

Abstract
Early piscine life stages are sensitive to polycyclic aromatic hydrocarbon (PAH) exposure, which can cause pericardial effusion and craniofacial malformations. We previously reported that certain combinations of PAHs cause synergistic developmental toxicity, as observed with coexposure to the aryl hydrocarbon receptor agonist β-naphthoflavone (BNF) and cytochrome P4501A inhibitor α-naphthoflavone (ANF). Herein, we hypothesized that oxidative stress is a component of this toxicity. We examined induction of antioxidant genes in zebrafish embryos (Danio rerio) exposed to BNF or ANF individually, a BNF + ANF combination, and a prooxidant positive control, tert-butylhydroperoxide (tBOOH). We measured total glutathione (GSH) and attempted to modulate deformities using the GSH synthesis inhibitor L-buthionine (S,R)-sulfoximine (BSO) and increase GSH pools with N-acetyl cysteine (NAC). In addition, we used a morpholino to knockdown expression of the antioxidant response element transcription factor NRF2 to determine if this would alter gene expression or increase deformity severity. BNF + ANF coexposure significantly increased expressions of superoxide dismutase 1 and 2, glutathione peroxidase 1, pi class glutathione-s-transferase, and glutamate cysteine-ligase to a greater extent than tBOOH, BNF, or ANF alone. BSO pretreatment decreased some GSH levels, but did not worsen deformities, nor did NAC diminish toxicity. Knockdown of NRF2 increased mortality following tBOOH challenge, prevented significant upregulation of antioxidant genes following both tBOOH and BNF + ANF exposures, and exacerbated BNF + ANF–related deformities. Collectively, these findings demonstrate that antioxidant responses are a component of PAH synergistic developmental toxicity and that NRF2 is protective against prooxidant and PAH challenges during development.
Keywords: PAH, NRF2, redox, ROS, embryonic development, glutathione
Many pollutants cause perturbations in cellular redox and can generate reactive oxygen species (ROS) (Livingstone, 2001; Valavanidis et al., 2006). While ROS are produced endogenously via respiration and oxygenating enzymes (Droge, 2003), oxidative stress can result when there is “a disruption of redox signaling and control” (Jones, 2006b). This is a mechanism of toxicity of numerous xenobiotics, and contributes to maladies ranging from chemical carcinogenesis to neurodegenerative diseases (Droge, 2003; Valavanidis et al., 2006). Embryonic development may be especially sensitive to oxidative stress, as even a 15%–20% increase in ROS can tip progenitor cells into premature cell cycle arrest and differentiation (Li et al., 2007; Smith et al., 2000). In mammals, in utero oxidative stress can alter gene expression persisting through adulthood, affecting DNA methylation, and increasing cancer susceptibility (Wan and Winn, 2006). Thus, toxicant exposures that cause oxidative stress during embryonic development are a significant health risk.
Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous contaminants produced by the incomplete combustion of organic compounds, and concentrations are increasing with usage of fossil fuels (Van Metre and Mahler, 2005). Complex mixtures of PAHs commonly include four- and five-ring structures that can have variable interactions with the aryl hydrocarbon receptor (AHR) pathway. The AHR is a member of the basic helix-loop-helix per-ARNT-Sim transcription factor family (Gu et al., 2000); ligand binding to the AHR upregulates several detoxifying enzymes, including cytochrome P4501A (CYP1A), important in metabolism of PAHs. While some PAHs are excellent agonists for the AHR, others can inhibit the activity of CYP1A. Coexposures to PAHs with these characteristics cause synergistic embryotoxicity characterized by craniofacial malformations and cardiovascular deformities including pericardial effusion (Billiard et al., 2006; Hodson et al., 2007; Wassenberg and Di Giulio, 2004).
PAHs can generate ROS via production of PAH metabolites that include redox-cycling quinones and radical cations, the latter being associated with DNA damage (Cavalieri and Rogan, 1992; Melius, 1984; Miranda et al., 2006). Also, PAHs can damage mitochondrial DNA and perturb mitochondrial function, which may enhance oxidative stress in these organelles (Li et al., 2003; Zhu et al., 1995). Induction of CYP450s via the AHR by compounds such as chlorinated dioxins and PCBs can lead to ROS generation and oxidative damage (Nebert et al., 2000; Schlezinger and Stegeman, 2001), associated with the recalcitrance of these compounds to metabolism. Numerous PAHs also induce CYP450s via the AHR, but this induction has not been thought to generate ROS per se, due to the suitability of PAHs as CYP substrates. However, ROS generation may be enhanced in coexposures to PAHs which include both AHR agonists and CYP inhibitors, exposures which give rise to synergistic embryotoxicity (Billiard et al., 2006; Wassenberg and Di Giulio, 2004).
Several studies have observed indications of oxidative stress in fish exposed to PAHs. Rainbow trout larvae exposed to the alkylated-PAH retene had reduced glutathione (GSH): glutathione disulfide ratios and whole-body vitamin E concentrations (Bauder et al., 2005). Killifish from a PAH-contaminated Superfund site had elevated total GSH and mitochondrial lipid peroxidation (Bacanskas et al., 2004). Laboratory-raised progeny from this site was better equipped than reference site F1s to withstand oxidative stress, with increased survival times to tert-butylhydroperoxide (tBOOH), total GSH, MnSOD protein, and GSH reductase activity (Meyer et al., 2003). Altered antioxidant defenses, an indication of oxidative stress, appear to be acting in response to PAH exposure.
Many antioxidant defenses respond to the antioxidant response element (ARE) transcription factor, NF-E2 p45–related factor 2 (NRF2), a member of the cap‘n’collar basic region-leucine zipper transcription factors. Under unstressed conditions, NRF2 is constitutively bound to Kelch-like ECH–associated protein 1 (KEAP1) in the cytoplasm and targeted for degradation (Nguyen et al., 2003a). Upon ROS detected by KEAP1 or by redox-responsive signaling pathways (Cullinan and Diehl, 2006; Cullinan et al., 2003; Huang et al., 2000; Kong et al., 2001; Zipper and Mulcahy, 2003), NRF2 translocates to the nucleus, associates with small MAF proteins, and binds to AREs in the promoter regions of a set of cytoprotective genes (Kobayashi et al., 2006). This pathway is conserved in zebrafish (Kobayashi et al., 2002; Li et al., 2008), and NRF2 has been shown to be upregulated by AHR activation in mice (Miao et al., 2005).
We hypothesized that oxidative stress is a component of the developmental toxicity of PAH mixtures. Zebrafish embryos were exposed to a combination of two oxygen-substituted model PAHs shown to result in synergistic toxicity, the AHR agonist β-naphthoflavone (BNF), and CYP1A inhibitor α-naphthoflavone (ANF) (Billiard et al., 2006). We examined expression of several antioxidant genes, total GSH, and whether redox state could modulate pericardial effusion. We also knocked down NRF2 and asked whether this exacerbated deformities. Our results support oxidative stress as a component of the synergistic developmental toxicity of PAH mixtures and identify a protective role for NRF2 in embryonic oxidative stress. However, the precise role of oxidative stress in cardiovascular deformities due to BNF + ANF coexposures remains unclear.
