

Abstract
The assembly and maintenance of the cardiac sarcomere, which contains the basic contractile components of actin and myosin, are essential for cardiac function. While often described as a static structure, the sarcomere is actually dynamic and undergoes constant turnover, allowing it to adapt to physiological changes while still maintaining function. A host of new factors have been identified that play a role in the regulation of protein quality control in the sarcomere, including chaperones that mediate the assembly of sarcomere components and ubiquitin ligases that control their specific degradation. There is clear evidence of sarcomere disorganization in animal models lacking muscle-specific chaperone proteins, illustrating the importance of these molecules in sarcomere structure and function. Although ubiquitin ligases have been found within the sarcomere structure itself, the role of the ubiquitin proteasome system in cardiac sarcomere regulation, and the factors that control its activity, are only just now being elucidated. The number of ubiquitin ligases identified with specificity for sarcomere proteins, each with distinct target substrates, is growing, allowing for tight regulation of this system. In this review, we highlight the dynamic interplay between sarcomere-specific chaperones and ubiquitin-dependent degradation of sarcomere proteins that is necessary in order to maintain structure and function of the cardiac sarcomere.
Keywords: Chaperones, Ubiquitin, Proteasome, Ubiquitin ligase, Muscle ring finger, Atrogin-1, MAFbx, C-terminal of Hsp70 interacting protein, MDM2, GimC, TriC, αB-crystallin, UPD-2, UNC-45, Protein quality control
1. Introduction
Cardiac contractility is regulated at the levels of calcium homeostasis, cell signalling, and through the maintenance of the sarcomere, the smallest contractile unit of cardiac muscle. The assembly and turnover of the cardiac sarcomere is essential to cardiac performance because it contains the basic contractile components of actin and myosin necessary for cardiac function. Given the importance of this basic component of cardiac function, it is surprising that very little is known about the actual mechanisms responsible for its turnover. Most accounts of the sarcomere describe it as a static structure used to generate force. However, the sarcomere is actually a dynamic structure constantly assembled and degraded by carefully regulated molecular mechanisms that we are only just now beginning to identify and understand.1 During the continuous contraction of the cardiac sarcomere, new proteins are exchanged into the structure via a carefully orchestrated process of synthesis and degradation. This continual remodelling allows adaptation to stressors, including exercise, metabolic influences, or disuse and must occur without affecting the integrity of the contractile force necessary for the heart to continue to function. Since this dynamic turnover of proteins becomes more necessary in the face of cardiac pathologies such as ischaemic heart disease and heart failure, it is essential to understand the mechanisms regulating the homeostasis of the sarcomere.
As we understand it currently, the quality control system of the sarcomere involves two main components: (1) chaperones, which protect proteins from mis-folding and are necessary for the assembly of specific major sarcomere components; and (2) the ubiquitin proteasome system (UPS), which recognizes specific proteins and targets them for degradation (Figure 1). The specificity of the UPS machinery is afforded by the ubiquitin ligases, which interact with E1 and E2 enzymes to place poly-ubiquitin chains on the substrates. It is these poly-ubiquitin chains that the 26S proteasome recognizes prior to substrate degradation (Figure 1). Several ubiquitin ligases have been identified that are integrated into the sarcomere of both skeletal and cardiac muscle, highlighting the physical integration of the UPS and sarcomere. These ligases include MuRF family proteins (MuRF1, MuRF2, and MuRF3) and MAFbx/atrogin-1. Since the study of protein quality control in the sarcomere is relatively new, much of what we know on the subject has come from studies looking at development of skeletal muscle. Insight into sarcomeric dysregulation has been gleaned from examination of muscle biopsy specimens taken from patients suffering from various myopathies. In addition, the assembly and behaviour of individual proteins making up the sarcomere have been studied biochemically. In this review, we will take data obtained from these various studies and use it to discuss the dynamic process of cardiac sarcomere protein quality control, including chaperones involved in the assembly of the sarcomere, and the UPS and autophagy involved in the degradation of the sarcomere.
Figure 1.

Sarcomere-specific chaperones and ubiquitin ligases are necessary for the assembly and degradation of sarcomere proteins and constitute the protein quality control system in the heart. The protein quality control of the sarcomere involves the continuous assembly (left side) and degradation (right side) of specific sarcomere proteins. In this example, the co-chaperones UNC-45 and Hsp90 are required for the assembly of myosin. This is balanced by the specific ubiquitination and degradation of proteins by the ubiquitin proteasome system. This involves specific ubiquitin ligases (designated E3) that place poly-ubiquitin tails on targets for degradation by the 26S proteasome. In this example, both MuRF1 and MuRF3 have shown to specifically ubiquitinate and degrade myosin in a proteasome-dependent manner. In the heart, this dynamic process of protein quality control occurs amid continuous use in order to maintain the fundamental construct necessary for contractility. Proteasome graphic courtesy of the U.S. Department of Energy Genome Programs (http://genomics.energy.gov).
2. Assembling the sarcomere
The basic components of the contractile apparatus of muscle include myosin thick filaments (Figure 1, in red), which act as molecular motors ratcheting down the actin light chain (Figure 1, light green) during contraction. There are many dozens of additional proteins that support this basic interaction, such as the troponins and tropomyosin that associate with actin and support myosin/actin interactions, and proteins such as α-actinin that link adjacent sarcomeres at the z-disk (Figure 1, dark green). Other essential structural proteins like titin (Figure 1, blue) link the Z-disk to the M-line to stabilize myosin and detect mechanical stress. In the established sarcomere, integration and exchange of new proteins occurs continuously as indicated by constituent t1/2 life of sarcomeric proteins. Troponin subunits (T/I/C) have t1/2 lives of approximately 3–5 days; actin and tropomyosin ∼7–10 days, while the t1/2 of myosin is approximately 5–8 days.2–4 The details of this replacement of contractile proteins with newly synthesized proteins are not fully understood; however, several recent studies have offered some insight. For example, using epitope-tagged tropomyosin (Tm) and troponin I (TnI) in adult cardiomyocytes, investigators have been able to identify the incorporation of newly synthesized myofilaments in differentiated cardiomyocytes using confocal microscopy.5 This approach revealed that TnI is initially detected along the entire thin filament, whereas epitope-tagged Tm replaces endogenous Tm only at the end of the thin filament. These observations demonstrate that the movement of myofilament proteins within the sarcomere is not a random event, but instead is a highly ordered process, tailored individually for each specific myofilament subtype.5 As will be discussed in the following sections, it is the job of chaperones and the muscle-specific ubiquitin ligases to ensure that the exchange of these sarcomere proteins occurs seamlessly and without interruption to normal contractile function.
2.1. Chaperones and actin assembly into the sarcomere
Actin is a highly dynamic molecule subject to both polymerization (into actin filaments) and aggregation. Correct folding and prevention of aggregation of actin are essential for the formation of thin filaments and their subsequent interaction with myosin in the sarcomere. Two molecular chaperones, GimC (Prefoldin) and TRiC (TCP-1 Ring Complex), play a synergistic role in the folding and assembling of actin. GimC attaches to actin as it is first translated and assists in its initial folding, preventing the nascent actin molecules from aggregating with one another.6 GimC then acts as a co-chaperone and passes the actin filaments onto the general chaperone TRiC, which mediates actin's maturation into filamentous actin.7–9 The subsequent process of actin assembly into the thin filaments of the sarcomere is not clear. However, it is known that TriC remains associated with actin throughout this process—possibly to prevent aggregation—and is able to modulate filament elongation in vitro.9 Other chaperones, including αB-crystallin10–12 and Hsp2713 also reportedly associate with actin. Very little is known about the functional significance of these associations, however, in the case of Hsp27, the interaction with actin does appear to be vital to cardiac and skeletal muscle development in the assembly of actin filament and myofibril assembly.13 These overlapping roles of chaperones and targets suggest that a highly cooperative relationship exists between various chaperones during the assembly of the actin thin filaments of the sarcomere.
