

Abstract
Background
Retinoblastoma (RB) is a childhood ocular malignancy associated with mutations in RB1, a tumor susceptibility gene. Inactivation of both copies of the RB1 gene in a retinal cell is followed by the sequential acquisition of additional genetic changes that define the course to tumor formation.
Methods
To identify the genetic events that cooperate with loss of the RB1 gene function, we performed a whole genome sampling assay (WGSA) based on SNP genotyping. We used DNA isolated from 25 sporadic, unilateral RB tumors and matched blood samples.
Results
Genomic profiles were analyzed to identify regions of loss of heterozygosity (LOH) and/or amplification. Two major subclasses of RB tumors were defined by the presence (n=18) or absence (n=7) of LOH of chromosome 13. LOH in most cases was due to copy neutral events caused by mitotic recombination and mitotic non-disjunction. Tumors harbored novel regions of amplification at 1q44, 3p25, 11q14, 11q25, 14q23, 15q21, 16p13, 17p11.2, 19q13, and 20q13 while regions of loss included 6q22, 7q21and 21q2.
Conclusion
WGSA-based analysis of unilateral RB tumors revealed novel regions as significant. These minimum critical regions that are lost or amplified are expected to harbor genes that aid the process of tumorigenesis.
Keywords: Molecular Karyotype, Retinoblastoma, SNP-array, Whole Genome Sampling Assay
Introduction
Retinoblastoma (RB) is a tumor of the eye that occurs in children and can lead to compromised vision or possibly death if not diagnosed and treated properly. RB is associated with mutations in RB1, a tumor susceptibility gene [1]. Cell cycle exit and terminal differentiation of retinal cells are serial events that ensure proper retinal development [2]. The RB1 gene encodes pRB, a nuclear protein that plays a critical role in this regulated series of events. RB tumors represent a deregulation of this process. Inactivation of both copies of the RB1 gene in a retinal cell, through mutations or epigenetic modifications, initiates the onset of RB. This event is followed, as in other cancers, by the sequential acquisition of additional genetic abnormalities that define the course leading to tumor formation and metastasis [3–8].
Genomic instability contributes to the progression of retinoma to malignant retinoblastoma [9–11]. In humans, this progression is characterized by loss of both copies of the RB1 gene in retinoma, followed by changes in the copy number of oncogenes such as MYCN (2p24.3), E2F3 and DEK (6p22), KLF14 (7q32) and MDM4 (1q32), as well as tumor suppressor genes CDH11 (16q21) and p75NTR (17q21). It has also been shown that when RB1 and TP53 are inactivated in mice, retinoblastoma develops [12, 13]. Similarly, inactivation of RB1 and RBL1 (p107) or RB1 and RBL2 (p130) give rise to retinoblastoma in mice, where RBL1 and RBL2 are members of the RB1 gene family with redundant but unique functions [14–16]. Collectively, these observations indicate that, beyond biallelic inactivation of RB1, a third and additional “hits” are required for the development of RB tumors in humans and mice [13, 17–19].
Comparative Genomic Hybridization (CGH) is an analytical tool used to identify regions of chromosomal loss and gain within tumors by comparing pooled normal DNA with tumor DNA. Several studies have used CGH to characterize genomic abnormalities in RB tumors [20–31]. Previously identified recurrent abnormalities include gain of chromosomal material at 6p, 1q, 2p, 13q and 19 and loss of material on chromosome16, 16q and 13q. Each of these abnormalities was observed in at least 10% of tumors studied in more than one series [20–22]. The gain of 6p, as in isochromosome 6p, was particularly common and seen in 44% to 69% of tumors. Some abnormalities, such as gain of 6p or 13q and loss of 13q, were more common in tumors with high-risk histological features; however, only the association with loss of 13q was statistically significant [20]. In three patients who developed extra-ocular relapse, tumors showed loss of 13q and two out of three also had loss of 5q, suggesting that loss of genetic material at these loci may be associated with metastasis [32]. Older children (> 36 months of age at enucleation) tend to have more CGH abnormalities per tumor than younger children (<12 months; median numbers 11 vs. 3). In addition, +1q, +13q, −16, and −16q were more frequent in children with an older age at enucleation. Recently it has been shown that loss or retention of specific regions of 1q correlated with the degree of differentiation of RB tumors [33].
Therefore, it is clear that identification of these genomic alterations serve as clues to better understand the molecular basis of RB tumor formation, and could pave the way to novel and more directed therapies.
In this report, we describe the molecular karyotype of 25 sporadic unilateral RB tumors as determined by the use of a whole genome sampling assay (WGSA), which incorporated the SNP genotyping of matched normal and tumor tissue. The WGSA method involves parallel genotyping of many bi-allelic single nucleotide polymorphisms located across all 23 chromosomes of the human genome. Comparison of the genotype calls and intensities of the different alleles from matched normal and tumor tissue samples allows one to determine regions of loss of heterozygosity or amplification in the tumor with high resolution.
Methods
Unilateral RB tumor specimens (italics, like other sections)
Primary unilateral RB tumors were collected at the Genetic Diagnostic Laboratory, University of Pennsylvania, and at Wills Eye Institute. For inclusion in this study, the tumors could not have received any therapy before enucleation. The protocol for genetic analysis of tumors was approved by the IRB of the University of Pennsylvania (Protocol number 706577, original approval date 09/23/05). Tumor samples were flash frozen and transferred on dry ice. Blood samples from affected individuals were collected in EDTA-containing tubes and transferred at room temperature.
Isolation of DNA from blood and frozen tumor samples
Genomic DNA was isolated from 1 to 3 ml of blood and frozen tumors using a commercial DNA isolation kit (Gentra, CA) following the manufacturer’s instructions.
Whole Genome Sampling Assay (WGSA)
The data for this report was generated using the 10K Single Nucleotide Polymorphism (SNP) chips from Affymetrix, CA. These Chips provided a platform for the parallel query of 10,000 SNPs selected from the human genome that are gene-centered and located within 10KB of the flanking sequences of any gene. The average distance between SNPs is 250KB and the average rate of heterozygosity is 0.37. A single array analysis was performed for each DNA sample (250 ng each) using the protocol defined by the manufacturer. In brief, DNA samples were digested with Xba1, ligated to an adaptor and amplified using a set of universal primers (available as components of Assay Kits P/N 900520 and 900521). The amplified DNA was fragmented, labeled with a fluorescent dye and hybridized to a chip. Hybridization and post hybridization washes were done in the Affymetrix fluidics station followed by scanning and analysis.
Data Analysis
Affymetrix GeneChip DNA Analysis Software (GTYPE V3.0), was used to perform automatic base calls with high base-calling accuracy, reproducibility and automatic generation of SNP summary reports [34]. The output data from GTYPE was analyzed using dCHIPSNP (http://www.biostat.harvard.edu/complab/dchip/snp/), a program that allows visualization of regions of loss or gain along the length of a chromosome [35, 36]. A “call” for a region of loss or gain is based on compiled data for at least 10 consecutive SNPs genotyped within a chromosomal region spanning 10 MB. The genome build of July 2003 human reference sequence (NCBI Build 34) was used for defining genomic regions.