MATERIALS AND METHODS
Chemicals.
The chemicals ANF, BNF, N-acetyl cysteine (NAC), L-buthionine (S,R)-sulfoximine (BSO), GSH, naphthalene dicarboxaldehyde (NDA), n-ethylmorpholine, triscarboxyethylphosphine, and tBOOH were obtained from Sigma Aldrich (St, MO). Dimethyl sulfoxide (DMSO), potassium hydroxide, and sodium hydroxide were purchased from Mallinckrodt Baker (Phillipsburg, NJ). Sulfosalicyclic acid was obtained from Ricca Chemical Company (Arlington, TX). ANF and BNF were dissolved in DMSO and kept protected from light at − 20°C and vortexed prior to use. NAC and BSO were dissolved in 30% Danieau water, and NAC was pH adjusted to 7.0 and stored at − 20°C; BSO was stored at 4°C.
Animals.
Zebrafish embryos were collected within 1 h following mating of AB strain adults (From the laboratory of Dr Elwood Linney at Duke University and the Zebrafish International Research Consortium in Eugene, OR) maintained at 28°C on a 14-h light, 10-h dark light cycle. Adults were fed with TetraMin flake food and brine shrimp. Embryos were maintained in 30% Danieau water (Nasevicius and Ekker, 2000) under the same temperature and photoperiod conditions.
Exposure for gene expression experiments.
At 24 hours post fertilization (hpf), embryos were dosed in replicate pools of five embryos, with three replicates per treatment, in 20-ml glass vials containing 7.5 ml 30% Danieau water. Final DMSO concentration was 0.03%. Coexposure experiments consisted of exposures to DMSO, 1 μg/l BNF alone, 100 μg/l ANF alone, and 1 μg/l BNF + 100 μg/l ANF in combination, sampled for RNA at 48 or 96 hpf. Coexposure experiments were repeated at least four times, and each data point represents an n of 12 pools of five embryos. The dose-response analysis for BNF was sampled at 48 hpf and included concentrations of 0, 1, 10, 50, and 100 μg/l; the concentration of 50 μg/l caused a slight increase in pericardial effusion, while 100 μg/l embryos appeared similar to those in the 1 μg/l BNF + 100 μg/l ANF coexposures at 96 hpf (Billiard et al., 2006). Dose-response experiments were repeated at least twice, and each data point represents an n of at least six pools of five embryos. Embryos were left in original dosing solutions for the duration of the experiments. The model prooxidant tBOOH was used as a positive control. Three pools of five embryos were dosed at 24 hpf in glass scintillation vials in 7.5 ml of 30% Danieau water containing either 0 or 500μM tBOOH; this exposure was repeated 3 h prior to fixation at 48 hpf in order to run in parallel to tBOOH survival experiments and allow sufficient time for a transcription response after the second dose. No death or deformities were observed at this time point. Before sampling, embryos were rinsed with 30% Danieau water and manually dechorionated where necessary, then fixed in RNA later (Ambion, Foster City, CA), and stored at − 80°C.
Total RNA extraction and reverse transcription.
RNA extractions were carried out according to the RNA easy MicroKit Protocol (Qiagen, Valencia, CA). RNA quantity and quality were analyzed spectrophotometrically using a NanoDrop ND-1000 (NanoDrop Technologies, Wilmington, DE). The Omniscript complementary DNA (cDNA) synthesis kit for Reverse Transcription (Qiagen) was used according to the manufacturer's instructions using 500 ng of RNA, random hexamers, and RNAse inhibitor and carried out in a thermocycler for 1 h at 37°C. Resulting cDNA was diluted to a working concentration of 2 ng/μl.
Quantitative real-time PCR.
β-Actin primers were designed using Light Cycler Probe Design software (Roche, Indianapolis, IN). Primers for gstp1, gstp2, and gclc were designed using PrimerQuest software (Integrated DNA Technologies, Inc., www.idtdna.com). Due to high sequence similarity between gstp1 and gstp2 (90.4% sequence similarity), conserved regulatory elements, and inconsistent results with previously published primers (Suzuki et al., 2005), we designed primers that would amplify both of these homologs at once, henceforth referred to as gstp. Sod2, Sod1, and gpx1 primers were published previously (Malek et al., 2004). Primer efficiencies were also tested to ensure the housekeeping and target genes amplified at the same rate. The sequences employed are listed in Table 1.
TABLE 1.
Primer and GenBank Accession Numbers for QRT-PCR
| Gene | GenBank ID | Forward primer (5′–3′) | Reverse primer (5′–3′) |
| β-Actin | AF057040 | ACATCCGTAAGGACCTG | GGTCGTTCGTTTGAATCTC |
| Sod1 | Y12236 | CGCATGTTCCCAGACATCTA | GAGCGGAAGATTGAGGATTG |
| Sod2 | AW07696 | CTAGCCCGCTGACATTACATC | TTGCCCACATAGAAATGCAC |
| Gpx1 | AW232474 | AGATGTCATTCCTGCACACG | AAGGAGAAGCTTCCTCAGCC |
| Gstp | NM_131734; NM_199277 | TCTGGACTCTTTCCCGTCTCTCAA | ATTCACTGTTGCCGTTGCCGT |
| Gclc | NM_199277 | AACCGACACCCAAAGATTCAGCACT | CCATCATCCTCTGGAAACACCTCC |
A 25 μl reaction contained 200nM of each primer, 12.5 μl 2× Sybrgreen Lightcycler Master Mix (Applied Biosystems, Foster City, CA), 9.5 μl dH2O, and 4 ng cDNA template. quantitative real-time PCR (QRT-PCR) was carried out on an ABI 7300 quantitative real-time PCR machine under the following thermal cycle: 10 min at 95°C, followed by 40 replicates of 15 s at 95°C, then 1 min at 60°C, followed by a dissociation curve. All samples were run in duplicate.
QRT-PCR data were analyzed using the ABI PRISM 7000 Sequence Detection System, Version 1.1 (Applied Biosystems). The comparative CT method (Livak and Schmittgen, 2001) was used to determine average fold induction of messenger RNA (mRNA) by comparing the CT of the target gene to that of the reference gene (β-actin). Reference gene expression was not altered by treatment. Each technical replicate was averaged prior to data analysis. The fold change obtained for each biological replicate pool of five embryos was averaged for treatments; 6–15 replicates were used to obtain final induction averages and standard error per treatment per time point.
GSH quantification.
