

Abstract
Perfluorooctanoic acid and perfluorodecanoic acid (PFDA) are commonly used as emulsifiers and surfactants in fluoropolymer manufacturing and are known peroxisome proliferator–activated receptor alpha (PPARα) agonists. PPARα activation induces β- and ω-oxidation enzymes such as Cyp4a14 and acyl-CoA oxidase, which are a likely cause of subsequent oxidative stress and peroxisome proliferation. Conversely, NF-E2-related factor-2 (Nrf2) is a transcription factor that protects against oxidative stress and inflammation by regulating several detoxification and xenobiotic transporter genes. Because PFDA markedly induces hepatic metabolism and oxidative stress, we hypothesized that PFDA exposure would increase expression of hepatic efflux multidrug resistance–associated protein (Mrp) transporters. A single PFDA dose (80 mg/kg) administered to mice increased hepatic Mrp3 (fourfold) and Mrp4 (31-fold) mRNA expression. Upregulation of Mrp3 and Mrp4 correlated with elevated serum-conjugated bilirubin and bile acids, respectively. To determine the mechanism of Mrp3 and Mrp4 induction, PFDA was administered to Nrf2-null mice, PPARα-null mice, and mice pretreated with gadolinium chloride, a Kupffer cell–depleting chemical capable of inhibiting the inflammatory cytokine response. In both PPARα- and Nrf2-null mice, maximal induction of Mrp3 and Mrp4 mRNA after PFDA administration was attenuated. Gadolinium chloride pretreatment reduced serum and hepatic tumor necrosis factor-α levels after PFDA treatment, as well as Mrp4 mRNA expression by 30%, suggesting that Kupffer cell–derived mediators may contribute to Mrp induction. Thus, after PFDA administration, the liver mounts a compensatory hepatoprotective response via PPARα and Nrf2, markedly increasing Mrp3 and Mrp4 expression, with corresponding increases in serum of known Mrp3 and Mrp4 substrates.
Keywords: Multidrug resistance-associated transporters, Mrp, Oxidative stress, PPARa, Nrf2, perfluorinated carboxylic fatty acids, transport
Perfluorinated carboxylic fatty acids (PFCAs) are straight-chain fatty acid analogs that have fluorines substituting for aliphatic hydrogen atoms. The PFCA class of chemicals includes perfluorodecanoic acid (PFDA), perfluorooctanoic acid (PFOA), and perfluorooctanesulfonate. Some PFCAs such as PFOA have been used extensively as industrial surfactants and emulsifiers in the manufacturing of fluoropolymers and fluorotelomers, which both are renowned for their ability to repel water and grease. The majority of environmental PFCAs come from these manufacturing processes, and residual amounts of PFOA have been detected in the commercial end products (Prevedouros et al., 2006). PFOA and PFDA are thought to have long half-lives and bioaccumulate and have thus become environmentally persistent. In humans, there is evidence that PFOA has a markedly longer half-life of 4.4 years in humans (Burris et al., 2002) than rats, suggesting the possibility for marked bioaccumulation in the human species. Such perfluorinated compounds can be detected in >98% of random human blood samples from persons not traditionally exposed by occupation to such chemicals, yet significant reductions in serum concentrations were noted in recent reports due to reduced usage (Calafat et al., 2007).
PFDA and PFOA both have key observed toxicities in rodents. PFOA has been reported to lead to wasting, neonatal mortality, developmental retardation, and immunosuppression and has been classified as a suspected carcinogen. Whereas there is far less literature for PFDA, more chronic, sustained wasting is seen with this compound, yet no teratogenic or developmental impairments have been observed at doses not maternally toxic (Harris and Birnbaum, 1989; Calafat et al., 2007). Although PFDA is also a suspected carcinogen, no 2-year carcinogenesis studies with this compound have been conducted.
PFDA profoundly affects lipid metabolism. PFDA inhibits hepatic mitochondrial β-oxidation in rodents, which subsequently increases long-chain fatty acid concentrations (Vahden Heuvel, 1996). These long-chain fatty acids activate peroxisome proliferator–activated receptor alpha (PPARα), which heterodimerizes with retinoid X receptor α and drives expression of microsomal ω-oxidation (Cyp4a14) and peroxisomal β-oxidation (fatty acyl-CoA oxidase) (Adinehzadeh et al., 1999; Brewster and Birnbaum, 1989; Chinje et al., 1994). This phenomenon is commonly referred to as peroxisomal proliferation (PP) due to the increased numbers of peroxisomes and accompanying hepatomegaly (George and Andersen, 1986; Harris and Birnbaum, 1989; Harris et al., 1989; Olson and Andersen, 1983; Van Rafelghem et al., 1987). Unlike other peroxisome proliferators, PFDA exposure in rodents induces anorexia, leading to hypophagia and subsequently to wasting. This weight loss and inappetance are similar to the effects observed with dioxins, although the mechanisms of PFDA and dioxin toxicity were shown to be different in these studies (Brewster and Birnbaum, 1989; George and Andersen, 1986; Goecke et al., 1992; Harris and Birnbaum, 1989; Olson and Andersen, 1983; Van Rafelghem et al., 1987). Whereas plasma concentrations of free fatty acids are not altered, liver triglycerides are markedly increased by PFDA in rats (Van Rafelghem et al., 1988). Similarly, hepatic accumulation of triglycerides was observed in both mouse and rat species after PFOA treatment (Kudo and Kawashima, 1997; Kudo et al., 1999) and fatty liver is observed in both rodent species as a consequence.
Following extended exposure, most peroxisome proliferators promote liver cancer in rodents but seemingly not in humans (Borges et al., 1992; Gonzalez, 2002). Furthermore, in rats treated with the cancer initiator 2-acetylaminofluorene, additional supplementation with PFOA led to increased incidence of malignant hepatocellular carcinoma (Abdellatif et al., 1991). Hepatocarcinogenesis induced by peroxisome proliferators has been postulated to be due in part to oxidative stress from persistent overexpression of enzymes involved in fatty acid β- and ω-oxidation and the subsequent generation of hydrogen peroxide as a by-product of oxidative reactions (Chinje et al., 1994; Glauert et al., 1992; Yeldandi et al., 2000). In addition to accumulation of fatty acids and hydrogen peroxide, PFDA produces oxidative DNA damage, as assessed by increased levels of 8-hydroxy-deoxyguanosine (Takagi et al., 1991).
PPARα-mediated activation of β- and ω-oxidation also has the potential for activating redox-sensitive pathways involved in cellular protection. The NF-E2-related factor 2 (Nrf2)/kelch-like ECH-associated protein (glutamic acid-cysteine-histidine) 1 pathway senses redox stress and initiates a corresponding induction of cytoprotective enzymes, including genes such as NAD(P)H:quinone oxidoreductase 1 (Nqo1) (Jaiswal, 2000), glutamate cysteine ligase catalytic and modifier subunits (Wild and Mulcahy, 2000), and glutathione S-transferase A2 (Daniel et al., 1989). A primary function of this stress response is to increase synthesis of the antioxidant glutathione (GSH), along with enzymes required for GSH transfer. Previous studies have demonstrated increased GSH levels after PFDA treatment in rodents, suggesting Nrf2 activation (Chen et al., 2001). In addition to controlling hepatic detoxification and GSH levels, Nrf2 also regulates the expression of several multidrug resistance–associated protein (Mrp) efflux transporters via Nrf2-mediated responses to oxidative stress (Maher et al., 2007). These transporters aid in general hepatic detoxification by the prompt removal of toxic metabolites into both the bile and the blood.
