

Abstract
Sodium methyldithiocarbamate (SMD) is the third most abundantly used conventional pesticide in the United States, and hundreds of thousands of persons are exposed to this compound or its major breakdown product, methylisothiocyanate, at levels greater than recommended by the Environmental Protection Agency. A previous study suggests three mechanisms of action involved to some degree in the inhibition of inflammation and decreased resistance to infection caused by exposure of mice to the compound. One of these mechanisms is oxidative stress. The purpose of the present study was to confirm that this mechanism is involved in the effects of SMD on cytokine production by peritoneal macrophages and to further characterize its role in altered cytokine production. Results indicated that SMD significantly decreased the intracellular concentration of reduced glutathione (GSH), suggesting oxidative stress. This was further indicated by the upregulation of genes involved in the “response to oxidative stress” as determined by microarray analysis. These effects were associated with the inhibition of lipopolysaccharide (LPS)-induced production of several proinflammatory cytokines. Experimental depletion of GSH with buthionine sulfoximine (BSO) partially prevented the decrease in LPS-induced interleukin (IL)-6 production caused by SMD and completely prevented the decrease in IL-12. In contrast, BSO plus SMD substantially enhanced the production of IL-10. These results along with results from a previous study are consistent with the hypothesis that SMD causes oxidative stress, which contributes to modulation of cytokine production. However, oxidative stress alone cannot explain the increased IL-10 production caused by SMD.
Sodium methyldithiocarbamate (SMD) is widely used as a preemergence herbicide, fungicide, and insecticide in the United States (∼60,000,000 pounds per year), and the most recent Environmental Protection Agency (EPA) market survey indicates that it is the third most abundantly used conventional pesticide (Donaldson et al., 2004). Extensive exposure of human beings to this compound has been documented both as a result of a transport accident and normal agricultural use. In fact, a school and parts of a town had to be evacuated due to high concentrations in the air after the compound was used to treat nearby fields (O'Malley et al., 2004). In addition, quantitative air sampling for the major breakdown product of SMD, methylisothiocyanate (MITC), revealed concentrations greater than the EPA's estimated safe level over an area which would result in exposure of well over 100,000 people (Lee et al., 2002). The estimated safe levels were determined on the basis of studies in rats, which revealed a lowest observable adverse effect level of 1700 ppb (Rubin, 2002). In this case, the most sensitive end point was irritation of the upper respiratory tract and eyes. Other effects such as decreased body weight gain or development of cancer were noted at dosages of ∼20 mg/kg in rats (Weiss and Lowit, 2004). Effects reported in previous studies and from the present study indicate a roughly linear dose-response effect with significant effects on expression of many genes at the lowest dosage evaluated (50 mg/kg).
Exacerbation of asthma was observed more frequently than expected by chance (Cone et al., 1994) in people exposed to high levels of SMD following derailment of a train car containing this pesticide. Asthma is associated with a shift from the predominance of a T helper (Th)1 phenotype to a Th2 phenotype. The concentration of the reduced form of glutathione (GSH) in dendritic cells influences Th1/Th2 balance toward Th2 (allergic) responses (Murata, Ohteki, et al., 2002). As discussed below, there is evidence suggesting that SMD may deplete GSH. This suggests that immunological effects, perhaps involving a shift away from Th1 cytokines (e.g., interferon [IFN]-γ and interleukin [IL]-12) and toward Th2 cytokines (e.g., IL-10), can be mediated by SMD exposure.
One report indicates that SMD can deplete the reduced form of GSH and cause oxidative stress (Thompson et al., 2002) in the liver of rats. On the contrary, it has also been reported that SMD can act as a free radical scavenger (Motohashi and Mori, 1986; Zanocco et al., 1989). In either case, the balance of reduction versus oxidation in cells can strongly influence cytokine production (Haddad, 2002). Therefore, if SMD alters this balance in macrophages, this alteration may be at least partly responsible for the changes in cytokine production reported previously in mice treated with SMD (Pruett et al., 2005). In a previous study, pretreatment of mice with a GSH precursor, N-acetyl cysteine (NAC), altered the effects of SMD by further suppressing production of key proinflammatory cytokines (Pruett et al., 2006). There is general agreement that production of cytokines by macrophages is affected by oxidative stress, but the results have been confusing, with some studies indicating that decreased GSH decreases production of proinflammatory cytokines (Murata, Shimamura, and Hamuro, 2002; Lehmann et al., 2007; Song et al., 2004; Wang et al., 1999) and some studies indicating that decreased GSH increases production of proinflammatory cytokines (Haddad, 2002; Murray et al., 2007; Utsugi et al., 2003). The basis for these contradictory results is not clear, but it is possible that a biphasic relationship exists such that a small decrease in GSH may be consistent with suppression of proinflammatory cytokine production, whereas a substantial decrease could be consistent with enhancement of proinflammatory cytokines or protection from decreases in cytokine production caused by other mechanisms. Some of the results presented here address this issue.
Previous studies in a mouse model of peritonitis demonstrate that SMD substantially inhibits innate resistance to sepsis caused by intraperitoneal administration of Escherichia coli (Pruett et al., 2006). This is associated with a decrease in proinflammatory cytokine production and an increase in IL-10 production in the early stages of infection. Mechanisms that have been implicated in the alteration of peritoneal macrophage cytokine production by SMD include the following: increased concentrations of stress mediators such as corticosterone, copper chelation, and altered cellular redox status (Pruett et al., 2006). The present study focuses on oxidative stress as a mechanism by which SMD inhibits inflammation and resistance to infection.
MATERIALS AND METHODS
Mice
Female B6C3F1 mice were purchased through the National Cancer Institute Animal Program. They were specific pathogen free and were allowed to recover from shipping stress for at least 2 weeks before being used in experiments at 8–14 weeks of age. Sentinel mice housed in the same room as these mice were negative for common infectious agents of mice throughout the period of this study. Mice were given access to food (Purina Laboratory Chow) and water ad libitum and were maintained on a 12 h light/dark cycle. Care and use of mice were consistent with the National Institutes of Health Guide and University regulations. The animal facility in which the mice were housed is accredited by the American Association for Accreditation of Laboratory Animal Care.