Total GSH (GSH + glutathione disulfide [GSSG]) levels were quantified in pools of 25 embryos at 72 hpf that had been exposed to DMSO, 1 μg/l BNF, 100 μg/l ANF, or BNF + ANF from 48 to 72 hpf and included replicates of these doses that had been pretreated at 24 hpf with either 100μM NAC or 1000μM BSO. Embryos were washed with Danieau water and snap frozen in liquid nitrogen and stored at − 80°C until analysis. We used a plate reader assay based on the reaction of NDA with GSH or γ-GC to form highly fluorescent cyclized products to quantify total GSH (White et al., 2003). Fluorescence was read on a plate reader (FLUOstar OPTIMA, BMG Labtech, Offenburg, Germany) at an excitation of 470 nm and emission of 520 nm, quantified based on a linear standard curve and corrected for protein content. Protein was measured using the BioRad Protein microtiter plate assay with BioRad Protein Assay dye reagent concentrate (Hercules, CA), bovine serum albumin standards, and absorbance at 595 nm.
Redox modulation and deformity experiments.
These experiments tested whether pretreatment with a GSH-modulating chemical, NAC or BSO, would alter severity of pericardial effusion resulting from BNF + ANF coexposures. Embryos were pretreated from 24 to 48 hpf and supplemented daily with BSO or NAC during a static 1 μg/l BNF + 100 μg/l ANF coexposure from 48 to 120 hpf. While the PAH exposure in these experiments is 24 h later than in the gene expression experiments, with respect to deformities, there is no difference between whether the embryos are dosed at 24 or 48 hpf (Timme-Laragy, unpublished results). Dosing was conducted in triplicate 20 ml glass scintillation vials containing pools of five embryos in a total volume of 7.5 ml 30% Danieau water, with a final DMSO concentration of 0.03% in all groups. At 120 hpf, embryos were washed with Danieau water, anesthetized in MS-222, mounted in 3% methylcellulose, and imaged in a left lateral orientation under ×50 magnification (Zeiss Axioskop, Thornwood, NY.). While other deformities including craniofacial malformations such as truncation of Meckel's cartilage are also observed and can be measured, we have found that pericardial effusion is a robust and corollary indicator of the severity of all related deformities (Billiard et al., 2006). Pericardial effusion area was quantified using IP Lab software (Scanalytics Inc., Fairfax, VA) and normalized to control pericardial area as described previously (Billiard et al., 2006). BSO experiments also included nontoxic BNF + ANF combination with a lower dose of ANF (50 instead of 100 μg/l) to determine whether there would be exacerbation of deformities; these were analyzed at 72 and 120 hpf.
Embryo morpholino injection.
We used a morpholino (Gene Tools, Philomath, OR) that had been designed previously to block initiation of translation of zebrafish NRF2 mRNA, (5′-CATTTCAATCTCCATCATGTCTCAG-3′) (Kobayashi et al., 2002). Gene Tools’ standard control morpholino (5′-CCTCTTACCTCAGTTACAATTTATA-3′) was used as the control morpholino (CO-MO) in experiments. Morpholinos were diluted to working stocks of 0.1mM in 30% Danieau's solution for injection (Nasevicius and Ekker, 2000). Injection working stocks were stored at 4°C and heated for 5 min at 65°C prior to use. Both morpholinos were tagged with a 3′-end carboxyfluorescein modification in order to monitor injection success.
Newly fertilized embryos at the one- to four-cell stage (up to approximately 1 hpf) were injected with approximately 3 nl of morpholino using a Narishige IM300 Microinjector (Tokyo, Japan). Only healthy embryos exhibiting strong, uniform fluorescence at 24 hpf were subsequently used in experiments.
Prooxidant survival exposures.
For tBOOH survival experiments, embryos were sorted into individual wells in a 96-well plate at 24 hpf in 200 μl of 30% Danieau water. Noninjected, control morpholino–injected, and NRF2 morpholino–injected embryos were exposed to concentrations of 0, 0.1, 0.5, 0.75, 1, and 2mM tBOOH at 48 and 72 hpf. Cumulative mortality was analyzed at 96 hpf.
Statistical analysis.
Data were analyzed with Statview for Windows (version 5.0.1; SAS Institute, Cary, NC). ANOVA was used to determine significant factors. When ANOVA yielded significance (p < 0.05), Fisher's protected least-significant differences was used as a post hoc test. Data are presented as mean ± SEM, and n defined as number of pools of five or 25 embryos. LC50 and 95% confidence intervals for tBOOH exposures were calculated using probit analysis in SPSS software (SPSS 15.0, SPPS Inc, Chicago, IL).
RESULTS
Upregulation of Antioxidant Genes
We examined induction of several antioxidant genes by quantitative real-time PCR, both prior to onset of deformities at 48 hpf, and after deformity development at 96 hpf, in zebrafish embryos exposed to a dose of BNF + ANF that results in synergistic developmental toxicity. We observed a significant fold-change induction (p ≤ 0.003) in the BNF + ANF coexposure groups compared to vehicle controls for gclc, gstp, gpx1, sod1, and sod2 at 48 hpf but not at 96 hpf (Fig. 1). At 48 hpf, gstp exhibited the highest induction of 10.7-fold, followed by gclc at 4.6-fold; both of these inductions were synergistic (two-factor ANOVA interaction terms 0.02 and 0.03). The next highest response was a 4.1-fold increase of gpx1. Sod2 and sod1 were the least responsive, yet still induced 2.9- and 2.6-fold, respectively. Individual doses of BNF (1 μg/l) and ANF (100 μg/l) did not result in a significant induction of any of these genes. No significant differences in antioxidant gene induction were observed after deformity onset at the 96 hpf time point, unlike the constant activation of the AHR, which continues to upregulate cyp1a, cyp1b1, cyp1c1, and ahrr2 at 96 hpf time points (Timme-Laragy et al., 2007).
FIG. 1.

Increase in expression at 48 but not 96 hpf of antioxidant genes gclc, gstp, gpx1, sod1, and sod2 in zebrafish embryos coexposed to BNF and ANF, but not either compound alone, compared to DMSO controls. All exposures began at 24 hpf. *p ≤ 0.003. Data are presented as mean + SEM, n ≥12.
Significant inductions of gstp, gpx1, and gclc were also observed at 48 hpf in embryos exposed to a sublethal dose of tBOOH (0.5mM; no mortality at this time point), and induction levels were similar to or less than those observed in the 48 hpf BNF + ANF embryos (Table 2). The induction of antioxidant genes is indicative of redox signaling perturbations, ROS, and/or oxidative damage.
TABLE 2.
Fold Changes of mRNA at 48 hpf by tBOOH (0.5mM) Compared to Induction by the BNF + ANF (1 + 100 μg/l) Coexposure
| Gene | tBOOH | BNF + ANF |
| gclc | 2.8 ± 0.5 | 4.6 ± 1.8 |
| gstp | 6.7 ± 1.3 | 10.7 ± 2.8 |
| gpx1 | 3.2 ± 0.6 | 4.1 ± 1.3 |
| sod2 | 1.6 ± 0.3 | 2.9 ± 0.7 |
| sod1 | 1.4 ± 0.3 | 2.6 ± 0.8 |
Bold text indicates significant inductions compared to the respective vehicle controls (p < 0.05). n ≥12 pools of five embryos.
