

Abstract
Exposure to cigarette smoke is a risk factor contributing to the severity of respiratory tract infections associated with respiratory syncytial virus (RSV). Stimulation of airway epithelial cells by either RSV or cigarette smoke condensate (CSC) has been shown to induce secretion of the proinflammatory chemokines. However, the effect of coexposure of airway epithelial cells to CSC and RSV on inducible chemokine production has not been previously investigated. The results of this study indicate that CSC costimulation significantly increased RSV-induced interleukin-8 (IL-8) and monocyte chemoattactant protein-1 gene and protein expression when compared with each stimulus alone. Promoter deletion studies identified the interferon stimulatory response element (ISRE) of the IL-8 promoter as a critical region responsible for the synergistic increase of IL-8 gene transcription during mixed exposure. CSC costimulation enhanced RSV-induced activation of interferon regulatory factor (IRF)-1 and IRF-7, which bind to the ISRE site. CSC also furthered RSV-induced activation of the transcription factor nuclear factor kappa B (NF-κB), as shown by increased NF-κB DNA binding to its specific site of the IL-8 promoter and increased NF-κB–driven gene transcription. Therefore, our data demonstrate that a combined exposure to CSC and RSV synergistically increases chemokine expression in airway epithelial cells, suggesting that CSC contributes to an exuberant immune response to RSV by stimulating overlapping signal transduction pathways.
Keywords: RSV, cigarette smoke condensate, chemokines, IRF, NF-κB
Respiratory syncytial virus (RSV) is a major cause of lower respiratory infection in infants, elderly, immunocompromised patients (Couch et al., 1997; Falsey et al., 2005) By the age of two nearly all children have been infected with RSV (Denny and Clyde, 1986). An effective vaccine for RSV has yet to be developed and immunity to infection is incomplete. Therefore, repeated attacks of RSV infection occur, causing symptoms ranging from mild nasal discharge to symptoms of greater morbidity, such as RSV-induced bronchiolitis and pneumonia requiring hospitalization. In addition to the acute morbidity of RSV infection, the long-term consequences include the development of hyperractive airway disease (Hall et al., 1984) and recurrent wheezing, both risk factors for the development of asthma (Sigurs et al., 1995).
Although the mechanisms of acute RSV-induced lung disease have not been clearly elucidated, increasing experimental evidence suggests that early inflammatory and immune events play a critical role. Therefore, exposure to external stimuli that either contribute to or enhance the inflammatory response to RSV infection may consequently increase the severity of RSV-induced lung disease and/or the long-term consequences associated with RSV infection. One such external stimulus is exposure to environmental tobacco smoke (ETS). Several epidemiological studies have reported that ETS exposure is associated with an increased frequency of lower respiratory tract infection (LRTI), persistent wheezing, increased incidence and severity of asthma episodes (Cook et al., 1999) and overall decreased pulmonary function (Chilmonczyk et al., 1993; Cook et al., 1999). In the United States, exposure to ETS may occur in up to 60% of the infants with RSV bronchiolitis and several studies have suggested that ETS exposure contributes to the severity of RSV brochiolitis and LRTI (Bauman et al., 2002; Bradley et al., 2005). Mechanisms associated with ETS exposure that could worsen RSV-induced acute lung inflammation and delay viral clearance include mucosal edema, decreased mucocilliary clearance, and bronchial inflammation (Cook and Strachan, 1999).
The airway epithelial cell, a primary target of RSV infection, plays a key role in regulating pulmonary inflammation by producing potent immunomodulatory and inflammatory mediators, such as chemokines. Chemokines are small cytokines that recruit and activate leukocytes into the airway mucosal. Therefore, chemokines play a pivotal role in regulating acute inflammatory and infectious processes (Zlotnik and Yoshie, 2000). Monocyte chemoattactant protein (MCP-1) and interleukin (IL)-8 are two chemokines that have been found in samples derived from the airways of hospitalized infants with naturally acquired RSV infection (Garofalo et al., 2001; Sheeran et al., 1999) and in the bronchoalveolar lavage of mice following exposure to ETS (D'hulst et al., 2005). MCP-1 is a major recruiter of monocytes and macrophages, whereas IL-8 promotes primarily the migration and activation of neutrophils.
The pathological mechanism underlying the epidemiologic association between cigarette exposure and the severity of RSV infection is unclear. In various experimental model systems, independent stimulation with either RSV or cigarette smoke condensate (CSC), a tar phase extract of cigarette smoke, has been linked to the activation of nuclear factor kappa B (NF-κB) (Anto et al., 2002; Garofalo et al., 1996; Shen et al., 1996). NF-κB is a ubiquitous multifunctional transcription factor involved in the expression of RSV-induced chemokines IL-8 and MCP-1 (Carpenter et al., 2002; Garofalo et al., 1996). However, the impact of a mixed exposure of CSC during RSV infection on chemokine secretion, as well as on activation of NF-κB and other transcription factors that regulate chemokine gene expression, has not been investigated.
In this study, we show that the presence of CSC increased the production of IL-8 and MCP-1 in RSV-infected airway epithelial cells. The synergistic induction of IL-8 gene transcription by RSV and CSC coexposure required the participation of the IL-8 promoter interferon stimulatory response element (ISRE) site, which binds transcription factors belonging to the IRF family. RSV and CSC coexposure induced increased nuclear translocation and binding of IRF proteins to the ISRE site. Furthermore, we observed enhanced NF-κB activation and NF-κB–driven gene transcription during RSV and CSC costimulation. These studies reveal important new mechanistic information on how cigarette smoke is an associative factor capable of contributing to the severity of RSV-induced inflammatory response.
MATERIALS AND METHODS
RSV preparation.
The RSV A2 strain was grown in HEp-2 cells and purified by centrifugation on discontinuous sucrose gradients as described elsewhere (Ueba, 1978). The virus titer of the purified RSV ranged form 8–9 log10 plaque forming units (PFU)/ml using a methylcellulose plaque assay. No contaminating cytokines were found in these sucrose-purified viral preparations (Patel et al., 1995). LPS, assayed using the limulus hemocyanin agglutination assay, was not detected. Virus pools were aliquoted, quick-frozen in liquid nitrogen, and stored at −70°C until used.
CSC treatment and infection of epithelial cells with RSV.
A549, human alveolar type II–like epithelial cells, and 293, a human embryonic kidney epithelial cell line (ATCC, Manassas, VA), were maintained in F12K and minimum essential medium respectively, containing 10% (vol/vol) fetal bovine serum, 10mM glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin. The CSC (Murty Pharmaceuticals, Lexington, Kentucky) was prepared by smoking University of Kentucky's Standard Research Cigarettes on an FTC Smoke Machine. The total particulate matter collected was extracted with dimethyl sulfoxide (DMSO) to generate a 4% solution. Cells monolayers were serum-starved for 4 h prior to infection. CSC exposure, at 1 μg/ml was performed 2 h prior to infection. Cells were subsequently infected with sucrose-purified RSV at a multiplicity of infection (MOI) of 1. A549 cells, not stimulated CSC, were treated with equivalent volumes of DMSO, the CSC diluent vehicle, and uninfected cells treated with 30% sucrose solution. Cigarette smoke extract (CSE) was prepared as described in (Vassallo et al., 2005). Briefly, CSE was prepared by drawing 35 ml of cigarette smoke into the syringe and then slowly bubbling the smoke into the medium. The cigarette was smoked up to 1-cm butt length. One cigarette was smoked per 10 ml of medium. Following preparation of CSE, the solution was filtered through 0.2-μm filters and used immediately. No cell toxicity was observed in measuring total cell number and viability following treatment with CSC or CSE at the used concentrations, as determined by trypan blue exclusion.