Administration of Drugs and Chemicals
Mice were treated with SMD (ChemService, West Chester, PA) by intranasal administration. All groups of mice were anesthetized with pentobarbital (ip) at 45 mg/kg, and then 50 μl of SMD solution (in sterile PBS) was placed on the nostrils to yield the desired dosage rate. This mode of administration was tested in a preliminary experiment in which Evans blue was administered and the lungs were evaluated. This indicated uniform distribution of the dye throughout the lungs as described in a study by Karol and colleagues (Ebino et al., 1999). This mode of administration was selected because this mode and dermal are the two most relevant modes of human exposure (Thongsinthusak, 2000; Weiss and Lowit, 2004). Dermal exposure has been evaluated previously and causes similar effects as oral administration (Padgett et al., 1992), so this study was done using intranasal administration to assess the effects of this relevant mode of exposure. The dosages used here (50–300 mg/kg) are within the range of dosages used in risk assessment for this compound during reregistration by the U.S. EPA (Weiss and Lowit, 2004). In addition, dermal dosing of mice with the commercial preparation of this compound required administration of only ∼20 μl to mice, and this would represent ∼3 ml for a human being. Exposures at this level or greater are undoubtedly very common for those involved in manufacturing and applying this compound, and at this exposure level, substantial immunological changes were observed in mice. This model in which the inflammatory response and the concentrations of GSH are measured (quickly) ex vivo after in vivo stimulus and SMD treatment is relatively uncommon. Studies of the role of GSH in inflammation are often conducted in cell culture. Unfortunately, the partial pressure of oxygen is much greater in cell culture than in vivo, and the potential effects of this are typically not considered. In addition, most studies in which a GSH depleter or enhancer is used do not include an assay of GSH to confirm that the effects were as expected. Including this analysis increases confidence in the relationships between GSH and inflammatory response effects.
An inflammatory response was stimulated via TLR4 using lipopolysaccharide (LPS, 0128:B4, Sigma Chemical Co., St Louis, MO) at 60 micrograms per mouse in PBS administered iv. SMD or vehicle was administered (intranasally), and LPS or vehicle was administered iv 30 min later. GSH was evaluated 1 h (as well as 2 and 4 h in a time course experiment) after LPS administration, and cytokines were measured 2 h after LPS administration.
Buthionine sulfoximine (BSO) was dissolved in drinking water at 1333 mg/l (to yield an approximate dosage of 400 mg/kg/day). This was provided to some groups of mice as the sole source of water for 16 days prior to administration of LPS. Results shown here indicate that this treatment effectively depleted GSH.
Cytokine and GSH Assays
Cytokines were quantified using ELISA kits from BD Pharmingen (San Diego, CA) or using a BioPlex multiplexed bead array system (BioRad, Hercules, CA). Standards were included with the kit, and samples were compared to standards using the standard curve with the best fit (usually quadratic). Total (total GSH), oxidized GSH (GSSG), and reduced GSH were quantified using a colorimetric, enzyme amplified kit from Dojindo Molecular Technologies (Gaithersburg, MD). The samples to be analyzed for cytokines were obtained by injecting 1 ml of PBS with 10% fetal bovine serum (FBS) into the peritoneal cavity, moving the mouse to mix and distribute the liquid, and withdrawing the liquid using a 23-g needle and syringe. Typically, 0.7 ml was recovered. To obtain peritoneal cells (mostly macrophages) to analyze for GSH, the sample obtained for cytokine analysis was centrifuged and the cell pellet was separated from the supernatant. An additional 7.0 ml of PBS with 10% FBS was injected into the peritoneal cavity and withdrawn to remove the remaining cells. These were combined with the cell pellet from the smaller sample. The concentration of GSH was expressed as the concentration (micrograms per milliliter) divided by the number of cells in the sample × 106. This was done to compensate for changes in cell number related to treatment or normal biological variability. The cell number was typically 1–3 × 106, so the concentrations shown are 1- to 3-fold less than the actual measured values.
Cytokine production over time in vivo was measured by iv administration of an anti-cytokine antibody followed 30 min later by administration of SMD and followed after an additional 30 min by LPS. A sample was then taken 6 h after LPS administration, and the sample was added to a microplate coated with an antibody specific for the anti-cytokine antibody. The final step was addition of a peroxidase-labeled anti-cytokine antibody followed by substrate solution. This technique was performed using a set of antibodies and standards designed for this purpose from BD Pharmingen according the manufacturer's instructions.
Statistical Analysis of Cytokine and GSH Data
The results for cytokine concentrations and for GSH concentrations were compared using one-way ANOVA followed by the SNK post hoc test to compare each mean with all other means. This was done using Prism 4.0 (GraphPad, San Diego, CA), and results shown are generally values that significantly differ from the positive control (LPS only).
Microarray Analysis
Macrophages were isolated from the peritoneal cavity of mice treated with SMD at 100, 200, and 300 mg/kg 15 min before administration of LPS (60 micrograms per mouse). Cells were harvested 2 h after LPS treatment. As in our previous studies, the peritoneal cells obtained were >85% macrophages (Pruett, Schwab, et al., 2004). Sample preparation and microarray analysis were performed exactly as described previously except Affymetrix mouse 430A chips were used (Pruett, Schwab, et al., 2004). Data were analyzed using Genesifter software (www.genesifter.net, Geospiza Inc., Seattle, WA). Cells from three mice are required to yield sufficient mRNA for a single microarray analysis, so nine mice were used to obtain three samples per group, and the values shown in this study are means of these three values for each treatment group.
RESULTS
The initial experiments in this study revealed that SMD consistently decreased the concentration of GSH in peritoneal cells (Fig. 1). GSSG was either unaffected or increased. Thus, the GSH to GSSG ratio was consistently decreased in mice treated with LPS plus SMD. The effects were generally dose responsive and were greater at 1 h after LPS than at later times. The administration of SMD without LPS did not affect GSH concentrations at 1 h compared to naive control but significantly decreased GSH at 2 h. Other investigators have reported that LPS increases production of reactive oxygen species, so it is not surprising that LPS in addition to SMD would be more effective in reducing GSH levels than SMD alone. It is also not surprising that this relatively short-lived effect (GSH returned to normal by 4 h) is associated with altered cytokine production because signaling (which occurs within the initial 2 h) is affected by oxidative stress. These results indicate that this particular mechanism of action would be operational for less than 4 h after dosing. Nevertheless, the observation of increased incidence of asthma in humans acutely exposed to SMD (Cone et al., 1994) suggests that brief exposure can modulate the immune system in an adverse manner.
FIG. 1.

Treatment of mice with SMD decreases GSH in peritoneal macrophages. Groups of five mice were treated with SMD intranasally at the indicated dosages 30 min before challenge with LPS (iv at 60 micrograms per mouse), and peritoneal macrophages were harvested at the indicated times after LPS treatment for analysis of GSH and GSSG using a colorimetric assay kit. Values shown are means ± SEM, and values significantly different from those for mice treated with LPS only are shown by * (p < 0.05) or ** (p < 0.01), as determined by ANOVA followed by the SNK post hoc test.