Significance Testing for Aberrant Copy-number (STAC)
The output from the dCHIPSNP analysis offers a simple multi-sample p-value for LOH, which tests the null hypothesis that there is no aberration at a given location in any of the samples. Once the significant aberrations are identified, it is important to define concordant aberrations across multiple samples. To address this need, we used an algorithm called STAC (Significance Testing for Aberrant Copy-number) (www.cbil.upenn.edu/STAC) [37], which defines two statistics, frequency (f) and foot-print (fp) to measure concordance across multiple samples. For each statistic and each location a multiple testing corrected permutation p-value is computed under a null permutation distribution. Frequency measures the number of aberrations present at a specific chromosomal location across samples. Footprint measures tightness of alignment of a set of aberration intervals that cover a given location [37].
Results
Sporadic unilateral RB occurs in a child without any family history, affects only one eye and has a later age of onset (average 2.5 years) compared to bilateral disease (average <1 year). It is caused by post-zygotic, somatic mutations of the RB1 gene in a retinal progenitor cell. However, in 10% of sporadic unilateral RB, a germline mutation is present. The latter class of mutations is generally associated with ‘low penetrance’ in that they are predicted to have subtle effects on the function or relative amounts of the expressed pRB protein. In addition mosaicism, germline or somatic, for de novo mutations have also been observed for unilateral RB.
The 25 sporadic unilateral RB samples included in this report were collected as part of a limited study for genetic testing of the mutation spectrum at the RB1 locus of unilateral tumors. However, the clinical information on the RB tumors was not collected as part of this study and is thus unavailable for comparison with genomic profile. There were 17 females and 8 males with ages of onset ranging from 0.3 years to 7.4 years. Using complementary assays, we were able to identify inactivating bialelic RB1 gene mutations in 100% of these tumors (Table 1). Of the 15 point mutations detected, 5 were C>T transitions in CpG dinucleotides. These data are consistent with prior studies demonstrating that methylation at cytosines within CpG islands generates hot spots for mutation in the RB1 gene. Among the tumor-associated mutations identified, 10 out of 15 (67%) were protein-truncating mutations. We also observed methylation of the promoter region as the mode of RB1 gene inactivation in 7 out of 25 (28%) tumors analyzed.
Table 1.
Description of 25 sporadic unilateral RB tumors
| ID | Sex | Age at enucleation | LOH of Chromosome 13 | Coding Sequence Mutation at RB1 locus | Change (amino acid)/Splicing | Number of Aberrations |
|---|---|---|---|---|---|---|
| 014 | female | 2.0 | No | g. 2162C>T, exon 1 | p.Q35X | 7 |
| 020 | female | 2.1 | No | ND* | 1 | |
| 042 | female | 0.5 | No | g. 78238C > T, exon 17 | p.R552X | 15 |
| 044 | male | 1.1 | Yes | ND | 25 | |
| 142 | female | 2.4 | Yes | g. 61797G>A, exon 9 | p.G310E | 10 |
| 223 | male | 5.5 | Yes | ND | 3 | |
| 233 | female | 3.2 | Yes | DEL EXON 15,16 and 20 | 25 | |
| 443 | male | 2.6 | Yes | ND | 25 | |
| 490 | female | 0.2 | Yes | ND | 1 | |
| 503 | female | 1.9 | No | g. 76898C>T, exon 15, g. 156833G> A, exon 20 | p.R467X, p.W681X | 2 |
| 501 | female | 2.4 | Yes | g. 76460C> T, exon 14 | p.R455X | 2 |
| 510 | male | 2.5 | Yes | ND | 1 | |
| 520 | female | 0.9 | Yes | ND | 5 | |
| 530 | female | 0.9 | Yes | ND | 3 | |
| 610 | male | 3.2 | Yes | ND | 25 | |
| 620 | male | 1.0 | No | g. 335InsTA, exon 2; g. 2364InsTTGA, exon 22 | p.76X, p.750X | 1 |
| 630 | female | 4.5 | Yes | g. 16184T>A, at −13 position of intron 21 | Novel splice acceptor | 25 |
| 710 | male | 7.4 | Yes | g. 150037C>T, exon 18 | p.R579X | 16 |
| 791 | female | 1.7 | Yes | g. 73750A>G, at −3 position of intron 12. | Novel splice acceptor | 4 |
| 840 | female | 0.7 | Yes | g. 65386C>T, exon 11 | p.R358X, | 25 |
| 860 | female | 2.5 | Yes | g. 150117 G>A, at +1 position of intron 18 | Splice donor | 1 |
| 902 | female | 2.5 | Yes | g. 160740 G>A, exon 21 | p.C706Y | 25 |
| 912 | female | 0.3 | Yes | ND | 0 | |
| 913 | male | 0.4 | No | ND | 0 | |
| 920 | male | 0.9 | Yes | g. 153198 A> G, at −12 position of intron 18 | Novel splice acceptor | 5 |
ND – Not detected
WGSA for determination of copy number variation
We used Affymetrix 10K SNP chip-based WGSA to generate genomic profiles of loss and/or gain of chromosomal material in unilateral RB tumors. The SNP call rates for DNA isolated from peripheral blood ranged between 89% and 99% while that for DNA isolated from tumor samples ranged between 86% and 99%. In all cases, the SNP calls from peripheral blood DNA were considered the “normal” standards, against which the SNP calls from tumor-derived DNA were compared. Loss of heterozygosity (LOH) or retention of heterozygosity (ROH) in the tumor was determined by comparing the blood and tumor SNP genotype calls.
Figure 1 demonstrates the output from the dCHIPSNP analysis as a whole genome LOH profile for the set of 25 paired normal/tumor samples (each indicated as separate columns). The summarized data indicate the LOH profile for each individual chromosome (indicated by the respective numbers on the left). The regions marked in yellow indicate retention of heterozygosity (ROH) and blue indicates regions of LOH. The clustering indicated on the top is obtained based on the co-occurrence of regions of loss in the tumors. The distances are based on the extent of discordance between the regions of ‘significant’ LOH between the individual tumors. The classification is unsupervised and can lead to unbiased schemes of identification of the sub-classes of tumors.
Figure 1.
