic myocardial injury.29 However, the specific role of MPO in neutrophil-initiated myocardial reperfusion injury has not been identified. Limited literature suggests that extravasated and activated neutrophils release MPO to the extracellular space, where it uses hydrogen peroxide that is also released by the neutrophils to form the toxic agent hypochlorous acid (HOCl) and other oxidizing species,28 thus contributing to oxidative cardiovascular injury. An unexpected finding of the current study is that in the ischaemic/reperfused heart, MPO immunostaining is not only present in the perivascular area, but also within ischaemic/reperfused cardiomyocytes, and this cardiomyocyte-containing MPO is further intensified in hypercholesterolaemic rabbits. In a recent study, Eiserich et al.30 reported that in aortic tissues obtained from rats injected with lipopolysaccharide, MPO was not only observed on the endothelial surface but also within endothelial cells. Moreover, they also provided direct evidence indicating that MPO functions as an NO oxidase and that endothelial-associated MPO is responsible for endothelial dysfunction. Our current study extends this observation and demonstrated that in the ischaemic/reperfused heart, leucocyte-secreted MPO not only enters vascular endothelial cells as previously reported, but also enters ischaemic/reperfused cardiomyocytes.
Given the recent discovery that the severity of free radical-induced cellular injury is critically affected by intracellular compartmentation of oxidant/antioxidant molecules,31 translocation of MPO into the cardiomyocyte may markedly facilitate MPO-induced cardiomyocyte injury. Intracardiomyocyte MPO can directly utilize cardiomyocyte-generated H2O2 (which is known to be markedly increased after ischaemia and reperfusion) to generate HOCl inside cardiomyocytes, thus initiating oxidative cardiomyocyte death. Moreover, recent studies have demonstrated that MPO utilizes H2O2 and nitrite to generate nitrogen dioxide and results in nitrative cell death.32–36 Two recent studies have demonstrated that H2O2-induced apoptosis and caspase-3 activation in HL-60 human leukaemia cells are MPO-dependent. However, this MPO-induced apoptosis is mediated neither by H2O2 itself nor by hydroxyl radicals.21,22 These results strongly suggest that MPO may increase the production of reactive nitrogen species and thus stimulate apoptotic cell death by nitrative inactivation of anti-apoptotic molecules, such as thioredoxin. In this connection, we have provided direct evidence here that treatment with an MPO inhibitor significantly inhibited MPO activity in the ischaemic/reperfused heart and reduced caspase-3 activation.
4.2. Anti-inflammation as a mechanism for rosiglitazone’s anti-apoptotic and cardioprotective effects
Another novel finding of the present study is that in vivo administration of RSG, a PPARγ agonist, blocked hypercholesterolaemia-induced leucocyte MPO overexpression and reduced MPO activity in ischaemic reperfused hearts. Several previous studies have demonstrated that PPARγ agonists block lipopolysaccharide-induced iNOS expression and inhibit NO production.37,38 In a recent study, we have demonstrated that hypercholesterolaemia-induced overexpression of gp91phox, a critical component of NADPH oxidase, is significantly blocked by in vivo treatment of RSG.39 Given the fact that MPO-catalysed, H2O2/NO-dependent protein nitration is increasingly recognized as a critical mechanism responsible for tissue injury,21,22,32,34,35 the triple inhibitory effect of PPARγ agonists (i.e. iNOS, NADPH oxidase and MPO) highly suggests that the anti-inflammatory property of the PPAR family may play a critical role in their cardioprotective effect.
The mechanisms by which PPARγ agonists inhibit hypercholesterolaemia-induced MPO overexpression cannot be answered by the present study. In a recent report, Kumar et al.16 demonstrated that PPARγ agonists strongly regulate MPO gene expression in cultured macrophages. An unusual aspect of this regulation is the opposite effects of PPARγ ligands in macrophages cultured in GMCSF vs. MCSF. PPARγ agonists upregulated MPO expression in MCSF-stimulated macrophages and downregulated MPO expression in GMCSF-stimulated macrophages. Moreover, this study provided three pieces of evidence that strongly suggest that PPARγ regulates MPO expression by binding to an Alu element encoding four hexamer repeats recognized by nuclear receptors (AluRRE). First, there is the PPARγ binding site in the AluRRE. Secondly, the −463GA polymorphism in the AluRRE strongly affects PPARγ regulation. Thirdly, the mouse MPO gene lacks the primate-specific AluRRE and is completely unresponsive to PPARγ ligands. Our present study demonstrated that in vivo administration of RSG significantly downregulates hypercholesterolaemia-induced MPO overexpression in neutrophils. This result is similar to that observed by Kumar et al. in GMCSF-stimulated macrophages. However, whether hypercholesterolaemia upregulates neutrophil MPO expression by a similar molecular mechanism remains unknown. Moreover, whether RSG may inhibit neutrophil MPO expression by an AluRRE-dependent mechanism needs to be further studied.