The effects of SMD on cytokine concentrations in the peritoneal cavity were generally consistent with the effects on GSH (i.e., dosages of SMD that altered cytokine production also altered GSH concentration) (Fig. 2). Production of MIP-1α, IL-1β, IL-12 (p40), and GM-CSF were significantly decreased by SMD. There was a trend toward decreased IFN-γ and TNF-α production as well, and this was revealed to be significant over time in a subsequent experiment. In contrast, the concentration of IL-10 was significantly increased by SMD.
FIG. 2.

Treatment of mice with SMD markedly alters production of some cytokines, chemokines, and growth factors in the peritoneal cavity in response to LPS. Mice were treated with SMD intranasally at the indicated dosages followed 30 min later by LPS (60 micrograms per mouse, iv). Peritoneal lavage was performed 3 h later, a time previously shown to be near optimum for most responses. Cytokines were quantified using multiplexed bead array analysis. Values shown are means ± SEM, and values significantly different from those for mice treated with LPS only are shown by * (p < 0.05), ** (p < 0.01), or *** (p < 0.001), as determined by ANOVA followed by the SNK post hoc test.
There were indications that SMD may decrease production of TNF-α and IFN-γ at one time point after stimulation, but the differences were not significant. To determine if this was due to selection of a time point that was less than optimal for assessment, evaluation of TNF-α and IFN-γ was performed using an in vivo, antibody capture method during the first 6 h after LPS administration. This demonstrated that SMD at 200 mg/kg significantly decreased production of both of these proinflammatory cytokines (Fig. 3). The decrease in TNF-α was similar to that noted at a single time point (Fig. 2), but the decrease in IFN-γ was substantially greater than that observed at a single time point. The SMD-induced increase in IL-10 production was observed at a single time point and was also significant in vivo during the 6 h after LPS administration (Fig. 3).
FIG. 3.
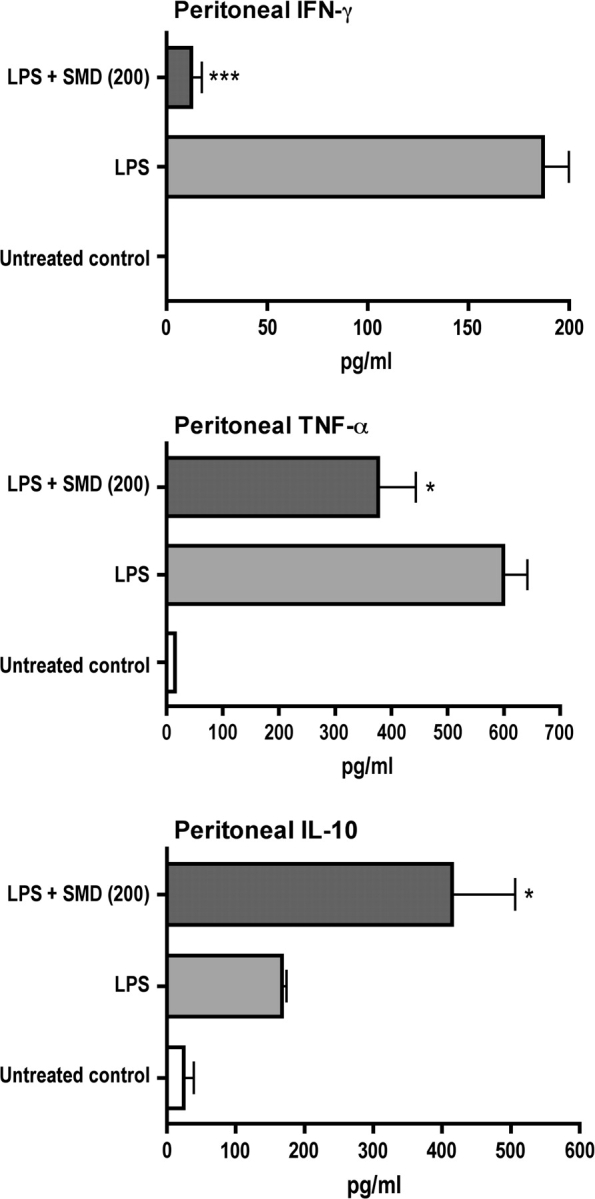
Cumulative production of selected cytokines over a 6-h period is substantially altered by SMD. Results shown indicate values from mice treated with SMD (200 mg/kg, intranasally) followed 30 min later by LPS (60 micrograms per mouse, iv). At the time of LPS injection, a monoclonal anti-cytokine antibody was also injected, and samples were harvested 6 h later. Cytokine attached to the monoclonal antibody was measured using an indirect ELISA (see Materials and Methods section). Values shown are means ± SEM, and values significantly different from those for mice treated with LPS only are shown by * (p < 0.05), ** (p < 0.01), or *** (p < 0.001), as determined by ANOVA followed by the SNK post hoc test.
To determine if there is a cause-effect relationship between SMD-induced changes in GSH and cytokine production, a GSH depleter, BSO, was administered to mice in drinking water for 16 days prior to experiments. This treatment effectively depleted GSH (Fig. 4). Treatment of mice with SMD depleted GSH to a lesser extent, and the combination of SMD and BSO had no greater effect than BSO alone. Evaluating the effects of these treatments on LPS-induced cytokine production indicated that BSO did not significantly affect the production of IL-10 or IL-12, but it significantly decreased IL-6 production (Fig. 5). The combination of BSO and SMD indicated that BSO partially prevented the inhibition of IL-6 production caused by SMD and completely prevented the decrease in IL-12 production caused by SMD. The concentration of GSH was similar in cells from mice treated with BSO or SMD plus BSO. Similarly, these treatments yielded comparable levels of IL-6 and IL-12. A smaller decrease in GSH was caused by SMD alone, and this was associated with substantial inhibition of IL-6 and IL-12 production. These results may indicate a role for GSH depletion in the effects of SMD on production of these cytokines, but the effects are more complex than anticipated. Our interpretation of these results is presented in the Discussion section.
FIG. 4.
