

Abstract
Smoke inhalation injury continues to increase morbidity and mortality in burn patients in both the third world and industrialized countries. The lack of uniform criteria for the diagnosis and definition of smoke inhalation injury contributes to the fact that, despite extensive research, mortality rates have changed little in recent decades. The formation of reactive oxygen and nitrogen species, as well as the procoagulant and antifibrinolytic imbalance of alveolar homeostasis, all play a central role in the pathogenesis of smoke inhalation injury. Further hallmarks include massive airway obstruction owing to cast formation, bronchospasm, the increase in bronchial circulation and transvascular fluid flux. Therefore, anticoagulants, antioxidants and bronchodilators, especially when administered as an aerosol, represent the most promising treatment strategies. The purpose of this review article is to provide an overview of the pathophysiological changes, management and treatment options of smoke inhalation injury based on the current literature.
Keywords: acute lung injury, anticoagulants, antioxidants, β2-agonists, carbon monoxide, cyanide, nitric oxide, reactive oxygen species
Smoke inhalation injury is generally defined as the inhalation of thermal or chemical irritants. With more than 23,000 injuries and 5000–10,000 deaths per year in the USA alone, smoke inhalation injury represents a major cause of morbidity and mortality in burn patients [1]. In addition, disasters such as the Station Nightclub Fire in Rhode Island in 2005 [2], or the devastating terrorism attacks on the World Trade Center in New York (NY, USA) [3,4] and the Pentagon in Washington (DC, USA) [5] in 2001 are associated with a high incidence of inhalation injuries. Among the 790 injured survivors from the World Trade Center attack, for example, 49% suffered from inhalation injury caused by toxic compounds in the smoke and dust [3,4]. Compared with isolated burns, a combined injury with smoke inhalation is associated with increases in fluid requirements [6], the incidence of pulmonary complications [7] and mortality [7–9]. It is important to remember however that smoke inhalation injury is not ‘only’ an adjunct to burn trauma; it is an independent injury in and of itself.
According to the WHO, more than 1 billion people develop airway and pulmonary inflammation as a result of inhaled smoke from indoor cooking fires, forest fires and burning of crops [10,11]. In addition, smoke toxicity is increasing because industrial products have shifted from woods and natural materials towards lighter construction materials, synthetics and petrochemicals, which ignite and burn two- to three-times hotter and faster. Thus, the probability that fire victims will breath in smoke and toxic gases is increased, because they have less time to escape.
While there has been remarkable progress in the treatment of cutaneous burns in recent decades, mortality rates of patients with inhalation injury have changed little in the past 20 years [12]. The heterogeneity of smoke and the resulting differences in clinical symptoms may be one reason why a definition and specific diagnostic criteria of smoke inhalation injury are still lacking. The purpose of this review article is to provide an overview of the pathophysiological changes, management and treatment options of smoke inhalation injury based on the current literature.
Pathophysiology & clinical symptoms
Smoke is composed of a gas phase and a particle phase. Particle size and tidal volume determine their distribution in the lung. Physiologically, the nasopharynx clears the inspiratory air of the majority of particles with a diameter larger than 5 μm [13]. During a fire, however, victims (both conscious and unconscious) breathe through the mouth owing to the nasopharyngeal irritation. As a result, particle deposition in the airway is much greater, causing progressive cellular injury and severe lung injury. The gas phase causes predominantly proximal airway and local damage, although some long-acting oxidants are able to reach distal lung tissues.
Smoke inhalation injury can be divided into three different types of injury [12]:
Thermal injury, which is mostly restricted to the upper airway (exception: blast injury or steam inhalation)
Chemical irritation of the respiratory tract
Systemic toxicity owing to toxic gases
The location of the injury depends on the ignition source, the size of the particles in the smoke, the duration of exposure and the solubility of the gases. Based on the predominant localization, smoke inhalation injury is classified into injuries of [14]:
The upper airway
The tracheobronchial system or lower airway
The lung parenchyma
Systemic toxicity
This classification will be used in the following sections to represent the different effects of smoke inhalation injury. Figure 1 summarizes the pathogenesis of lung injury following smoke inhalation.