Our previous work showed that exposure to 1 μg/l BNF + 100 μg/l ANF mimicked the AHR-regulated gene induction of higher, single doses of BNF (50 and 100 μg/l), suggesting alterations in metabolism may occur in this mixture toxicity (Timme-Laragy et al., 2007). Increasing BNF exposure resulted in a dose-response induction of gclc, gstp, gpx1, and sod2 (p ≤ 0.024), but not sod1 (Fig. 2). The highest dose of BNF (100 μg/l) yielded either similar (for gstp and gpx1) or lower levels than the combination dose.
FIG. 2.

The antioxidant genes gstp, gpx1, gclc, and sod2 are induced in a dose-dependent manner following 24-h exposure to BNF in 48 hpf zebrafish embryos. The inductions of gclc, sod1, and sod2 have been additionally plotted on an appropriate viewing scale (right). Following significant ANOVAs (p ≤ 0.03), Fisher's protected least-significant differences significance above control is noted by (*p ≤ 0.04). Data are presented as mean ± SEM where n ≥6.
Total GSH Quantification
Total GSH was measured at 72 hpf, a time period which followed pretreatment with NAC or BSO (24–48 hpf) and was 24 h postadministration of BNF + ANF at 48 hpf. Exposures to BNF, ANF, and BNF + ANF elicited no significant differences in GSH concentrations among groups (Fig. 3). However, all three of these exposures trended toward an increase in total GSH levels from approximately 30–40 μmol GSH/μg protein compared to DMSO controls (Fig. 3, p = 0.075–0.093). Pretreatment with 100μM NAC, an antioxidant that increases available pools of cysteine and can increase GSH levels, did increase total GSH concentrations in control embryos, but did not elevate levels above those observed by PAH treatment. As GSH is normally present in high concentrations, this may present an inherent difficulty in detecting a statistically significant increase in these samples. Also, these experiments measured total GSH in whole embryos, which could mask important tissue-specific changes.
FIG. 3.

Total GSH quantification at 72 hpf, with and without 100μM NAC and 1mM BSO. NAC and BSO treatments spanned 24–72 hpf and PAH and vehicle control treatments spanned 48–72 hpf. A three-factor ANOVA for pretreatment, BNF, and ANF showed a significant effect of NAC (p = 0.03) and BSO (p < 0.0001). *indicates a significant difference (p ≤ 0.05) of either NAC or BSO pretreatment on GSH levels compared to no pretreatment controls within an exposure group; mean + SEM; n = 3 pools of 25 embryos.
BSO inhibits the rate-limiting enzyme in GSH synthesis, glutamate-cysteine ligase, thereby preventing replacement of GSH pools and hindering the GSH response to oxidative stress. BSO alone did not decrease GSH in the control treatments, but significant lower total GSH levels were observed with BSO and BNF and BNF + ANF groups compared with no pretreatment (Fig. 3, p = 0.001), indicating an accelerated depletion of GSH and increased utilization of this endogenous antioxidant peptide. This supports the hypothesis that oxidative events are occurring in this toxicity, although this may be a BNF effect that is not specific to the synergistic toxicity.
Redox Modulation and Deformities
In these experiments, embryos were pretreated with a redox-modulating chemical to determine whether a more reduced or oxidized redox level would alter deformity severity resulting from exposure to BNF + ANF. Pretreatment with NAC at nontoxic doses of 50 and 100μM did not prevent deformities, and 500μM worsened pericardial effusion in the BNF + ANF–treated embryos (Fig. 4). Controls treated with 500μM NAC began to show signs of toxicity as indicated by a slight increase in pericardial effusion and thinning of the lower jaw tissue. The highest dose used, 1000μM, was fatal (data not shown).
FIG. 4.
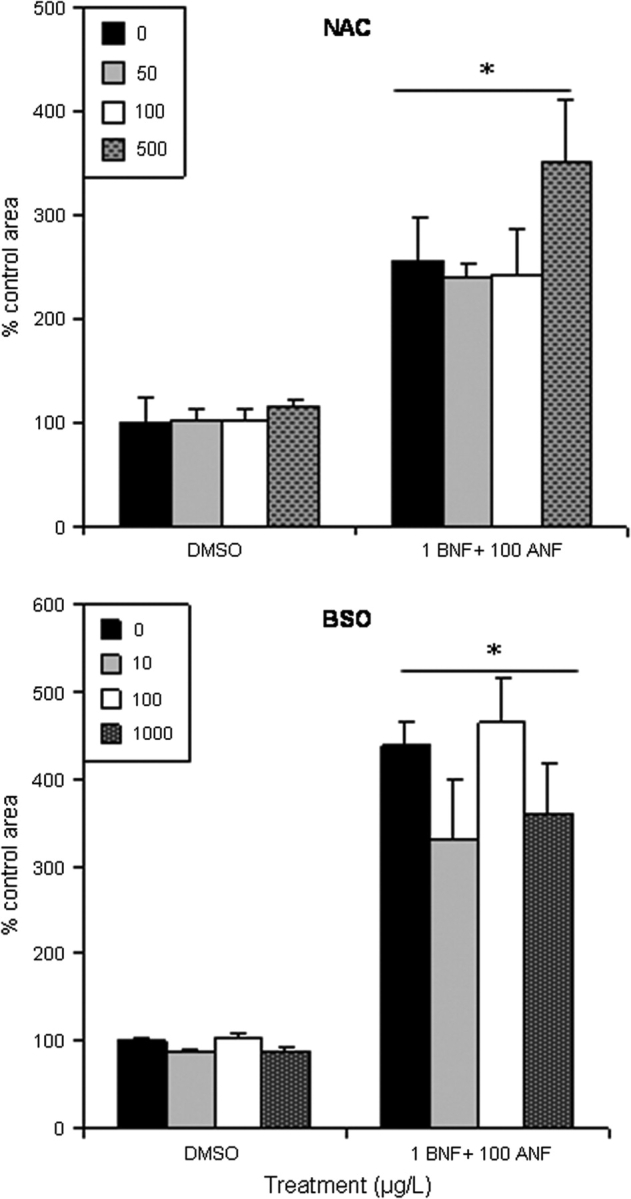
Pretreatment with varying doses (μM) of NAC did not rescue pericardial effusion, and BSO did not worsen it. Pericardial effusion areas in BNF + ANF exposures are significantly higher than DMSO controls (*p ≤ 0.05). Single doses of 1 μg/l BNF and 100 μg/l ANF showed no significant differences from DMSO controls and were excluded from graphs for simplicity, but included in statistical analyses. Data are presented as mean + SEM; n = 3 pools of five embryos, and each data point is representative of at least 30 embryos.