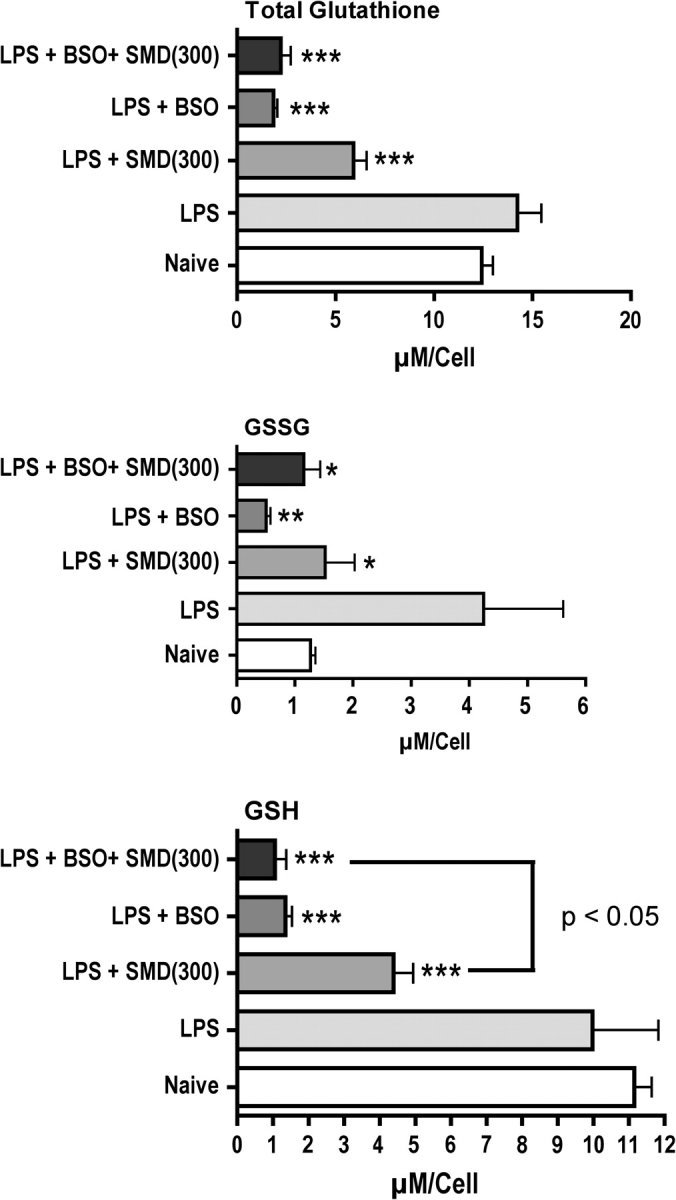
Treatment of mice with BSO (at 400 mg/kg/day for 16 days in the drinking water) substantially depletes GSH. Mice were treated with BSO or with normal water and then treated as noted in the legend for Figure 3 with SMD and LPS. Peritoneal macrophages were harvested 1 h after LPS administration, and GSH and GSSG in these cells were quantified as described in Materials and Methods section. Values shown are means ± SEM (group size = 5), and values significantly different from those for mice treated with LPS only are shown by * (p < 0.05), ** (p < 0.01), or *** (p < 0.001), as determined by ANOVA followed by the SNK post hoc test.
FIG. 5.
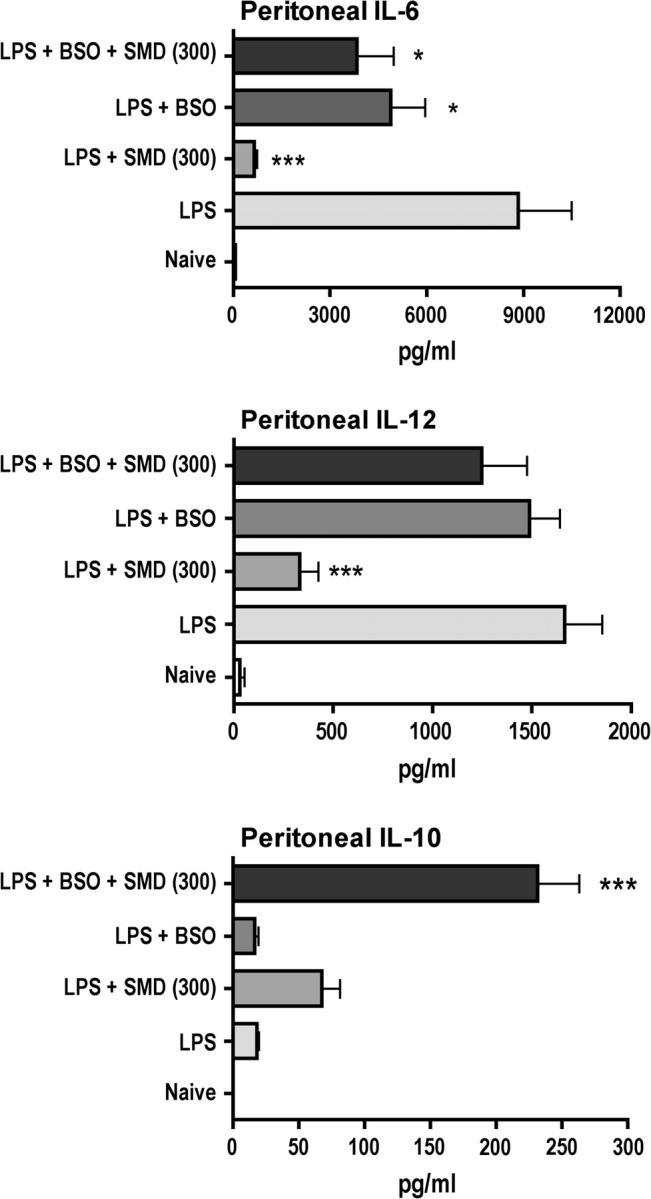
BSO reverses effects of SMD on LPS-induced IL-12 production and partially inhibits the effect of SMD on IL-6 but substantially enhances IL-10 production. The same peritoneal fluid samples from which macrophages were obtained for GSH analysis in Figure 4 were also evaluated for cytokines (using ELISA kits), and the results are illustrated here. Values shown are means ± SEM (group size = 5), and values significantly different from those for mice treated with LPS only are shown by * (p < 0.05), ** (p < 0.01), or *** (p < 0.001), as determined by ANOVA followed by the SNK post hoc test.
In contrast, BSO plus SMD produced a much greater increase in IL-10 concentration than either alone when administered before LPS. SMD plus LPS, but not BSO plus LPS, also tended to increase IL-10 production as previously reported. Thus, the production of IL-10 did not correspond well to changes in the concentration of GSH because BSO and BSO plus SMD caused similar depletion of GSH, but BSO plus SMD caused production of much more IL-10.
Decreases in GSH caused by SMD indicated that SMD causes oxidative stress, but it remained possible that the overall effects of SMD directly as a free radical scavenger balanced the depletion of GSH resulting in no net oxidative stress. However, the results shown in Table 1 indicate that the cells were exposed to a stimulus that induced an array of genes involved in protection from oxidative stress. The data were analyzed using Genesifter software (www.genesifter.net), and the gene ontology category “response to oxidative stress” was selected for analysis. The results indicate that LPS increased and decreased expression of a roughly equivalent number of genes in this category, as indicated by comparing naive mice with LPS-treated mice. However, comparing LPS-treated mice to LPS + SMD–treated mice reveals that many more of the genes necessary for protection from oxidative stress are upregulated than downregulated (Table 1). It should also be noted that a total of 108 Affymetrix probes (representing about 85 genes) were represented in the mRNA samples from naive mice, and nearly one-fourth of these were significantly up- or downregulated by SMD and/or LPS with most being upregulated. This is more than we observed in mice treated acutely with MITC (the major breakdown produce of SMD, data not shown), in which 22 genes were significantly altered and 15 of them were downregulated. It should also be noted that the annotations used to categorize genes in response to oxidative stress include every circumstance and every cell type in which the gene in question has ever been observed to be up- or downregulated in response to oxidative stress. Thus, it would not be expected that all the genes of this category would be activated by a particular stimulus in a particular cell type.