The effects of perfluorinated compounds on hepatic transport are almost completely unknown. Other peroxisomal proliferators such as clofibrate, ciprofibrate, and diethylhexylphthalate induce expression of Mrp3 (Maher et al., 2005a). Furthermore, with a different regimen of clofibrate dosing, mRNA and protein expression of Mrp3 and Mrp4 was increased in a PPARα-dependent manner (Moffit et al., 2006). Taken together, these observations suggest that PPARα is involved in regulation of these two transporters, but currently a direct role for PPARα in their transcriptional activation has yet to be proven. Thus, the purpose of this study is to determine whether Mrp transporter expression is increased during peroxisome proliferation and to determine the involvement of two transcription factors, namely PPARα and Nrf2, in mediating the regulation of these efflux transporters.
MATERIALS AND METHODS
Chemicals.
PFDA, PFOA, gadolinium chloride, and propylene glycol were purchased from Sigma-Aldrich (St Louis, MO). All chemicals were of the highest grade available (≥99.0%). PFCAs were dissolved in a propylene glycol:H20 mixture (1:1) for dosing. Antibodies for Mrp3 and Mrp4 were gifts from George Scheffer (Vrije Universiteit Medical Center, Amsterdam).
Mice.
Male C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA). The mice utilized were age matched from 8 to 10 weeks of age. All mice were maintained on automatically timed 12-h dark/light cycles in an American Animal Associations Laboratory Animal Care (AAALAC)–accredited facility at the University of Kansas Medical Center and allowed water and rodent chow ad libitum (Teklad; Harlan, Indianapolis, IN). Wild-type (WT) and Nrf2-null mice on a C57BL/6 background were bred and housed at this facility as well (Maher et al., 2007). PPARα-null mice on a C57BL6 background were housed and bred in an AAALAC-accredited facility at Penn State (Akiyama et al., 2001; Lee et al., 1995). All protocols were approved by the Institutional Animal Care and Use Committees of the respective institutions. C57BL/6 mice were administered ip (dosing volume of 5 ml/kg) several different doses ranging from 0 to 80 mg/kg of PFDA. As 80 mg/kg of PFDA elicited the highest induction of Mrp3 and Mrp4, the 80 mg/kg dosing level of PFDA or PFOA was used for all further experiments. A propylene glycol:H20 mixture (1:1) was utilized as the vehicle and as the vehicle control, with PFDA/PFOA dissolved into solution by stirring.
There was one notable exception—in the WT and PPARα-null mice study, the dose was lowered to 40 mg/kg due to some observed lethality at 80 mg/kg in PPARα-null mice before the 48-h time point.
Tissue preparation and RNA isolation.
Livers were harvested 48 h after PFCA dosing, rinsed in isotonic saline solution, weighed, and snap-frozen in liquid nitrogen, and stored at −80°C. Mice were anesthetized by pentobarbital overdose, serum was collected by cardiac puncture, and plasma was collected using Microtainer serum separator tubes (BD Biosciences, San Jose, CA). Tissue slices specific for immunohistochemistry were first placed in isopentane cooled on dry ice and placed in −80° storage.
Total RNA was isolated using RNA-Bee reagent (Tel-Test, Friendswood, TX) according to the manufacturer's protocol. The concentration of total RNA in each sample was quantified spectrophotometrically at 260 nm. The integrity of each RNA sample was evaluated by formaldehyde-agarose gel electrophoresis before analysis.
Branched DNA signal amplification assay.
Mouse Mrp transcripts were quantified using the branched signal amplification assay as previously described (QuantiGene, High Volume bDNA Signal Amplification Kit; Panomics, Fremont, CA; Maher et al., 2005b).
Alanine aminotransferase activity.
Plasma alanine aminotransferase (ALT) activity was determined as a biochemical indicator of hepatocellular injury using Infinity ALT Liquid Stable Reagent (Thermotrace, Melbourne, Australia) according to the manufacturer's protocol.
Direct bilirubin and bile acid assays.
Direct bilirubin was quantified by colorimetric assay using protocol 0245 from the Direct Bilirubin kit (Stanbio, Boerne, TX). Total bile acids were quantified using Procedure #450 from the total bile acid kit from Trinity Biotech (Wicklow, Ireland). All samples were quantified in a 96-well format at 540 and 530nM, respectively, for the two assays.
Preparation of crude liver membranes.
To analyze Mrp protein expression, crude membrane fractions were isolated from liver based on a previously described method (Trauner et al., 1997). Livers from mice were removed, placed in four volumes of ice-cold homogenization buffer (10mM Tris, 250mM sucrose, pH 7.4, containing 50mM phenylmethylsulfonyl fluoride), and homogenized by 20 up-down strokes with a dounce homogenizer. The homogenate was centrifuged at 100,000 × g at 4°C for 1 h. The resulting pellet was resuspended in 0.3mM sucrose, 20mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, pH 7.4, containing 50mM phenylmethylsulfonyl fluoride. Protein concentration of liver crude membrane fractions was determined with the bicinchoninic acid Protein Assay Reagent Kit (Pierce, Rockford, IL) as described by the manufacturer.
Western blotting.
Liver membrane preparations containing 60 μg of total protein were loaded per well and separated on 8% SDS-polyacrylamide gels. Proteins were transferred overnight (30 V) at 4°C to polyvinylidene difluoride membranes. Blots were blocked with 1% nonfat dry milk (NFDM) for 1 h in Tris-buffered saline with 0.5% Tween 20 (TBS-T), incubated for 2 h at room temperature in 1% NFDM-TBS-T containing a 1:2000 dilution of the primary antibodies (Mrp3:M3II-2; Mrp4:M4I-10) and washed three times in TBS-T. Blots were subsequently incubated in anti-rat secondary antibody (1:2000) in 1% NFDM-TBS-T (Sigma-Aldrich). Each blot was incubated in the enhanced chemiluminescence substrate reagent for horseradish peroxidase (Amersham Biosciences, Piscataway, NJ), and immunopositive interactions were visualized by exposure of the blot to X-ray film (Fuji Photo Film, Japan). Intensity of protein bands was quantified using the Discovery Series Quantity One 1-D Analysis software (Bio-Rad Laboratories, Hercules, CA). Blots were stripped and reprobed for β-actin (ab8227; Abcam, Cambridge, UK) for use as an internal loading control.
Immunofluorescence staining and confocal laser scanning microscopy.