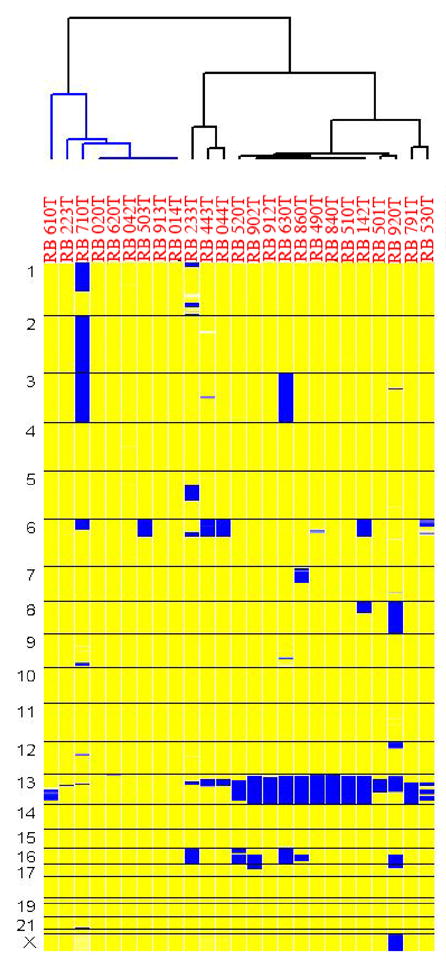
This picture describes the clustering of 25 RB tumor samples based on their whole genome loss of heterozygosity (LOH) profile. Each column represents a matched normal/tumor pair and each horizontal block indicates a chromosome. The yellow regions indicate retention of heterozygosity (ROH) and blue regions indicate LOH.
One of the advantages of WGSA is its ability to simultaneously derive SNP genotype as well as the relative copy number of each SNP based on information gained from the signal intensity of hybridized DNA. The upper panel in Figure 2A indicates the LOH profiles for chromosome 13 for direct comparison with the copy number profiles, which are shown in the lower panel. For copy number, the shades indicate values ranging from 0 (white) to ≥ 6 (dark red). More than half of the RB tumors had loss of heterozygosity at 13q (indicated by blue regions on the upper panel), the most common form of inactivation of RB1. Three tumors (710T, 443T and 044T) exhibited homozygous loss of the region encompassing RB1 (indicated by white regions and the arrows). For two tumors, 610T and 710T, the LOH region (<250 kb) was small and below the level of detection using the 10K SNP chips, where the average distance between consecutive SNPs is approximately 250 KB.
Figure 2.
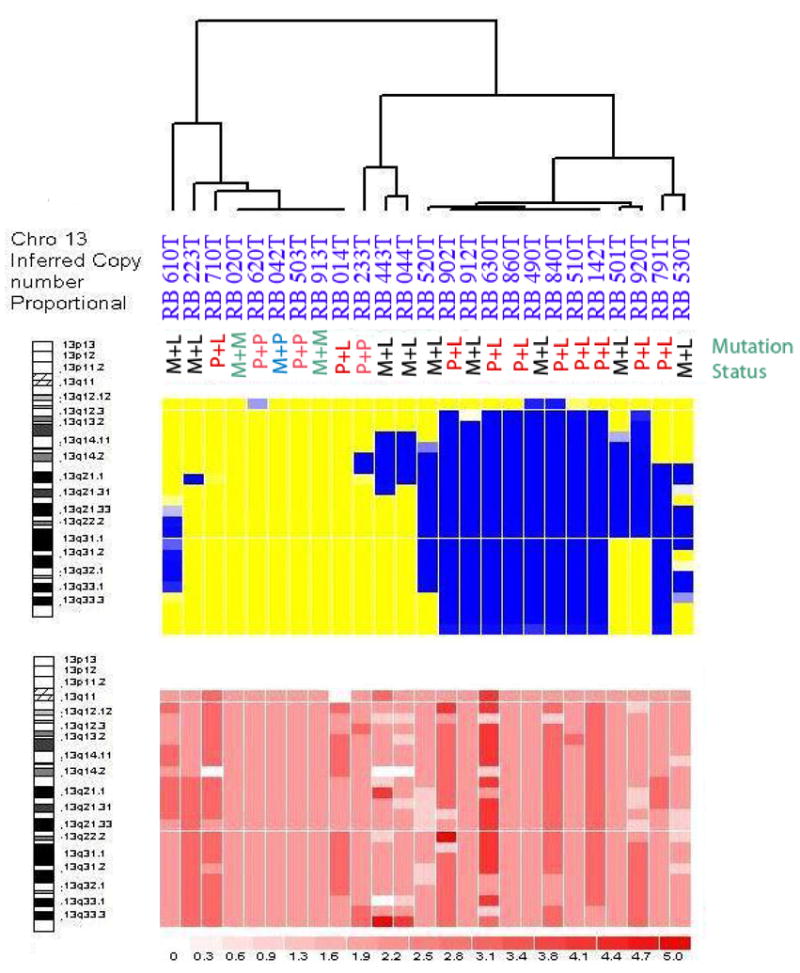
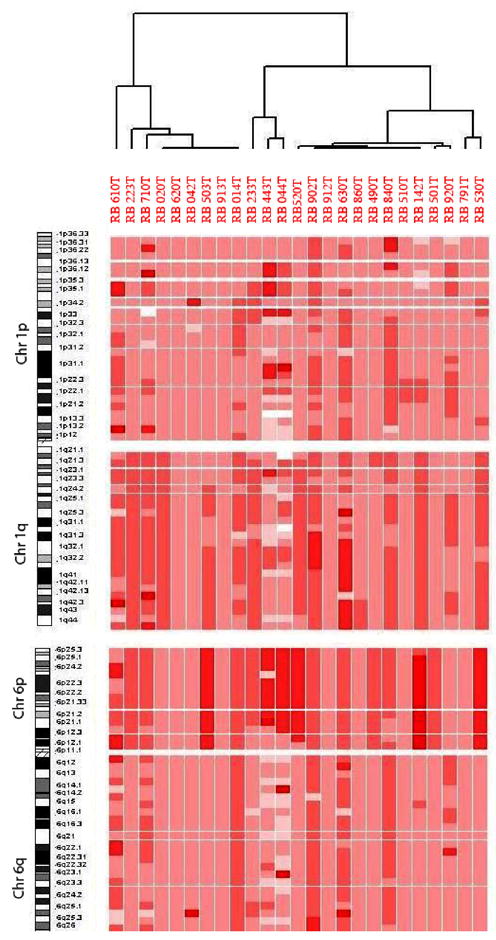
Figure 2A. This figure represents the results for chromosome 13, which harbors the RB1 gene on 13q14.2 [8].
The different shades of pink indicate different copy numbers white for 0 copy, pink for 2 copies and red for 6 or more copies. The RB tumors cluster according to LOH (upper panel). Abbreviations: M: Methylation of RB1 promoter; P: Point mutations in the coding sequence of RB1 gene; L: LOH.
Figure 2B. This figure represents chromosomes 1 and 6 aberrations. The amplifications in chromosomes 1q and 6q are correlated. Tumors in the right cluster (12 out of 25) have dark red color for 6p and most likely indicate isochromosome 6p.
Aberrations on other chromosomes
Figure 2B shows the copy number profiles for chromosomes 1 and 6. We observe that amplifications of chromosome 1q and 6p are highly correlated– in 12 out of 14 tumors with 1q amplification (86 %), we also found amplification of 6p. Fifteen out of 25 tumors exhibited amplification of almost the entire 6p arm (60%). These data most likely reflect the tumors with isochromosome 6p, a very common aberration seen in RB tumors.