It should be indicated that, in the present study, the profound anti-apoptotic effect of RSG treatment did not proportionally translate into infarct-sparing effect. A great deal of evidence exists that apoptosis is the major form of cell death after MI/R and that treatment of normal animals subjected to MI/R with numerous anti-apoptotic molecules have been proven to reduce infarct size proportionally. It is possible that the pathologic stress sustained by cells in our study (i.e. hypercholesterolaemia plus MI/R) was of too great extent, such that some of the apoptotic cells rescued by RSG may die through other mechanisms such as necrosis. This possibility will be directly investigated in our future studies.
In summary, we have demonstrated that hypercholesterolaemia, a well-recognized risk factor for ischaemic heart disease, causes significant overexpression of leucocyte MPO, a newly identified risk factor for coronary artery disease. Treatment with RSG, a drug that is currently used in the treatment of type-2 diabetes, almost completely abolished hypercholesterolaemia-induced MPO overexpression, markedly inhibited MPO activity in the ischaemic/reperfused heart, and significantly reduced myocardial reperfusion injury. These novel results are not only scientifically important because they provide direct evidence to link hypercholesterolaemia with MPO overexpression, but are also clinically significant because they demonstrate that PPARγ agonists inhibit an inflammatory response through multiple mechanisms and might be the therapeutic choice for ischaemic heart disease patients with multiple risk factors such as hypercholesterolaemia and diabetes.
Conflict of interest: none declared.
Funding
This research was supported by the following grants: NIH 2R01HL-63828, American Diabetes Association 7-08-RA-98, American Heart Association (GIA0855554D), Commonwealth of Pennsylvania—Department of Health (to X.-L.M.) and American Diabetes Association 7-06-JF59 (to L.T.).
References
- 1.Eisenberg DA. Cholesterol lowering in the management of coronary artery disease: the clinical implications of recent trials. Am J Med. 1998;104:2S–5S. doi: 10.1016/s0002-9343(98)00038-2. [DOI] [PubMed] [Google Scholar]
- 2.Nicholls SJ, Hazen SL. The role of myeloperoxidase in the pathogenesis of coronary artery disease. Jpn J Infect Dis. 2004;57:S21–S22. [PubMed] [Google Scholar]
- 3.Sugiyama S, Kugiyama K, Aikawa M, Nakamura S, Ogawa H, Libby P. Hypochlorous acid, a macrophage product, induces endothelial apoptosis and tissue factor expression: involvement of myeloperoxidase-mediated oxidant in plaque erosion and thrombogenesis. Arterioscler Thromb Vasc Biol. 2004;24:1309–1314. doi: 10.1161/01.ATV.0000131784.50633.4f. [DOI] [PubMed] [Google Scholar]
- 4.Vita JA, Brennan ML, Gokce N, Mann SA, Goormastic M, Shishehbor MH, et al. Serum myeloperoxidase levels independently predict endothelial dysfunction in humans. Circulation. 2004;110:1134–1139. doi: 10.1161/01.CIR.0000140262.20831.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hazen SL. Myeloperoxidase and plaque vulnerability. Arterioscler Thromb Vasc Biol. 2004;24:1143–1146. doi: 10.1161/01.ATV.0000135267.82813.52. [DOI] [PubMed] [Google Scholar]
- 6.Baldus S, Heeschen C, Meinertz T, Zeiher AM, Eiserich JP, Munzel T, et al. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108:1440–1445. doi: 10.1161/01.CIR.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- 7.Zhang R, Brennan ML, Fu X, Aviles RJ, Pearce GL, Penn MS, et al. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 2001;286:2136–2142. doi: 10.1001/jama.286.17.2136. [DOI] [PubMed] [Google Scholar]
- 8.Stokes KY, Cooper D, Tailor A, Granger DN. Hypercholesterolemia promotes inflammation and microvascular dysfunction: role of nitric oxide and superoxide. Free Radic Biol Med. 2002;33:1026–1036. doi: 10.1016/s0891-5849(02)01015-8. [DOI] [PubMed] [Google Scholar]
- 9.Williams FM. Neutrophils and myocardial reperfusion injury. Pharmacol Ther. 1996;72:1–12. doi: 10.1016/s0163-7258(96)00090-3. [DOI] [PubMed] [Google Scholar]