Pretreatment with BSO failed to worsen deformities at all nontoxic doses used (10, 100, and 1000μM) (Fig. 4). Furthermore, studies which used a nontoxic combination of BNF + ANF (1 μg/l BNF and 50 μg/l ANF) previously shown to exhibit toxicity upon injection of a CYP1A-morpholino (Billiard et al., 2006), did not reveal deformity exacerbation by GSH depletion (Fig. 5).
FIG. 5.

Toxicity from a nontoxic combination of BNF + ANF is not exacerbated by pretreatment with 1000μM BSO at either 72 or 120 hpf. No statistically significant differences were observed in pericardial effusion between any of the treatment groups, including single exposure of 1 μg/l BNF and 50 μg/l ANF, which were excluded from the graph for simplicity. Data are presented as mean + SEM, n = 3 pools of five embryos.
NRF2 Knockdown and tBOOH Survival
Although the NRF2 morpholino sequence has been previously described and verified (Kobayashi et al., 2002), those experiments did not examine the effect of NRF2 knockdown on embryo survival to a prooxidant exposure. As did Kobayashi et al. (2002), we observed that injection of NRF2 morpholino at concentrations of 0.1 or 0.25mM had no effect on normal embryonic development (data not shown). In challenging NRF2- versus control morpholino–injected embryos with the model prooxidant tBOOH, we observed a statistically significant shift in the 48-h LC50 at 96 hpf from 891μM in control morpholino to 586μM in NRF2 morpholino–injected embryos (Table 3). This indicates that knockdown of NRF2 at this concentration (0.1mM) and injection volume (3–5 nl) is sufficient to render the fish more sensitive to the toxic effects of a prooxidant.
TABLE 3.
Forty-Eight Hour LC50s at 96 hpf and Confidence Intervals for Noninjected, Control Morpholino–Injected, and NRF2 Knockdown Embryos Exposed to the Prooxidant tBOOH at 48 and 72 hpf
| LC50 (mM tBOOH) | 95% Confidence intervals | |
| Noninjected | 0.816 | (0.675, 0.948) |
| Control morpholino | 0.891 | (0.735, 1.095) |
| NRF2 morpholino | 0.586* | (0.459, 0.705) |
Significant difference (*) from injected control.
NRF2 Knockdown and Gene Expression
We also examined the effectiveness of the NRF2 morpholino to knock down several antioxidant genes following chemical exposure. Following one dose of 500μM tBOOH at 24 hpf and then again 3 h prior to sampling at 48 hpf (no death or deformities were observed at this time point), noninjected and control morpholino embryos exposed to tBOOH showed significant upregulation of gstp, gpx1, and gclc at levels indistinguishable from one another (Fig. 6). None of these genes were induced in tBOOH-exposed NRF2-MO embryos (Fig. 6). Following exposure of control and NRF2 morpholino–injected embryos to DMSO, 1 μg/l BNF, 100 μg/l ANF, or a BNF + ANF combination, there was a significant interaction between treatment and morpholino (p ≤ 0.003) and significant induction of all five genes examined in the control morpholino groups exposed to BNF + ANF (sod1, sod2, gstp, gpx1, and gclc; Fig. 7, p ≤ 0.0035). The induction of these genes was also significantly inhibited by NRF2 knockdown (Fig. 7, p ≤ 0.0008).
FIG. 6.

Induction of gene expression by tBOOH is inhibited by NRF2 knockdown in 48 hpf embryos exposed to tBOOH at 24 hpf and again 3 h prior to fixation. *p ≤ 0.05.
FIG. 7.

Inhibited upregulation of antioxidant genes at 48 hpf in NRF2 knockdown (NRF2-MO) compared to control morpholino (CO-MO)–injected embryos after a 24-h exposure to DMSO, 1 μg/l BNF, 100 μg/l ANF, or a combination of BNF + ANF. There was a significant interaction between injection and treatment (p ≤ 0.003) in two-way ANOVAs. (*p ≤ 0.004; mean + SEM, n = 3).
NRF2 Knockdown and Deformities
Having confirmed the effectiveness of the morpholino in its ability to render the embryo more sensitive to oxidative stress and inhibit antioxidant gene induction, we then tested the hypothesis that NRF2 plays a protective role in the synergistic developmental toxicity of PAHs. We predicted that knockdown of NRF2 would exacerbate deformity severity caused by toxicity of BNF + ANF. Using a combination of BNF (1 μg/l) and ANF (50 μg/l) that does not cause pericardial effusion in noninjected embryos, we examined whether knockdown of NRF2 was able to exacerbate toxicity. NRF2 knockdown exacerbated pericardial effusion by 35% with the lower dose of ANF in combination with BNF (p ≤ 0.05, Fig. 8), indicating that a fully functional antioxidant response utilizing NRF2 is protective against synergistic developmental toxicity of BNF + ANF.
FIG. 8.

Quantified pericardial effusion at 96 hpf. This dose does not cause pericardial effusion in noninjected (NI) fish, but in NRF2 morpholino (NRF2-MO) embryos, toxicity is exacerbated and results in moderate pericardial effusion. Significant differences noted between both DMSO controls and NI treatment–matched group (*p ≤ 0.003). Data are presented as mean + SEM, n = 20–30 fish.
DISCUSSION
We previously demonstrated that the AHR mediates the synergistic embryotoxicity of BNF + ANF and that CYP1A is protective (Billiard et al., 2006). To further understand the mechanism of this toxicity, we looked for genes downstream of the AHR besides cytochrome P450s that could play a role. NRF2 is one such candidate, with three AHR-binding xenobiotic response elements (XREs) in its promoter in the mouse, and it has been shown to be upregulated by TCDD (Miao et al., 2005). The current study demonstrates that antioxidant responses are acomponent of PAH synergistic developmental toxicity and that NRF2 is protective against prooxidant and PAH exposures during development.
We have shown that antioxidant genes sod1, sod2, gstp, gpx1, and gclc are upregulated at 48 hpf after coexposure to 1 μg/l BNF + 100 μg/l ANF, but not affected by either of these doses alone. These data indicate that there are signaling pathways sensitive to redox perturbations that are responding to BNF + ANF prior to deformity onset at 48 hpf. This appears to be a transient response since at 96 hpf, following onset of pericardial effusion and craniofacial malformations in the BNF + ANF treatments, expression of these genes had returned to control levels. This is in contrast to AHR-regulated genes which remain elevated at this time point (Timme-Laragy et al., 2007).
The induction of antioxidant genes in this synergistic toxicity occurred without a significant change in total GSH, one of the most abundant cellular defenses against oxidative stress and an important cofactor in phase II metabolism. Other studies have observed increased GSH by PAH exposure in fish (Meyer et al., 2003; Wassenberg, 2004). We did significantly increase total GSH levels with pretreatment of NAC in controls, although NAC pretreatment did not exceed the levels of induction by the treatments with BNF and ANF. This could be due to the fact that GSH itself is a negative regulator of the modifier subunit of glutamate-cysteine ligase, the rate-limiting enzyme of GSH synthesis (Di Giulio and Meyer, 2008), thus maintaining a limit on maximum GSH content. Pericardial effusion could also not be reduced with NAC. Rather, at high concentrations, NAC appeared to actually exacerbate deformities.