Chemokine protein determination.
The supernatants from treated and infected cells were harvested at 24-h postinfection (p.i.) to measure the release of inflammatory mediators. Both IL-8 (DuoSet) and MCP-1 (Quantikine) proteins were quantified by enzyme-linked immunosorbent assay (ELISA) following the manufacturer's protocol (R&D Systems, Minneapolis, MN). The production of chemokines and cytokines were measured using the Bio-Plex Cytokine Mouse Multi-Plex panel (Bio-Rad Laboratories, Hercules, CA), according to the manufacture instructions as previously described (Guerrero-Plata et al., 2005).
Quantitative real-time polymerase chain reaction.
Total RNA was extracted from A549 cells harvested at 6, 12, and 24 h p.i. using the RNeasy Mini Kit (Qiagen, Valencia CA). RNA integrity was assessed by gel electrophoresis and quantitated with absorbance A260 > 1.5. For IL-8 and MCP-1 gene transcript amplification by quantitative real-time polymerase chain reaction(Q-RT-PCR), Taqman Gene Expression Assay singleplex containing a 20× mix of primers and FAM TaqMan MGB probes for target genes and 18S rRNA VIC TaqMan assay reagent were used (Applied Biosystems, Foster City, CA). Separate tubes (singleplex) one-step RT-PCR was performed with 100 ng RNA for both target genes and endogenous control. The cycling parameters using TaqMan master mix one-step RT-PCR are as follows: reverse transcription 48°C for 30 min, AmpliTaq activation 95°C for 10 min, denaturation 95°C for 15 s and annealing/extension 60°C for 1 min (repeat 30 times) on ABI7000. Duplicate CT values were analyzed in Microsoft Excel using the comparative CT (ΔΔCT) method as described by the manufacturer (Applied Biosystems Foster City, CA). The amount of target (2−ΔΔCT) was obtained by normalizing to endogenous reference (18S) sample.
Plasmid construction and cell transfections.
Luciferase reporter gene plasmid constructs containing 5′ deletions of the human IL-8 promoter at upstream positions −162, −132, and −99, multimeric ISRE and NF-κB binding sites and mutations within the ISRE (RSVRE) site have been previously described (Casola et al., 2000). Luciferase reporter constructs were transiently transfected into A549 and 293 cells using Fugene 6 (Roche Applied Science, Branford, CT). Logarithmically growing cells were transfected with 1 μg of the reporter gene plasmid and 0.5 μg of β-galactosidase premixed with Fugene 6 in a 1:3 ratio (μg/μl). After 18 h of transfection, triplicate plates were treated with either control solvent (DMSO), CSC (1 μg/ml), RSV (MOI 1), or RSV + CSC. At the indicated time, cells were harvested to measure luciferase and β-galactosidase activity, as previously described (Garofalo et al., 1996). Luciferase activity was normalized to the internal control β-galactosidase activity and results reported as fold induction over baseline control values. Results are representative of three independent experiments.
Electrophoretic mobility shift assay.
Nuclear extracts were prepared from A549 cells infected or treated with either CSC or RSV or a combination of the two at 6 and 12 p.i. using the hypotonic/nonionic detergent lysis as previously described (Casola et al., 2000). The costimulation treatment group is designated as R + C for simplicity. To ensure equal loading, nuclear extracts were quantitated by protein assay (Bio-Rad) and incubated with oligonucleotides corresponding to the ISRE and the NF-κB wild-type (WT) and mutated (MUT) binding sites of the IL-8 promoter. Sequences for the oligonucleotides are shown below with mutated nucleotides that disrupt NF-κB binding shown in bold and underlined. Oligonucleotides were synthesized by Integrated DNA Technologies, Coralville, IA. The DNA-binding reaction for the NF-κB probe contained 10 μg of nuclear protein, 1 μg of polydeoxyadenylicthymidylic acid (polydA-dT), 5% glycerol, and 50,000 cpm α32P –labeled double stranded oligonucleotide in a total volume of 20 μl. Additional reagents for the ISRE-binding reactions included 5mM dithiothreitol. For competition assays, 2 pmol of unlabeled WT or MUT competitor was included in the binding reaction. The resulting hybridized DNA–protein complexes were resolved using a 6% nondenaturing polyacrylamide gel and visualized by autoradiography.
ISRE-IL-8: 5′-GAT CCA CCG TAT TTG ATA AGG AAC AAA TAG GAG TGT TA-3′; 3′-CTA GGT GGC ATA AAC TAT TCC TTG TTT ATC CTC ACA AT-5′
NF-κB-IL-8 (WT): 5′-GATCCATCAGTTGCAAATCGTGGAATTTCCTCTA-3′; 3′-GTAGTCAACGTTTAGCACCTTAAAGGAGATCTAG-5′
NF-κB-IL-8 (MUT): 5′-GATCCATCAGTTGCAAATCGTTTAATTTAATCTA-3′; 3′-GTAGTCAACGTTTAGCAAATTAAATTAGATCTAG-5′
In the gel mobility supershift, commercially available antibodies against p65, p50, IRF-1, -3, or -7 (Santa Cruz Biotech, Inc., Santa Cruz, CA) were added to the binding reactions and incubated on ice for one hour prior to fractionation on 6% polyacrylamide gel electrophoresis (PAGE). Preimmune serum was used as a control for any nonspecific effects of the immune antisera.
Western immunoblot.
Nuclear extracts from treated A549 cells were fractionated by sodium dodecyl sulfate (SDS)-PAGE, transferred to a polyvinylidene fluoride (PVDF) membrane, and probed with commercially available primary rabbit polyclonal IgG antibodies against IRF-1, IRF-3, IRF-7 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), and anti-p65 (Upstate, Lake Placid, NY) and Lamin B (Zymed Laboratories, Inc., San Francisco, CA). Following probing with horseradish peroxidase-secondary antibody, proteins were detected using an Enhanced Chemiluminescence system (Amersham Life Science, Arlington Heights, IL) and visualized through autoradiography. Protein bands were quantitated using Alpha Imager (AlphaInnotech, SanLeandro, CA).
Viral titration.
Supernatants from treated A549 cells were obtained at 24 h p.i., snap frozen in liquid nitrogen, and stored at −70°C. Serial dilutions of the supernatants were added to confluent HEp-2 cells grown in 24-well plates and viral titers were determined by the methylcellulose plaque assay, as previously described (Shen et al., 1996).
Statistical analysis.
The two treatment groups RSV versus RSV + CSC were analyzed using an unpaired, two-tailed, Student's t-test (GraphPad Prism; GraphPad Software, Inc., San Diego, CA). Results are expressed as mean ± SD.