2.2. UNC-45, Hsp90, and Hsp70 participate in myosin integration into the sarcomere
A detailed understanding of how myosin is incorporated into the sarcomere spatially and temporally is not known. However, it is known that—in contrast to the spontaneous polymerization of actin in solution—myosin does not self-assemble without additional factors.14 Much of what we know about myosin assembly and its role in sarcomere organization comes from studies involving skeletal muscle development in Caenorhabditis elegans. The assembly of myosin requires multiple chaperones including UNC-45, Hsp90, and Hsp70. UNC-45, the first member of the UNC family to be identified, was named for the un-coordinated phenotype that was seen in C. elegans when this protein was mutated.15 The UNC-45 protein has two distinct binding domains; on the N-terminal end UNC-45 has tetratricopeptide repeat (TPR) clamp domains while the C-terminal end has a UCS (UNC-45/Cro1/She4) domain. Myosin binding to UNC-45 through its UCS domain prevents myosin aggregation,16,17 whereas the N-terminal of UNC-45 contains a region that binds to the more general chaperone proteins Hsp90 and Hsp70,17,18 and is essential for the organization of the thick filament experimentally.18–21 Mutations in conserved regions of the unc-45 UCS domain result in reduced numbers of myofilaments with severe disorganization.22
Vertebrates have UNC-45 homologues named UNC-45A and UNC-45B, which may have conserved function. In mice and humans, UNC-45A is found in many tissues, while the UNC-45B homologue is found only in cardiac and skeletal muscle.23 Anti-sense experiments in C2C12 skeletal myogenic cells demonstrate the general cell isoform UNC-45A has a role in proliferation and cell fusion, while UNC-45B has a role in sarcomere organization.23 This is complementary to other studies in zebrafish that have identified that UNC-45B and Hsp90a co-localize with myosin during muscle development and associate with the Z-line upon myofibril assembly.19,24 Consistent with the aforementioned studies in C. elegans, vertebrate UNC-45B has a significant role in sarcomere organization. Mammalian UNC-45B selectively binds unfolded conformations of the myosin motor domain and promotes de novo folding, indicating a fundamental role in myofibrillogenesis.25,26 While no definitive associations have been made with UNC-45B and human disease, a statistical analyses has identified UNC-45B (a.k.a. CMYA4) as a cardiomyopathy-associated gene,27 although the role of UNC-45B in cardiomyopathy and other cardiac diseases remains to be elucidated.
Whereas UNC-45 regulates the process of folding and assembly of myosin, the regulation of UNC-45 itself is carefully tuned by the UPS. The degradation of UNC-45 is mediated by two ubiquitin ligases: (1) UFD-2 (Ubiquitin fusion degradation-2); and (2) CHIP (C-terminal of Hsp70 interacting protein).28 CHIP is both a molecular chaperone and ubiquitin ligase, co-operating with Hsp70 and Hsp90 to target abnormal proteins for degradation by ubiquitination.29,30 CHIP exerts both chaperone and ligase activities on myosin and, as such, CHIP is central to coordinating the folding and degradation of this molecule. The stoichiometry of UFD-2 and CHIP to UNC-45 appears to regulate the degradation of UNC-45. In the presence of UFD-2 or CHIP individually, UNC-45 is ubiquitinated with only 1–3 ubiquitin molecules, which is not sufficient to target UNC-45 for degradation by the 26S proteasome.28,31 In the presence of both UFD-2 and CHIP, UNC-45 is markedly more ubiquitinated in vitro.28 Interestingly, increasing UNC-45 in the presence of non-functional UFD-2 and CHIP mutants results in defects in sarcomere assembly.28,31 Although the exact process behind the sarcomere defect in this case is not known, it is possible that the increase in UNC-45 may enhance the stabilization of myosin, such that it is not available to form thick filaments necessary for the assembly of the sarcomere.32 This stabilized yet unassembled myosin may also be targeted for degradation,33 once again leading to defects in sarcomere assembly.
2.3. Desmin assembly requires αB-crystallin
Desmin is an intermediate filament found at the Z-line of the sarcomere that is necessary for sarcomere integrity. The assembly of desmin requires the chaperone αB-crystallin, a 20 kDa peptide belonging to the family of small heat shock proteins that is abundant in cardiomyocytes. αB-crystallin interacts with several cytoskeletal proteins, including desmin,10,34 to prevent protein mis-folding. In addition, similar to the chaperones associated with actin and myosin, αB-crystallin can prevent desmin aggregation from occurring.35,36 Mutations in either desmin or αB-crystallin are the cause of numerous pathologies. Desmin-related myopathies (DRM) are sporadic and familial myopathies caused by mutations in either desmin or αB-crystallin. Patients with these mutations have a striking loss of sarcomere organization, resulting in the loss of the myofibrillar integrity and accumulation of desmin, αB-crystallin, and actin in the skeletal muscle and heart.37–43 Although the exact cause of this family of myopathies has not been elucidated, evidence for the mis-folding of desmin as the underlying cause of these diseases has been reported both in vitro and in vivo.44–46 Mutations in either desmin or its chaperone αB-crystallin also lead to accumulations of desmin-positive sarcomeric inclusions that are associated with dilated cardiomyopathy.42,43,47 Interestingly, these inclusions may reflect impairments in both the chaperone and proteasome components of protein quality control. Evidence suggests that improper folding of desmin leads to toxic accumulations,48 that in turn inhibit the proteasome, resulting in a failure to clear the mis-folded proteins.26,49–51 It is interesting to note that αB-crystallin interacts with FBX4, an F-box-containing protein component of the SCF complex (SKP1/Cul1/F-box) that has ubiquitin ligase activity.52 Mutant αB-crystallin (i.e. R120G) has been reported to have enhanced interaction with FBX4, stimulating the ubiquitination of yet to be identified proteins.52 Together, these data suggest that αB-crystallin may play a key role in both the assembly and turnover of sarcomeres, thereby linking chaperone activity with protein refolding and protein degradation through its interactions with the UPS. αB-crystallin also interacts with other central sarcomere proteins, such as titin53–56 and the aforementioned actin. This may suggest a larger role for αB-crystallin in the formation of the sarcomere.
3. The calpain and ubiquitin proteasome systems mediate sarcomere degradation
The concept of UPS degradation of sarcomere proteins and mediation of sarcomere turnover is relatively new. A recent study in Drosophila using a conditional transgenic system to disrupt proteasome function demonstrated widespread muscle disorganization and induction of atrophy in as little as 12 h after proteasome inhibition.57 By 24 h, all movement was suspended and a loss of sarcomere organization and parallel increases in autophagosomes and expression of the chaperone GRP78 were identified.57 Therefore, global proteasome function appears necessary for sarcomere stability.
3.1. Targeting proteins for proteasome degradation: cardiac ubiquitin ligases
Several muscle-specific ubiquitin ligases have recently been identified, including Muscle Ring Finger (MuRF) family proteins, CHIP, murine double minute 2 (MDM2), and MAFbx/atrogin-1.58,59 Their known and putative role in maintaining the cardiac sarcomere is discussed below.