GSH levels in another fish model, the channel catfish, have been shown to be difficult to modulate by BSO alone, requiring an additional stressor to deplete total GSH concentrations (Gallagher et al., 1992). Similarly, only in our BSO pretreatment groups that were exposed to the additional stressor of BNF or BNF + ANF showed depletion of GSH, evidence that oxidative events are occurring. This may be a BNF effect that is not specific to the synergistic toxicity, further suggested by the lack of GSH depletion on deformity severity.
Some prooxidants have been shown to cause similar deformities as those produced by BNF + ANF in early life stages of fish, such as cumene hydroperoxide in killifish (Wassenberg, 2004) and ethanol in zebrafish (Reimers et al., 2004). Yet the absence of deformities in our nontoxic NAC and BSO pretreatment experiments does not mean that oxidative stress is not an important factor in BNF + ANF toxicity. Oxidative stress does not always result in gross deformities. For example, the prooxidant paraquat failed to produce deformities in rainbow trout larvae (Bauder et al., 2005), and we also did not observe any deformities with the prooxidant tBOOH.
Perturbed redox signaling could contribute to deformities in ways that are independent of thiol status (Jones, 2006b). Beyond GSH status, specifically that of GSH:GSSG which was not a measurement parameter in our experiments, other important thiol/disulfide antioxidant compartments exist: cysteine/cystine in the extracellular space, and thioredoxin-1 (-SH2/-SS-). All three of these are independently controlled and do not necessarily equilibrate amongst each other (Hansen et al., 2006; Jones, 2006a,b; Jones et al., 2004). Methods by which to evaluate these redox ratios in zebrafish embryos would be very useful, but to our knowledge, are not yet available. We would predict that as we were able to detect changes in the coarse measurement of total GSH with BSO pretreatment and BNF exposure, if we were able to examine these three redox compartment ratios we may yet see more significant changes indicative of oxidative stress. However, since ARE signaling can occur independent of GSH:GSSG status and of Trx-1 (Go et al., 2004), changes in antioxidant gene expression are a sufficient method by which to capture significant perturbations in redox status.
To understand how antioxidant responses factor into deformities of this toxicity, we cast a wider net over the redox-responding capability of the embryo by knocking down the ARE transcription factor NRF2. In doing so, we prevented the upregulation of all antioxidant genes examined and also exacerbated pericardial effusion. However, this deformity was only mildly enhanced, suggesting that while oxidative stress is a component of this synergistic toxicity, its precise role in contributing to deformities remains unclear.
AREs are present in many phase II genes; XREs are found in both phase I and phase II genes (Rushmore and Kong, 2002). These two mediators of the xenobiotic detoxification system were previously thought to function independently, but recent identification of three functional XREs in the promoter region of NRF2 (Miao et al., 2005) has caused this assumption to be reevaluated. Knockdown of AHR in mouse cell lines abolished NRF2 induction by TCDD. Site-directed mutagenesis showed that both the ARE and XRE in the NRF2 promoter region can result in mRNA induction of NRF2 by TCDD, and the AHR was shown to directly bind to the NRF2 promoter (Miao et al., 2005). Mouse, rat, and human NRF2 promoters all have multiple copies of XREs (Miao et al., 2005). Little is known about this cross-talk in fish models.
There are several genes with both XRE and AREs including sod1, nqo1, ugt1a, and gstα (Nguyen et al., 2003b). In addition, some chemicals, including the PAHs BNF and benzo(a)pyrene, have been designated as bifunctional inducers, by which metabolism via AHR-XRE–regulated induction of CYP1A can form reactive metabolites that lead to induction of NRF2-ARE–regulated genes (Nguyen et al., 2003b). Our gene expression data showed that the antioxidant genes examined were not induced in NRF2 knockdown embryos; similar results have also been shown in the mammalian and piscine literature (Kobayashi and Yamamoto, 2005; Kobayashi et al., 2004; Rushmore and Kong, 2002; Zhu et al., 2005). The fact that NRF2 knockdown prevented upregulation of these genes with exposure to both PAHs and a prooxidant suggests that the ARE is the more important response element in these promoters.
Knocking down NRF2 increased sensitivity to the tBOOH (decreased LC50) as well as to synergistic PAH toxicity (enhanced pericardial effusion). Other studies also support a protective role for NRF2 in ROS and PAH toxicity. In NRF2−/− mice, cardiac fibroblasts from newly born pups have been shown to have lower expression/antioxidant activity of catalase, glutathione-s-transferase, NAD(P)H quinine oxidoreductase 1, glutathione reductase, and lower GSH levels (Zhu et al., 2005). These cells were also more sensitive to cytotoxic effects of ROS and reactive nitrogen species. Further evidence is seen in adult NRF2−/− mice, which are more susceptible to oxidative damage from PAHs and diesel exhaust. These mice have been shown to have increased rates of stomach neoplasia following exposure to benzo(a)pyrene (Ramos-Gomez et al., 2001) and accelerated 8-oxo-DG DNA adduct formation in lungs of mice exposed to diesel exhaust (Aoki et al., 2001).
In conclusion, we show antioxidant responses, which can be taken as an indication of oxidative stress, are a component of the synergistic developmental toxicity of BNF + ANF and also specify a protective role for NRF2 in prooxidant and PAH exposures. However, the contribution of oxidative stress to the development of related deformities remains unclear. Additional studies are necessary to further an understanding of the role of oxidative stress in the developmental toxicity of PAHs in early life stages of fishes and possibly other vertebrates.
FUNDING
National Institute for Environmental Health Sciences–supported Duke University Superfund Basic Research Program (P42 ES10356), National Institute for Environmental Health Sciences–supported Duke University Integrated Toxicology & Environmental Health Program (TS ES07031), United States Environmental Protection Agency STAR fellowship to A.T.-L., Duke University RJR-Leon Golberg Memorial Postdoctoral Training Program in Toxicology to A.T.-L., and the Postdoctoral Scholar Program at the Woods Hole Oceanographic Institution, with funding provided by the J. Seward Johnson Fund and The Walter A. and Hope Noyes Smith Chair to A.T-L.
Acknowledgments
The authors acknowledge helpful discussions and statistical and technical assistance from Dr Cole Matson and Mr Bryan Clark.