RESULTS
Effect of CSC Exposure on RSV-Induced Chemokine Production in Airway Epithelial Cells
To study the effect of CSC on RSV-induced chemokine secretion in airway epithelial cells we used A549 cells, which resemble type II alveolar epithelial cells, a model system we have previously shown to be suitable for investigating RSV infection (Casola et al., 2001b; Garofalo et al., 1996). Because RSV is a potent inducer of inflammatory mediators, we tested increasing concentrations of CSC (0.1, 0.5, 1, 10 μg/ml) in combination with low MOIs of RSV infection (0.1, 0.5, and 1) to identify a potential effect of CSC on RSV infection. Cell-free supernatants of treated and infected cells were obtained at 12, 24, and 48 h p.i. and IL-8 and MCP-1 secretion was measured by ELISA. As shown in Figure 1A, the combination of RSV infection at an MOI of 1 in the presence of CSC at a concentration 1 μg/ml consistently led to a synergistic increase in IL-8 and MCP-1 protein secretion at 24 h p.i., with a trend in increased chemokine secretion also at earlier time points, although not statistically different (data not shown). Therefore, the treatment conditions of RSV (MOI of 1) and CSC (1 μg/ml) were used for all future experiments unless otherwise noted. To identify the potential release of additional inflammatory mediators, supernatants were also analyzed by Bio-Plex Multiplex Suspension Array (Bio-Rad), as previously described (Guerrero-Plata et al., 2005). However, no significant additive or synergistic increase of other chemokine or cytokine was observed in the combination treatment versus RSV alone (data not shown). Similar results were obtained when we used CSE alone or in combination with RSV infection to stimulate A549 cells (Fig. 1B).
FIG. 1.
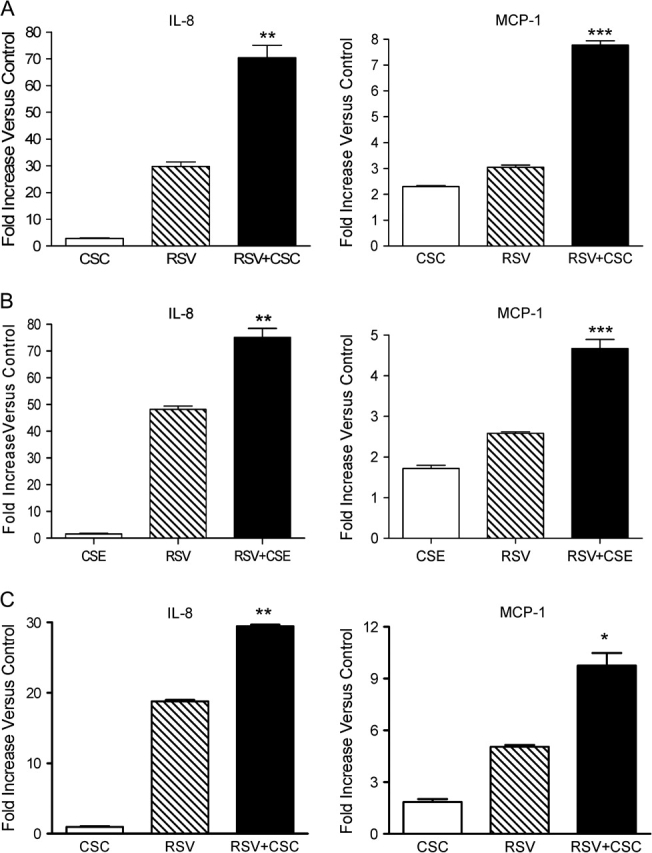
RSV infection and CSC treatment of A549 cells synergistically stimulates IL-8 and MCP-1 protein release and gene expression. A549 cells were treated with either CSC 1 μg/ml (A) or 2% CSE (B) and infected with RSV at an MOI of 1. Supernatants were harvested at 24 h p.i. and assayed for IL-8 and MCP-1 protein secretion by ELISA. IL-8 and MCP-1 gene expression was quantified by real-time PCR at 12 h p.i. (C). Data are presented as fold increases over control and are representative of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 RSV relative to RSV + CSC.
To evaluate whether CSC potentiates RSV-induced chemokine production at the level of gene expression, IL-8 and MCP-1 mRNA abundance was determined by Q-RT-PCR. A549 cells were treated with control solvent or CSC and either mock- or RSV-infected, as previously described. Cells were harvested at 6, 12, and 24 h p.i. to extract total RNA. CSC treatment alone increased only slightly the levels of IL-8 and MCP-1 mRNA, whereas there was a significant increase in expression for both genes in RSV-infected cells compared with uninfected cells at 12 h (Fig. 1C) and a trend toward a statistical significant difference at 24 h but not at 6 h p.i. CSC and RSV coexposure led to a synergistic increase in the level of IL-8 and MCP-1 mRNA compared with CSC exposure or RSV infection alone (Fig. 1C).
To determine whether the synergistic effect of chemokine induction following RSV and CSC costimulation was due to an increased viral replication, we assessed viral titer in supernatants from treated and infected A549 cells harvested at 24 h p.i. As shown in Figure 2, exposure of RSV-infected cells to CSC did not alter RSV viral replication, supporting the hypothesis that stimulation of overlapping signaling pathways, and not enhanced viral replication could be responsible for the observed increase in chemokine expression.
FIG. 2.

Effect of CSC on RSV replication. Supernatants of A549 cells exposed to CSC 1 μg/ml and infected with RSV at MOI of 1 were harvested at 24 h p.i. to measure RSV viral replication using the methylcellulose plaque assay. Data are representative of two independent experiments.
CSC Enhances RSV-Induced IL-8 Transcription through IRF Activation
In order to identify the molecular mechanism regulating the increased chemokine production in RSV + CSC treated cells, we investigated the role of specific cis-regulatory elements in activation of the IL-8 promoter and the transcription factors binding to them in response to RSV and CSC costimulation. A549 cells were transiently transfected with plasmids containing 5′ serial deletions, site mutations, or modifications of the human IL-8 promoter linked to the luciferase (LUC) reporter gene. The constructs were previously generated and detailed description provided in Casola et al. (2000). We have previously shown that nucleotides starting at −162 nt upstream the transcription start site of the IL-8 promoter are sufficient for RSV-induced promoter activation (Garofalo et al., 1996). The diagram in Figure 3A depicts the 5′ flanking region of the IL-8 promoter starting with the full-length WT construct designated as the −162/+44 hIL-8 LUC reporter plasmid. The promoter region contains transcription factor binding sites identified as ISRE (an IRF-1 binding site), AP-1, NF-IL6, NF-κB, upstream the transcriptional initiation site. All four binding sites play an important role in RSV-induced IL-8 promoter activation in A549 cells (Casola et al., 2000, 2001b; Garofalo et al., 1996).
FIG. 3.