TABLE 1.
SMD-Induced Changes in the Expression of Genes in the Category “Response to Oxidative Stress” from www.genesifter.net. Values Shown Are Means of Absolute Fluorescence Values (n = 3). Statistical Analysis Was Done by Genesifter Software Using an ANOVA with Benjamini-Hochberg Correction. The Dosage of SMD in milligrams/kilogram Body Weight Is Indicated in Parentheses. Values that Are Increased in Expression Are Indicated by Bold Print, and the Others Are Decreased
| Affymetrix ID | Gene Name | LPS | LPS + SMD (100) | LPS + SMD (200) | LPS + SMD (300) | p value |
| 1426875_s_at | Sulfiredoxin 1 homolog (Saccarhomyces cerevisiae) | 376.0 | 1454.5 | 2582.2 | 2583.3 | 0.002272 |
| 1451680_at | Sulfiredoxin 1 homolog (Saccarhomyces cerevisiae) | 127.7 | 350.6 | 719.1 | 618.6 | 0.001677 |
| 1422433_s_at | Isocitrate dehydrogenase 1 (NADP+), soluble | 781.9 | 1770.4 | 2760.1 | 6781.6 | 0.000479 |
| 1419821_s_at | Isocitrate dehydrogenase 1 (NADP+), soluble | 520.5 | 1271.0 | 1521.3 | 4139.4 | 0.000197 |
| 1418627_at | Glutamate-cysteine ligase, modifier subunit | 294.3 | 702.2 | 1313.0 | 968.8 | 0.002926 |
| 1429972_s_at | Thioredoxin reductase 2 | 61.6 | 143.8 | 130.9 | 210.9 | 0.028776 |
| 1416430_at | Catalase | 6100.5 | 8235.3 | 10584.0 | 12164.6 | 0.00019 |
| 1433866_x_at | Peroxiredoxin 1 | 8446.9 | 11910.1 | 14555.7 | 14674.3 | 0.003185 |
| 1417491_at | Cathepsin B | 1883.9 | 2446.8 | 3083.6 | 3312.7 | 0.006226 |
| 1460725_at | Xeroderma pigmentosum, complementation group A | 347.1 | 353.7 | 517.1 | 728.6 | 0.003233 |
| 1417492_at | Cathepsin B | 2634.8 | 3132.5 | 3655.1 | 4585.3 | 0.003054 |
| 1417694_at | Growth factor receptor bound protein 2–associated protein 1 | 1134.2 | 1733.4 | 1665.0 | 1376.6 | 0.034644 |
| 1416000_a_at | Peroxiredoxin 1 | 17857.0 | 21490.3 | 28237.0 | 24461.1 | 0.023674 |
| 1416429_a_at | Catalase | 4223.1 | 5524.8 | 4793.3 | 6584.7 | 0.016235 |
| 1426798_a_at | Protein phosphatase 1, regulatory (inhibitor) subunit 15b | 10400.8 | 13307.9 | 15165.4 | 12681.2 | 0.020064 |
| 1436691_x_at | Peroxiredoxin 1 | 20726.7 | 24007.5 | 28504.5 | 28376.3 | 0.041459 |
| 1417784_at | Amyotrophic lateral sclerosis 2 (juvenile) homolog (human) | 49.8 | 17.4 | 81.9 | 106.8 | 0.018596 |
| 1454976_at | Superoxide dismutase 2, mitochondrial | 7040.7 | 8671.7 | 7479.4 | 3676.7 | 0.005291 |
| 1418333_at | Metal response element–binding transcription factor 1 | 377.2 | 398.2 | 283.2 | 259.4 | 0.013446 |
| 1428979_at | Metal response element–binding transcription factor 1 | 859.1 | 847.7 | 720.1 | 402.8 | 0.001567 |
| 1415676_a_at | Proteasome (prosome, macropain) subunit, beta type 5 | 3411.3 | 2939.0 | 2392.8 | 1720.0 | 0.041332 |
| 1449106_at | GSH peroxidase 3 | 384.5 | 320.3 | 214.5 | 188.6 | 0.031448 |
| 1416130_at | Prion protein | 242.1 | 179.7 | 110.8 | 122.0 | 0.028782 |
| 1431981_at | HIF-1, alpha subunit | 60.3 | 32.8 | 8.9 | 7.2 | 0.039661 |
Note. NADP: nicotinamide adenine dinucleotide phosphate.
Among key genes that were significantly upregulated were catalase, a regulator subunit of cysteine-glutamate ligase (which catalyzes GSH synthesis), sulfiredoxin, peroxiredoxin, and isocitrate dehydrogenase. Like catalase, peroxiredoxin acts to reduce hydrogen peroxide, and sulfiredoxin reduces the oxidized form of peroxiredoxin (Abbas et al., 2008). Thus, it is not surprising that the genes for these proteins are coordinately regulated. Isocitrate dehydrogenase protects activated macrophages from nitric oxide (Maeng et al., 2004). Thioredoxin reductase 2 has previously been reported to decrease the response and activity of hypoxia-inducible factor 1 (HIF-1) in activated macrophages (Zhou et al., 2008), and upregulation of thioredoxin reductase 2 was associated with decreased expression of HIF-1 in the present study (Table 1). The decrease in expression of mitochondrial superoxide dismutase may seem inconsistent with the hypothesis that this represents a response to oxidative stress, but the response to oxidative stress may be distinct in the mitochondria as compared to the cytoplasm and can be complex. Collectively, these results provide substantial evidence that SMD increases the level of oxidative stress in macrophages from treated mice.
DISCUSSION
A previous report indicates that SMD depletes GSH in the liver in rats (Thompson et al., 2002). However, there was no reason to assume that this would also be the case in macrophages. Direct measurement of GSH in the present study demonstrated that SMD decreases the concentration of reduced GSH in peritoneal cells in mice. Thus, the basic observation of decreased GSH is consistent between the present study in mice and a previous study in rats. The role of the major breakdown product of SMD (MITC) in this GSH depletion will be the topic of a future report.