References
- Aoki Y, Sato H, Nishimura N, Takahashi S, Itoh K, Yamamoto M. Accelerated DNA adduct formation in the lung of the Nrf2 knockout mouse exposed to diesel exhaust. Toxicol. Appl. Pharmacol. 2001;173(3):154–160. doi: 10.1006/taap.2001.9176. [DOI] [PubMed] [Google Scholar]
- Bacanskas LR, Whitaker J, Di Giulio RT. Oxidative stress in two populations of killifish (Fundulus heteroclitus) with differing contaminant exposure histories. Mar. Environ. Res. 2004;58(2–5):597–601. doi: 10.1016/j.marenvres.2004.03.048. [DOI] [PubMed] [Google Scholar]
- Bauder MB, Palace VP, Hodson PV. Is oxidative stress the mechanism of blue sac disease in retene-exposed trout larvae? Environ. Toxicol. Chem. 2005;24(3):694–702. doi: 10.1897/04-23r.1. [DOI] [PubMed] [Google Scholar]
- Billiard SM, Timme-Laragy AR, Wassenberg DM, Cockman C, Di Giulio RT. The role of the aryl hydrocarbon receptor pathway in mediating synergistic developmental toxicity of polycyclic aromatic hydrocarbons to zebrafish. Toxicol. Sci. 2006;92(2):526–536. doi: 10.1093/toxsci/kfl011. [DOI] [PubMed] [Google Scholar]
- Cavalieri EL, Rogan EG. The approach to understanding aromatic hydrocarbon carcinogenesis. The central role of radical cations in metabolic activation. Pharmacol. Ther. 1992;55(2):183–199. doi: 10.1016/0163-7258(92)90015-r. [DOI] [PubMed] [Google Scholar]
- Cullinan SB, Diehl JA. Coordination of ER and oxidative stress signaling: The PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006;38(3):317–332. doi: 10.1016/j.biocel.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003;23(20):7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giulio RT, Meyer JN. Reactive oxygen species and oxidative stress. In: Di Giulio RT, Hinton DE, editors. The Toxicology of Fishes. Boca Raton, FL: Taylor and Francis; 2004. [Google Scholar]
- Droge W. Oxidative stress and aging. Adv. Exp. Med. Biol. 2003;543:191–200. doi: 10.1007/978-1-4419-8997-0_14. [DOI] [PubMed] [Google Scholar]
- Gallagher EP, Hasspieler BM, Di Giulio RT. Effects of buthionine sulfoximine and diethyl maleate on glutathione turnover in the channel catfish. Biochem. Pharmacol. 1992;43(10):2209–2215. doi: 10.1016/0006-2952(92)90180-q. [DOI] [PubMed] [Google Scholar]
- Go YM, Gipp JJ, Mulcahy RT, Jones DP. H2O2-dependent activation of GCLC-ARE4 reporter occurs by mitogen-activated protein kinase pathways without oxidation of cellular glutathione or thioredoxin-1. J. Biol. Chem. 2004;279(7):5837–5845. doi: 10.1074/jbc.M307547200. [DOI] [PubMed] [Google Scholar]
- Gu Y-Z, Hogenesch JB, Bradfield CA. The PAS superfamily: Sensors of environmental and developmental signals. Annu. Rev. Pharmacol. Toxicol. 2000;40(1):519–561. doi: 10.1146/annurev.pharmtox.40.1.519. [DOI] [PubMed] [Google Scholar]
- Hansen JM, Go YM, Jones DP. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu. Rev. Pharmacol. Toxicol. 2006;46:215–234. doi: 10.1146/annurev.pharmtox.46.120604.141122. [DOI] [PubMed] [Google Scholar]
- Hodson PV, Qureshi K, Noble CA, Akhtar P, Brown RS. Inhibition of CYP1A enzymes by alpha-naphthoflavone causes both synergism and antagonism of retene toxicity to rainbow trout (Oncorhynchus mykiss) Aquat. Toxicol. 2007;81(3):275–285. doi: 10.1016/j.aquatox.2006.12.012. [DOI] [PubMed] [Google Scholar]
- Huang HC, Nguyen T, Pickett CB. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc. Natl Acad. Sci. U.S.A. 2000;97(23):12475–12480. doi: 10.1073/pnas.220418997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DP. Disruption of mitochondrial redox circuitry in oxidative stress. Chem. Biol. Interact. 2006a;163(1–2):38–53. doi: 10.1016/j.cbi.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Jones DP. Redefining oxidative stress. Antioxid. Redox Signal. 2006b;8(9–10):1865–1879. doi: 10.1089/ars.2006.8.1865. [DOI] [PubMed] [Google Scholar]
- Jones DP, Go YM, Anderson CL, Ziegler TR, Kinkade JM, Jr, Kirlin WG. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. Faseb. J. 2004;18(11):1246–1248. doi: 10.1096/fj.03-0971fje. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Itoh K, Suzuki T, Osanai H, Nishikawa K, Katoh Y, Takagi Y, Yamamoto M. Identification of the interactive interface and phylogenic conservation of the Nrf2-Keap1 system. Genes Cells. 2002;7(8):807–820. doi: 10.1046/j.1365-2443.2002.00561.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi A, Kang MI, Watai Y, Tong KI, Shibata T, Uchida K, Yamamoto M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 2006;26(1):221–229. doi: 10.1128/MCB.26.1.221-229.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi A, Ohta T, Yamamoto M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Meth. Enzymol. 2004;378:273–286. doi: 10.1016/S0076-6879(04)78021-0. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid. Redox Signal. 2005;7(3–4):385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- Kong AN, Owuor E, Yu R, Hebbar V, Chen C, Hu R, Mandlekar S. Induction of xenobiotic enzymes by the MAP kinase pathway and the antioxidant or electrophile response element (ARE/EpRE) Drug Metab. Rev. 2001;33(3–4):255–271. doi: 10.1081/dmr-120000652. [DOI] [PubMed] [Google Scholar]
- Li Z, Dong T, Proschel C, Noble M. Chemically diverse toxicants converge on Fyn and c-Cbl to disrupt precursor cell function. PLoS Biol. 2007;5(2):e35. doi: 10.1371/journal.pbio.0050035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Li, Kobayashi M, Kaneko H, Nakajima-Takagi Y, Nakayama Y, Yamamoto M. Molecular evolution of Keap1: Two Keap1 molecules with distinctive intervening region structures are conserved among fish. J. Biol. Chem. 2008;283(6):3248–3255. doi: 10.1074/jbc.M708702200. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Livingstone DR. Contaminant-stimulated reactive oxygen species production and oxidative damage in aquatic organisms. Mar. Pollut. Bull. 2001;42(8):656–666. doi: 10.1016/s0025-326x(01)00060-1. [DOI] [PubMed] [Google Scholar]
- Malek RL, Sajadi H, Abraham J, Grundy MA, Gerhard GS. The effects of temperature reduction on gene expression and oxidative stress in skeletal muscle from adult zebrafish. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2004;138(3):363–373. doi: 10.1016/j.cca.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Melius P. Comparative benzo[a]pyrene metabolite patterns in fish and rodents. National Cancer Institute Monograph. 1984;65:387–390. [PubMed] [Google Scholar]