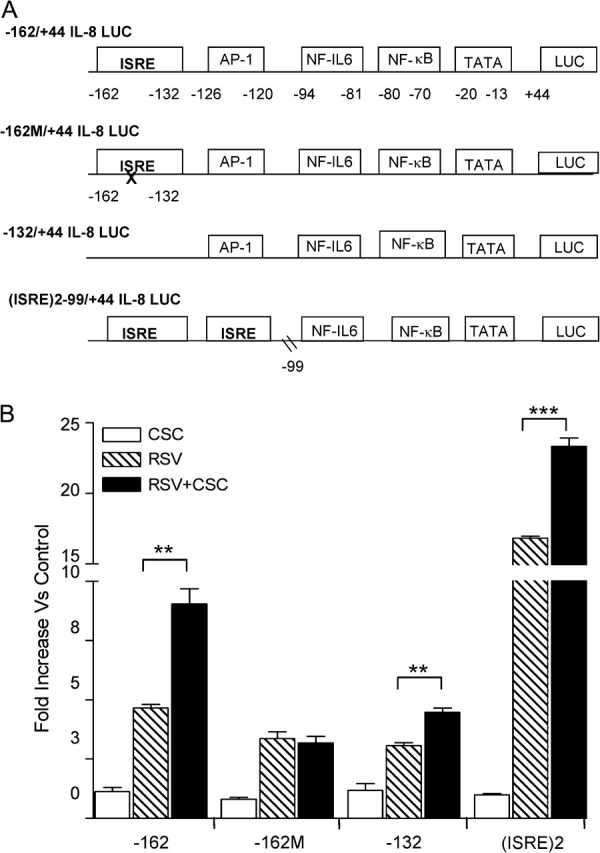
Effect of CSC treatment on RSV-induced IL-8 promoter activation. (A) Schematic representation of plasmid constructs containing 5′ flanking region of the IL-8 promoter region, starting upstream at −162 bp, either WT or containing modification of the ISRE-binding site including ISRE deletion (−132 IL-8), ISRE mutation (−162M), two multiple ISRE-binding sites, linked to a luciferase (LUC) reporter gene. Additional regulatory binding sites corresponding to the AP-1, NF-IL6, and NF-κB sites are also shown. The nucleotide numbers indicated their position in relationship to the IL-8 promoter transcriptional initiation site. (B) A549 cells were transfected with various hIL-8 LUC plasmids and infected with RSV alone and in combination with CSC. Treated and infected cells were harvested at 6 h p.i. to measure luciferase activity measured by luminometer. Data are presented as fold increases over control and are representative of three independent experiments. **p < 0.01, ***p < 0.001 RSV relative to RSV + CSC.
A549 cells were transfected with the various IL-8 plasmid constructs and 18 h later treated with CSC and subsequently infected with RSV. Cells were harvested and assayed for luciferase reporter gene activity at 6, 12, and 24 h p.i. For all constructs, no significant increase of luciferase activity was observed in CSC stimulated versus control treated cells (Fig. 3B). RSV infection led to a fivefold increase in luciferase activity of the full-length promoter (−162/+44 hIL-8 LUC) versus control, as previously observed (Garofalo et al., 1996). On the other hand, costimulation with RSV + CSC resulted in a heightened synergistic increase (ninefold) in luciferase activity in comparison to RSV infection alone.
RSV infection in cells transfected with the 162M/+44 hIL-8 LUC construct, containing a site mutation in the ISRE site, led to a slightly decreased luciferase induction (fourfold) in comparison to the −162 hIL-8 promoter. As a consequence of ISRE mutation, costimulation with RSV + CSC failed to enhance IL-8 promoter activation as opposed to the synergistic induction observed with the full-length promoter. The −132/+44 hIL-8 LUC construct is a deleted mutant that lacks the ISRE site (Casola et al., 2000). Similarly to the −162M promoter, the −132 deleted mutant showed decreased promoter activation (fourfold) in response to RSV infection and only a slight increase after CSC and RSV coexposure, in comparison to RSV alone, which was not synergistic (Fig. 3B).
We have previously shown that the −99/+44 hIL-8 LUC, which contains only the NF-IL6 and the NF-κB sites of the IL-8 promoter, is not inducible in response to RSV infection (Casola et al., 2001b). Costimulation with RSV + CSC also was not able to induce the reporter activity of −99/+44 hIL-8 promoter in A549 cells (data not shown). To validate the role of the ISRE-binding site in RSV + CSC enhanced IL-8 promoter activity, a construct containing two ISRE sites and lacking the AP-1 site, linked to the nonresponsive −99/+44 promoter region, designated as 2(ISRE)−99/+44 IL-8 LUC was transfected into A549 cells. Inserting two copies of the ISRE site into the −99/+44 hIL-8 LUC restored RSV inducibility of the −99/+44 IL-8 promoter (17 fold) and re-established the synergistic promoter activation observed following RSV + CSC costimulation (24-fold) (Fig. 3B). In summary, these results indicate that the IL-8 promoter sequence spanning from −162 to −132, corresponding to the ISRE site, is necessary for enhanced IL-8 gene transcription following RSV and CSC costimulation.
To further investigate the role of the ISRE site in RSV + CSC–induced enhanced IL-8 gene transcription, we determined whether RSV + CSC coexposure produced changes in the abundance of ISRE-binding proteins by electrophoretic mobility shift assay (EMSA), using nuclear extracts prepared from uninfected and RSV-infected cells in the presence or absence of CSC. As shown in Figure 4A, three binding complexes, C1, C2, and C3, were detected in control A549 cells. There was a slight induction of C1 complex when stimulated with CSC alone, and more in response to RSV infection. CSC + RSV coexposure resulted in a significant increase of C1 complex formation, compared with each stimulus alone (Fig. 4A). The inducible C1 complex was sequence specific, as demonstrated by its competition by unlabeled WT. The enhanced induction of C1 complex during costimulation was observed starting at 6 h p.i. and was sustained until 12 h p.i. (shown in Fig. 4A).
FIG. 4.
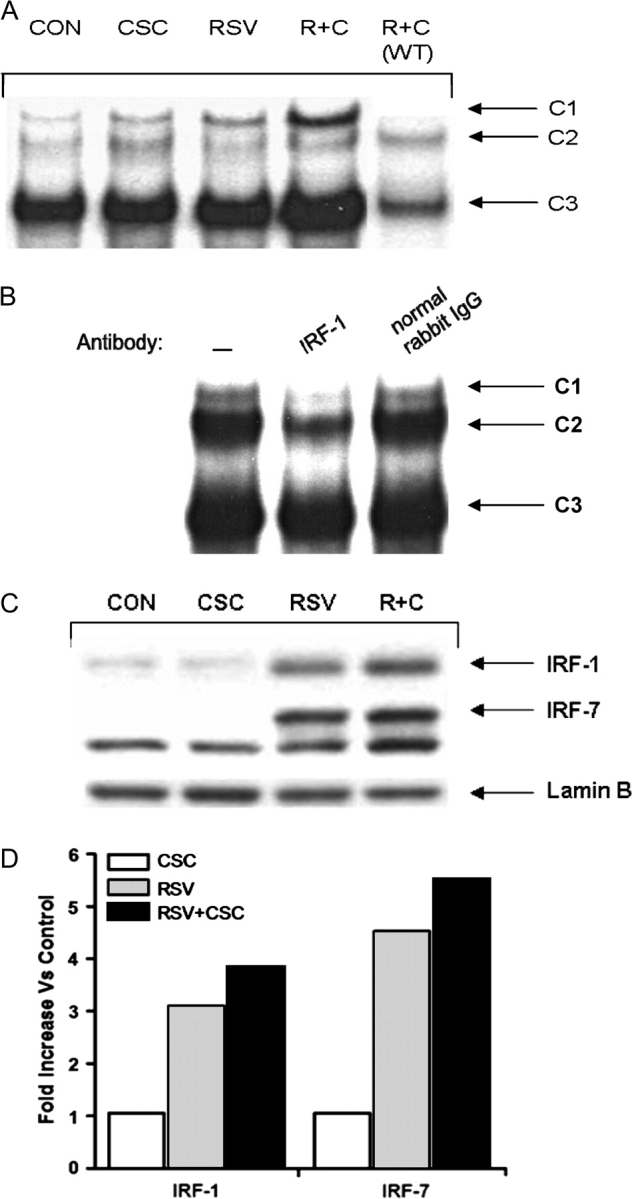
CSC costimulation enhanced RSV-induced IRF protein activation. (A) EMSA. Nuclear extracts were prepared from uninfected (CON), RSV-infected, CSC-treated, and CSC + RSV (R + C) exposed cells at 12 h p.i. and used for EMSA using a radiolabeled oligonucleotide corresponding to the IL-8 promoter ISRE site. For the competition assay, an excess of 2 pmol unlabeled WT ISRE probe was included in the binding reactions of RSV and CSC nuclear extracts. (B) Supershift assay. Nuclear extracts of A549 cells infected for 12 h were used in the EMSA in the presence of preimmune (PI) serum, anti-IRF-1, IRF-3 or IRF-7 antibodies. (C) Western blot. Nuclear extracts from treated and infected A549 cells were fractionated by 10% SDS-PAGE, transferred to PVDF membranes and incubated with the appropriate polyclonal primary antibody. Proteins were detected by enhanced chemiluminescence and autoradiography. (D) Densitometric analysis of protein bands of Western blot.