3.1.1. MuRF family proteins
MuRF1, found mainly in the M-line of the sarcomere where it interacts with the giant protein titin, specifically recognizes and degrades troponin I in a proteasome-dependent manner.60 MuRF1 is reported to also interact with troponin T, myosin light chain-2, myotilin, and telethonin.61 Interestingly, the closely related MuRF2 protein also interacts with these aforementioned proteins (at least experimentally), suggesting that a redundant system may exist for turning over these proteins.61 Supporting this theory is the finding that, while mice deficient in either MuRF1 or MuRF2 develop normally, mice lacking both MuRF1 and MuRF2 develop cardiac hypertrophy during development.62 The MuRF family of proteins has also been implicated in the development of cardiac hypertrophy that occurs as a result of mechanical stress. Mice lacking MuRF1 develop an exaggerated cardiac hypertrophy in response to transaortic constriction compared with wild-type mice, whereas, MuRF2 null mice develop cardiac hypertrophy to the same extent as wild-type control mice.63 Despite the apparent lack of involvement of MuRF2 in stress-induced cardiac hypertrophy in vivo, Lange et al.64 demonstrated that MuRF2 can mediate degradation of serum response factor and the inhibition of cardiac hypertrophy signaling pathways64 in rat neonatal cardiomyocytes following mechanical stress. However, the in vivo significance of this finding is not yet clear. Taken together, these findings suggest that while MuRF1 and MuRF2 may have similar specificities in vitro, their redundancy may be physiologically relevant during cardiac development, and less important in the process of cardiac hypertrophy induced in the adult heart.
MuRF1 and MuRF3 also interact cooperatively, in this case with E2 enzymes (UbcH5a, b, and c), to degrade beta/slow myosin heavy chain and MHCIIa in a proteasome-dependent manner in the heart and skeletal muscle.65 Mice lacking both MuRF1 and MuRF3 develop a hypertrophic cardiomyopathy and skeletal muscle myopathy with MHC accumulation, myofibre fragmentation, and impaired muscle performance.65 Although the exact nature of how MuRF family proteins target proteins for degradation is not known, it has been suggested that MuRF may regulate the turnover of specific proteins that have been worn or damaged over time with use. While this hypothesis is currently untested, there is evidence that MuRF1 does play a role in degrading damaged proteins. The enzyme creatine kinase, which is essential for the delivery of ATP from the mitochondria to all parts of cardiomyocytes, has recently been suggested to be a MuRF1 substrate. Specifically, MuRF1 is able to poly-ubiquitinate creatine kinase.66,67 Interesting, however, is the finding that MuRF1 preferentially poly-ubiquitinates oxidized creatine kinase, a post-translational modification that occurs after many types of cellular stresses indicative of ‘cellular wear’.66,67 Because creatine kinase isoforms are found at the M-line, where MuRF1 is mainly localized in the sarcomere, this observation supports the hypothesis that the ubiquitin ligase MuRF1 is ensuring protein quality control by detecting damaged proteins, perhaps through enhanced affinities, to allow the essential continuous function of the heart.
3.1.2. C-terminus of Hsp70-interacting protein
The ubiquitin ligase CHIP has been recognized as an important factor involved in ischaemia reperfusion injury in the heart59 and in aging.68 Recent studies implicate CHIP in the degradation of myosins during skeletal muscle loss in a C. elegans model of Duchenne muscular dystrophy.69 CHIP may therefore play an essential role in the turnover of myosin as both a regulator of the UNC-45 co-chaperone (discussed previously), and as a ubiquitin ligase for myosin directly.69
3.1.3. MAFbx/Atrogin-1 (muscle atrophy F-box)
Like MuRF1, MAFbx/Atrogin-1 was initially identified as a ubiquitin ligase essential to the process of skeletal muscle atrophy (i.e. degradation of the sarcomere).58 Mice lacking MAFbx/atrogin-1 do not undergo atrophy to the same extent as wild-type mice, suggesting that MAFbx/atrogin-1 may directly degrade specific sarcomere proteins.58 However, unlike MuRF1, specific sarcomere protein substrates of MAFbx/Atrogin-1 have not been identified. MAFbx/atrogin-1 has also been described as a regulator of several transcription factors essential to regulation of cardiac hypertrophy.70,71 The specific role of MAFbx/atrogin-1 in the regulation of yet-identified sarcomere substrates has yet to be determined but remains a subject of intense scrutiny.
3.1.4. Murine double minute 2
MDM2 is a ubiquitin ligase that recognizes the N-terminal activation domain of p53 to inhibit its transcriptional activation. In the heart, MDM2 is a critical regulator of apoptosis through its ubiquitin-dependent degradation of ARC (Apoptosis Repressor with Caspase recruitment domain).72 In the context of the sarcomere, MDM2 interacts with and down-regulates the sarcomeric protein telethonin, in a proteasome-dependent manner.73 The specific role MDM2 plays in the regulation of telethonin degradation in the maintenance of the sarcomere has yet to be determined.
3.2. The calpain system is necessary for ubiquitin ligases to reach sarcomere proteins
Calpains are a group of calcium-dependent, non-lysosomal cysteine proteases expressed ubiquitously in all cells. As a whole, calpains are not well understood, but it is known that they are involved in a variety of cellular processes, including cell-cycle control and cell fusion.74 Various members of the calpain family have been localized to skeletal muscle where they are purported to be involved in both muscle growth and atrophy.75,76 Calpain 1 has been found in tight association with skeletal muscle myofibrils where it binds tightly to the giant protein titin in a calcium-dependent manner.77 Recently, it was discovered that calpain-1 is required to mediate the dissociation of sarcomere proteins from the assembled myofibril before the UPS system is able to degrade them (Figure 2). The UPS is capable of degrading myofibrillar components, but only when they are in their monomeric state; sarcomere proteins found in multimeric complexes cannot be degraded by the proteasome in vivo.78 When the calpain system is inhibited in the heart, as in mice over-expressing calpastatin (an endogenous calpain-specific inhibitor), morphological evidence of widespread protein aggregation has been identified along with increased autophagy (discussed in more detail in the following section).79 This aggregation may represent proteins not able to be degraded by the UPS in the absence of calpain-1 intervention. This coordinated effort by calpain and ubiquitin ligases is also illustrated in models of skeletal muscle atrophy. Ubiquitin ligases, including MuRF1 and MAFbx/atrogin-1, have proved to be essential in the atrophic process. When calpain inhibitors are introduced into the system, sarcomere degradation is inhibited, thereby inhibiting muscular atrophy, without reducing MuRF1 or MAFbx/atrogin-1 levels.80 This is consistent with a pivotal role of the calpain system in allowing ubiquitin ligases to mediate the degradation of the sarcomere.
Figure 2.

Cardiac calpain-1 is necessary for cardiac proteins to be ubiquitinated. In order for sarcomere proteins to be ubiquitinated by ubiquitin ligases, calpain-1 activated release of the sarcomere appears necessary (left). Calpain-1 is also necessary for the regular turnover of aggregated proteins, which if not cleared, can result in increased autophagy. Adapted from Galvez et al.79
Beyond its critical role in monitoring normal cardiac function, calpain-1 may also mediate pathological effects by allowing enhanced ubiquitination of proteins to occur. In cardiac-specific conditional calpain-1 transgenic mice, widespread myocytolysis, cardiac hypertrophy, and inflammation leading to heart failure was identified in mice expressing increased calpain-1 activity levels.79 Since several of the ubiquitin ligases that have been described in the heart (i.e. MuRF1, MuRF3) recognize sarcomeric proteins, understanding the role of the calpain system will be critical to determine how they function in the heart.