Livers were embedded in optimal cutting temperature compound and brought to −20°C. Cryosections (5 μm) were thaw-mounted onto Superfrost glass slides (Fisher Scientific, Pittsburgh, PA) and stored at −80°C with desiccant until use. Tissue sections were fixed with 4% paraformaldehyde for 5 min. All antibody solutions were filtered through 0.22-micron membrane syringe-driven filter units (Osmonics, Minnetonka, MN) prior to use. Sections were blocked with 5% goat serum/PBS with 0.1% Triton X (PBS-Tx) for 1 h and then incubated with M3II-2 or M4I-10 primary antibody diluted 1:100 in 5% goat serum/PBS-Tx for 2 h at room temperature. After incubation, the sections were washed three times in PBS-Tx and incubated for 1 h with goat anti-rat IgG Alexa 488 IgG (Invitrogen Corporation, Carlsbad, CA) diluted 1:200 in 5% goat serum/PBS-Tx. Slides were washed in PBS and then rinsed twice in distilled deionized water. The sections were air dried and mounted in Prolong Gold (Invitrogen Corporation). Images were acquired on a Leica SP2 Spectral Confocal Microscope (Leica Microsystems, Exton, PA) equipped with an argon laser, which allowed excitation at 488-nm wavelength for detection of Alexa 488. Images were pseudocolored red. Negative control staining was performed by incubating sections without primary antibody.
Gadolinium chloride depletion of macrophages.
Mice were pretreated with either an intravenous tail vein injection of saline or gadolinium chloride (20 mg/kg; iv) 6 h before treatment with PFDA (80 mg/kg) or 50% propylene glycol vehicle. At 48 h after dosing with vehicle or PFDA, livers from all mice were removed and processed according to the above specifications. Tumor necrosis factor-α (TNF-α) levels were measured in serum and liver to assess the effectiveness of GdCl3 in depleting and/or inactivating liver macrophages. TNF-α concentrations were quantified by sandwich antibody enzyme-linked immunosorbent assay (R&D systems; Minneapolis, MN).
Statistics.
For differences between two groups, a two-tailed t-test with significance set at p < 0.05 was utilized. For comparisons between multiple groups, data were analyzed by one-way ANOVA, followed by Tukey's post hoc test. Bars represent SEM.
RESULTS
Hepatic Mrp Induction in Response to PFDA and PFOA Treatment
A single ip dose of 80 mg/kg PFDA induced Mrp3 mRNA by fourfold and Mrp4 mRNA by 31-fold at 48 h (Fig. 1). A single ip dose of 80 mg/kg PFOA induced expression of Mrp3 to virtually the same extent as PFDA, with lower, but still marked induction of Mrp4.
FIG. 1.

Mrps are induced by various PFCAs. Expression of Mrp3 and Mrp4 mRNA in liver 48 h after treatment with vehicle or 80 mg/kg PFDA or PFOA. Total RNA from livers of chemically treated male C57BL/6 mice (n = 5 per treatment) was analyzed by the branched DNA assay. Data are presented as mean relative light units (RLU) ± SEM. Asterisks (*) represent statistically significant differences between vehicle- or PFDA-treated mice (p < 0.05).
PFDA Induces Mrp3 and Mrp4 over a Wide Range of Doses
PFDA was administered ip to mice (n = 5 per group) at 0.25, 0.50, 1, 10, 20, 40, and 80 mg/kg. PFDA statistically increased the liver/body weight ratios at every dose tested (0.25–80 mg/kg; Fig. 2A). Mice treated with only vehicle had liver/body weight ratios of 4.8%, and at the highest dose of PFDA (80 mg/kg), that percentage increased to 8.3%, nearly doubling the relative liver weight. At 48 h, PFDA increased mRNA expression of the PPARα target gene Cyp4a14 at every dose (0.25–80 mg/kg; Fig. 2B). Mrp3 and Mrp4 were not induced at lower doses, with the lowest statistically significant induction occurring at 10 and 20 mg/kg, respectively.
FIG. 2.

Mrp transporter expression is induced over a range of PFDA doses. (A) Corresponding increases in the liver-to-body weight ratio over the same dosing range. Data are presented as a percentage liver weight to total body weight. (B) Hepatic Mrp3, Mrp4, and Cyp4a14 mRNA expression was quantified from mouse livers 48 h after PFDA dosages ranging from 0 to 80 mg/kg. Data are presented as mean relative light units (RLU) ± SEM. Asterisks (*) represent statistically significant differences between vehicle- and PFDA-treated mice (p < 0.05).
Zonal Expression of Mrp3 and Mrp4 in WT Mice after PFDA Treatment
Immunohistochemical localization of basolateral transporters Mrp3 and Mrp4 was examined in frozen liver sections from mice administered vehicle or 80 mg/kg PFDA (Fig. 3). Mrp3 exhibited basolateral staining throughout the liver in untreated sections (periportal to centrilobular regions), with significant induction in all zones observed after treatment with PFDA. Mrp3 staining tended to be higher in centrilobular hepatocytes, with expression decreasing in intensity from the central vein outward to the portal vein, as observed previously (Aleksunes et al., 2005). Minimal Mrp4 expression was detected in livers from mice treated with vehicle. However, tremendous induction of Mrp4 was observed in centrilobular hepatocytes after PFDA exposure. Intense basolateral staining occurred in zone 3 hepatocytes in the areas immediately surrounding the central vein that radiates 5–10 cells deep, with other regions of the liver exhibiting minimal expression.
FIG. 3.

Mrp localization and induction after PFDA exposure. Immunofluorescent staining of murine Mrp3 and Mrp4 from 5-μm frozen liver sections 48 h after administration of either vehicle or PFDA (80 mg/kg). Images of Mrp3 and Mrp4 staining in centrilobular regions were obtained at ×40 magnification and pseudocolored red.
Parallel Increases in Serum-conjugated Bilirubin and Bile Acids after PFDA Administration
Mrp3-null mice have decreased serum levels of conjugated bilirubin after bile duct ligation, thus demonstrating that Mrp3 aids in the maintenance of serum bilirubin levels by transporting bile acids from liver back into the plasma (Zelcer et al., 2006). Mrp4 transports conjugated bile acids in a similar manner, with Km values in the low micromolar range (Rius et al., 2006). Elevated serum-conjugated bilirubin and bile acids would be expected with increased Mrp3 and Mrp4 expression and a vectoral shift from biliary to sinusoidal export. Consistent with this notion, a 3.5- and 4-fold increase in serum levels of conjugated bilirubin and bile acids, respectively, were observed 48 h after PFDA administration (Fig. 4). Thus, elevations in basolateral Mrp expression correspond with enhanced hepatic efflux of conjugated endogenous Mrp substrates after PFDA administration.
FIG. 4.

PFDA increases serum levels of representative substrates for the basolateral efflux transporters Mrp3 and Mrp4. Conjugated bilirubin and bile acid levels in mice treated with vehicle or 80 mg/kg PFDA were quantified. Asterisks (*) represent a statistically significant difference between vehicle- and PFDA-treated mice (p < 0.05).
PPARα and Nrf2 Marker Gene Induction after PFDA Exposure
Perhaps, the most well-characterized Nrf2 target gene is the quinone-reducing enzyme Nqo1, which is often used as a marker for Nrf2 activation (Maher et al., 2007). Markers of oxidative stress and H2O2 have been demonstrated after PFDA administration in previous studies, with clear increases in H2O2 generation and lipid peroxidation (Takagi et al., 1991). After PFDA exposure to WT mice, hepatic Nqo1 mRNA expression increases; yet this response was attenuated in Nrf2-null mice (Fig. 5). Conversely, no attenuation of Nqo1 induction by PFDA was observed in PPARα-null mice. These data suggest that PFDA activates Nrf2, and in agreement with previous reports, oxidative stress is likely the cause of Nrf2 activation and the subsequent Nqo1 induction.