- 10.Plutzky J. Peroxisome proliferator-activated receptors in vascular biology and atherosclerosis: emerging insights for evolving paradigms. Curr Atheroscler Rep. 2000;2:327–335. doi: 10.1007/s11883-000-0067-3. [DOI] [PubMed] [Google Scholar]
- 11.Plutzky J. Inflammatory pathways in atherosclerosis and acute coronary syndromes. Am J Cardiol. 2001;88:10K–15K. doi: 10.1016/s0002-9149(01)01924-5. [DOI] [PubMed] [Google Scholar]
- 12.Pasceri V, Wu HD, Willerson JT, Yeh ET. Modulation of vascular inflammation in vitro and in vivo by peroxisome proliferator-activated receptor-gamma activators. Circulation. 2000;101:235–238. doi: 10.1161/01.cir.101.3.235. [DOI] [PubMed] [Google Scholar]
- 13.Marx N, Kehrle B, Kohlhammer K, Grub M, Koenig W, Hombach V, et al. PPAR activators as antiinflammatory mediators in human T lymphocytes: implications for atherosclerosis and transplantation-associated arteriosclerosis. Circ Res. 2002;90:703–710. doi: 10.1161/01.res.0000014225.20727.8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shiomi T, Tsutsui H, Hayashidani S, Suematsu N, Ikeuchi M, Wen J, et al. Pioglitazone, a peroxisome proliferator-activated receptor-{gamma} agonist, attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2002;106:3126–3132. doi: 10.1161/01.cir.0000039346.31538.2c. [DOI] [PubMed] [Google Scholar]
- 15.Liu HR, Tao L, Gao E, Lopez BL, Christopher TA, Willette RN, et al. Anti-apoptotic effects of rosiglitazone in hypercholesterolemic rabbits subjected to myocardial ischemia and reperfusion. Cardiovasc Res. 2004;62:135–144. doi: 10.1016/j.cardiores.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 16.Kumar AP, Piedrafita FJ, Reynolds WF. Peroxisome proliferator-activated receptor {gamma} ligands regulate myeloperoxidase expression in macrophages by an estrogen-dependent mechanism involving the -463GA promoter polymorphism. J Biol Chem. 2004;279:8300–8315. doi: 10.1074/jbc.M311625200. [DOI] [PubMed] [Google Scholar]
- 17.Gao F, Yue TL, Shi DW, Christopher TA, Lopez BL, Ohlstein EH, et al. p38 MAPK inhibition reduces myocardial reperfusion injury via inhibition of endothelial adhesion molecule expression and blockade of PMN accumulation. Cardiovasc Res. 2002;53:414–422. doi: 10.1016/s0008-6363(01)00488-6. [DOI] [PubMed] [Google Scholar]
- 18.Weyrich AS, Buerke M, Albertine KH, Lefer AM. Time course of coronary vascular endothelial adhesion molecule expression during reperfusion of the ischemic feline myocardium. J Leukoc Biol. 1995;57:45–55. doi: 10.1002/jlb.57.1.45. [DOI] [PubMed] [Google Scholar]
- 19.Ledbetter TK, Paape MJ, Douglass LW. Cytotoxic effects of peroxynitrite, polymorphonuclear neutrophils, free-radical scavengers, inhibitors of myeloperoxidase, and inhibitors of nitric oxide synthase on bovine mammary secretory epithelial cells. Am J Vet Res. 2001;62:286–293. doi: 10.2460/ajvr.2001.62.286. [DOI] [PubMed] [Google Scholar]
- 20.Kettle AJ, Gedye CA, Hampton MB, Winterbourn CC. Inhibition of myeloperoxidase by benzoic acid hydrazides. Biochem J. 1995;308(Pt 2):559–563. doi: 10.1042/bj3080559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Myzak MC, Carr AC. Myeloperoxidase-dependent caspase-3 activation and apoptosis in HL-60 cells: protection by the antioxidants ascorbate and (dihydro)lipoic acid. Redox Rep. 2002;7:47–53. doi: 10.1179/135100002125000181. [DOI] [PubMed] [Google Scholar]
- 22.Wagner BA, Buettner GR, Oberley LW, Darby CJ, Burns CP. Myeloperoxidase is involved in H2O2-induced apoptosis of HL-60 human leukemia cells. J Biol Chem. 2000;275:22461–22469. doi: 10.1074/jbc.M001434200. [DOI] [PubMed] [Google Scholar]