4. Autophagy-dependent maintenance of the sarcomere
In addition to the UPS-dependent sarcomere proteins (summarized in Table 1), autophage-mediated degradation also contributes to protein quality control of the sarcomere. Autophagy removes invading pathogens, damaged organelles (such as mitochondria), and mis-folded proteins through lysosomal-mediated degradation of these materials (Figure 3). Cardiac autophagy is initiated in response to energy stress during periods of nutrient deprivation or high metabolic demand, ischaemia/reperfusion injury, and heart failure.81 Genetic models targeting the Atg family of genes, more than 20 evolutionary conserved genes essential to autophagy,82 demonstrate a fundamental role for autophagy in cardiac function. Cardiac-specific deletion of a critical autophagy gene involved in vesicle elongation (Atg5) in mice leads to global derangements in the sarcomere.83 Although cardiac organogenesis and development are not perturbed in Atg5(−/−) mice, cardiac hypertrophy, left ventricular dilation, and contractile dysfunction develop in adulthood. Electron-micrographs of Atg5(−/−) hearts reveal both a global derangement in sarcomere structure and the misalignment and aggregation of mitochondria.83 Although cardiac abnormalities are not seen until adulthood in these mice, induction of pressure overload in younger animals does reveal a role for Atg in managing the response to cardiac stress. Young Atg5(−/−) mice subjected to transaortic constriction suffer a significant decrease in function and increase in left ventricular dilation after only 1 week of pressure overload, suggesting that autophagy is cardio-protective during pathological stimulation. Furthermore, using an in vitro model of ischaemia, over-expression of another gene that functions in the elongation phase of autophagy, Beclin 1 (also known as Atg 6), decreased, whereas a dominant-negative form of Atg5 increased cellular injury,84 consistent with an adaptive/protective role for autophagy.
Table 1.
Contributors to protein quality control of the sarcomere
| Thin filament | |
| Actin assembly | GimC, TRiC, αB-crystallin, Hsp27 |
| Troponin I degradation | MuRF1 |
| Thick filament | |
| Myosin assembly | UNC-45, Cro1, She4p, Hsp90, and Hsp70 |
| Myosin degradation | CHIP, MuRF1, MuRF3 |
| UNC-45 regulation | CHIP, UFD-2, p97 |
| Intermediate filament | |
| Desmin assembly | αB-crystallin |
| Telethonin degradation | MDM2 |
| Sarcomere organization and remodelling | |
| Calpain system | Calpain-1 |
| Autophagy | Atg5, Beclin 1 |
Figure 3.
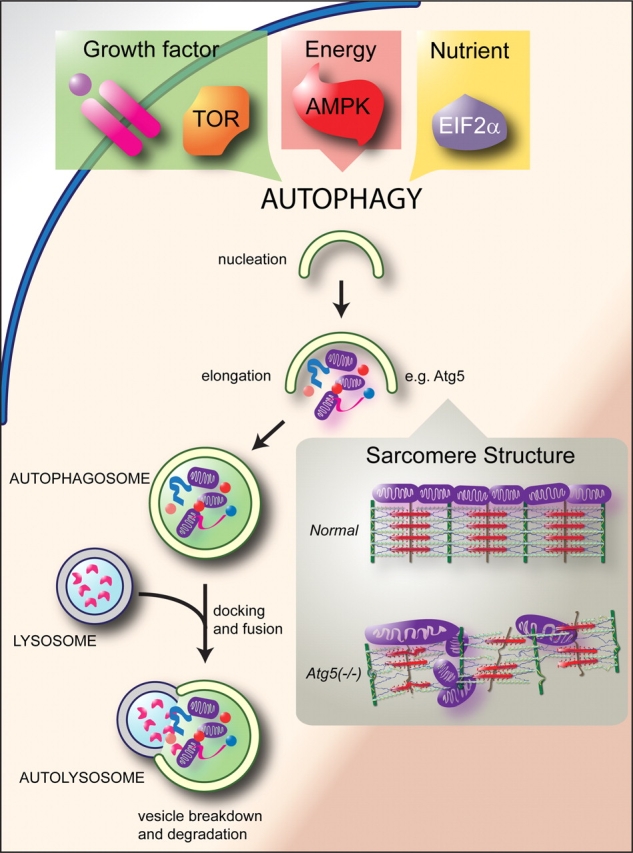
Regulation of protein quality control by autophagy. The best defined regulator of autophagy to date is the target of rapamycin (TOR) kinase, which signals upstream of the Atg family of genes, which include more than 20 evolutionary conserved genes essential to autophagy.82 Target of rapamycin kinase is regulated by growth factor signalling, such as Akt signalling, thereby completing a nutrient-sensitive autophagy regulatory circuit. Other ‘sensing’ molecules contribute to autophagy activation, such as the energy sensor 5′-AMP-activated protein kinase (AMPK) and the nutrient-sensitive eukaryotic initiation factor 2alpha (eIF2alpha). Autophagy describes a multi-step process (nucleation, elongation, and completion) of a double-membrane vesicle forming around cytoplasmic cargo developing into an autophagosome. Subsequent docking and fusion of the autophagosome with a lysosome forms an autolysosome and exposes the cargo to lysosomal proteases leading to cargo degradation. Given the variety of stimuli that can activate autophagy, it is not surprising that multiple regulators of autophagy have been identified. For an exhaustive discussion of these and other regulatory proteins with a focus on disease pathogenesis, readers are encouraged to read excellent recent reviews on this topic.107,108 Inset: Inhibiting autophagy, in this case by knocking out cardiac Atg5 in mice, leads to prominent defects in the sarcomere structure, leading to sarcomere and mitochondrial disarray in the heart.83 Adapted from Levine and Kroemer.108
Despite the somewhat overlapping roles of autophagy and the UPS, these two pathways have only recently been suggested to function cooperatively, particularly in the heart. When autophagy is inactivated in Atg5 (−/−) mouse hearts, there is an increase in poly-ubiquitinated proteins,83 which is surprising as the hearts of these mice also exhibit increased proteasome activity that should ensure the clearance of these tagged proteins. This suggests that the increase in toxic protein aggregates caused by a lack of autophagy overwhelms the UPS so that in spite of its compensatory increase in activity, it is unable to clear the poly-ubiquitinated proteins.85,86 This theory is supported by a more recent study demonstrating that inhibition of proteasome activity in the heart leads to the accumulation of poly-ubiquitinated proteins and subsequent activation of autophagy.87 These findings outline a cooperative role between the UPS and autophagy in maintaining protein quality control in the heart through a combination of targeting of monomeric proteins via the UPS and degradation of protein aggregates and damaged mitochondria via autophagy.
Accumulating data demonstrating a link between the UPS and autophagy prompted studies looking at common regulatory circuits, thus far limited to skeletal muscle. One emerging pathway is the Akt-FOXO pathway, linking growth factor signalling to the coordinated regulation of the proteasomal and lysosomal systems (reviewed in refs. 88–90). FOXO3 mediates both cardiac and skeletal muscle atrophy through its direct activation of the muscle-specific ubiquitin ligase Atrogin-1/MAFbx, promoting degradation of proteins via the UPS (described earlier). Focusing on skeletal muscle, studies from two independent groups demonstrated an mTOR-independent circuit involving Akt-mediated regulation of FOXO3.67,91 FOXO3 positively regulates the expression of a cadre of autophagy-related genes, as seen in mouse models of muscle atrophy such as denervation or fasting, and is necessary and sufficient to activate autophagy in skeletal muscle in vivo. The ability of Foxo3 to up-regulate autophagy is also independent of the UPS system. Thus Foxo3 has a unique role in its regulation of both proteasomal and lysosomal systems.
5. Disruption of sarcomere protein quality control disrupts sarcomere integrity
Although we have focused on the role of chaperones and ligases in this review, malfunction of the assembly and turnover of proteins may result from mutations in sarcomere proteins themselves. Mutations in sarcomeric proteins, including TnT, TnI, β-myosin heavy chain, and cardiac myosin binding protein-C (recently reviewed by Morimoto92), lead to a diverse array of cardiomyopathies. Cardiac dysfunction in familial hypertrophic cardiomyopathies results from defects in calcium signalling and delayed relaxation.93–96 Recent studies have identified that truncated cMyBP-c mutations have an accelerated proteasome-mediated degradation, leading to proteasome impairment and toxic accumulation of proteins.96,97 So in addition to the known biochemical defects resulting from sarcomere mutations, specific genetic cardiomyopathies may involve defects in cardiac protein quality control to maintain the folding, assembly, and degradation necessary for the proper maintenance of the heart.