FIG. 5.

PFDA activates both the PPARα and the Nrf2 pathways. Representative target genes for PPARα and Nrf2, Cyp4a14, and Nqo1, respectively, were quantified in WT, Nrf2-null, or PPARα-null mice. Data are presented as mean relative light units (RLU) ± SEM. Pound signs (#) represent statistically lower expression in null mice in either the vehicle- or the PFDA-treated groups (p < 0.05).
Cyp4a14 is known to be regulated by PPARα (Sterchele et al., 1996), and a modest difference in basal Cyp4a14 expression in liver was observed between WT and PPARα-null mice. After PFDA exposure, expression of Cyp4a14 increases approximately 26-fold in WT mice, and this effect was not observed in PPARα-null mice (Fig. 5). Conversely, no attenuation of Cyp4a14 induction was observed in Nrf2-null mice. Thus, PFDA activates the PPARα pathway, subsequently leading to PPARα-mediated induction of Cyp4a14.
Nrf2 and PPARα Aid in the Induction of Hepatic Mrp Transporters after PFDA Administration
To examine the effects of Nrf2 disruption on Mrp3 and Mrp4 after PFDA, WT and Nrf2-null mice were administered a single dose of PFDA (80 mg/kg) and mRNA expression was examined in livers at 48 h. Mrp3 (2.5-fold) and Mrp4 (31-fold) were induced by PFDA in WT mice (Fig. 6). In Nrf2-null mice, Mrp3 and Mrp4 induction, while not completely absolved, was significantly attenuated.
FIG. 6.

PPARα and Nrf2 both play a role in hepatic Mrp regulation after PFDA exposure. Expression of Mrp3 and Mrp4 mRNA was quantified in WT, Nrf2-, or PPARα-null mice treated with vehicle or PFDA in liver 48 h after PFDA administration. Data are presented as mean relative light units (RLU) ± SEM. Pound signs (#) represent statistically lower expression in null mice in either the vehicle- or the PFDA-treated groups (p < 0.05).
PFDA induction of Mrps mRNA in PPARα-null mice was not as robust as observed in other treatment sets, primarily due to a decrease in the dose of PFDA from 80 to 40 mg/kg to avoid toxicity in PPARα-null mice. However, Mrp3 (fourfold) and Mrp4 (sevenfold) were still markedly increased by PFDA in WT mice, but this induction was significantly attenuated in PPARα-null mice (Fig. 6).
Mrp3 and Mrp4 proteins were both markedly induced by PFDA in crude membrane fractions (3- and 15-fold, respectively) and to nearly the same extent as seen at the mRNA level (Fig. 7). Induction of Mrp3 and Mrp4 protein was significantly attenuated in both Nrf2-null and PPARα-null mice, and thus Nrf2 and PPARα appear to contribute to overall Mrp3 and Mrp4 regulation.
FIG. 7.

PPARα and Nrf2 alter hepatic Mrp protein expression. Western immunoblots for Mrp3 and Mrp4 were performed using liver membrane fractions (60 μg protein/lane) from WT, Nrf2-null, and PPARα-null mice 48 h following treatment with PFDA (80 mg/kg) or vehicle. Data are presented as individual blots (A) and as mean relative protein expression ± SE (B). Equal protein loading was confirmed by detection of β-actin. Pound signs (#) represent statistically lower expression in the null mice in either the vehicle- or the PFDA-treated groups (p < 0.05).
Gadolinium Chloride Depletion of Macrophages Leads to Minor Changes in Mrp Expression
Kupffer cell release of cytokines and inflammatory mediators is a known means of stimulation and cell signaling in several hepatic disease models, such as cholestasis and fibrosis, with the most common mechanism being TNF-α–mediated inflammation and apoptosis. Serum and hepatic TNF-α levels were utilized as a marker for Kupffer cell activation. Previous investigations have reported more than eightfold induction of TNF-α at 24-h postadministration with PFDA and PFOA at doses within the range used in the current study (Adinehzadeh and Reo, 1998). However, we noted only a twofold induction in both serum and liver 48 h after PFDA administration (Fig. 8A). Gadolinium chloride has been used extensively to inactivate or deplete Kupffer cell function in liver (Husztik et al., 1980). GdCl3 was fully successful in tempering PFDA-mediated TNF-α induction, suggesting that GdCl3-mediated inactivation or destruction of Kupffer cells was effective. Gadolinium chloride pretreatment (20 mg/kg; iv) decreased the induction of Mrp4 after PFDA administration by approximately 30% (Fig. 8B). In contrast, Mrp3 mRNA expression was unaffected by GdCl3 pretreatment. Thus, Kupffer cell activation has no influence on Mrp3 levels, yet the decrease in Mrp4 expression suggests that Mrp4 induction by PFDA occurs in part through a Kupffer cell–mediated cytokine or oxidative stress response.
FIG. 8.
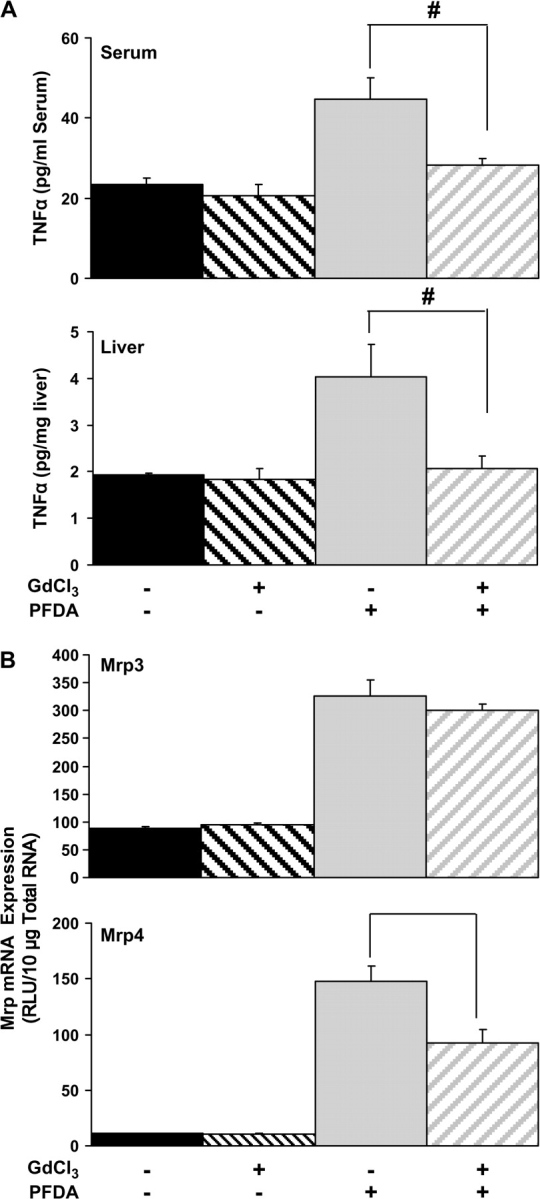
Mrp transporter expression after PFDA administration in a GdCl3 model of Kupffer cell depletion. (A) Serum and liver TNF-α cytokine levels were measured by ELISA in vehicle- or PFDA-treated mice pretreated with saline or GdCl3 (20 mg/kg; iv). TNF-α levels were utilized as confirmation that Kupffer cell activation occurred after PFDA administration and that inactivation of Kupffer cells occurred with GdCl3 pretreatment. Pound signs (#) represent statistically lower expression in GdCl3-treated mice as compared with saline-treated controls. (B) Mrp3 and Mrp4 mRNA levels were quantified in livers from vehicle- or PFDA-treated mice pretreated with saline or GdCl3 (iv).