Figure 1. Proposed pathophysiology of smoke inhalation injury.

iNOS: Inducible nitric oxide synthase; NF: Nuclear factor; nNOS: Neuronal nitric oxide synthase; PARP: Poly(ADP ribose) polymerase; RNS: Reactive nitrogen species; ROS: Reactive oxygen species.
Reproduced with kind permission from Frontiers in Bioscience [27].
Upper airway
Owing to highly efficient heat exchange in the oro- and nasopharynx [15], the leading injury in the upper airway, above the vocal cords, is caused by thermal injury. In accordance to burns of other body areas, heat destroys the epithelial layer, denatures proteins and activates the complement cascade leading to the release of histamine and the formation of xanthine oxidase [16]. This enzyme catalyses the breakdown of purins to uric acid and thereby releases reactive oxygen species (ROS) such as superoxide [17]. Superoxide represents a highly reactive molecule, which is physiologically stabilized by its formation to hydrogen peroxide catalyzed by superoxide dismutase [18]. At the same time, nitric oxide (NO) formation by endothelial cells is increased by histamine stimulation. In this case, the reaction with NO to form reactive nitrogen species (RNS), such as peroxynitrite, is faster [18]. Both ROS and RNS cause an increased permeability of endothelium for proteins, resulting in edema formation. In addition, eicosanoids and IL-8 are released after injury, leading to the attraction of polymorphonuclear cells [19,20], which amplify the inflammatory process, for example by ROS production. In parallel, activation of pulmonary C-fiber receptors by inflammation and irritants of smoke causes vasodilation by increasing NO production, further aggravating edema formation [21].
While the immediate injury results in erythema, ulceration and edema (Figure 2), clinical symptoms, such as stridor, dyspnea or increased work of breathing might not be obvious until the edema is sufficiently large enough to impair the airway diameter significantly. This time difference can amount up to 18 h or even longer [22]. In the case of combined burn and smoke inhalation injury, however, the aggressive fluid administration necessary to treat the burn shock promotes edema formation [23]. In addition, accompanying face and neck burns might cause anatomic distortion or external compression of the upper airway, further complicating proper airway management [22]. In addition to the inflammation, damage of ciliary function impairs the physiological cleaning process of the airway, resulting in an increased risk of bacterial infections for several weeks. Furthermore, the increased production of viscous secretions causes distal airway obstruction and atelectasis, thereby impairing pulmonary gas exchange [22,24].
Figure 2. Bronchoscopic finding after smoke inhalation injury.
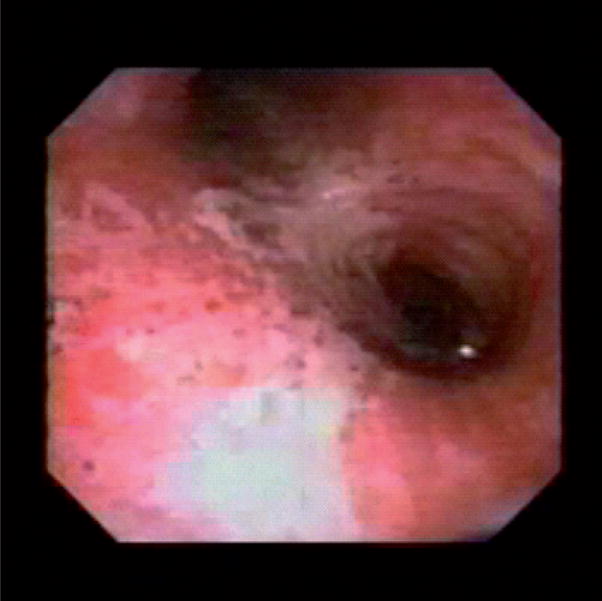
Erythema, edema and ulceration (view into the left and right main bronchi).