- 23.Bekesi G, Heinle H, Kakucs R, Pazmany T, Szombath D, Dinya M, et al. Effect of inhibitors of myeloperoxidase on the development of aortic atherosclerosis in an animal model. Exp Gerontol. 2005;40:199–208. doi: 10.1016/j.exger.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 24.Ohara Y, Peterson TE, Harrison DG. Hypercholesterolemia increases endothelial superoxide anion production. J Clin Invest. 1993;91:2546–2551. doi: 10.1172/JCI116491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osborne JA, Lento PH, Siegfried MR, Stahl GL, Fusman B, Lefer AM. Cardiovascular effects of acute hypercholesterolemia in rabbits. Reversal with lovastatin treatment. J Clin Invest. 1989;83:465–473. doi: 10.1172/JCI113905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung O, Jung W, Malinski T, Wiemer G, Schoelkens BA, Linz W. Ischemic preconditioning and infarct mass: the effect of hypercholesterolemia and endothelial dysfunction. Clin Exp Hypertens. 2000;22:165–179. doi: 10.1081/ceh-100100070. [DOI] [PubMed] [Google Scholar]
- 27.Sakamoto S, Kashiki M, Imai N, Liang CS, Hood WB., Jr Effects of short-term, diet-induced hypercholesterolemia on systemic hemodynamics, myocardial blood flow, and infarct size in awake dogs with acute myocardial infarction. Circulation. 1991;84:378–386. doi: 10.1161/01.cir.84.1.378. [DOI] [PubMed] [Google Scholar]
- 28.Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 29.Ma XL, Tsao PS, Lefer AM. Antibody to CD-18 exerts endothelial and cardiac protective effects in myocardial ischemia and reperfusion. J Clin Invest. 1991;88:1237–1243. doi: 10.1172/JCI115427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eiserich JP, Baldus S, Brennan ML, Ma W, Zhang C, Tousson A, et al. Myeloperoxidase, a leukocyte-derived vascular NO oxidase. Science. 2002;296:2391–2394. doi: 10.1126/science.1106830. [DOI] [PubMed] [Google Scholar]
- 31.Noctor G, Gomez L, Vanacker H, Foyer CH. Interactions between biosynthesis, compartmentation and transport in the control of glutathione homeostasis and signalling. J Exp Bot. 2002;53:1283–1304. doi: 10.1093/jexbot/53.372.1283. [DOI] [PubMed] [Google Scholar]
- 32.Hazen SL, Zhang R, Shen Z, Wu W, Podrez EA, MacPherson JC, et al. Formation of nitric oxide-derived oxidants by myeloperoxidase in monocytes: pathways for monocyte-mediated protein nitration and lipid peroxidation in vivo. Circ Res. 1999;85:950–958. doi: 10.1161/01.res.85.10.950. [DOI] [PubMed] [Google Scholar]
- 33.Robinson AJ, Dickenson JM. Regulation of p42/p44 MAPK and p38 MAPK by the adenosine A(1) receptor in DDT(1)MF-2 cells. Eur J Pharmacol. 2001;413:151–161. doi: 10.1016/s0014-2999(01)00761-0. [DOI] [PubMed] [Google Scholar]
- 34.Greenacre SA, Rocha FA, Rawlingson A, Meinerikandathevan S, Poston RN, Ruiz E, et al. Protein nitration in cutaneous inflammation in the rat: essential role of inducible nitric oxide synthase and polymorphonuclear leukocytes. Br J Pharmacol. 2002;136:985–994. doi: 10.1038/sj.bjp.0704798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaut JP, Byun J, Tran HD, Lauber WM, Carroll JA, Hotchkiss RS, et al. Myeloperoxidase produces nitrating oxidants in vivo. J Clin Invest. 2002;109:1311–1319. doi: 10.1172/JCI15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA, Halliwell B, et al. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- 37.Kim EJ, Kwon KJ, Park JY, Lee SH, Moon CH, Baik EJ. Effects of peroxisome proliferator-activated receptor agonists on LPS-induced neuronal death in mixed cortical neurons: associated with iNOS and COX-2. Brain Res. 2002;941:1–10. doi: 10.1016/s0006-8993(02)02480-0. [DOI] [PubMed] [Google Scholar]
- 38.Kwon G, Xu G, Marshall CA, McDaniel ML. Tumor necrosis factor alpha-induced pancreatic beta-cell insulin resistance is mediated by nitric oxide and prevented by 15-deoxy-Delta12,14-prostaglandin J2 and aminoguanidine. A role for peroxisome proliferator-activated receptor gamma activation and inos expression. J Biol Chem. 1999;274:18702–18708. doi: 10.1074/jbc.274.26.18702. [DOI] [PubMed] [Google Scholar]
- 39.Tao L, Liu HR, Gao E, Teng ZP, Lopez BL, Christopher TA, et al. Antioxidative, antinitrative, and vasculoprotective effects of a peroxisome proliferator-activated receptor-{gamma} agonist in hypercholesterolemia. Circulation. 2003;108:2805–2811. doi: 10.1161/01.CIR.0000097003.49585.5E. [DOI] [PubMed] [Google Scholar]