Figure 3 shows the clustering of the RB tumors based on copy number aberration (CNA) as opposed to LOH as in Figure 1. The cluster on the left includes 3 tumors, 912T, 913T and 620T, with the least number of aberrations and the cluster on the right includes 7 tumors with aberrations along the entire genome. The cluster in the middle includes tumors with specific aberrations: one subset with amplification of 1q, one subset with 6p amplification and a third subset with co-amplification of 1q and 6p.
Figure 3.
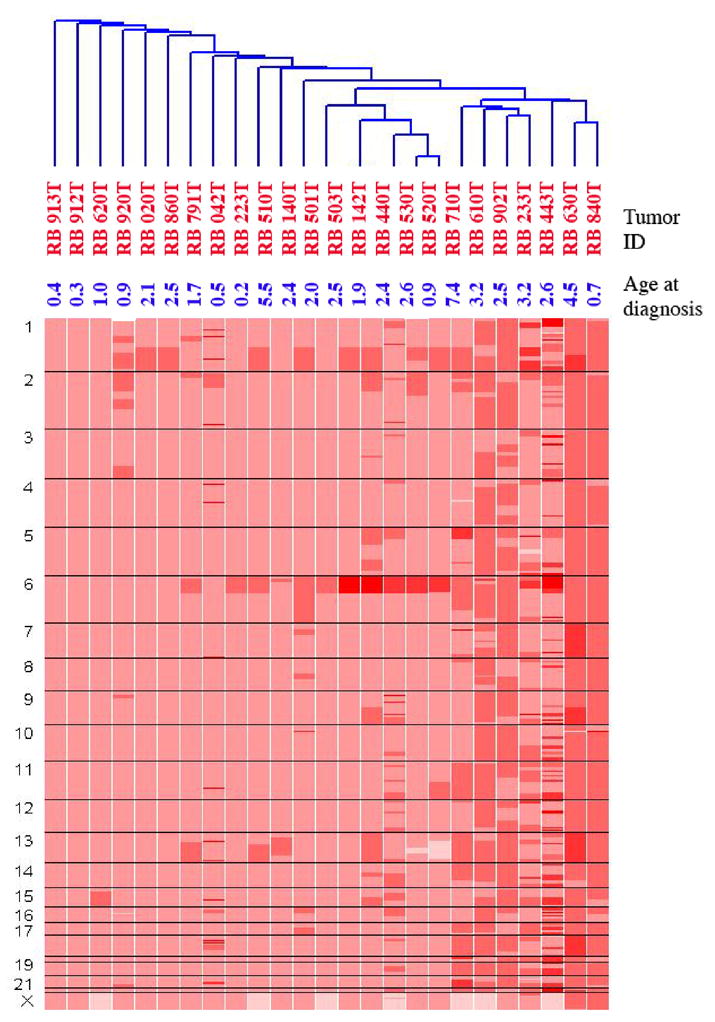
This figure represents the clustering of RB tumors based on genome wide copy number profiles. The total number of aberrations drives the clustering of tumor subgroups.
Significance Testing for Aberrant Copy-number (STAC)
We applied STAC to the copy number output from dCHIPSNP on the cohort of 25 pairs of normal/tumor DNA samples to identify statistically significant regions of concurrent amplification/loss. Figure 4 shows the STAC output for the shared regions of gain or loss along the lengths of individual chromosomes (Table 2). The regions indicating gain (green bars) or loss (red bars) across the whole genome are those with the most significant changes in copy number (p values 0.05). STAC is designed to localize regions of concurrent aberration across multiple samples, within a chromosome arm, down to 1Mb resolution or less. Therefore, if an entire chromosome is gained or lost across all, or most samples, STAC will generally not give a significant p-value. This is the case for chromosome 6p, where the entire arm is amplified in 60% of the tumors. Such gross aberrations are easily identified by other means. STAC is designed to highlight the concordant local aberrations, and to distinguish them from the aberrations unique to a single sample.
Figure 4.
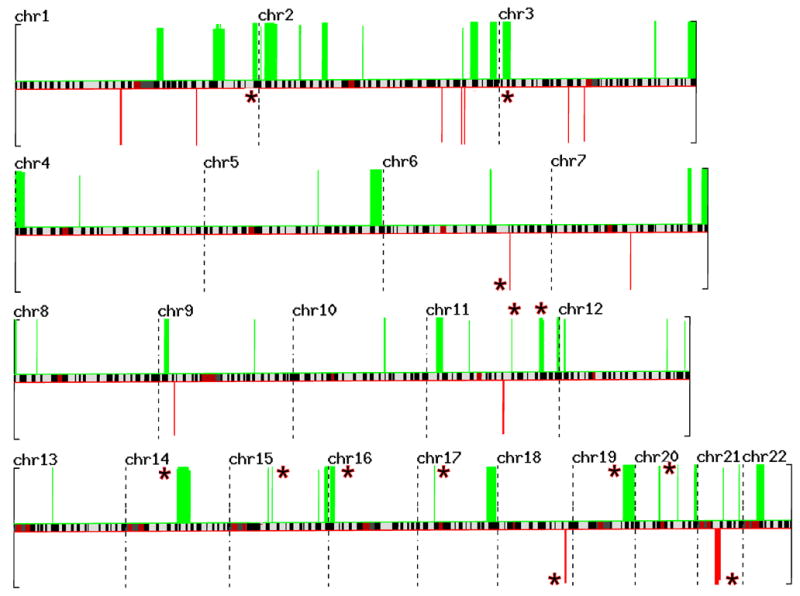
This figure represents the STAC output for the whole genome displayed across individual chromosomes– the green bars indicate gains and red bars indicate losses. Each bar represents an aberration that has a p-value less than 0.05. The red asterisks indicate 13 novel regions identified in this analysis.
Table 2.