Tracheobronchial system
With rare exceptions, such as the inhalation of steam, injury to the tracheobronchial area is usually caused by chemicals in smoke. The airway is richly innervated by vasomotor and sensory nerve endings [25]. Smoke inhalation stimulates these nerves to release neuropeptids [26], which are potent bronchoconstrictors. Under physiological conditions, the mucosa produces neutral endopeptidases, which neutralize these toxic agents. Owing to severe cellular damage, this neutralization is lost [22]. Neuropeptidases also attract and activate neutrophils, resulting in the production of ROS [20]. Simultaneously, the activity of neuronal NO synthases (nNOS, NOS-1) is upregulated by neuropeptides [27]. NOS catalyze the formation of NO and L-citrulline from the amino acid L-arginine in a complex five-electron redox reaction. Three different isoforms of NOS have been identified in mammals: nNOS and endothelial NOS (eNOS, NOS-3) are constitutive isoforms, while the inducible isoenzyme (iNOS, NOS-2) can be upregulated by oxidative stress and systemic inflammation [28]. Notably, in case of substrate (L-arginine) or cofactor limitation, for example under pathophysiological conditions, NOS can also produce superoxide [29]. NO reacts with ROS such as superoxide to form RNS, for example peroxinitrite. Although this highly cytotoxic RNS represents the most cited example, most of the ROS and RNS are able to interact with proteins, DNA or lipids by oxidation, nitration or nitrosylation, causing the inactivation of key enzymes such as the glycolytic enzyme glyceraldehydes-3-phosphate dehydrogenase or produce a single-strand breakage in DNA [30]. DNA damage leads to the formation of poly(ADP ribose) polymerase (PARP), a nuclear enzyme involved in DNA repair [31,32]. It has been shown that PARP activation can be induced by NO and its toxic products such as RNS [33].
Although the physiological function of PARP is beneficial, PARP activation depletes cellular ATP levels by forming ADP, possibly resulting in cellular dysfunction and apoptosis [32,34]. In addition, PARP activates nuclear factor κB (NF-κB), independent of its action to form PAR [35,36]. NF-κB is known to upregulate IL-8 [37], resulting in the attraction and activation of polymorphonuclear cells and consecutively in the production of ROS. NF-κB also stimulates iNOS formation [38], thereby accelerating the production of NO. NO and ROS react to form RNS, leading to DNA damage and PARP activation, and thereby a positive feedback loop is established [27].
Histologically, there is evidence of damage to the mucosal lining and peribronchial inflammation [14,22,39–42]. Loss of bronchial epithelium results in a profuse transudate with a high protein content. While these secretions form foam material during the early response, this transudate/exudate mixture solidifies during the pathogenesis, forming obstructive material, which might occlude lower airways. These airway ‘casts’ (Figure 3) are composed of mucus secretions, denuded airway epithelial cells, inflammatory cells and fibrin [24]. Especially when volume-controlled ventilator settings are selected, obstruction can promote a barotrauma of still-ventilated areas. Besides massive airway obstruction, further hallmarks of smoke inhalation injury include bronchospasm, increases in bronchial circulation and transvascular fluid flux [43]. A tenfold increase in bronchial blood flow within 20 min after smoke inhalation was reported in an experimental study. In addition, the same animals demonstrated a sixfold increase in pulmonary transvascular fluid flux [44]. Both of these changes promote the development of airway edema. Further clinical symptoms include persistent coughing and wheezing, soot-containing airway secretions, increased work of breathing resulting in hypoventilation, erythema, hyperemia and increased pulmonary shunting [22]. The latter is caused by the loss of hypoxic pulmonary vasoconstriction owing to high NO production [14].
Figure 3. Isolated airway cast.

The formation of mucus secretions, denuded airway epithelial cells, inflammatory cells and fibrin leads to subtotal or total occlusion of distal as well as main airways.