DISCUSSION
In the current study, marked changes in basolateral efflux transporter expression are observed after PFDA and PFOA exposure. This induction pattern of Mrp3 and Mrp4 is similar, yet distinct from those observed during carbon tetrachloride (CCl4) and acetaminophen-induced liver injury (Aleksunes et al., 2005). For example, the hepatocyte degeneration and necrosis and the increases in serum ALT activity that occurs subsequently to CCl4 and acetaminophen administration are not observed with PFDA (plasma ALT activity for vehicle 14.5 ± 1.6, PFDA 23.82 ± 2.9, and CCl4 3306.1 ± 809 units/l). Furthermore, CCl4 and acetaminophen are bioactivated to reactive intermediates, whereas PFDA is minimally biotransformed (Vanden Heuvel et al., 1991). In cholestatic liver disease models, Mrp3 and Mrp4 induction is often observed, presumably serving to protect the liver from accumulation of hydrophobic bile acids, bilirubin, and other potentially toxic endogenous and exogenous chemicals (Donner and Keppler, 2001; Wagner et al., 2003; Zollner et al., 2003). Taken together, Mrp3 and Mrp4 induction may be an adaptive response necessary for increasing basolateral excretion due to PFDA-induced toxicity or cholestasis.
In the current study, marked changes in Mrp3 and Mrp4 expression correlated well with increased serum-conjugated bilirubin and bile acid levels (Fig. 4). Although changes in metabolism or synthesis cannot be fully discounted, there are several precedents that suggest a functional correlation between changes in disposition of conjugated bile acids and bilirubin levels and induction of Mrp proteins. In vivo cholestasis models suggest that Mrps serve as low-affinity, high-capacity transporters that are inducible when the liver is damaged and/or canalicular efflux is compromised to protect the hepatocyte from endo- and xenobiotics (Belinsky et al., 2005). This can be readily observed after bile duct ligation of Mrp3-null mice, where serum-conjugated bile acids and bilirubin were lower in sera compared with WT with increased retention of bile acids and other normal biliary constituents in liver (Belinsky et al., 2005). Mrp4 can transport bile acids with high affinity in vitro, and its expression is markedly increased during cholestasis (Donner et al., 2004; Maher et al., 2005b; Rius et al., 2003; Zelcer et al., 2003). Similarly, Mrp3 and Mrp4 are thought to transport many other endogenous substrates, including nucleotides and arachidonic acid metabolites, such as leukotrienes and prostaglandins (Jedlitschky et al., 1996; Reid et al., 2003; Rius et al., 2003). Furthermore, this induction may serve to rid the cell of lipid peroxidation products or damaged nucleotides, such as 4-hydroxynonenal or 8-hydroxy-deoxyguanosine (Reichard et al., 2003; Renes et al., 2000).
The mechanisms of how PFCAs are transported and distributed in the body are currently unknown. Besides modifying the expression of Mrp3 and Mrp4, it is plausible that PFCAs could serve as competitive inhibitors for transporters. Because increased expression of Mrp3 and Mrp4 is observed along with increases in serum bile acids and bilirubin, PFCAs may serve as competitive inhibitors of biliary transport as the resultant condition resembles the genetic and biochemical changes indicative of cholestasis. Further exploration of the nature of PFCA transport may aid in the broader picture of the effects of PFCAs on toxicokinetics.
PPARα, a critical transcriptional mediator of the pleotropic response during hepatic peroxisome proliferation, also plays an important role in regulating basolateral Mrp3 and Mrp4 expression in liver. Even at doses as low as 0.25 mg/kg body weight, PFDA induced expression of the PPARα target gene Cyp4a14 (Fig. 2). Induction of Mrp transporters was statistically significant at higher PFDA concentrations of 10 mg/kg for Mrp3 and 20 mg/kg for Mrp4. The differing thresholds for induction between Cyp4a14 and Mrp3/Mrp4 are not completely clear but could involve (1) differences in promoter access or chromatin remodeling, (2) a need for gene-dependent transcriptional cofactors, or (3) a subsequent, secondary regulation of Mrps by PPARα-independent transcription factors. However, induction of Mrp3 and Mrp4 after PFDA administration was markedly attenuated in PPARα-null mice, demonstrating that PPARα is required in some capacity for induction of both Mrps by PFDA (Fig. 7). The presence of several direct repeat-1 elements in the Mrp3 and Mrp4 5′ regulatory regions (Jonathan M. Maher, Lauren M. Aleksunes, Matthew Z. Dieter, Yuji Tanaka, Jeffrey M. Peters, Jose E. Manautou, and Curtis D. Klaassen, unpublished data) generally supports the idea that PPARα can directly regulate these two Mrp transporters.
Currently, the mechanism of Nrf2 activation by PFDA is unknown. One recent report suggests that oxidized fatty acids (n-3) can lead to direct Nrf2 activation, suggesting that Nrf2 may be directly activated by PFDA (Gao et al., 2007). Alternatively, activation of Nrf2 could be due to PPARα-dependent activation of drug-metabolizing enzymes, leading to secondary changes in oxidative or inflammatory conditions that could result in Nrf2 activation. Antioxidant and redox signaling pathways could be responding to marked alterations in hepatic lipid chemistry, increases in H2O2, or a variety of other physiological parameters that occur during PP. Interestingly, both Nrf2 and PPARα share intrinsic anti-inflammatory activities and overlapping but distinct profiles of downstream gene expression (Anderson et al., 2004; Hill et al., 1999). Both PPARα and Nrf2 have potential regulatory elements in both Mrp gene promoter regions, and Nrf2 has been shown to bind the regulatory regions of the Mrp3 and Mrp4 genes via 5′ antioxidant response elements during Nrf2 activation (Hayashi et al., 2003; Maher et al., 2007; Vollrath et al., 2006).
Resident liver macrophages, or Kupffer cells, are an important source of pro-inflammatory cytokines, hydrogen peroxide, and superoxide radicals (Higuchi et al., 1990). Eightfold increases in serum TNF-α levels have been observed in rats after PFDA exposure. However, only modest TNF-α induction (approximately twofold) in serum and liver by PFDA treatment was observed in the current experiments. Kupffer cell inactivation by GdCl3 pretreatment (Frankenberg et al., 2005) leads to decreased levels of inflammatory cytokines, such as TNF-α, as reported here with PFDA (Fig. 8). This suggests that macrophages may aid in the PFDA-mediated transcriptional changes in hepatocytes via inflammatory or oxidative mediators, which may occur through PPARα- and Nrf2-mediated pathways or through other cytokine signaling pathways.