We have previously shown that IRF-1 binds to the ISRE IL-8 promoter in response to RSV infection (Casola et al., 2000); RSV infection also induces the activation of IRF-3 and IRF-7 in airway epithelial cells (Casola et al., 2001a). To determine the composition of the RSV-inducible complex, antibodies recognizing IRF-1, IRF-3, and IRF-7, or preimmune serum were added to the binding reaction. The anti-IRF-1 antibody affected the C1 binding, inducing the disappearance of the complex (Fig. 4B), whereas there was no change after addition of the anti-IRF-3 and -7 antibodies (data not shown), indicating that IRF-1 is a major component of the RSV + CSC–inducible complex. Because the ability of an antibody to produce a supershift depends on its affinity and the availability of the epitope within the DNA–protein complex, the absence of a supershift cannot be used to exclude the presence of IRF-3 and -7 binding to the ISRE.
To determine the effect of CSC on IRF activation during RSV infection, nuclear translocation of IRF1, -3, and -7 was analyzed by Western blot following various treatments at 6 h p.i. As shown in Figures 4C and 4D, costimulation led to an increase in IRF-1 and -7 nuclear translocation compared with RSV alone. A modest increase in IRF-3 phosphorylation, a marker of activation Indukuri et al. (2006), but not in nuclear abundance, was also observed (RSV vs. R + C; 1.26 vs. 1.2, data not shown).
CSC Enhances NF-κB Activation in Response to RSV Infection
NF-κB is a ubiquitous multifunctional transcription factor that plays an important role in proinflammatory gene expression Barnes (1997). In various experimental systems both RSV and CSC, each as an independent stimulus, have been shown to induce signaling pathways leading to the NF-κB activation (Anto et al., 2002; Garofalo et al., 1996; Shen et al., 1996). We have previously shown that NF-κB is necessary for RSV-induced IL-8 promoter activation (Garofalo et al., 1996). To determine whether NF-κB activation was enhanced by RSV + CSC costimulation, an NF-κB–driven artificial promoter, containing three copies of the IL-8 NF-κB binding site, was used to investigate NF-κB activation following RSV+CSC stimulation. Cells were transfected with the NF-κB–driven luciferase reporter plasmid and either infected with RSV alone or in combination with CSC. Cells were harvested at various time points to measure luciferase activity. As shown in Figure 5, CSC alone did not induce luciferase activity, in contrast to RSV which induced a nine fold increase at 6 h, but not at 12 or 24 h p.i., compared with uninfected cells. Costimulation of RSV+CSC produced a synergistic increase in promoter activation, compared with RSV alone at all time points.
FIG. 5.

CSC costimulation enhanced RSV-induced NF-κB–driven gene transcription. 293 cells were transiently transfected with a luciferase reporter construct containing multiple IL-8 NF-κB binding sites. Cells were then treated with CSC alone or in combination with RSV infection. At various times p.i. cells were harvested to measure luciferase activity. Data are presented as fold increases over control and are representative of three independent experiments. **p < 0.01, ***p < 0.001 RSV relative to RSV + CSC.
Based on the observation that CSC contributed to RSV-induced NF-κB activation, we investigated whether CSC enhanced RSV-induced NF-κB DNA-binding activity by EMSA. Nuclear extracts were incubated with a radiolabeled oligonucleotide corresponding to the IL-8 promoter NF-κB site. RSV infection induced the formation of two DNA-binding complexes, C1 and C2, which were enhanced by RSV and CSC coexposure, both at 6 and 12 h p.i. C3 was constitutively present in uninfected and infected cells (Fig. 6A). C1and C2 binding was specific, as demonstrated by competition assays with WT or MUT NF-κB binding site.
FIG. 6.
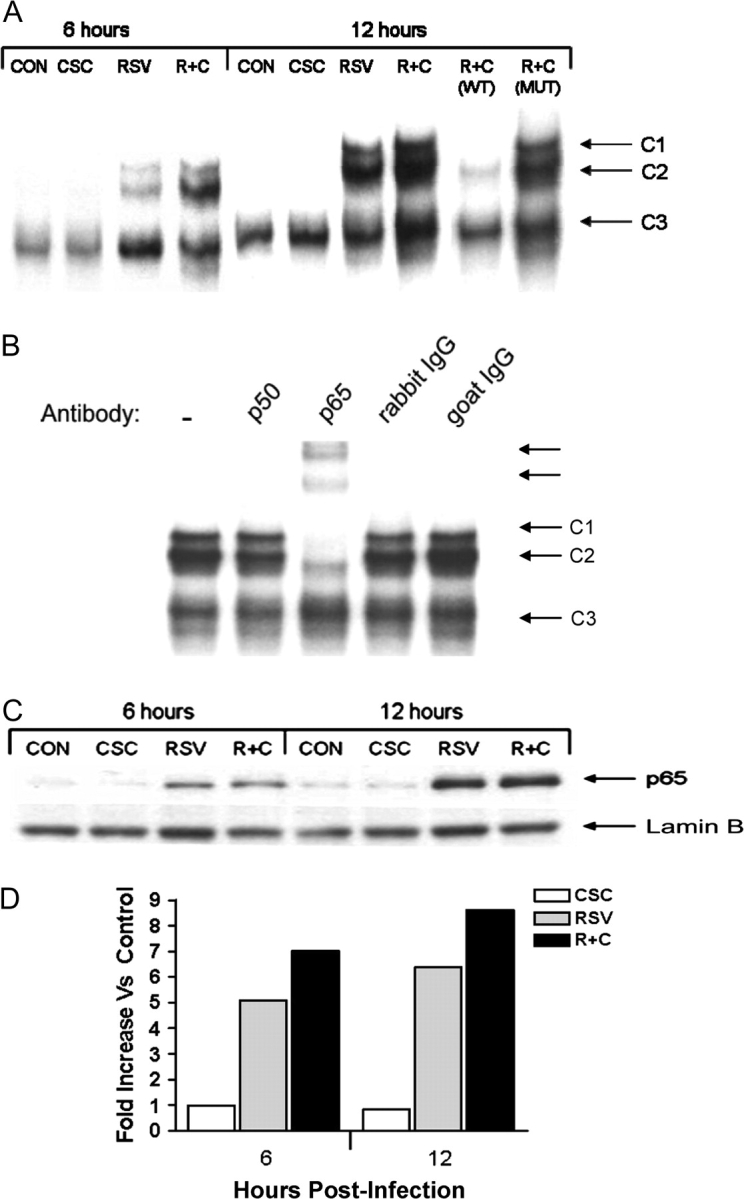
CSC costimulation enhanced RSV-induced NF-κB activation. (A) EMSA. Nuclear extracts were prepared from uninfected (CON), RSV-infected, CSC-treated, and CSC + RSV (R + C) exposed cells at 6 and 12 h p.i. and used for EMSA using a radiolabeled oligonucleotide corresponding to the IL-8 promoter NF-κB site. For the competition assay, an excess of 2 pmol unlabeled WT and MUT probe was included in the binding reactions of RSV and CSC nuclear extracts. (B) Supershift assay. Nuclear extracts of A549 cells infected for 12 h were used in the EMSA in the presence of preimmune (PI) serum, anti-p50 or anti-p65 antibodies. Supershifted bands are indicated by the arrows. (C) Western blot. Nuclear extracts from treated and infected A549 cells were fractionated by 10% SDS-PAGE, transferred to PVDF membranes and incubated with the appropriate polyclonal primary antibody. Proteins were detected by enhanced chemiluminescence and autoradiography. (D) Densitometric analysis of protein bands of Western blot.