STAC Output for Significant regions of Copy Number Gain in RB Tumors
| Chromosomal Gain Regions (based on July 2003 map of UCSC browser) | ||||||
|---|---|---|---|---|---|---|
| arm | aberration | fp (chr) | Start (bp) | Stop (bp) | fp_pvalue | fp_confidence (1- fp_pvalue) |
| chr1q | Gain | chr1q21.1 | 143,000,001 | 150,000,000 | 0.047952048 | 0.952047952 |
| chr1q | Gain | chr1q32.1 | 200,000,001 | 203,000,000 | 0.053946054 | 0.946053946 |
| chr1q | Gain | chr1q32.1 | 203,000,001 | 206,000,000 | 0.01998002 | 0.98001998 |
| chr1q | Gain | chr1q32.2 | 206,000,001 | 207,000,000 | 0.053946054 | 0.946053946 |
| chr1q | Gain | chr1q32.3 | 207,000,001 | 208,000,000 | 0.01998002 | 0.98001998 |
| chr1q | Gain | chr1q32.3 | 208,000,001 | 212,000,000 | 0.053946054 | 0.946053946 |
| chr1q | gain | chr1q44 | 240,000,001 | 245,000,000 | 0.00999001 | 0.99000999 |
| chr2p | gain | chr 2p25.3 | 2,000,001 | 3,000,000 | 0.037 | 0.963 |
| chr2p | gain | chr 2p25.3 | 6,000,001 | 16,000,000 | 0.005 | 0.995 |
| chr2p | gain | chr 2p24.3 | 16,000,001 | 19,000,000 | 0.018 | 0.982 |
| chr2p | gain | chr 2p25.3 | 41,000,001 | 43,000,000 | 0.027 | 0.973 |
| chr2p | gain | chr 2p14 | 64,000,001 | 70,000,000 | 0.005 | 0.995 |
| chr2q | gain | chr 2q12 | 105,000,001 | 106,000,000 | 0.051948052 | 0.948051948 |
| chr2q | gain | chr 2q33 | 206,000,001 | 207,000,000 | 0.051948052 | 0.948051948 |
| chr2q | gain | chr 2q34 | 214,000,001 | 220,000,000 | 0.001998002 | 0.998001998 |
| chr2q | gain | chr 2q35 | 220,000,001 | 222,000,000 | 0.006993007 | 0.993006993 |
| chr2q | gain | chr 2q36–37 | 221,000,001 | 241,000,000 | 0.006993007 | 0.993006993 |
| chr3p | gain | chr3p25.1 | 4,000,001 | 7,000,000 | 0.001998002 | 0.998001998 |
| chr3p | gain | chr3p25.3 | 9,000,001 | 12,000,000 | 0.001998002 | 0.998001998 |
| chr3q | gain | chr3p25.31 | 157,000,001 | 159,000,000 | 0.001998002 | 0.998001998 |
| chr3q | gain | chr3q25 | 191,000,001 | 200,000,000 | 0.01998002 | 0.98001998 |
| chr4p | gain | chr4p16 | 1 | 10,000,000 | 0.003996004 | 0.996003996 |
| chr4q | gain | chr4q13.1 | 65,000,001 | 66,000,000 | 0.045954046 | 0.954045954 |
| chr5q | gain | chr5q23.1 | 115,000,001 | 116,000,000 | 0.005994006 | 0.994005994 |
| chr5q | gain | chr5q35.2 | 168,000,001 | 180,000,000 | 0.005994006 | 0.994005994 |
| chr6q | gain | chr6q21 | 108,000,001 | 109,000,000 | 0.007992008 | 0.992007992 |
| chr6q | gain | chr6q21 | 109,000,001 | 110,000,000 | 0.015984016 | 0.984015984 |
| chr7q | gain | chr7q34 | 138,000,001 | 142,000,000 | 0.006993007 | 0.993006993 |
| chr7q | gain | chr7q36.2-.3 | 152,000,001 | 159,000,000 | 0.002997003 | 0.997002997 |
| chr8p | gain | chr8p23.3 | 1,000,001 | 3,000,000 | 0.005994006 | 0.994005994 |
| chr8p | gain | chr8p21.2 | 23,000,001 | 24,000,000 | 0.005994006 | 0.994005994 |
| chr9p | gain | chr9p24.1-23 | 6,000,001 | 11,000,000 | 0.005994006 | 0.994005994 |
| chr9q | gain | chr9q31.1 | 97,000,001 | 98,000,000 | 0.016983017 | 0.983016983 |
| chr10q | gain | chr10q23.31 | 92,000,001 | 94,000,000 | 0.004995005 | 0.995004995 |
| chr11p | gain | chr11p15 | 10,000,001 | 17,000,000 | 0.005994006 | 0.994005994 |
| chr11p | gain | chr11p12-11 | 43,000,001 | 44,000,000 | 0.04995005 | 0.95004995 |
| chr11q | gain | chr11q14.2 | 86,000,001 | 87,000,000 | 0.000999001 | 0.999000999 |
| chr11q | gain | chr11q23.2-.3 | 114,000,001 | 119,000,000 | 0.000999001 | 0.999000999 |
| chr11q | gain | chr11q25 | 132,000,001 | 135,000,000 | 0.000999001 | 0.999000999 |
| chr12p | gain | chr12p13.31 | 5,000,001 | 7,000,000 | 0.026973027 | 0.973026973 |
| chr12q | gain | chr12q24.11 | 109,000,001 | 110,000,000 | 0.026973027 | 0.973026973 |
| chr12q | gain | chr12q24.32 | 127,000,001 | 128,000,000 | 0.044955045 | 0.955044955 |
| chr13q | gain | chr13q14.11 | 39,000,001 | 40,000,000 | 0.005994006 | 0.994005994 |
| chr14q | gain | chr14q23.1 | 52,000,001 | 64,000,000 | 0.002797202 | 0.997202798 |
| chr14q | gain | chr14q23.3 | 64,000,001 | 66,000,000 | 0.034965035 | 0.965034965 |
| chr15q | gain | chr15q15 | 39,000,001 | 40,000,000 | 0.013986014 | 0.986013986 |
| Arm | aberration | chr | Start (bp) | Stop (bp) | fp_pvalue | fp_confidence |
| chr15q | gain | chr15q21.1 | 43,000,001 | 44,000,000 | 0.001998002 | 0.998001998 |
| chr15q | gain | chr15q26.1 | 90,000,001 | 91,000,000 | 0.031968032 | 0.968031968 |
| chr15q | gain | chr15q26.3 | 96,000,001 | 100,000,000 | 0.013986014 | 0.986013986 |
| chr16p | gain | chr16p13 | 2,000,001 | 7,000,000 | 0.003996004 | 0.996003996 |
| chr17p | gain | chr17p11.2 | 17,000,001 | 18,000,000 | 0.002997003 | 0.997002997 |
| chr17q | gain | chr17q24.3-25.2 | 70,000,001 | 80,000,000 | 0.015984016 | 0.984015984 |
| chr19q | gain | chr19q13.3 | 51,000,001 | 63,000,000 | 0.003996004 | 0.996003996 |
| chr20p | gain | chr 20p11.21 | 24,000,001 | 26,000,000 | 0.015984016 | 0.984015984 |
| chr20q | gain | chr 20q13.12 | 43,000,001 | 44,000,000 | 0.000999001 | 0.999000999 |
| chr20q | gain | chr 20q13.33 | 60,000,001 | 63,000,000 | 0.000999001 | 0.999000999 |
| chr21q | gain | chr 21q21.3 | 26,000,001 | 27,000,000 | 0.03996004 | 0.96003996 |
| chr21q | gain | chr 21q22.3 | 42,000,001 | 43,000,000 | 0.003996004 | 0.996003996 |
| chr22q | gain | chr 22q11.1-.22 | 14,000,001 | 22,000,000 | 0.008991009 | 0.991008991 |
| Chromosomal Loss Regions | ||||||
| chr1p | loss | chr1p21.1-13.3 | 106,000,001 | 108,000,000 | 0.017982018 | 0.982017982 |
| chr1q | loss | chr1q31.1 | 183,000,001 | 184,000,000 | 0.005994006 | 0.994005994 |
| chr2q | loss | chr 2q32.1 | 185,000,001 | 186,000,000 | 0.024975025 | 0.975024975 |
| chr2q | loss | chr 2q33.1 | 205,000,001 | 206,000,000 | 0.001998002 | 0.998001998 |
| chr2q | loss | chr 2q33.3 | 208,000,001 | 209,000,000 | 0.001998002 | 0.998001998 |
| chr3p | loss | chr3p13 | 70,000,001 | 71,000,000 | 0.010989011 | 0.989010989 |
| chr3p | loss | chr3p12.1 | 86,000,001 | 87,000,000 | 0.024975025 | 0.975024975 |
| chr6q | loss | chr6q22.33 | 128,000,001 | 129,000,000 | 0.000999001 | 0.999000999 |
| chr7q | loss | chr7q21.11 | 80,000,001 | 81,000,000 | 0.002997003 | 0.997002997 |
| chr9p | loss | chr9p22.3-22.2 | 16,000,001 | 17,000,000 | 0.032967033 | 0.967032967 |
| chr11q | loss | chr11q13.5-14.1 | 77,000,001 | 79,000,000 | 0.034965035 | 0.965034965 |
| chr18q | loss | chr18q22.2 | 68,000,001 | 70,000,000 | 0.021978022 | 0.978021978 |
| chr21q | loss | chr 21q21.1 | 18,000,001 | 22,000,000 | 0.00999001 | 0.99000999 |
| chr21q | loss | chr 21q21.1 | 22,000,001 | 24,000,000 | 0.047952048 | 0.952047952 |
Using STAC, we identified several recurrent regions of amplification or loss that are in agreement with previous studies. For example, STAC correctly identified 2p24.3 as an amplified region that harbors the MYCN gene, a known region of amplification in RB tumors. The complete list of chromosomal regions amplified or lost with p-values smaller than 0.05 is included in Table 2. In addition to previously identified regions, we also observed that there were several novel loci exhibiting gain or loss. These novel chromosomal regions are indicated by the asterisk next to the cytoband and include amplifications at 1q44, 3p25, 11q14 and 11q25, 14q23, 15q21, 16p13, 17p11.2, 19q13, and 20q13.33. Novel regions of loss included 6q22, 7q21and 21q2.