PFDA and PFOA have a marked effect on hepatic basolateral transporters, likely resulting in functional increases in serum levels of bilirubin and bile acids via a compensatory hepatoprotective mechanism, which is similar to the response observed after CCl4 and acetaminophen exposure (Aleksunes et al., 2006, 2008). Induction of Mrp3 and Mrp4 in response to PFDA is clearly multifactorial and involves signaling from both PPARα and Nrf2. The current study utilizes pharmacologically exaggerated, acute dosing in the typical ranges for experimental purposes. Thus, the implications to humans, which typically endure much lower exposures and respond differently to peroxisomal proliferators than rodents, are unclear. However, PFDA has “high toxic potency” for a fluorohydrocarbon (Olson and Andersen, 1983) and leads to dramatic wasting at doses greater than 40 mg/kg. With strong mechanistic activation of lipid and redox signaling pathways at these doses, marked induction of Mrps may be part of the toxicological or the adaptive response to fluorohydrocarbons, with these efflux transporters having the ability to markedly alter the disposition of xenobiotic or endogenous substrates.
Acknowledgments
The authors thank Drs. Xingguo Cheng, Angela Slitt, and David Buckley for their contributions to this manuscript. National Institutes of Health grant numbers (ES013714, ES09649, ES09716, and RR021940).
References
- Abdellatif AG, Preat V, Taper HS, Roberfroid M. The modulation of rat liver carcinogenesis by perfluorooctanoic acid, a peroxisome proliferator. Toxicol. Appl. Pharmacol. 1991;111:530–537. doi: 10.1016/0041-008x(91)90257-f. [DOI] [PubMed] [Google Scholar]
- Adinehzadeh M, Reo NV. Effects of peroxisome proliferators on rat liver phospholipids: Sphingomyelin degradation may be involved in hepatotoxic mechanism of perfluorodecanoic acid. Chem. Res. Toxicol. 1998;11:428–440. doi: 10.1021/tx970155t. [DOI] [PubMed] [Google Scholar]
- Adinehzadeh M, Reo NV, Jarnot BM, Taylor CA, Mattie DR. Dose-response hepatotoxicity of the peroxisome proliferator, perfluorodecanoic acid and the relationship to phospholipid metabolism in rats. Toxicology. 1999;134:179–195. doi: 10.1016/s0300-483x(99)00038-4. [DOI] [PubMed] [Google Scholar]
- Akiyama TE, Nicol CJ, Fievet C, Staels B, Ward JM, Auwerx J, Lee SS, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-alpha regulates lipid homeostasis, but is not associated with obesity: Studies with congenic mouse lines. J. Biol. Chem. 2001;276:39088–39093. doi: 10.1074/jbc.M107073200. [DOI] [PubMed] [Google Scholar]
- Aleksunes LM, Scheffer GL, Jakowski AB, Pruimboom-Brees IM, Manautou JE. Coordinated expression of multidrug resistance-associated proteins (Mrps) in mouse liver during toxicant-induced injury. Toxicol. Sci. 2006;89:370–379. doi: 10.1093/toxsci/kfi332. [DOI] [PubMed] [Google Scholar]
- Aleksunes LM, Slitt AL, Maher JM, Augustine LM, Goedken MJ, Chan JY, Cherrington NJ, Klaassen CD, Manautou JE. Induction of Mrp3 and Mrp4 transporters during acetaminophen hepatotoxicity is dependent on Nrf2. Toxicol. Appl. Pharmacol. 2008;226:74–83. doi: 10.1016/j.taap.2007.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksunes LM, Slitt AM, Cherrington NJ, Thibodeau MS, Klaassen CD, Manautou JE. Differential expression of mouse hepatic transporter genes in response to acetaminophen and carbon tetrachloride. Toxicol. Sci. 2005;83:44–52. doi: 10.1093/toxsci/kfi013. [DOI] [PubMed] [Google Scholar]
- Anderson SP, Howroyd P, Liu J, Qian X, Bahnemann R, Swanson C, Kwak MK, Kensler TW, Corton JC. The transcriptional response to a peroxisome proliferator-activated receptor alpha agonist includes increased expression of proteome maintenance genes. J. Biol. Chem. 2004;279:52390–52398. doi: 10.1074/jbc.M409347200. [DOI] [PubMed] [Google Scholar]
- Belinsky MG, Dawson PA, Shchaveleva I, Bain LJ, Wang R, Ling V, Chen ZS, Grinberg A, Westphal H, Klein-Szanto A, et al. Analysis of the in vivo functions of Mrp3. Mol. Pharmacol. 2005;68:160–168. doi: 10.1124/mol.104.010587. [DOI] [PubMed] [Google Scholar]
- Borges T, Robertson LW, Peterson RE, Glauert HP. Dose-related effects of perfluorodecanoic acid on growth, feed intake and hepatic peroxisomal beta-oxidation. Arch. Toxicol. 1992;66:18–22. doi: 10.1007/BF02307265. [DOI] [PubMed] [Google Scholar]
- Brewster DW, Birnbaum LS. The biochemical toxicity of perfluorodecanoic acid in the mouse is different from that of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol. 1989;99:544–554. doi: 10.1016/0041-008x(89)90161-0. [DOI] [PubMed] [Google Scholar]
- Burris J, Lundberg JK, Olsen G, Simpson C, Mandel J. Determination of serum half-lives of several fluorochemicals. 2002 Interim Report #2 3M Company, Corporate Occupational Medicine Department, US EPA AR226-1086. [Google Scholar]
- Calafat AM, Wong LY, Kuklenyik Z, Reidy JA, Needham LL. Polyfluoroalkyl chemicals in the U.S. population: Data from the National Health and Nutrition Examination Survey (NHANES) 2003–2004 and comparisons with NHANES 1999–2000. Environ. Health Perspect. 2007;115:1596–1602. doi: 10.1289/ehp.10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LC, Tatum V, Glauert HP, Chow CK. Peroxisome proliferator perfluorodecanoic acid alters glutathione and related enzymes. J. Biochem. Mol. Toxicol. 2001;15:107–113. doi: 10.1002/jbt.6. [DOI] [PubMed] [Google Scholar]
- Chinje E, Kentish P, Jarnot B, George M, Gibson G. Induction of the CYP4A subfamily by perfluorodecanoic acid: The rat and the guinea pig as susceptible and non-susceptible species. Toxicol. Lett. 1994;71:69–75. doi: 10.1016/0378-4274(94)90200-3. [DOI] [PubMed] [Google Scholar]
- Daniel V, Sharon R, Bensimon A. Regulatory elements controlling the basal and drug-inducible expression of glutathione S-transferase Ya subunit gene. DNA. 1989;8:399–408. doi: 10.1089/dna.1.1989.8.399. [DOI] [PubMed] [Google Scholar]