We have previously shown that the p65 NF-κB subunit is the major protein constituting RSV-induced C1 and C2 complexes (Garofalo et al., 1996). To determine the composition of the RSV + CSC–inducible complexes, we performed supershift assays, adding specific antibodies to p50 and p65 in EMSA. The addition of anti-p65 antibody supershifted both C1 and C2, indicating that p65 is a major component of both complexes (Fig. 6B).
To confirm that RSV + CSC costimulation enhanced NF-κB activation, nuclear extracts were prepared from A549 cells at 6 and 12 h p.i. and NF-κB nuclear translocation by analyzed by Western blot. Although there was no significant increase in p65 nuclear translocation after stimulation of A549 cells with CSC alone, nuclear abundance of p65 was higher in RSV + CSC stimulated cells in comparison to RSV alone (Figs. 6C and 6D). All together these results indicate that CSC enhances RSV-induced NF-κB activation and subsequent NF-κB–dependent gene transcription.
DISCUSSION
In the past few decades, there has been increasing evidence that passive smoking or exposure to ETS increases the incidence and severity of respiratory viral infections. In a recent study, children of smoking parents who were hospitalized with RSV bronchiolitis had a higher level of serum cotinine, a metabolite of nicotine and biomarker of ETS exposure, than children of smoking parents hospitalized with nonrespiratory symptoms (Gurkan et al., 2000). After one month, children diagnosed with RSV bronchiolitis still had higher cotinine levels than controls suggesting that heavy exposure to ETS may contribute to the development of a more severe respiratory infection (Gurkan et al., 2000). The study of Mario Castro et al. has clearly shown that the severity of RSV bronchiolitis early in life seems to be modified by postnatal maternal cigarette smoke exposure (Bradley et al., 2005). An additional investigation, using a neonatal mouse model of RSV infection exposed daily to cigarette smoke, found a priming effect of smoke on RSV replication, diminished Th1 and enhanced Th2 T-cell responses following secondary RSV infection, suggesting that cigarette smoke exposure alters immune responses to RSV infection (Phaybouth et al., 2006). These investigations on the interactions between RSV and cigarette smoke indicate that cigarette smoke exposure has the potential to modulate and exacerbate the host inflammatory/immune response to RSV infection.
A fundamental event in RSV-induced pulmonary inflammation is thought to be the response of the airway epithelial cell to RSV infection. Although the pathophysiology of RSV-induced airway disease is not clearly understood, an initial step in RSV-induced pulmonary inflammation is the release of inflammatory chemokines from infected epithelial cells (Olszewska-Pazdrak et al., 1998; Saito et al., 1997). IL-8 is a potent chemokine in recruiting and activating immune cells, in particular neutrophils. Previous studies have shown that RSV is a potent inducer of IL-8 airway epithelial cells (Jamaluddin et al., 1996) and high levels of IL-8 protein have been detected in the nasal lavage washes of children with RSV upper respiratory infections (Noah et al., 1995). Therefore increased IL-8 chemokine expression by external environmental stimuli, such as cigarette smoke could be a critical factor in modulating the severity of RSV-induced lung disease.
In this study, we used CSC, which represents the insoluble tar component of mainstream cigarette smoke. Cigarette smoke is a highly complex mixture consisting of approximately 4000 chemicals distributed within the gas or particulate phases (Hoffmann and Wynder, 1986). CSC, which provides a close semblance to the mixture of mainstream cigarette smoke, was used as a feasible and readily accessible in vitro representative of cigarette smoke. CSC has been shown to activate epithelial and immune cells and stimulate the secretion of inflammatory mediators into the airway mucosa (Hellermann et al., 2002; Shen et al., 1996). Although others have noted a stimulatory and proinflammatory effect of CSC on airway epithelial cells, there are currently no studies that have examined whether cigarette smoke exposure is capable of modulating the response of human respiratory epithelial cells to RSV infection.
Our results show that CSC and RSV costimulation led to an increase in both MCP-1 and IL-8 chemokine gene and protein expression in airway epithelial cells. Increased IL-8 expression was due to enhanced promoter activation following RSV and CSC costimulation. IL-8 promoter deletion and mutation analysis identified the region between −162 nt and −132 nt as a critical regulatory region contributing to the synergistic transcriptional activation of the IL-8 promoter. We have previously shown that this region contains an ISRE-binding site which is an important regulatory site for IL-8 gene transcription not only in response to RSV infection, but also to other stimuli such as H. pylori and hepatitis C virus (Wagoner et al., 2007; Yamaoka et al., 2004). Costimulation of a promoter containing a single nucleotide mutation of the ISRE site (−162M IL-8) failed to enhance IL-8 promoter activation, in comparison to RSV stimulation alone. Similarly, costimulation of the −132 nt IL-8 promoter, which lacks the ISRE site, resulted in a loss of synergistic induction of promoter activation. On the other hand, the presence of multiple copies of the ISRE upstream an inert IL-8 promoter restored the enhanced IL-8 promoter activation observed in response to CSC and RSV costimulation. In agreement with the promoter studies, we found that CSC and RSV costimulation enhanced nuclear translocation and DNA binding of IRF proteins, which are known to bind to the IL-8 promoter ISRE site.