The direct measurement of GSH concentration suggests that SMD causes oxidative stress in peritoneal macrophages (Fig. 1). However, it could be argued that a moderate decrease in GSH concentration is not sufficient evidence to determine if a state of elevated oxidative stress was present. Therefore, microarray analysis was used to determine if SMD caused a change of gene expression, indicating a response to oxidative stress. The results in Table 1 indicate that the many of the genes identified by Genesifter software as involved in response to oxidant stress were significantly upregulated by SMD. Expression of a smaller set of genes was downregulated, and in at least one case (HIF-1), downregulation is consistent with a response to oxidant stress (HIF-1 is induced by hypoxia, not oxidant stress per se, so decreased HIF would be consistent with a high oxygen environment, not hypoxia) (Jantsch et al., 2008). Isocitrate dehydrogenase 1 was substantially upregulated by SMD. This enzyme produces much of the NADH in cells, and it has recently been shown to play an important role in protecting macrophages from LPS-induced oxidative stress (Maeng et al., 2004). Sulfiredoxin 1 is highly upregulated by SMD as well, and it participates with peroxiredoxins (also upregulated) in a cyclic reaction series leading to detoxification of peroxides induced by inflammatory stimuli in macrophages (Diet et al., 2007).
The suppression of LPS-induced proinflammatory cytokines was somewhat selective with regard to amount of suppression, but all proinflammatory cytokines tended to be decreased. In addition, in vivo assessment over a 6-h period of two of these cytokines that were not significantly decreased at a single time point (IFN-γ and TNF-α) indicated significant suppression over a 6-h time course in vivo (Fig. 3). This suggests that the overall suppressive effect on proinflammatory cytokine production is greater than that suggested by results obtained at a single time point (Fig. 2). A somewhat unexpected observation was the substantial decrease, even at the lowest SMD dosage, of GM-CSF (Fig. 2). The induction of this key growth factor by LPS is also inhibited by other immunotoxicants, such as ethanol, and this can profoundly decrease host defense functions (Joshi et al., 2005), an effect already reported for SMD (Pruett et al., 2005).
Comparing the present results with results from our previous study in which NAC was used to diminish oxidative stress (Pruett et al., 2006) further supports a role for oxidative stress in modulation of IL-12 and IL-6. Treatment of mice with SMD plus NAC caused a further suppression of the IL-12 production observed in mice treated with SMD (Pruett et al., 2006). Thus, a moderate decrease in GSH caused by SMD is associated with a decrease in IL-12 and IL-6 production (present study); a large decrease in GSH (caused by BSO or BSO plus SMD) prevents this suppression of IL-12 and partially prevents suppression of IL-6 (Fig. 5). Preventing the decrease in GSH caused by SMD (with NAC) is associated with greater suppression of IL-12 (Pruett et al., 2006). Thus, modulation of IL-6 and IL-12 could be explained entirely by changes in oxidative stress if a moderate decrease in GSH decreases IL-12 and IL-6 production and a substantial decrease in GSH has no effect (IL-12) or slightly decreases (IL-6) cytokine production. In this study, BSO moderately decreased IL-6 but not IL-12 production, suggesting that a severe decrease in GSH is not sufficient to enhance IL-6 or IL-12 production. This differs from results of some previous studies, in which depletion of GSH was associated with increased IL-12 or IL-6 production (Haddad, 2002; Murray et al., 2007; Utsugi et al., 2003). However, the present study has the advantage of evaluation of a relevant cell type very quickly after the cells were obtained following treatments in vivo.
A previous study demonstrated that cytokines detected in the peritoneal cavity are mostly derived from local production, not transfer from the circulation (Pruett, Zheng, et al., 2004). Expression of the three cytokines evaluated in detail in this study is regulated in part by NF-κB (Sarady et al., 2002), and NF-κB activation can be sensitive to oxidative stress (Haddad, 2002; Komatsu et al., 2003). However, our previous study revealed that SMD did not inhibit LPS-induced activation of NF-κB, but it did inhibit activation of the mitogen-activated protein (MAP) kinases JNK, p38, and ERK (Pruett et al., 2005). Other investigators have found that increasing reduced GSH concentrations prevents LPS-induced phosphorylation of p38, ERK, and JNK in macrophages (Chung et al., 2008; Sun et al., 2006). As would be expected, we found that inhibition of these upstream kinases decreases the activation of AP-1 (Pruett et al., 2005). The expression of IL-12 and IL-6 is mediated in part by AP-1 (Nicholson et al., 1996; Ma et al., 2004). In contrast, IL-10 gene expression is not mediated by AP-1 (Brightbill et al., 2000). Thus, it is not surprising that inhibition of AP-1 activation is associated with decreased expression of IL-12 and IL-6, but not IL-10.
Apparently, the relationship between oxidative stress and MAP kinase activation must be complex because SMD substantially decreased production of IL-6 and IL-12 but only moderately decreased GSH (Figs. 1 and 2). Serum cytokines were also measured, and the patterns were very similar to those observed in the peritoneal fluid, providing confirmation of these patterns (data not shown). There is evidence from many other studies that oxidative stress can affect cytokine production. Both enhancement and inhibition of proinflammatory cytokine production have been reported in cells or animals exposed to oxidative stress (Wang et al., 1999; Haddad, 2002). The results presented here suggest a biphasic dose-response curve in which moderate decreases in GSH suppress proinflammatory cytokine production and larger decreases in GSH are less suppressive or even stimulatory for proinflammatory cytokine production. However, it remains possible that other mechanisms are also involved and that oxidative stress simply modulates the effect of these mechanisms.
Mechanisms other than oxidative stress are probably responsible for the increase in IL-10 production caused by SMD. In the present study, BSO plus LPS substantially decreased GSH and LPS plus SMD caused a smaller decrease. However, neither treatment had a significant effect on IL-10 production. In contrast, LPS plus SMD plus BSO caused a similar decrease in GSH as LPS plus BSO, but it caused a substantial increase in IL-10. Thus, IL-10 production did not correlate with GSH concentration, even considering the possibility of a complex concentration-response curve. Other mechanisms that seem to contribute to the effects of SMD on cytokine production were suggested in a previous study, in which copper chelation and neuroendocrine stress mediators induced by SMD affected cytokine production in SMD-treated mice (Pruett et al., 2006). The reciprocal regulation of IL-10 and IL-12 has been reported previously and can be mediated by other chemicals as well as SMD plus BSO (Pruett et al., 2003).