Gains along chromosomes 1q and 14q as revealed by significant fp-value (Table 2) are shown in Figures 5A and 5B. There are three regions of significant gain on chromosome 1 with the shade of grey indicating the level of significance. Thus the region between nucleotides 200, 000, 001 – 206,000,001 (chromosome 1q32.1–32.3) has significant regions of gain with sub-regions of highly significant gain (p-value less than 0.019; adjusted for multiple testing). Similarly, the significance of gain in the region on 1q44 is 0.0099 and includes SMYD3, a histone methyltransferase that plays a role in transcriptional regulation as a member of an RNA polymerase complex. While chromosome 1q gains are known, gain on 14q22.3 –23.1 is novel. One of the regions resides on chromosome14q23.1–23.3 and includes the gene encoding CEP170, a protein involved in centriole architecture, which when deregulated gives rise to chromosomal non-disjunction during mitosis.
Figure 5.
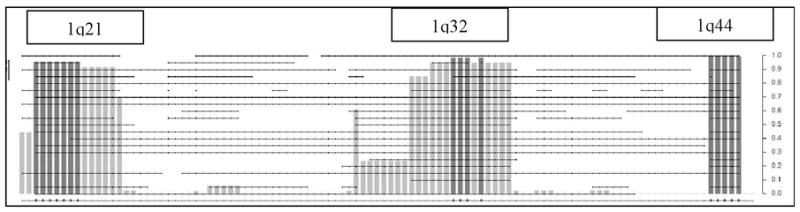
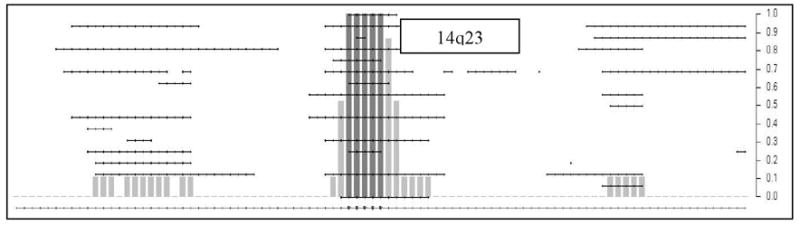
Figure 5A and 5B. This figure represents the expanded STAC output for chromosomes 1 and 14. The dotted horizontal lines indicate individual tumor profiles. The X-axis included the footprint (fp)-values and Y-axis includes the frequency (f) values.
Discussion
This report describes the molecular karyotype of primary unilateral RB tumors as determined by use of a SNP-array based WGSA on matched DNA samples from normal and tumor tissue. This study is limited to unilateral RB tumors as bilateral RB tumors are usually not enucleated before therapy. The aim of this project was to identify minimal critical genomic regions that are gained or lost in multiple samples of primary RB. The advantages of the SNP array based genomic approaches include confirmation of previously reported chromosomal gains/losses, detection of copy neutral LOH and higher resolution mapping of novel genomic regions of loss or gain leading to the identification of target oncogenes and tumor suppressor genes.
The results included in Figure 1 indicate that LOH on chromosome 13 is the major genetic alteration driving the clustering of RB tumors. Excluding losses on chromosomes 6 and 16, there are only infrequent regions of LOH or homozygous loss across the genome of RB tumors. Upon closer inspection in Figure 2, two different classes of RB tumors are identified, including those with and without LOH for chromosome 13. The lower panel indicates the CNA output for the same set of paired samples. It is evident that for the right cluster of tumors with LOH (top panel), the copy number data does not indicate any loss of genetic material (lower panel). This finding supports the known concept that RB1 inactivation leads to mitotic non-disjunction and LOH due to duplication of the mutant allele [38]. For 7 out of 16 tumors with LOH around the RB1 gene locus, there is amplification of the retained allele (as indicated by the darker red color in 014, 902, 840 and 142), which may represent double strand breaks during DNA replication followed by aberrant DNA repair. There is also a subclass of RB tumors where the LOH/aberrations are limited to segments of chromosome 13. In these cases, mitotic recombination rather than non-disjunction, likely explains the development of LOH. Therefore, these events reflect aberrant sister-chromatid exchange and compromised double strand break repair machinery. Both of these modes of copy-neutral LOH attest to the known properties of pRB protein in maintaining genomic stability.
STAC is designed to localize regions of concurrent aberration across multiple samples, within a chromosome arm, down to less than 1Mb resolution [37]. Using this method, we found that the recurrent regions of amplification or loss, as indicated in Figure 4, include all regions identified in previous studies and as well as 13 novel chromosomal regions (indicated by the asterisk next to the cytoband) [7, 33, 39, 40]. For example, one of well-established hallmarks of RB tumors is the presence of MYCN amplification (Reference). Indeed, we observed this event in 15 out of 25 tumors (60%) (Figures 3 and 4; Table 2).
The novel regions of amplification or loss with highest level of significance are indicated in bold letters in Table 2. We confirmed the validity of these regions by repeating WGSA using 250K SNP chips and reproducing the same results (data not shown). The novel regions amplified included 1q44, 3p25, 11q14 and 11q25, 14q23, 15q21, 16p13, 17p11.2, 19q13, and 20q13.33. The regions of significant loss included 6q22, 7q21and 21q2. The total number of genes in these chromosomal regions is large due to the limited resolution of the SNP-chip platform used in this report. The use of higher resolution platforms is expected to further narrow these minimum regions of chromosomal aberration.