- Donner MG, Keppler D. Up-regulation of basolateral multidrug resistance protein 3 (Mrp3) in cholestatic rat liver. Hepatology. 2001;34:351–359. doi: 10.1053/jhep.2001.26213. [DOI] [PubMed] [Google Scholar]
- Donner MG, Warskulat U, Saha N, Haussinger D. Enhanced expression of basolateral multidrug resistance protein isoforms Mrp3 and Mrp5 in rat liver by LPS. Biol. Chem. 2004;385:331–339. doi: 10.1515/BC.2004.029. [DOI] [PubMed] [Google Scholar]
- Frankenberg MV, Weimann J, Fritz S, Fiedler J, Mehrabi A, Buchler MW, Kraus TW. Gadolinium chloride-induced improvement of postischemic hepatic perfusion after warm ischemia is associated with reduced hepatic endothelin secretion. Transpl. Int. 2005;18:429–436. doi: 10.1111/j.1432-2277.2004.00058.x. [DOI] [PubMed] [Google Scholar]
- Gao L, Wang J, Sekhar KR, Yin H, Yared NF, Schneider SN, Sasi S, Dalton TP, Anderson ME, Chan JY, et al. Novel n-3 fatty acid oxidation products activate Nrf2 by destabilizing the association between Keap1 and Cullin3. J. Biol. Chem. 2007;282:2529–2537. doi: 10.1074/jbc.M607622200. [DOI] [PubMed] [Google Scholar]
- George ME, Andersen ME. Toxic effects of nonadecafluoro-n-decanoic acid in rats. Toxicol. Appl. Pharmacol. 1986;85:169–180. doi: 10.1016/0041-008x(86)90110-9. [DOI] [PubMed] [Google Scholar]
- Glauert HP, Srinivasan S, Tatum VL, Chen LC, Saxon DM, Lay LT, Borges T, Baker M, Chen LH, Robertson LW, et al. Effects of the peroxisome proliferators ciprofibrate and perfluorodecanoic acid on hepatic cellular antioxidants and lipid peroxidation in rats. Biochem. Pharmacol. 1992;43:1353–1359. doi: 10.1016/0006-2952(92)90513-i. [DOI] [PubMed] [Google Scholar]
- Goecke CM, Jarnot BM, Reo NV. A comparative toxicological investigation of perfluorocarboxylic acids in rats by fluorine-19 NMR spectroscopy. Chem. Res. Toxicol. 1992;5:512–519. doi: 10.1021/tx00028a009. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ. The peroxisome proliferator-activated receptor alpha (PPARalpha): Role in hepatocarcinogenesis. Mol. Cell. Endocrinol. 2002;193:71–79. doi: 10.1016/s0303-7207(02)00098-9. [DOI] [PubMed] [Google Scholar]
- Harris MW, Birnbaum LS. Developmental toxicity of perfluorodecanoic acid in C57BL/6N mice. Fundam. Appl. Toxicol. 1989;12:442–448. doi: 10.1016/0272-0590(89)90018-3. [DOI] [PubMed] [Google Scholar]
- Harris MW, Uraih LC, Birnbaum LS. Acute toxicity of perfluorodecanoic acid in C57BL/6 mice differs from 2,3,7,8-tetrachlorodibenzo-p-dioxin. Fundam. Appl. Toxicol. 1989;13:723–736. doi: 10.1016/0272-0590(89)90330-8. [DOI] [PubMed] [Google Scholar]
- Hayashi A, Suzuki H, Itoh K, Yamamoto M, Sugiyama Y. Transcription factor Nrf2 is required for the constitutive and inducible expression of multidrug resistance-associated protein 1 in mouse embryo fibroblasts. Biochem. Biophys. Res. Commun. 2003;310:824–829. doi: 10.1016/j.bbrc.2003.09.086. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Higashi N, Taki H, Osawa T. Cytolytic mechanisms of activated macrophages. Tumor necrosis factor and L-arginine-dependent mechanisms act synergistically as the major cytolytic mechanisms of activated macrophages. J. Immunol. 1990;144:1425–1431. [PubMed] [Google Scholar]
- Hill MR, Clarke S, Rodgers K, Thornhill B, Peters JM, Gonzalez FJ, Gimble JM. Effect of peroxisome proliferator-activated receptor alpha activators on tumor necrosis factor expression in mice during endotoxemia. Infect. Immun. 1999;67:3488–3493. doi: 10.1128/iai.67.7.3488-3493.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husztik E, Lazar G, Parducz A. Electron microscopic study of Kupffer-cell phagocytosis blockade induced by gadolinium chloride. Br. J. Exp. Pathol. 1980;61:624–630. [PMC free article] [PubMed] [Google Scholar]
- Jaiswal AK. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic. Biol. Med. 2000;29:254–262. doi: 10.1016/s0891-5849(00)00306-3. [DOI] [PubMed] [Google Scholar]
- Jedlitschky G, Leier I, Buchholz U, Barnouin K, Kurz G, Keppler D. Transport of glutathione, glucuronate, and sulfate conjugates by the MRP gene-encoded conjugate export pump. Cancer Res. 1996;56:988–994. [PubMed] [Google Scholar]
- Kudo N, Kawashima Y. Fish oil-feeding prevents perfluorooctanoic acid-induced fatty liver in mice. Toxicol. Appl. Pharmacol. 1997;145:285–293. doi: 10.1006/taap.1997.8186. [DOI] [PubMed] [Google Scholar]
- Kudo N, Mizuguchi H, Yamamoto A, Kawashima Y. Alterations by perfluorooctanoic acid of glycerolipid metabolism in rat liver. Chem. Biol. Interact. 1999;118:69–83. doi: 10.1016/s0009-2797(99)00002-2. [DOI] [PubMed] [Google Scholar]
- Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol. Cell. Biol. 1995;15:3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher JM, Cheng X, Slitt AL, Dieter MZ, Klaassen CD. Induction of the multidrug resistance-associated protein family of transporters by chemical activators of receptor-mediated pathways in mouse liver. Drug Metab. Dispos. 2005a;33:956–962. doi: 10.1124/dmd.105.003798. [DOI] [PubMed] [Google Scholar]
- Maher JM, Dieter MZ, Aleksunes LM, Slitt AL, Guo G, Tanaka Y, Scheffer GL, Chan JY, Manautou JE, Chen Y, et al. Oxidative and electrophilic stress induces multidrug resistance-associated protein transporters via the nuclear factor-E2-related factor-2 transcriptional pathway. Hepatology. 2007;46:1597–1610. doi: 10.1002/hep.21831. [DOI] [PubMed] [Google Scholar]
- Maher JM, Slitt AL, Cherrington NJ, Cheng X, Klaassen CD. Tissue distribution and hepatic and renal ontogeny of the multidrug resistance-associated protein (Mrp) family in mice. Drug Metab. Dispos. 2005b;33:947–955. doi: 10.1124/dmd.105.003780. [DOI] [PubMed] [Google Scholar]