6. Summary/Conclusions
The simple elegance of the static sarcomere structure has given way to a theory of a complex, dynamic structure that is in constant flux. The necessity of this turnover in the heart is self-evident given its constant activity, periodic stress, and vital necessity. In this review, we describe how the two parallel processes of protein assembly and protein degradation are necessary for the maintenance of the sarcomere. The process of sarcomere assembly is aided by chaperones such as GimC, TRiC and αβ-crystallin that associate with the various proteins that make up the myofilaments. The degradative process of sarcomere maintenance is mediated in part by the UPS. Specifically, ubiquitin ligases with sarcomere-specific interactions have been identified, incl uding CHIP, MuRF1, MuRF2, MuRF3, and MDM2 (see Table 1). In addition, recent studies have identified the need for calpain-1 activation in the heart for ubiquitin ligases, such as those just mentioned, to access the sarcomere. Given the fact that each sarcomere may have its own chaperone/ubiquitin ligase regulators, the complexity of the turnover of the entire sarcomere is certainly just beginning to be elucidated.
The maintenance of cardiac sarcomere function becomes even more essential in the context of disease, where adequate contractility is necessary for survival. Likewise, understanding these fundamental protein quality control mechanisms in the heart will assist in the development of specific therapies for conditions where maintenance and/or recovery of sarcomere function is essential, such as in ischaemic heart disease and heart failure. Indeed, a major focus of future research will be directed at identifying how the UPS system balances the protein chaperone system in cardiac disease, the importance of which is exemplified by the contradictory effects of proteasome inhibitors on the heart. While a number of studies have identified that proteasome inhibition is beneficial in the face of ischaemia in animal models,98–103 unexpected cardiotoxicity has been reported in humans.104 A recent study identified that the proteasome induced-cardiotoxicity in culture was due to a disequilibrium between proteasome activity and ER chaperones.105,106 Overexpressing ER chaperones reversed the proteasome inhibitor-induced cardiomyocyte death.106 Both chaperone and proteasome systems appear to be necessary to balance the dynamics of protein synthesis and degradation in the heart. Therapeutic strategies that take advantage of the constant building and remodelling of the heart may help in both the structural and energetic changes that occur in disease.
Conflict of interest: none declared.
Funding
This work was supported by Children’s Cardiomyopathy Foundation Grant (M.S.W.), AHA Scientist Development Grant (to M.S.W.), American Heart Association Pre-Doctoral Fellowship (to J.C.S.), and R01 HL65619 (to C.P.).
References
- 1.Linke WA. Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res. 2008;77:637–648. doi: 10.1016/j.cardiores.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 2.Martin AF, Rabinowitz M, Blough R, Prior G, Zak R. Measurements of half-life of rat cardiac myosin heavy chain with leucyl-tRNA used as precursor pool. J Biol Chem. 1977;252:3422–3429. [PubMed] [Google Scholar]
- 3.Everett AW, Prior G, Zak R. Equilibration of leucine between the plasma compartment and leucyl-tRNA in the heart, and turnover of cardiac myosin heavy chain. Biochem J. 1981;194:365–368. doi: 10.1042/bj1940365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morkin E, Kimata S, Skillman JJ. Myosin synthesis and degradation during development of cardiac hypertrophy in the rabbit. Circ Res. 1972;30:690–702. doi: 10.1161/01.res.30.6.690. [DOI] [PubMed] [Google Scholar]
- 5.Michele DE, Albayya FP, Metzger JM. Thin filament protein dynamics in fully differentiated adult cardiac myocytes: toward a model of sarcomere maintenance. J Cell Biol. 1999;145:1483–1495. doi: 10.1083/jcb.145.7.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hansen WJ, Cowan NJ, Welch WJ. Prefoldin-nascent chain complexes in the folding of cytoskeletal proteins. J Cell Biol. 1999;145:265–277. doi: 10.1083/jcb.145.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vainberg IE, Lewis SA, Rommelaere H, Ampe C, Vandekerckhove J, Klein HL, et al. Prefoldin, a chaperone that delivers unfolded proteins to cytosolic chaperonin. Cell. 1998;93:863–873. doi: 10.1016/s0092-8674(00)81446-4. [DOI] [PubMed] [Google Scholar]
- 8.Siegers K, Waldmann T, Leroux MR, Grein K, Shevchenko A, Schiebel E, et al. Compartmentation of protein folding in vivo: sequestration of non-native polypeptide by the chaperonin-GimC system. EMBO J. 1999;18:75–84. doi: 10.1093/emboj/18.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grantham J, Ruddock LW, Roobol A, Carden MJ. Eukaryotic chaperonin containing T-complex polypeptide 1 interacts with filamentous actin and reduces the initial rate of actin polymerization in vitro. Cell Stress Chaperones. 2002;7:235–242. doi: 10.1379/1466-1268(2002)007<0235:ecctcp>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bennardini F, Wrzosek A, Chiesi M. Alpha B-crystallin in cardiac tissue. Association with actin and desmin filaments. Circ Res. 1992;71:288–294. doi: 10.1161/01.res.71.2.288. [DOI] [PubMed] [Google Scholar]
- 11.Melkani GC, Cammarato A, Bernstein SI. alphaB-crystallin maintains skeletal muscle myosin enzymatic activity and prevents its aggregation under heat-shock stress. J Mol Biol. 2006;358:635–645. doi: 10.1016/j.jmb.2006.02.043. [DOI] [PubMed] [Google Scholar]
- 12.Singh BN, Rao KS, Ramakrishna T, Rangaraj N, Rao Ch M. Association of alphaB-crystallin, a small heat shock protein, with actin: role in modulating actin filament dynamics in vivo. J Mol Biol. 2007;366:756–767. doi: 10.1016/j.jmb.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 13.Brown DD, Christine KS, Showell C, Conlon FL. Small heat shock protein Hsp27 is required for proper heart tube formation. Genesis. 2007;45:667–678. doi: 10.1002/dvg.20340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barral JM, Epstein HF. Protein machines and self assembly in muscle organization. Bioessays. 1999;21:813–823. doi: 10.1002/(SICI)1521-1878(199910)21:10<813::AID-BIES3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 15.Epstein HF, Thomson JN. Temperature-sensitive mutation affecting myofilament assembly in Caenorhabditis elegans. Nature. 1974;250:579–580. doi: 10.1038/250579a0. [DOI] [PubMed] [Google Scholar]
- 16.Kachur T, Ao W, Berger J, Pilgrim D. Maternal UNC-45 is involved in cytokinesis and colocalizes with non-muscle myosin in the early Caenorhabditis elegans embryo. J Cell Sci. 2004;117:5313–5321. doi: 10.1242/jcs.01389. [DOI] [PubMed] [Google Scholar]