The results presented here demonstrate that altering the GSH concentration in peritoneal macrophages can modulate the effects of SMD on IL-6 and IL-12 production and can directly alter LPS-induced IL-6 production. However, other mechanisms of SMD action must also be involved in modulating IL-10 production, and these mechanisms could also play a role in modulation of IL-12 and IL-6. However, the modulation of IL-6 and IL-12 is also consistent with a scenario in which moderate oxidative stress substantially inhibits IL-12 and IL-6 production but greater oxidative stress causes less (for IL-6) or no (for IL-12) inhibition and can counteract the effects of SMD on both cytokines. Further studies are planned to inhibit the other mechanisms by which SMD may act (copper chelation and increased stress hormones) and evaluate the role of GSH changes when those mechanisms are eliminated.
FUNDING
National Institute for Environmental Health Sciences (R01ES013708).
References
- Abbas K, Breton J, Drapier JC. The interplay between nitric oxide and peroxiredoxins. Immunobiology. 2008;213:815–822. doi: 10.1016/j.imbio.2008.07.029. [DOI] [PubMed] [Google Scholar]
- Brightbill HD, Plevy SE, Modlin RL, Smale ST. A prominent role for Sp1 during lipopolysaccharide-mediated induction of the IL-10 promoter in macrophages. J. Immunol. 2000;164:1940–1951. doi: 10.4049/jimmunol.164.4.1940. [DOI] [PubMed] [Google Scholar]
- Chung J, Lee HS, Chung HY, Yoon TR, Kim HK. Salicylideneamino-2-thiophenol inhibits inflammatory mediator genes (RANTES, MCP-1, IL-8 and HIF-1alpha) expression induced by tert-butyl hydroperoxide via MAPK pathways in rat peritoneal macrophages. Biotechnol. lett. 2008;30:1553–1558. doi: 10.1007/s10529-008-9744-z. [DOI] [PubMed] [Google Scholar]
- Cone JE, Wugofski L, Blames JR, Das R, Bowler R, Alexeeff G, Shusterman D. Persisent respiratory health effects after a metam sodium pesticide spill. Chest. 1994;106:500–508. doi: 10.1378/chest.106.2.500. [DOI] [PubMed] [Google Scholar]
- Diet A, Abbas K, Bouton C, Guillon B, Tomasello F, Fourquet S, Toledano MB, Drapier JC. Regulation of peroxiredoxins by nitric oxide in immunostimulated macrophages. J. Biol. Chem. 2007;282:36199–36205. doi: 10.1074/jbc.M706420200. [DOI] [PubMed] [Google Scholar]
- Donaldson D, Kiely T, Grube A. 2000–2001 Pesticide Market Estimates. 2004. Pesticides Industry Sales and Usage: 2000 and 2001 Market Estimates. Biological and Economic Analysis, Division Office of Pesticide Programs, Office of Prevention, Pesticides, and Toxic Substances, U.S. Environmental Protection Agency (Washington, DC) [Google Scholar]
- Ebino K, Lemus R, Karol MH. The importance of the diluent for airway transport of toluene diisocyanate following intranasal dosing of mice. Inhal. Toxicol. 1999;11:171–185. doi: 10.1080/089583799197131. [DOI] [PubMed] [Google Scholar]
- Haddad JJ. Redox regulation of pro-inflammatory cytokines and IkappaB-alpha/NF-kappaB nuclear translocation and activation. Biochem. Biophys. Res. Commun. 2002;296:847–856. doi: 10.1016/s0006-291x(02)00947-6. [DOI] [PubMed] [Google Scholar]
- Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG, Volke M, Glasner J, Warnecke C, Wiesener MS, et al. Hypoxia and hypoxia-inducible factor-1{alpha} modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 2008;180:4697–4705. doi: 10.4049/jimmunol.180.7.4697. [DOI] [PubMed] [Google Scholar]
- Joshi PC, Applewhite L, Ritzenthaler JD, Roman J, Fernandez AL, Eaton DC, Brown LA, Guidot DM. Chronic ethanol ingestion in rats decreases granulocyte-macrophage colony-stimulating factor receptor expression and downstream signaling in the alveolar macrophage. J. Immunol. 2005;175:6837–6845. doi: 10.4049/jimmunol.175.10.6837. [DOI] [PubMed] [Google Scholar]
- Komatsu H, Hoshino A, Funayama M, Kawahara K, Obata F. Oxidative modulation of the glutathione-redox couple enhances lipopolysaccharide-induced interleukin 12 P40 production by a mouse macrophage cell line, J774A.1. Free Radic. Res. 2003;37:293–299. doi: 10.1080/1071576021000046613. [DOI] [PubMed] [Google Scholar]
- Lee S, McLaughlin R, Harnly M, Gunier R, Kreutzer R. Community exposures to airborne agricultural pesticides in California: Ranking of inhalation risks. Environ. Health Perspect. 2002;110:1175–1184. doi: 10.1289/ehp.021101175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann JC, Listopad JJ, Rentzsch CU, Igney FH, von Bonin A, Hennekes HH, Asadullah K, Docke WD. Dimethylfumarate induces immunosuppression via glutathione depletion and subsequent induction of heme oxygenase 1. J. Invest. Dermatol. 2007;127:835–845. doi: 10.1038/sj.jid.5700686. [DOI] [PubMed] [Google Scholar]
- Ma W, Gee K, Lim W, Chambers K, Angel JB, Kozlowski M, Kumar A. Dexamethasone inhibits IL-12p40 production in lipopolysaccharide-stimulated human monocytic cells by down-regulating the activity of c-Jun N-terminal kinase, the activation protein-1, and NF-kappa B transcription factors. J. Immunol. 2004;172:318–330. doi: 10.4049/jimmunol.172.1.318. [DOI] [PubMed] [Google Scholar]
- Maeng O, Kim YC, Shin HJ, Lee JO, Huh TL, Kang KI, Kim YS, Paik SG, Lee H. Cytosolic NADP(+)-dependent isocitrate dehydrogenase protects macrophages from LPS-induced nitric oxide and reactive oxygen species. Biochem. Biophys. Res. Commun. 2004;317:558–564. doi: 10.1016/j.bbrc.2004.03.075. [DOI] [PubMed] [Google Scholar]
- Motohashi N, Mori I. Thiol-induced hydroxyl radical formation and scavenger effect of thiocarbamides on hydroxyl radicals. J. Inorg. Biochem. 1986;26:205–212. doi: 10.1016/0162-0134(86)80042-3. [DOI] [PubMed] [Google Scholar]