Copy number variations have been detected as a natural variation of human genomic DNA [41, 42]. Some of the identified regions of amplification or deletion in Table 2 overlap previously identified copy number polymorphism regions. However the data for this report has been generated using matched DNA from normal and tumor tissue. Therefore, only copy number changes that are different between normal and tumor tissue are detected as significant.
Gains along chromosomes 1q and 14q as revealed by significant fp-value (Table 2) are shown in Figure 5. Figure 5A demonstrates gains on chromosome 1. It has been established that mouse models of RB require inactivation of TP53 in addition to RB1. Mutations in TP53 gene were not detected in this set of RB tumors (data not shown) and these results confirm what is known in the literature [43]. However amplification of MDM4 has been observed as an alternate pathway for inactivation of TP53 in the mouse model. Consistent with this model, in our dataset, the amplified region of 1q32 includes MDM4, GAC1, and others, which encode proteins involved in the TP53 pathway and regulate p53 activity [43].
Figure 5B demonstrate gains on chromosome 14. While chromosome 1q gains are known, significant gain on 14q23.1–23.2 is novel. The list of genes present in 14q23.1 includes CEP170 and SIX1 and SIX4 (genes associated with bilateral anophthalmia). The clustering of the RB tumors based on CNA (Figure 3) identifies tumors with different degrees of genomic instability. It is known that tumors from younger children have fewer numbers of aberrations [22]. Tumors 913T, 912T and 620T have the least number of aberrations genome wide and belong to children ages 0.4y, 0.3y, and 1 y respectively. In contrast, the 7 tumors belonging to the cluster on the extreme right with the most aberrations are derived from older children age 7.4y, 3.2y, 2.5y, 3.2y, 2.6y, 4.5y and 0.7y(the only exception) respectively (last column, Table 2). The tumors in the middle have co-amplifications of chromosome 1q and 6p.
In conclusion, the use of WGSA to examine unilateral RB tumors revealed novel features of the genomic architecture. The major advantage of the SNP based analysis was the identification of regions of copy neutral LOH. The regions identified as significant are anticipated to establish the minimum critical regions harboring genes that contribute to RB tumorigenesis.
Acknowledgments
The authors acknowledge the technical assistance of Courtney MacMullen and Lori Swanson for generation of this data. In addition the assistance of Sharon Diskin, PhD in the initial phase of this study is gratefully acknowledged. This work was supported in part by a grant from NIH/NCI (R21 CA123196-01).
Footnotes
There is no conflict of interest for any one of the authors.
References
- 1.Lee WH, Bookstein R, Lee EY. Studies on the human retinoblastoma susceptibility gene. J Cell Biochem. 1988;38(3):213–27. doi: 10.1002/jcb.240380309. [DOI] [PubMed] [Google Scholar]
- 2.Dyer MA, Bremner R. The search for the retinoblastoma cell of origin. Nat Rev Cancer. 2005;5(2):91–101. doi: 10.1038/nrc1545. [DOI] [PubMed] [Google Scholar]
- 3.Honavar SG, Singh AD, Shields CL, et al. Postenucleation adjuvant therapy in high-risk retinoblastoma. Arch Ophthalmol. 2002;120(7):923–31. doi: 10.1001/archopht.120.7.923. [DOI] [PubMed] [Google Scholar]
- 4.Makimoto A. Results of treatment of retinoblastoma that has infiltrated the optic nerve, is recurrent, or has metastasized outside the eyeball. Int J Clin Oncol. 2004;9(1):7–12. doi: 10.1007/s10147-003-0364-2. [DOI] [PubMed] [Google Scholar]
- 5.Chantada G, Fandino A, Davila MT, et al. Results of a prospective study for the treatment of retinoblastoma. Cancer. 2004;100(4):834–42. doi: 10.1002/cncr.11952. [DOI] [PubMed] [Google Scholar]
- 6.Bowles E, Corson TW, Bayani J, et al. Profiling genomic copy number changes in retinoblastoma beyond loss of RB1. Genes Chromosomes Cancer. 2007;46(2):118–29. doi: 10.1002/gcc.20383. [DOI] [PubMed] [Google Scholar]
- 7.Corson TW, Gallie BL. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosomes Cancer. 2007;46(7):617–34. doi: 10.1002/gcc.20457. [DOI] [PubMed] [Google Scholar]
- 8.Hong FD, Huang HJ, To H, et al. Structure of the human retinoblastoma gene. Proc Natl Acad Sci U S A. 1989;86(14):5502–6. doi: 10.1073/pnas.86.14.5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dimaras H, Khetan V, Halliday W, et al. Loss of RB1 induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet. 2008;17(10):1363–72. doi: 10.1093/hmg/ddn024. [DOI] [PubMed] [Google Scholar]
- 10.Dimaras H, Gallie BL. The p75 NTR neurotrophin receptor is a tumor suppressor in human and murine retinoblastoma development. Int J Cancer. 2008;122(9):2023–9. doi: 10.1002/ijc.23356. [DOI] [PubMed] [Google Scholar]
- 11.Dimaras H, Coburn B, Pajovic S, Gallie BL. Loss of p75 neurotro phin receptor expression accompanies malignant progression to human and murine retinoblastoma. Mol Carcinog. 2006;45(5):333–43. doi: 10.1002/mc.20179. [DOI] [PubMed] [Google Scholar]
- 12.Windle JJ, Albert DM, O’Brien JM, et al. Retinoblastoma in transgenic mice. Nature. 1990;343(6259):665–9. doi: 10.1038/343665a0. [DOI] [PubMed] [Google Scholar]
- 13.Gallie BL, Campbell C, Devlin H, et al. Developmental basis of retinal-specific induction of cancer by RB mutation. Cancer Res. 1999;59(7 Suppl):1731s–1735s. [PubMed] [Google Scholar]
- 14.Chen D, Livne-bar I, Vanderluit JL, et al. Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death-resistant cell-of-origin in retinoblastoma. Cancer Cell. 2004;5(6):539–51. doi: 10.1016/j.ccr.2004.05.025. [DOI] [PubMed] [Google Scholar]
- 15.MacPherson D, Sage J, Kim T, et al. Cell type-specific effects of Rb deletion in the murine retina. Genes Dev. 2004;18(14):1681–94. doi: 10.1101/gad.1203304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dannenberg JH, Schuijff L, Dekker M, et al. Tissue-specific tumor suppressor activity of retinoblastoma gene homologs p107 and p130. Genes Dev. 2004;18(23):2952–62. doi: 10.1101/gad.322004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amare Kadam PS, Ghule P, Jose J, et al. Constitutional genomic instability, chromosome aberrations in tumor cells and retinoblastoma. Cancer Genet Cytogenet. 2004;150(1):33–43. doi: 10.1016/j.cancergencyto.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 18.Knudson AG. Hereditary cancer: two hits revisited. J Cancer Res Clin Oncol. 1996;122(3):135–40. doi: 10.1007/BF01366952. [DOI] [PubMed] [Google Scholar]