- 17.Barral JM, Hutagalung AH, Brinker A, Hartl FU, Epstein HF. Role of the myosin assembly protein UNC-45 as a molecular chaperone for myosin. Science. 2002;295:669–671. doi: 10.1126/science.1066648. [DOI] [PubMed] [Google Scholar]
- 18.Srikakulam R, Winkelmann DA. Chaperone-mediated folding and assembly of myosin in striated muscle. J Cell Sci. 2004;117:641–652. doi: 10.1242/jcs.00899. [DOI] [PubMed] [Google Scholar]
- 19.Etard C, Behra M, Fischer N, Hutcheson D, Geisler R, Strahle U. The UCS factor Steif/Unc-45b interacts with the heat shock protein Hsp90a during myofibrillogenesis. Dev Biol. 2007;308:133–143. doi: 10.1016/j.ydbio.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 20.Du SJ, Li H, Bian Y, Zhong Y. Heat-shock protein 90alpha1 is required for organized myofibril assembly in skeletal muscles of zebrafish embryos. Proc Natl Acad Sci USA. 2008;105:554–559. doi: 10.1073/pnas.0707330105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hawkins TA, Haramis AP, Etard C, Prodromou C, Vaughan CK, Ashworth R, et al. The ATPase-dependent chaperoning activity of Hsp90a regulates thick filament formation and integration during skeletal muscle myofibrillogenesis. Development. 2008;135:1147–1156. doi: 10.1242/dev.018150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barral JM, Bauer CC, Ortiz I, Epstein HF. Unc-45 mutations in Caenorhabditis elegans implicate a CRO1/She4p-like domain in myosin assembly. J Cell Biol. 1998;143:1215–1225. doi: 10.1083/jcb.143.5.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Price MG, Landsverk ML, Barral JM, Epstein HF. Two mammalian UNC-45 isoforms are related to distinct cytoskeletal and muscle-specific functions. J Cell Sci. 2002;115:4013–4023. doi: 10.1242/jcs.00108. [DOI] [PubMed] [Google Scholar]
- 24.Etard C, Roostalu U, Strahle U. Shuttling of the chaperones Unc45b and Hsp90a between the A band and the Z line of the myofibril. J Cell Biol. 2008;180:1163–1175. doi: 10.1083/jcb.200709128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srikakulam R, Liu L, Winkelmann DA. Unc45b forms a cytosolic complex with Hsp90 and targets the unfolded myosin motor domain. PLoS ONE. 2008;3:e2137. doi: 10.1371/journal.pone.0002137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu L, Srikakulam R, Winkelmann DA. Unc45 activates Hsp90-dependent folding of the myosin motor domain. J Biol Chem. 2008;283:13185–13193. doi: 10.1074/jbc.M800757200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker MG. Pharmaceutical target identification by gene expression analysis. Mini Rev Med Chem. 2001;1:197–205. doi: 10.2174/1389557013407034. [DOI] [PubMed] [Google Scholar]
- 28.Hoppe T, Cassata G, Barral JM, Springer W, Hutagalung AH, Epstein HF, et al. Regulation of the myosin-directed chaperone UNC-45 by a novel E3/E4-multiubiquitylation complex in C. elegans. Cell. 2004;118:337–349. doi: 10.1016/j.cell.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 29.Rosser MF, Washburn E, Muchowski PJ, Patterson C, Cyr DM. Chaperone functions of the E3 ubiquitin ligase CHIP. J Biol Chem. 2007;282:22267–22277. doi: 10.1074/jbc.M700513200. [DOI] [PubMed] [Google Scholar]
- 30.Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, Fan CY, et al. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126:571–582. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 31.Janiesch PC, Kim J, Mouysset J, Barikbin R, Lochmuller H, Cassata G, et al. The ubiquitin-selective chaperone CDC-48/p97 links myosin assembly to human myopathy. Nat Cell Biol. 2007;9:379–390. doi: 10.1038/ncb1554. [DOI] [PubMed] [Google Scholar]
- 32.Landsverk ML, Li S, Hutagalung AH, Najafov A, Hoppe T, Barral JM, et al. The UNC-45 chaperone mediates sarcomere assembly through myosin degradation in Caenorhabditis elegans. J Cell Biol. 2007;177:205–210. doi: 10.1083/jcb.200607084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J, Lowe T, Hoppe T. Protein quality control gets muscle into shape. Trends Cell Biol. 2008;18:264–272. doi: 10.1016/j.tcb.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 34.Djabali K, de Nechaud B, Landon F, Portier MM. AlphaB-crystallin interacts with intermediate filaments in response to stress. J Cell Sci. 1997;110:2759–2769. doi: 10.1242/jcs.110.21.2759. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Klevitsky R, Huang W, Glasford J, Li F, Robbins J. AlphaB-crystallin modulates protein aggregation of abnormal desmin. Circ Res. 2003;93:998–1005. doi: 10.1161/01.RES.0000102401.77712.ED. [DOI] [PubMed] [Google Scholar]
- 36.Wang X, Osinska H, Gerdes AM, Robbins J. Desmin filaments and cardiac disease: establishing causality. J Card Fail. 2002;8:S287–S292. doi: 10.1054/jcaf.2002.129279. [DOI] [PubMed] [Google Scholar]
- 37.Bar H, Strelkov SV, Sjoberg G, Aebi U, Herrmann H. The biology of desmin filaments: how do mutations affect their structure, assembly, and organisation? J Struct Biol. 2004;148:137–152. doi: 10.1016/j.jsb.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 38.Bova MP, Yaron O, Huang Q, Ding L, Haley DA, Stewart PL, et al. Mutation R120G in alphaB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function. Proc Natl Acad Sci USA. 1999;96:6137–6142. doi: 10.1073/pnas.96.11.6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldfarb LG, Vicart P, Goebel HH, Dalakas MC. Desmin myopathy. Brain. 2004;127:723–734. doi: 10.1093/brain/awh033. [DOI] [PubMed] [Google Scholar]
- 40.Selcen D, Engel AG. Myofibrillar myopathy caused by novel dominant negative alpha B-crystallin mutations. Ann Neurol. 2003;54:804–810. doi: 10.1002/ana.10767. [DOI] [PubMed] [Google Scholar]
- 41.Treweek TM, Rekas A, Lindner RA, Walker MJ, Aquilina JA, Robinson CV, et al. R120G alphaB-crystallin promotes the unfolding of reduced alpha-lactalbumin and is inherently unstable. FEBS J. 2005;272:711–724. doi: 10.1111/j.1742-4658.2004.04507.x. [DOI] [PubMed] [Google Scholar]
- 42.Vicart P, Caron A, Guicheney P, Li Z, Prevost MC, Faure A, et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998;20:92–95. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- 43.Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, et al. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- 44.Bar H, Mucke N, Ringler P, Muller SA, Kreplak L, Katus HA, et al. Impact of disease mutations on the desmin filament assembly process. J Mol Biol. 2006;360:1031–1042. doi: 10.1016/j.jmb.2006.05.068. [DOI] [PubMed] [Google Scholar]
- 45.Kaminska A, Strelkov SV, Goudeau B, Olive M, Dagvadorj A, Fidzianska A, et al. Small deletions disturb desmin architecture leading to breakdown of muscle cells and development of skeletal or cardioskeletal myopathy. Hum Genet. 2004;114:306–313. doi: 10.1007/s00439-003-1057-7. [DOI] [PubMed] [Google Scholar]
- 46.Munoz-Marmol AM, Strasser G, Isamat M, Coulombe PA, Yang Y, Roca X, et al. A dysfunctional desmin mutation in a patient with severe generalized myopathy. Proc Natl Acad Sci USA. 1998;95:11312–11317. doi: 10.1073/pnas.95.19.11312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med. 2000;342:770–780. doi: 10.1056/NEJM200003163421104. [DOI] [PubMed] [Google Scholar]