Although IL-8 transcriptional regulation is modulated by a variety of transcription factors including IRF proteins, NF-κB is essential for IL-8 gene expression in response to many different kinds of stimuli. NF-κB has been shown to regulate IL-8 gene expression in response to either cigarette smoke or RSV alone (Garofalo et al., 1996; Hellermann et al., 2002). Our gene reporter studies, using multimers of the IL-8-NF-κB site showed that, whereas CSC alone did not strongly induce NF-κB–driven gene transcription, a heightened response was observed with CSC and RSV costimulation. Furthermore, CSC and RSV costimulation enhanced nuclear translocation and DNA binding of NF-κB proteins, suggesting that CSC likely enhanced RSV-induced IL-8 promoter activation also by contributing to NF-κB activation. The lack of an effect of CSC stimulation alone on NF-κB activation observed in our study is in agreement with a previous study, showing that cigarette smoke plays a more important role in inducing NF-κB activation in normal cells compared with cancerous (Fields et al., 2005) cell lines, (Hellermann et al., 2002). However, the amount of CSC used in our study was not able to induce IL-8 production also in normal human bronchial cells, in which only amounts fifty to one hundred times higher were able to elicit IL-8 secretion (Fields et al., 2005). On the other hand, CSE has been shown to induce IL-8 and IL-6 production in primary small alveolar epithelial cells, but not in A549 cells (Kode et al., 2006). Differential levels of IL-8 gene expression are likely to play an important role in the pathogenesis of RSV-induced lung disease, as recently shown by the association of single gene polymorphisms of the IL-8 promoter with increased disease severity in children with RSV infection (Hull et al., 2001; Puthothu et al., 2006). The finding that ETS exposure contributes to the severity of RSV LRTI (Bauman et al., 2002), could be in part explained by a priming effect of smoke on RSV-induced secretion of proinflammatory mediators by infected airway epithelial cells, which results in enhanced pulmonary inflammation. More studies are needed to conclusively identify cigarette smoke as an important risk factor contributing to the severity of RSV-induced disease and to better understand the mechanisms of viral-induced enhanced pulmonary disease in order to implement efforts aimed at reducing cigarette exposure in infants, with the ideal objective of decreasing the severity and possibly long-term consequences, such as the development of asthma, of RSV infection.
FUNDING
National Institute of Environmental Health Science (ES06676); Flight Attendant Medical Research Institute Clinical Investigator Awards to A.C. and R.P.G.; National Institute of Environmental Health Science training grant (T32 ES07254) supported S.M.C.; and National Institutes of General Medical Sciences Ruth L. Kirschstein National Research Service Award (F31 GM072231).
Acknowledgments
A special thank you is attributed to Mary Treinen-Moslen, Ph.D., for her continuous support of this study, and to Cynthia Tribble for her assistance in the manuscript editing and submission.
References
- Anto RJ, Mukhopadhyay A, Shishodia S, Gairola CG, Aggarwal BB. Cigarette smoke condensate activates nuclear transcription factor-kappaB through phosphorylation and degradation of IkappaB(alpha): Correlation with induction of cyclooxygenase-2. Carcinogenesis. 2002;23:1511–1518. doi: 10.1093/carcin/23.9.1511. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Nuclear factor-kappa B. Int. J. Biochem. Cell Biol. 1997;29:867–870. doi: 10.1016/s1357-2725(96)00159-8. [DOI] [PubMed] [Google Scholar]
- Bauman LJ, Wright E, Leickly FE, Crain E, Kruszon-Moran D, Wade SL, Visness CM. Relationship of adherence to pediatric asthma morbidity among inner-city children. Pediatrics. 2002;110:e6. doi: 10.1542/peds.110.1.e6. [DOI] [PubMed] [Google Scholar]
- Bradley JP, Bacharier LB, Bonfiglio J, Schechtman KB, Strunk R, Storch G, Castro M. Severity of respiratory syncytial virus bronchiolitis is affected by cigarette smoke exposure and atopy. Pediatrics. 2005;115:e7–14. doi: 10.1542/peds.2004-0059. [DOI] [PubMed] [Google Scholar]
- Carpenter LR, Moy JN, Roebuck KA. Respiratory syncytial virus and TNF alpha induction of chemokine gene expression involves differential activation of Rel A and NF-kappa B1. BMC. Infect. Dis. 2002;2:5. doi: 10.1186/1471-2334-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola A, Burger N, Liu T, Jamaluddin M, Brasier AR, Garofal RP. Oxidant tone regulates RANTES gene transcription in airway epithelial cells infected with respiratory syncytial virus: Role in viral-induced interferon regulatory factor activation. J. Biol. Chem. 2001a;276:19715–19722. doi: 10.1074/jbc.M101526200. [DOI] [PubMed] [Google Scholar]
- Casola A, Garofalo RP, Haeberle H, Elliott TF, Lin A, Jamaluddin M, Brasier AR. Multiple cis regulatory elements control RANTES promoter activity in alveolar epithelial cells infected with respiratory syncytial virus. J. Virol. 2001b;75:6428–6439. doi: 10.1128/JVI.75.14.6428-6439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola A, Garofalo RP, Jamaluddin M, Vlahopoulos S, Brasier AR. Requirement of a novel upstream response element in RSV induction of interleukin-8 gene expression: Stimulus-specific differences with cytokine activation. J. Immunol. 2000;164:5944–5951. doi: 10.4049/jimmunol.164.11.5944. [DOI] [PubMed] [Google Scholar]
- Chilmonczyk BA, Salmun LM, Megathlin KN, Neveux LM, Palomaki GE, Knight GJ, Pulkkinen AJ, Haddow JE. Association between exposure to environmental tobacco smoke and exacerbations of asthma in children. N. Engl. J. Med. 1993;328:1665–1669. doi: 10.1056/NEJM199306103282303. [DOI] [PubMed] [Google Scholar]
- Cook DG, Strachan DP. Health effects of passive smoking-10: Summary of effects of parental smoking on the respiratory health of children and implications for research. Thorax. 1999;54:357–366. doi: 10.1136/thx.54.4.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch RB, Englund JA, Whimbey E. Respiratory viral infections in immunocompetent and immunocompromised persons. Am. J. Med. 1997;102:2–9. doi: 10.1016/S0002-9343(97)00003-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny FW, Clyde WA., Jr Acute lower respiratory tract infections in nonhospitalized children. J. Pediatr. 1986;108:635–646. doi: 10.1016/s0022-3476(86)81034-4. [DOI] [PubMed] [Google Scholar]
- D'hulst AI, Vermaelen KY, Brusselle GG, Joos GF, Pauwels RA. Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur. Respir. J. 2005;26:204–213. doi: 10.1183/09031936.05.00095204. [DOI] [PubMed] [Google Scholar]
- Falsey AR, Hennessey PA, Formica MA, Cox C, Walsh EE. Respiratory syncytial virus infection in elderly and high-risk adults. N. Engl. J. Med. 2005;352:1749–1759. doi: 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- Fields WR, Leonard RM, Odom PS, Nordskog BK, Ogden MW, Doolittle DJ. Gene expression in normal human bronchial epithelial (NHBE) cells following in vitro exposure to cigarette smoke condensate. Toxicol. Sci. 2005;86:84–91. doi: 10.1093/toxsci/kfi179. [DOI] [PubMed] [Google Scholar]