- 19.Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1(2):157–62. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- 20.Lillington DM, Kingston JE, Coen PG, et al. Comparative genomic hybridization of 49 primary retinoblastoma tumors identifies chromosomal regions associated with histopathology, progression, and patient outcome. Genes Chromosomes Cancer. 2003;36(2):121–8. doi: 10.1002/gcc.10149. [DOI] [PubMed] [Google Scholar]
- 21.Chen D, Gallie BL, Squire JA. Minimal regions of chromosomal imbalance in retinoblastoma detected by comparative genomic hybridization. Cancer Genet Cytogenet. 2001;129(1):57–63. doi: 10.1016/s0165-4608(01)00427-7. [DOI] [PubMed] [Google Scholar]
- 22.Herzog S, Lohmann DR, Buiting K, et al. Marked differences in unilateral isolated retinoblastomas from young and older children studied by comparative genomic hybridization. Hum Genet. 2001;108(2):98–104. doi: 10.1007/s004390000450. [DOI] [PubMed] [Google Scholar]
- 23.Mairal A, Pinglier E, Gilbert E, et al. Detection of chromosome imbalances in retinoblastoma by parallel karyotype and CGH analyses. Genes Chromosomes Cancer. 2000;28(4):370–9. [PubMed] [Google Scholar]
- 24.Chow SN, Lin MC, Shen J, et al. Analysis of chromosome abnormalities by comparative genomic hybridization in malignant peripheral primitive neuroectodermal tumor of the ovary. Gynecol Oncol. 2004;92(3):752–60. doi: 10.1016/j.ygyno.2003.11.027. [DOI] [PubMed] [Google Scholar]
- 25.Shattuck TM, Kim TS, Costa J, et al. Mutational analyses of RB and BRCA2 as candidate tumour suppressor genes in parathyroid carcinoma. Clin Endocrinol (Oxf) 2003;59(2):180–9. doi: 10.1046/j.1365-2265.2003.01814.x. [DOI] [PubMed] [Google Scholar]
- 26.Kujawski M, Rydzanicz M, Sarlomo-Rikala M, Szyfter K. Rearrangements involving the 13q chromosome arm committed to the progression of laryngeal squamous cell carcinoma. Cancer Genet Cytogenet. 2002;137(1):54–8. doi: 10.1016/s0165-4608(02)00545-9. [DOI] [PubMed] [Google Scholar]
- 27.Hui AB, Pang JC, Ko CW, Ng HK. Detection of chromosomal imbalances in growth hormone-secreting pituitary tumors by comparative genomic hybridization. Hum Pathol. 1999;30(9):1019–23. doi: 10.1016/s0046-8177(99)90218-6. [DOI] [PubMed] [Google Scholar]
- 28.Yeager TR, DeVries S, Jarrard DF, et al. Overcoming cellular senescence in human cancer pathogenesis. Genes Dev. 1998;12(2):163–74. doi: 10.1101/gad.12.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perry A, Nobori T, Ru N, et al. Detection of p16 gene deletions in gliomas: a comparison of fluorescence in situ hybridization (FISH) versus quantitative PCR. J Neuropathol Exp Neurol. 1997;56(9):999–1008. doi: 10.1097/00005072-199709000-00005. [DOI] [PubMed] [Google Scholar]
- 30.Godbout R, Pandita A, Beatty B, et al. Comparative genomic hybridization analysis of Y79 and FISH mapping indicate the amplified human mitochondrial ATP synthase alpha-subunit gene (ATP5A) maps to chromosome 18q12-->q21. Cytogenet Cell Genet. 1997;77(3–4):253–6. doi: 10.1159/000134588. [DOI] [PubMed] [Google Scholar]
- 31.Bayani J, Thorner P, Zielenska M, et al. Application of a simplified comparative genomic hybridization technique to screen for gene amplification in pediatric solid tumors. Pediatr Pathol Lab Med. 1995;15(6):831–44. doi: 10.3109/15513819509027020. [DOI] [PubMed] [Google Scholar]
- 32.Munier FL, Thonney F, Balmer A, et al. Prognostic factors associated with loss of heterozygosity at the RB1 locus in retinoblastoma. Ophthalmic Genet. 1997;18(1):7–12. doi: 10.3109/13816819709057878. [DOI] [PubMed] [Google Scholar]
- 33.Gratias S, Schuler A, Hitpass LK, et al. Genomic gains on chromosome 1q in retinoblastoma: consequences on gene expression and association with clinical manifestation. Int J Cancer. 2005;116(4):555–63. doi: 10.1002/ijc.21051. [DOI] [PubMed] [Google Scholar]
- 34.Cutler DJ, Zwick ME, Carrasquillo MM, et al. High-throughput variation detection and genotyping using microarrays. Genome Res. 2001;11(11):1913–25. doi: 10.1101/gr.197201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2(8):RESEARCH0032. doi: 10.1186/gb-2001-2-8-research0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin M, Wei LJ, Sellers WR, et al. dChipSNP: significance curve and clustering of SNP-array-based loss-of-heterozygosity data. Bioinformatics. 2004;20(8):1233–40. doi: 10.1093/bioinformatics/bth069. [DOI] [PubMed] [Google Scholar]
- 37.Diskin S, Eck T, Greshock J, Mosse YP, Naylor T, Stoeckert JC, BL Weber, Maris J, Grant G. STAC: A method for testing the significance of DNA copy-number aberrations across multiple array-CGH experiments. Genome Research. 2006 doi: 10.1101/gr.5076506. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hagstrom SA, Dryja TP. Mitotic recombination map of 13cen-13q14 derived from an investigation of loss of heterozygosity in retinoblastomas. Proc Natl Acad Sci U S A. 1999;96(6):2952–7. doi: 10.1073/pnas.96.6.2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kawakami M, Staub J, Cliby W, et al. Involvement of H-cadherin (CDH13) on 16q in the region of frequent deletion in ovarian cancer. Int J Oncol. 1999;15(4):715–20. doi: 10.3892/ijo.15.4.715. [DOI] [PubMed] [Google Scholar]
- 40.Zheng L, Lee WH. Retinoblastoma tumor suppressor and genome stability. Adv Cancer Res. 2002;85:13–50. doi: 10.1016/s0065-230x(02)85002-3. [DOI] [PubMed] [Google Scholar]
- 41.McCarroll SA, Altshuler DM. Copy-number variation and association studies of human disease. Nat Genet. 2007;39(7 Suppl):S37–42. doi: 10.1038/ng2080. [DOI] [PubMed] [Google Scholar]
- 42.Shelling AN, Ferguson LR. Genetic variation in human disease and a new role for copy number variants. Mutat Res. 2007;622(1–2):33–41. doi: 10.1016/j.mrfmmm.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 43.Laurie NA, Donovan SL, Shih CS, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444(7115):61–6. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]