- Meyer JN, Smith JD, Winston GW, Di Giulio RT. Antioxidant defenses in killifish (Fundulus heteroclitus) exposed to contaminated sediments and model prooxidants: Short-term and heritable responses. Aquat. Toxicol. 2003;65(4):377–395. doi: 10.1016/j.aquatox.2003.06.001. [DOI] [PubMed] [Google Scholar]
- Miao W, Hu L, Scrivens PJ, Batist G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: Direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem. 2005;280(21):20340–20348. doi: 10.1074/jbc.M412081200. [DOI] [PubMed] [Google Scholar]
- Miranda CL, Chung WG, Wang-Buhler JL, Musafia-Jeknic T, Baird WM, Buhler DR. Comparative in vitro metabolism of benzo[a]pyrene by recombinant zebrafish CYP1A and liver microsomes from [beta]-naphthoflavone-treated rainbow trout. Aquat. Toxicol. 2006;80(2):101–108. doi: 10.1016/j.aquatox.2006.07.018. [DOI] [PubMed] [Google Scholar]
- Nasevicius A, Ekker SC. Effective targeted gene ‘knockdown’ in zebrafish. Nat. Genet. 2000;26(2):216–220. doi: 10.1038/79951. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 2000;59(1):65–85. doi: 10.1016/s0006-2952(99)00310-x. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Sherratt PJ, Huang HC, Yang CS, Pickett CB. Increased protein stability as a mechanism that enhances Nrf2-mediated transcriptional activation of the antioxidant response element. Degradation of Nrf2 by the 26 S proteasome. J. Biol. Chem. 2003a;278(7):4536–4541. doi: 10.1074/jbc.M207293200. [DOI] [PubMed] [Google Scholar]
- Nguyen T, Sherratt PJ, Pickett CB. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 2003b;43:233–260. doi: 10.1146/annurev.pharmtox.43.100901.140229. [DOI] [PubMed] [Google Scholar]
- Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, Kensler TW. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc. Natl Acad. Sci. U.S.A. 2001;98(6):3410–3415. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimers MJ, Flockton AR, Tanguay RL. Ethanol- and acetaldehyde-mediated developmental toxicity in zebrafish. Neurotoxicol. Teratol. 2004;26(6):769–781. doi: 10.1016/j.ntt.2004.06.012. [DOI] [PubMed] [Google Scholar]
- Rushmore TH, Kong AN. Pharmacogenomics, regulation and signaling pathways of phase I and II drug metabolizing enzymes. Curr. Drug Metab. 2002;3(5):481–490. doi: 10.2174/1389200023337171. [DOI] [PubMed] [Google Scholar]
- Schlezinger JJ, Stegeman JJ. Induction and suppression of cytochrome P450 1A by 3,3′,4,4′,5-pentachlorobiphenyl and its relationship to oxidative stress in the marine fish scup (Stenotomus chrysops) Aquat. Toxicol. 2000;52(2):101–115. doi: 10.1016/s0166-445x(00)00141-7. [DOI] [PubMed] [Google Scholar]
- Smith J, Ladi E, Mayer-Proschel M, Noble M. Redox state is a central modulator of the balance between self-renewal and differentiation in a dividing glial precursor cell. Proc. Natl Acad. Sci. U.S.A. 2000;97(18):10032–10037. doi: 10.1073/pnas.170209797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Takagi Y, Osanai H, Li L, Takeuchi M, Katoh Y, Kobayashi M, Yamamoto M. Pi class glutathione S-transferase genes are regulated by Nrf 2 through an evolutionarily conserved regulatory element in zebrafish. Biochem. J. 2005;388(Pt 1):65–73. doi: 10.1042/BJ20041860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timme-Laragy AR, Cockman CJ, Matson CW, Di Giulio RT. Synergistic induction of AHR regulated genes in developmental toxicity from co-exposure to two model PAHs in zebrafish. Aquat. Toxicol. 2007;85(4):241–250. doi: 10.1016/j.aquatox.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valavanidis A, Vlahogianni T, Dassenakis M, Scoullos M. Molecular biomarkers of oxidative stress in aquatic organisms in relation to toxic environmental pollutants. Ecotoxicol. Environ. Saf. 2006;64(2):178–189. doi: 10.1016/j.ecoenv.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Van Metre PC, Mahler BJ. Trends in hydrophobic organic contaminants in urban and reference lake sediments across the United States, 1970–2001. Environ. Sci. Technol. 2005;39(15):5567–5574. doi: 10.1021/es0503175. [DOI] [PubMed] [Google Scholar]
- Wan J, Winn LM. In utero-initiated cancer: The role of reactive oxygen species. Birth Defects Res. C. 2006;78(4):326–332. doi: 10.1002/bdrc.20080. [DOI] [PubMed] [Google Scholar]
- Wassenberg DM. Interactive Effects of Polycyclic Aromatic Hydrocarbons on Cytochrome P4501A Activity and Embryonic Development in the Killifish Fundulus heteroclitus. Durham, NC: Nicholas School of the Environment and Earth Sciences, Integrated Toxicology Program, Duke University; 2004. [Google Scholar]
- Wassenberg DM, Di Giulio RT. Synergistic embryotoxicity of polycyclic aromatic hydrocarbon aryl hydrocarbon receptor agonists with cytochrome P4501A inhibitors in Fundulus heteroclitus. Environ. Health Perspect. 2004;112(17):1658–1664. doi: 10.1289/ehp.7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CC, Viernes H, Krejsa CM, Botta D, Kavanagh TJ. Fluorescence-based microtiter plate assay for glutamate-cysteine ligase activity. Anal. Biochem. 2003;318(2):175–180. doi: 10.1016/s0003-2697(03)00143-x. [DOI] [PubMed] [Google Scholar]
- Zhu H, Itoh K, Yamamoto M, Zweier JL, Li Y. Role of Nrf2 signaling in regulation of antioxidants and phase 2 enzymes in cardiac fibroblasts: Protection against reactive oxygen and nitrogen species-induced cell injury. FEBS Lett. 2005;579(14):3029–3036. doi: 10.1016/j.febslet.2005.04.058. [DOI] [PubMed] [Google Scholar]
- Zhu H, Li YB, Trush MA. Characterization of benzo[a]pyrene quinone-induced toxicity to primary cultured bone marrow stromal cells from DBA/2 mice: potential role of mitochondrial dysfunction. Toxicol. Appl. Pharmacol. 2003;103(1):108–120. doi: 10.1006/taap.1995.1015. [DOI] [PubMed] [Google Scholar]
- Zipper LM, Mulcahy RT. Erk activation is required for Nrf2 nuclear localization during pyrrolidine dithiocarbamate induction of glutamate cysteine ligase modulatory gene expression in HepG2 cells. Toxicol. Sci. 2003;73(1):124–134. doi: 10.1093/toxsci/kfg083. [DOI] [PubMed] [Google Scholar]