- 48.Rappaport L, Contard F, Samuel JL, Delcayre C, Marotte F, Tome F, et al. Storage of phosphorylated desmin in a familial myopathy. FEBS Lett. 1988;231:421–425. doi: 10.1016/0014-5793(88)80863-9. [DOI] [PubMed] [Google Scholar]
- 49.Liu J, Tang M, Mestril R, Wang X. Aberrant protein aggregation is essential for a mutant desmin to impair the proteolytic function of the ubiquitin-proteasome system in cardiomyocytes. J Mol Cell Cardiol. 2006;40:451–454. doi: 10.1016/j.yjmcc.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 50.Chen Q, Liu JB, Horak KM, Zheng H, Kumarapeli AR, Li J, et al. Intrasarcoplasmic amyloidosis impairs proteolytic function of proteasomes in cardiomyocytes by compromising substrate uptake. Circ Res. 2005;97:1018–1026. doi: 10.1161/01.RES.0000189262.92896.0b. [DOI] [PubMed] [Google Scholar]
- 51.Lin DI, Barbash O, Kumar KG, Weber JD, Harper JW, Klein-Szanto AJ, et al. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol Cell. 2006;24:355–366. doi: 10.1016/j.molcel.2006.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.den Engelsman J, Keijsers V, de Jong WW, Boelens WC. The small heat-shock protein alpha B-crystallin promotes FBX4-dependent ubiquitination. J Biol Chem. 2003;278:4699–4704. doi: 10.1074/jbc.M211403200. [DOI] [PubMed] [Google Scholar]
- 53.Inagaki N, Hayashi T, Arimura T, Koga Y, Takahashi M, Shibata H, et al. Alpha B-crystallin mutation in dilated cardiomyopathy. Biochem Biophys Res Commun. 2006;342:379–386. doi: 10.1016/j.bbrc.2006.01.154. [DOI] [PubMed] [Google Scholar]
- 54.Bullard B, Ferguson C, Minajeva A, Leake MC, Gautel M, Labeit D, et al. Association of the chaperone alphaB-crystallin with titin in heart muscle. J Biol Chem. 2004;279:7917–7924. doi: 10.1074/jbc.M307473200. [DOI] [PubMed] [Google Scholar]
- 55.Golenhofen N, Arbeiter A, Koob R, Drenckhahn D. Ischemia-induced association of the stress protein alpha B-crystallin with I-band portion of cardiac titin. J Mol Cell Cardiol. 2002;34:309–319. doi: 10.1006/jmcc.2001.1513. [DOI] [PubMed] [Google Scholar]
- 56.Golenhofen N, Htun P, Ness W, Koob R, Schaper W, Drenckhahn D. Binding of the stress protein alpha B-crystallin to cardiac myofibrils correlates with the degree of myocardial damage during ischemia/reperfusion in vivo. J Mol Cell Cardiol. 1999;31:569–580. doi: 10.1006/jmcc.1998.0892. [DOI] [PubMed] [Google Scholar]
- 57.Haas KF, Woodruff E, III, Broadie K. Proteasome function is required to maintain muscle cellular architecture. Biol Cell. 2007;99:615–626. doi: 10.1042/BC20070019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 59.Zhang C, Xu Z, He XR, Michael LH, Patterson C. CHIP, a cochaperone/ubiquitin ligase that regulates protein quality control, is required for maximal cardioprotection after myocardial infarction in mice. Am J Physiol Heart Circ Physiol. 2005;288:H2836–H2842. doi: 10.1152/ajpheart.01122.2004. [DOI] [PubMed] [Google Scholar]
- 60.Kedar V, McDonough H, Arya R, Li HH, Rockman HA, Patterson C. Muscle-specific RING finger 1 is a bona fide ubiquitin ligase that degrades cardiac troponin I. Proc Natl Acad Sci USA. 2004;101:18135–18140. doi: 10.1073/pnas.0404341102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Witt SH, Granzier H, Witt CC, Labeit S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: towards understanding MURF-dependent muscle ubiquitination. J Mol Biol. 2005;350:713–722. doi: 10.1016/j.jmb.2005.05.021. [DOI] [PubMed] [Google Scholar]
- 62.Witt CC, Witt SH, Lerche S, Labeit D, Back W, Labeit S. Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. EMBO J. 2008;27:350–360. doi: 10.1038/sj.emboj.7601952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Willis MS, Ike C, Li L, Wang DZ, Glass DJ, Patterson C. Muscle ring finger 1, but not muscle ring finger 2, regulates cardiac hypertrophy in vivo. Circ Res. 2007;100:456–459. doi: 10.1161/01.RES.0000259559.48597.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lange S, Xiang F, Yakovenko A, Vihola A, Hackman P, Rostkova E, et al. The kinase domain of titin controls muscle gene expression and protein turnover. Science. 2005;308:1599–1603. doi: 10.1126/science.1110463. [DOI] [PubMed] [Google Scholar]
- 65.Fielitz J, Kim MS, Shelton JM, Latif S, Spencer JA, Glass DJ, et al. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J Clin Invest. 2007;117:2486–2495. doi: 10.1172/JCI32827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koyama S, Hata S, Witt CC, Ono Y, Lerche S, Ojima K, et al. Muscle RING-finger protein-1 (MuRF1) as a connector of muscle energy metabolism and protein synthesis. J Mol Biol. 2008;376:1224–1236. doi: 10.1016/j.jmb.2007.11.049. [DOI] [PubMed] [Google Scholar]
- 67.Zhao TJ, Yan YB, Liu Y, Zhou HM. The generation of the oxidized form of creatine kinase is a negative regulation on muscle creatine kinase. J Biol Chem. 2007;282:12022–12029. doi: 10.1074/jbc.M610363200. [DOI] [PubMed] [Google Scholar]
- 68.Min JN, Whaley RA, Sharpless NE, Lockyer P, Portbury AL, Patterson C. CHIP deficiency decreases longevity, with accelerated aging phenotypes accompanied by altered protein quality control. Mol Cell Biol. 2008;28:4018–4025. doi: 10.1128/MCB.00296-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nyamsuren O, Faggionato D, Loch W, Schulze E, Baumeister R. A mutation in CHN-1/CHIP suppresses muscle degeneration in Caenorhabditis elegans. Dev Biol. 2007;312:193–202. doi: 10.1016/j.ydbio.2007.09.033. [DOI] [PubMed] [Google Scholar]
- 70.Li HH, Kedar V, Zhang C, McDonough H, Arya R, Wang DZ, et al. Atrogin-1/muscle atrophy F-box inhibits calcineurin-dependent cardiac hypertrophy by participating in an SCF ubiquitin ligase complex. J Clin Invest. 2004;114:1058–1071. doi: 10.1172/JCI22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li HH, Willis MS, Lockyer P, Miller N, McDonough H, Glass DJ, et al. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J Clin Invest. 2007;117:3211–3223. doi: 10.1172/JCI31757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Foo RS, Chan LK, Kitsis RN, Bennett MR. Ubiquitination and degradation of the anti-apoptotic protein ARC by MDM2. J Biol Chem. 2007;282:5529–5535. doi: 10.1074/jbc.M609046200. [DOI] [PubMed] [Google Scholar]
- 73.Tian LF, Li HY, Jin BF, Pan X, Man JH, Zhang PJ, et al. MDM2 interacts with and downregulates a sarcomeric protein, TCAP. Biochem Biophys Res Commun. 2006;345:355–361. doi: 10.1016/j.bbrc.2006.04.108. [DOI] [PubMed] [Google Scholar]
- 74.Dargelos E, Poussard S, Brule C, Daury L, Cottin P. Calcium-dependent proteolytic system and muscle dysfunctions: a possible role of calpains in sarcopenia. Biochimie. 2008;90:359–368. doi: 10.1016/j.biochi.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 75.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 76.Jackman RW, Kandarian SC. The molecular basis of skeletal muscle atrophy. Am J Physiol Cell Physiol. 2004;287:C834–C843. doi: 10.1152/ajpcell.00579.2003. [DOI] [PubMed] [Google Scholar]