- Moffit JS, Aleksunes LM, Maher JM, Scheffer GL, Klaassen CD, Manautou JE. Induction of hepatic transporters multidrug resistance-associated proteins (Mrp) 3 and 4 by clofibrate is regulated by peroxisome proliferator-activated receptor alpha. J. Pharmacol. Exp. Ther. 2006;317:537–545. doi: 10.1124/jpet.105.093765. [DOI] [PubMed] [Google Scholar]
- Olson CT, Andersen ME. The acute toxicity of perfluorooctanoic and perfluorodecanoic acids in male rats and effects on tissue fatty acids. Toxicol. Appl. Pharmacol. 1983;70:362–372. doi: 10.1016/0041-008x(83)90154-0. [DOI] [PubMed] [Google Scholar]
- Prevedouros K, Cousins IT, Buck RC, Korzeniowski SH. Sources, fate and transport of perfluorocarboxylates. Environ. Sci. Technol. 2006;40:32–44. doi: 10.1021/es0512475. [DOI] [PubMed] [Google Scholar]
- Reichard JF, Doorn JA, Simon F, Taylor MS, Petersen DR. Characterization of multidrug resistance-associated protein 2 in the hepatocellular disposition of 4-hydroxynonenal. Arch. Biochem. Biophys. 2003;411:243–250. doi: 10.1016/s0003-9861(03)00002-x. [DOI] [PubMed] [Google Scholar]
- Reid G, Wielinga P, Zelcer N, van der Heijden I, Kuil A, de Haas M, Wijnholds J, Borst P. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. U.S.A. 2003;100:9244–9249. doi: 10.1073/pnas.1033060100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renes J, de Vries EE, Hooiveld GJ, Krikken I, Jansen PL, Muller M. Multidrug resistance protein MRP1 protects against the toxicity of the major lipid peroxidation product 4-hydroxynonenal. Biochem. J. 2000;350(Pt 2):555–561. [PMC free article] [PubMed] [Google Scholar]
- Rius M, Hummel-Eisenbeiss J, Hofmann AF, Keppler D. Substrate specificity of human ABCC4 (MRP4)-mediated cotransport of bile acids and reduced glutathione. Am. J. Physiol. Gastrointest. Liver Physiol. 2006;290:G640–G649. doi: 10.1152/ajpgi.00354.2005. [DOI] [PubMed] [Google Scholar]
- Rius M, Nies AT, Hummel-Eisenbeiss J, Jedlitschky G, Keppler D. Cotransport of reduced glutathione with bile salts by MRP4 (ABCC4) localized to the basolateral hepatocyte membrane. Hepatology. 2003;38:374–384. doi: 10.1053/jhep.2003.50331. [DOI] [PubMed] [Google Scholar]
- Sterchele PF, Sun H, Peterson RE, Vanden Heuvel JP. Regulation of peroxisome proliferator-activated receptor-alpha mRNA in rat liver. Arch. Biochem. Biophys. 1996;326:281–289. doi: 10.1006/abbi.1996.0077. [DOI] [PubMed] [Google Scholar]
- Takagi A, Sai K, Umemura T, Hasegawa R, Kurokawa Y. Short-term exposure to the peroxisome proliferators, perfluorooctanoic acid and perfluorodecanoic acid, causes significant increase of 8-hydroxydeoxyguanosine in liver DNA of rats. Cancer Lett. 1991;57:55–60. doi: 10.1016/0304-3835(91)90063-n. [DOI] [PubMed] [Google Scholar]
- Trauner M, Arrese M, Soroka CJ, Ananthanarayanan M, Koeppel TA, Schlosser SF, Suchy FJ, Keppler D, Boyer JL. The rat canalicular conjugate export pump (Mrp2) is down-regulated in intrahepatic and obstructive cholestasis. Gastroenterology. 1997;113:255–264. doi: 10.1016/s0016-5085(97)70103-3. [DOI] [PubMed] [Google Scholar]
- Vahden Heuvel JP. Perfluorodecanoic acid as a useful pharmacologic tool for the study of peroxisome proliferation. Gen. Pharmacol. 1996;27:1123–1129. doi: 10.1016/0306-3623(95)00126-3. [DOI] [PubMed] [Google Scholar]
- Vanden Heuvel JP, Kuslikis BI, Van Rafelghem MJ, Peterson RE. Disposition of perfluorodecanoic acid in male and female rats. Toxicol. Appl. Pharmacol. 1991;107:450–459. doi: 10.1016/0041-008x(91)90308-2. [DOI] [PubMed] [Google Scholar]
- Van Rafelghem MJ, Mattie DR, Bruner RH, Andersen ME. Pathological and hepatic ultrastructural effects of a single dose of perfluoro-n-decanoic acid in the rat, hamster, mouse, and guinea pig. Fundam. Appl. Toxicol. 1987;9:522–540. [PubMed] [Google Scholar]
- Van Rafelghem MJ, Vanden Heuvel JP, Menahan LA, Peterson RE. Perfluorodecanoic acid and lipid metabolism in the rat. Lipids. 1988;23:671–678. doi: 10.1007/BF02535666. [DOI] [PubMed] [Google Scholar]
- Vollrath V, Wielandt AM, Iruretagoyena M, Chianale J. Role of Nrf2 in the regulation of the Mrp2 (ABCC2) gene. Biochem. J. 2006;395:599–609. doi: 10.1042/BJ20051518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner M, Fickert P, Zollner G, Fuchsbichler A, Silbert D, Tsybrovskyy O, Zatloukal K, Guo GL, Schuetz JD, Gonzalez FJ, et al. Role of farnesoid X receptor in determining hepatic ABC transporter expression and liver injury in bile duct-ligated mice. Gastroenterology. 2003;125:825–838. doi: 10.1016/s0016-5085(03)01068-0. [DOI] [PubMed] [Google Scholar]
- Wild AC, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression: Insights into transcriptional control of antioxidant defenses. Free Radic. Res. 2000;32:281–301. doi: 10.1080/10715760000300291. [DOI] [PubMed] [Google Scholar]
- Yeldandi AV, Rao MS, Reddy JK. Hydrogen peroxide generation in peroxisome proliferator-induced oncogenesis. Mutat. Res. 2000;448:159–177. doi: 10.1016/s0027-5107(99)00234-1. [DOI] [PubMed] [Google Scholar]
- Zelcer N, Reid G, Wielinga P, Kuil A, van der Heijden I, Schuetz JD, Borst P. Steroid and bile acid conjugates are substrates of human multidrug-resistance protein (MRP) 4 (ATP-binding cassette C4) Biochem. J. 2003;371:361–367. doi: 10.1042/BJ20021886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelcer N, van de Wetering K, de Waart R, Scheffer GL, Marschall HU, Wielinga PR, Kuil A, Kunne C, Smith A, van der Valk M, et al. Mice lacking Mrp3 (Abcc3) have normal bile salt transport, but altered hepatic transport of endogenous glucuronides. J. Hepatol. 2006;44:768–775. doi: 10.1016/j.jhep.2005.07.022. [DOI] [PubMed] [Google Scholar]
- Zollner G, Fickert P, Silbert D, Fuchsbichler A, Marschall HU, Zatloukal K, Denk H, Trauner M. Adaptive changes in hepatobiliary transporter expression in primary biliary cirrhosis. J. Hepatol. 2003;38:717–727. doi: 10.1016/s0168-8278(03)00096-5. [DOI] [PubMed] [Google Scholar]