- Garofalo RP, Patti J, Hintz KA, Hill V, Ogra PL, Welliver RC. Macrophage inflammatory protein 1-alpha, and not T-helper type 2 cytokines, is associated with severe forms of bronchiolitis. J. Infect. Dis. 2001;184:393–399. doi: 10.1086/322788. [DOI] [PubMed] [Google Scholar]
- Garofalo RP, Sabry M, Jamaluddin M, Yu RK, Casola A, Ogra PL, Brasier AR. Transcriptional activation of the interleukin-8 gene by respiratory syncytial virus infection in alveolar epithelial cells: Nuclear translocation of the RelA transcription factor as a mechanism producing airway mucosal inflammation. J. Virol. 1996;70:8773–8781. doi: 10.1128/jvi.70.12.8773-8781.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Plata A, Casola A, Garofalo RP. Human metapneumovirus induces a profile of lung cytokines distinct from that of respiratory syncytial virus. J. Virol. 2005;79:14992–14997. doi: 10.1128/JVI.79.23.14992-14997.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurkan F, Kiral A, Dagli E, Karakoc F. The effect of passive smoking on the development of respiratory syncytial virus bronchiolitis. Eur. J. Epidemiol. 2000;16:465–468. doi: 10.1023/a:1007658411953. [DOI] [PubMed] [Google Scholar]
- Hall CB, Hall WJ, Gala CL, MaGill FB, Leddy JP. Long-term prospective study in children after respiratory syncytial virus infection. J. Pediatr. 1984;105:358–364. doi: 10.1016/s0022-3476(84)80005-0. [DOI] [PubMed] [Google Scholar]
- Hellermann GR, Nagy SB, Kong X, Lockey RF, Mohapatra SS. Mechanism of cigarette smoke condensate-induced acute inflammatory response in human bronchial epithelial cells. Respir. Res. 2002;3:22. doi: 10.1186/rr172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann D, Wynder EL. Chemical constituents and bioactivity of tobacco smoke. IARC Sci. Publ. 1986;74:145–165. [PubMed] [Google Scholar]
- Hull J, Ackerman H, Isles K, Usen S, Pinder M, Thomson A, Kwiatkowski D. Unusual haplotypic structure of IL8, a susceptibility locus for a common respiratory virus. Am. J. Hum. Genet. 2001;69:413–419. doi: 10.1086/321291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indukuri H, Castro SM, Liao SM, Feeney LA, Dorsch M, Coyle AJ, Garofalo RP, Brasier AR, Casola A. Ikkepsilon regulates viral-induced interferon regulatory factor-3 activation via a redox-sensitive pathway. Virology. 2006;353:155–165. doi: 10.1016/j.virol.2006.05.022. [DOI] [PubMed] [Google Scholar]
- Jamaluddin M, Garofalo RP, Ogra PL, Brasier AR. Inducible translational regulation of the NF-IL6 transcription factor by respiratory syncytial virus infection in pulmonary epithelial cells. J. Virol. 1996;70:1554–1563. doi: 10.1128/jvi.70.3.1554-1563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kode A, Yang SR, Rahman I. Differential effects of cigarette smoke on oxidative stress and proinflammatory cytokine release in primary human airway epithelial cells and in a variety of transformed alveolar epithelial cells. Respir. Res. 2006;7:132. doi: 10.1186/1465-9921-7-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noah TL, Henderson FW, Wortman IA, Devlin RB, Handy J, Koren HS, Becker S. Nasal cytokine production in virus acute upper respiratory infection of childhood. J. Infect. Dis. 1995;171:584–592. doi: 10.1093/infdis/171.3.584. [DOI] [PubMed] [Google Scholar]
- Olszewska-Pazdrak B, Casola A, Saito T, Alam R, Crowe SE, Mei F, Ogra PL, Garofalo RP. Cell-specific expression of RANTES, MCP-1, and MIP-1alpha by lower airway epithelial cells and eosinophils infected with respiratory syncytial virus. J. Virol. 1998;72:4756–4764. doi: 10.1128/jvi.72.6.4756-4764.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JA, Kunimoto M, Sim TC, Garofalo R, Eliott T, Baron S, Ruuskanen O, Chonmaitree T, Ogra PL, Schmalstieg F. Interleukin-1 alpha mediates the enhanced expression of intercellular adhesion molecule-1 in pulmonary epithelial cells infected with respiratory syncytial virus. Am. J. Respir. Cell Mol. 1995;13:602–609. doi: 10.1165/ajrcmb.13.5.7576697. [DOI] [PubMed] [Google Scholar]
- Phaybouth V, Wang SZ, Hutt JA, McDonald JD, Harrod KS, Barrett EG. Cigarette smoke suppresses Th1 cytokine production and increases RSV expression in a neonatal model. Am. J. Physiol. Lung Cell Mol. Physiol. 2006;290:L222–L231. doi: 10.1152/ajplung.00148.2005. [DOI] [PubMed] [Google Scholar]
- Puthothu B, Krueger M, Heinze J, Forster J, Heinzmann A. Impact of IL8 and IL8-receptor alpha polymorphisms on the genetics of bronchial asthma and severe RSV infections. Clin. Mol. Allergy. 2006;4:2. doi: 10.1186/1476-7961-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Deskin RW, Casola A, Haeberle H, Olszewska B, Ernst PB, Alam R, Ogra PL, Garofalo R. Respiratory syncytial virus induces selective production of the chemokine RANTES by upper airway epithelial cells. J. Infect. Dis. 1997;175:497–504. doi: 10.1093/infdis/175.3.497. [DOI] [PubMed] [Google Scholar]
- Sheeran P, Jafri H, Carubelli C, Saavedra J, Johnson C, Krisher K, Sanchez PJ, Ramiliom O. Elevated cytokine concentrations in the nasopharyngeal and tracheal secretions of children with respiratory syncytial virus disease. Pediatr. Infect. Dis. J. 1999;18:115–122. doi: 10.1097/00006454-199902000-00007. [DOI] [PubMed] [Google Scholar]
- Shen Y, Rattan V, Sultana C, Kalra VK. Cigarette smoke condensate-induced adhesion molecule expression and transendothelial migration of monocytes. Am. J. Physiol. 1996;270:H1624–H1633. doi: 10.1152/ajpheart.1996.270.5.H1624. [DOI] [PubMed] [Google Scholar]
- Sigurs N, Bjarnason R, Sigurbergsson F, Kjellman B, Bjorksten B. Asthma and immunoglobulin E antibodies after respiratory syncytial virus bronchiolitis: A prospective cohort study with matched controls. Pediatrics. 1995;95:500–505. [PubMed] [Google Scholar]
- Ueba O. Respiratory syncytial virus: I. concentration and purification of the infectious virus. Acta Med. Okayama. 1978;32:265–272. [PubMed] [Google Scholar]
- Vassallo R, Tamada K, Lau JS, Kroening PR, Chen L. Cigarette smoke extract suppresses human dendritic cell function leading to preferential induction of Th-2 priming. J. Immunol. 2005;175:2684–2691. doi: 10.4049/jimmunol.175.4.2684. [DOI] [PubMed] [Google Scholar]
- Wagoner J, Austin M, Green J, Imaizumi T, Casola A, Brasier A, Khabar KS, Wakita T, Gale M, Jr, Polyak SJ. Regulation of CXCL-8 (interleukin-8) induction by double-stranded RNA signaling pathways during hepatitis C virus infection. J. Virol. 2007;81:309–318. doi: 10.1128/JVI.01411-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka Y, Kudo T, Lu H, Casola A, Brasier AR, Graham DY. Role of interferon-stimulated responsive element-like element in interleukin-8 promoter in Helicobacter pylori infection. Gastroenterology. 2004;126:1030–1043. doi: 10.1053/j.gastro.2003.12.048. [DOI] [PubMed] [Google Scholar]
- Zlotnik A, Yoshie O. Chemokines: A new classification system and their role in immunity. Immunity. 2000;12:121–127. doi: 10.1016/s1074-7613(00)80165-x. [DOI] [PubMed] [Google Scholar]