- 77.Raynaud F, Fernandez E, Coulis G, Aubry L, Vignon X, Bleimling N, et al. Calpain 1-titin interactions concentrate calpain 1 in the Z-band edges and in the N2-line region within the skeletal myofibril. FEBS J. 2005;272:2578–2590. doi: 10.1111/j.1742-4658.2005.04683.x. [DOI] [PubMed] [Google Scholar]
- 78.Solomon V, Goldberg AL. Importance of the ATP-ubiquitin-proteasome pathway in the degradation of soluble and myofibrillar proteins in rabbit muscle extracts. J Biol Chem. 1996;271:26690–26697. doi: 10.1074/jbc.271.43.26690. [DOI] [PubMed] [Google Scholar]
- 79.Galvez AS, Diwan A, Odley AM, Hahn HS, Osinska H, Melendez JG, et al. Cardiomyocyte degeneration with calpain deficiency reveals a critical role in protein homeostasis. Circ Res. 2007;100:1071–1078. doi: 10.1161/01.RES.0000261938.28365.11. [DOI] [PubMed] [Google Scholar]
- 80.Fareed MU, Evenson AR, Wei W, Menconi M, Poylin V, Petkova V, et al. Treatment of rats with calpain inhibitors prevents sepsis-induced muscle proteolysis independent of atrogin-1/MAFbx and MuRF1 expression. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1589–R1597. doi: 10.1152/ajpregu.00668.2005. [DOI] [PubMed] [Google Scholar]
- 81.Gustafsson AB, Gottlieb RA. Recycle or die: the role of autophagy in cardioprotection. J Mol Cell Cardiol. 2008;44:654–661. doi: 10.1016/j.yjmcc.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 83.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 84.Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem. 2006;281:29776–29787. doi: 10.1074/jbc.M603783200. [DOI] [PubMed] [Google Scholar]
- 85.Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007;6:352–361. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 86.Bennett EJ, Bence NF, Jayakumar R, Kopito RR. Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell. 2005;17:351–365. doi: 10.1016/j.molcel.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 87.Tannous P, Zhu H, Nemchenko A, Berry JM, Johnstone JL, Shelton JM, et al. Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation. 2008 doi: 10.1161/CIRCULATIONAHA.107.763870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mammucari C, Schiaffino S, Sandri M. Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy. 2008;4:524–526. doi: 10.4161/auto.5905. [DOI] [PubMed] [Google Scholar]
- 89.Zhao J, Brault JJ, Schild A, Goldberg AL. Coordinate activation of autophagy and the proteasome pathway by FoxO transcription factor. Autophagy. 2008;4:378–380. doi: 10.4161/auto.5633. [DOI] [PubMed] [Google Scholar]
- 90.Attaix D, Bechet D. FoxO3 controls dangerous proteolytic liaisons. Cell Metab. 2007;6:425–427. doi: 10.1016/j.cmet.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 91.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 92.Morimoto S. Sarcomeric proteins and inherited cardiomyopathies. Cardiovasc Res. 2008;77:659–666. doi: 10.1093/cvr/cvm084. [DOI] [PubMed] [Google Scholar]
- 93.Witt CC, Gerull B, Davies MJ, Centner T, Linke WA, Thierfelder L. Hypercontractile properties of cardiac muscle fibers in a knock-in mouse model of cardiac myosin-binding protein-C. J Biol Chem. 2001;276:5353–5359. doi: 10.1074/jbc.M008691200. [DOI] [PubMed] [Google Scholar]
- 94.Yang Q, Sanbe A, Osinska H, Hewett TE, Klevitsky R, Robbins J. A mouse model of myosin binding protein C human familial hypertrophic cardiomyopathy. J Clin Invest. 1998;102:1292–1300. doi: 10.1172/JCI3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gomes AV, Potter JD. Cellular and molecular aspects of familial hypertrophic cardiomyopathy caused by mutations in the cardiac troponin I gene. Mol Cell Biochem. 2004;263:99–114. doi: 10.1023/B:MCBI.0000041852.42291.aa. [DOI] [PubMed] [Google Scholar]
- 96.Pohlmann L, Kroger I, Vignier N, Schlossarek S, Kramer E, Coirault C, et al. Cardiac myosin-binding protein C is required for complete relaxation in intact myocytes. Circ Res. 2007 doi: 10.1161/CIRCRESAHA.107.158774. [DOI] [PubMed] [Google Scholar]
- 97.Sarikas A, Carrier L, Schenke C, Doll D, Flavigny J, Lindenberg KS, et al. Impairment of the ubiquitin-proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc Res. 2005;66:33–44. doi: 10.1016/j.cardiores.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 98.Bao J, Sato K, Li M, Gao Y, Abid R, Aird W, et al. PR-39 and PR-11 peptides inhibit ischemia-reperfusion injury by blocking proteasome-mediated I kappa B alpha degradation. Am J Physiol Heart Circ Physiol. 2001;281:H2612–H2618. doi: 10.1152/ajpheart.2001.281.6.H2612. [DOI] [PubMed] [Google Scholar]
- 99.Gao Y, Lecker S, Post MJ, Hietaranta AJ, Li J, Volk R, et al. Inhibition of ubiquitin-proteasome pathway-mediated I kappa B alpha degradation by a naturally occurring antibacterial peptide. J Clin Invest. 2000;106:439–448. doi: 10.1172/JCI9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Luss H, Schmitz W, Neumann J. A proteasome inhibitor confers cardioprotection. Cardiovasc Res. 2002;54:140–151. doi: 10.1016/s0008-6363(02)00232-8. [DOI] [PubMed] [Google Scholar]
- 101.Pye J, Ardeshirpour F, McCain A, Bellinger DA, Merricks E, Adams J, et al. Proteasome inhibition ablates activation of NF-kappa B in myocardial reperfusion and reduces reperfusion injury. Am J Physiol Heart Circ Physiol. 2003;284:H919–H926. doi: 10.1152/ajpheart.00851.2002. [DOI] [PubMed] [Google Scholar]
- 102.Stangl K, Gunther C, Frank T, Lorenz M, Meiners S, Ropke T, et al. Inhibition of the ubiquitin-proteasome pathway induces differential heat-shock protein response in cardiomyocytes and renders early cardiac protection. Biochem Biophys Res Commun. 2002;291:542–549. doi: 10.1006/bbrc.2002.6476. [DOI] [PubMed] [Google Scholar]
- 103.Stansfield WE, Moss NC, Willis MS, Tang R, Selzman CH. Proteasome inhibition attenuates infarct size and preserves cardiac function in a murine model of myocardial ischemia-reperfusion injury. Ann Thorac Surg. 2007;84:120–125. doi: 10.1016/j.athoracsur.2007.02.049. [DOI] [PubMed] [Google Scholar]
- 104.Enrico O, Gabriele B, Nadia C, Sara G, Daniele V, Giulia C, et al. Unexpected cardiotoxicity in haematological bortezomib treated patients. Br J Haematol. 2007;138:396–397. doi: 10.1111/j.1365-2141.2007.06659.x. [DOI] [PubMed] [Google Scholar]
- 105.Appelman YE, Doevendans PA. Proteasome inhibition and stress compromise the heart in chemotherapy. Cardiovasc Res. 2008;79:547–548. doi: 10.1093/cvr/cvn191. [DOI] [PubMed] [Google Scholar]
- 106.Fu HY, Minamino T, Tsukamoto O, Sawada T, Asai M, Kato H, et al. Overexpression of endoplasmic reticulum-resident chaperone attenuates cardiomyocyte death induced by proteasome inhibition. Cardiovasc Res. 2008;79:600–610. doi: 10.1093/cvr/cvn128. [DOI] [PubMed] [Google Scholar]
- 107.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]