- Murata Y, Ohteki T, Koyasu S, Hamuro J. IFN-gamma and pro-inflammatory cytokine production by antigen-presenting cells is dictated by intracellular thiol redox status regulated by oxygen tension. Eur. J. Immunol. 2002;32:2866–2873. doi: 10.1002/1521-4141(2002010)32:10<2866::AID-IMMU2866>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Murata Y, Shimamura T, Hamuro J. The polarization of T(h)1/T(h)2 balance is dependent on the intracellular thiol redox status of macrophages due to the distinctive cytokine production. Int. Immunol. 2002;14:201–212. doi: 10.1093/intimm/14.2.201. [DOI] [PubMed] [Google Scholar]
- Murray AR, Kisin E, Castranova V, Kommineni C, Gunther MR, Shvedova AA. Phenol-induced in vivo oxidative stress in skin: Evidence for enhanced free radical generation, thiol oxidation, and antioxidant depletion. Chem. Res. Toxicol. 2007;20:1769–1777. doi: 10.1021/tx700201z. [DOI] [PubMed] [Google Scholar]
- Nicholson WJ, Slight J, Donaldson K. Inhibition of the transcription factors NF-kappa B and AP-1 underlies loss of cytokine gene expression in rat alveolar macrophages treated with a diffusible product from the spores of Aspergillus fumigatus. Am. J. Respir. Cell Mol. Biol. 1996;15:88–96. doi: 10.1165/ajrcmb.15.1.8679226. [DOI] [PubMed] [Google Scholar]
- O'Malley M, Barry T, Verder-Carlos M, Rubin A. Modeling of methyl isothiocyanate air concentrations associated with community illnesses following a metam-sodium sprinkler application. Am. J. Ind. Med. 2004;46:1–15. doi: 10.1002/ajim.20037. [DOI] [PubMed] [Google Scholar]
- Padgett EL, Barnes DB, Pruett SB. Disparate effects of representative dithiocarbamates on selected immunological parameters in vivo and cell survival in vitro in female B6C3F1 mice. J. Toxicol. Environ. Health. 1992;37:559–571. doi: 10.1080/15287399209531693. [DOI] [PubMed] [Google Scholar]
- Pruett SB, Fan R, Zheng Q. Acute ethanol administration profoundly alters poly I:C-induced cytokine expression in mice by a mechanism that is not dependent on corticosterone. Life Sci. 2003;72:1825–1839. doi: 10.1016/s0024-3205(02)02507-9. [DOI] [PubMed] [Google Scholar]
- Pruett SB, Fan R, Zheng Q. Involvement of three mechanisms in the alteration of cytokine responses by sodium methyldithiocarbamate. Toxicol. Appl. Pharmacol. 2006;213:172–178. doi: 10.1016/j.taap.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Pruett SB, Schwab C, Zheng Q, Fan R. Suppression of innate immunity by ethanol: A global perspective and a new mechanism beginning with inhibition of signaling through Toll-like receptor 3. J. Immunol. 2004;173:2715–2724. doi: 10.4049/jimmunol.173.4.2715. [DOI] [PubMed] [Google Scholar]
- Pruett SB, Zheng Q, Fan R, Matthews K, Schwab C. Ethanol suppresses cytokine responses induced through Toll-like receptors as well as innate resistance to Escherichia coli in a mouse model for binge drinking. Alcohol. 2004;33:147–155. doi: 10.1016/j.alcohol.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Pruett SB, Zheng Q, Schwab C, Fan R. Sodium methyldithiocarbamate inhibits MAP kinase activation through Toll-like receptor 4, alters cytokine production by mouse peritoneal macrophages, and suppresses innate immunity. Toxicol. Sci. 2005;87:75–85. doi: 10.1093/toxsci/kfi215. [DOI] [PubMed] [Google Scholar]
- Rubin A. Evaluation of Methyl Isothiocyanate as a Toxic Air Contaminant. Part C—Human Health Assessment. Sacramento, CA: EPA; 2002. [Google Scholar]
- Sarady JK, Otterbein SL, Liu F, Otterbein LE, Choi AM. Carbon monoxide modulates endotoxin-induced production of granulocyte macrophage colony-stimulating factor in macrophages. Am. J. Respir. Cell Mol. Biol. 2002;27:739–745. doi: 10.1165/rcmb.4816. [DOI] [PubMed] [Google Scholar]
- Song M, Kellum JA, Kaldas H, Fink MP. Evidence that glutathione depletion is a mechanism responsible for the anti-inflammatory effects of ethyl pyruvate in cultured lipopolysaccharide-stimulated RAW 264.7 cells. J. Pharmacol. Exp. Ther. 2004;308:307–316. doi: 10.1124/jpet.103.056622. [DOI] [PubMed] [Google Scholar]
- Sun S, Zhang H, Xue B, Wu Y, Wang J, Yin Z, Luo L. Protective effect of glutathione against lipopolysaccharide-induced inflammation and mortality in rats. Inflamm. Res. 2006;55:504–510. doi: 10.1007/s00011-006-6037-7. [DOI] [PubMed] [Google Scholar]
- Thompson RW, Valentine HL, Valentine WM. In vivo and in vitro hepatotoxicity and glutathione interactions of N-methyldithiocarbamate and N,N-dimethyldithiocarbamate in the rat. Toxicol. Sci. 2002;70:269–280. doi: 10.1093/toxsci/70.2.269. [DOI] [PubMed] [Google Scholar]
- Thongsinthusak T. Evaluation of Methylisothiocyanate as an Air Contaminant. Part B. Exposure Assessment. Sacramento, CA: Department of Pesticide Regulation; 2000. p. 33. [Google Scholar]
- Utsugi M, Dobashi K, Ishizuka T, Endou K, Hamuro J, Murata Y, Nakazawa T, Mori M. c-Jun N-terminal kinase negatively regulates lipopolysaccharide-induced IL-12 production in human macrophages: Role of mitogen-activated protein kinase in glutathione redox regulation of IL-12 production. J. Immunol. 2003;171:628–635. doi: 10.4049/jimmunol.171.2.628. [DOI] [PubMed] [Google Scholar]
- Wang F, Wang LY, Wright D, Parmely MJ. Redox imbalance differentially inhibits lipopolysaccharide-induced macrophage activation in the mouse liver. Infect. Immun. 1999;67:5409–5416. doi: 10.1128/iai.67.10.5409-5416.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss S, Lowit A. Metam Sodium/Metam Potassium: The HED Chapter of the Reregistration Eligibility Decision Document (RED) Washington, DC: U.S. EPA; 2004. [Google Scholar]
- Zanocco AL, Pavez R, Videla LA, Lissi EA. Antioxidant capacity of diethyldithiocarbamate in a metal independent lipid peroxidative process. Free Radic. Biol. Med. 1989;7:151–156. doi: 10.1016/0891-5849(89)90006-3. [DOI] [PubMed] [Google Scholar]
- Zhou J, Eleni C, Spyrou G, Brune B. The mitochondrial thioredoxin system regulates nitric oxide-induced HIF-1alpha protein. Free Radic. Biol. Med. 2008;44:91–98. doi: 10.1016/j.freeradbiomed.2007.09.012. [DOI] [PubMed] [Google Scholar]