Lung parenchyma
The damage of lung parenchyma following smoke inhalation injury is delayed. The time difference between the initial trauma and a decrease in arterial oxygen tension to inspiratory oxygen fraction (PaO2:FiO2) is correlated with the severity of the lung injury, and represents a consequence of changes described for the upper airway and the tracheobronchial system [14]. Alveolar injury is characterized by alveolar collapse and atelectasis owing to increased transvascular fluid flux, a lack of surfactant and a loss of hypoxic pulmonary vasoconstriction, resulting in impaired oxygenation. In addition, a severe imbalance in alveolar hemostasis, increased procoagulatory and decreased antifibrinolytic activity, leads to massive fibrin deposition in the airway, which prevents regular ventilation and causes a ventilation/perfusion mismatch [43]. By inhibiting surfactant, fibrin itself leads to atelectasis [45] and can attract inflammatory cells [46].
Activated neutrophils play a central role in the pathogenesis of smoke inhalation injury. First, they initiate and continuously support the pathological pathway described previously by producing ROS and releasing proteases, such as elastase [27]. Second, neutrophils activated in the bronchial circulation are drained into the pulmonary microvasculature [14]. Since activated neutrophils are stiff owing to F-actin activation, they are not able to traverse the pulmonary capillaries as usual by changing their shape [14]. Instead, they adhere to the alveolar capillary membrane via L-selectin [47], causing direct damage by releasing proteases and ROS. Our group and our colleagues in Boston (MA, USA) were able to demonstrate that ligation of the bronchial artery reduces parenchymal injury following smoke inhalation [48–52]. The important role of activated neutrophils in the pathogenesis of smoke inhalation injury was verified by Basadre et al. [53]. In leucocyte-depleted sheep, a high percentage of the described response to smoke inhalation was prevented from occurring. However, since patients with smoke inhalation injury are at high risk for pulmonary infections, the depletion of leukocytes represents only a mechanistic rather than a therapeutic approach.
Systemic response & toxicity
Systemic response
The systemic response to smoke inhalation injury is characterized by a sytemic inflammatory response syndrome (SIRS), caused at least in part by systemic circulation carrying proinflammatory mediators via the lung through the bronchial and pulmonary vasculature to systemic organs [48–52]. The reduction in systemic oxygen delivery owing to elevated carboxyhemoglobin (COHb) levels and a decreased cardiac function might represent further potential mechanisms [54]. After combined burn and smoke inhalation injury, the hypermetabolic state is characterized by increased oxygen consumption and a shift of arterial blood flow from the intestine to soft tissues or muscles, thereby increasing the risk of organ failure owing to bacterial translocation [55]. In addition, inflammatory mediators released in the lung lead to increased systemic vascular permeability and oxidative stress [56]. In the absence of burn injury, these systemic effects might occur with a delay of 24–48 h [22].
The interaction between burn and smoke inhalation injury was investigated by our research group in an established ovine model [57]. Pulmonary vascular permeability as represented by increases in lung lymph flow and bloodless lung wet-to-dry ratio was caused by smoke inhalation injury. The combination with burn injury increased permeability, while isolated burn injury showed no statistically significant differences compared with sham animals. Another interesting observation of this study was that burn injury caused an immediate myocardial depression, which was also seen in combined burn and smoke inhalation injury and is probably related to hypovolemia owing to fluid losses through the burned area. By contrast, myocardial dysfunction occured approximately 18–24 h after isolated smoke inhalation and is mostly part of the SIRS.
The increased amounts of ROS after smoke inhalation injury originate from various origins [58]:
Metabolism of adenosine monophosphate in ischemic tissues to hypoxanthine and its subsequent reaction with xanthine oxidase, leading to excessive production of superoxide and hydrogen peroxide;
During oxidative phosphorylation in the mitochondrial respiratory chain;
From the nicotinamide adenine dinucleotide phosphate oxidase system in several different cell types; for example, in neutrophils;
Breakdown of arachidonic acid to form prostaglandins and leukotrienes [59].
Simultaneously, antioxidative protection mechanisms may be adversely affected by the disease process. Accordingly, we reported a marked reduction in antioxidant levels in severely burned patients [60]. Antioxidant deficiency potentially occurs for multiple reasons, including redistribution of antioxidants to immunoactive tissues, dilution owing to fluid administration, insufficient intake and losses through biological fluids (exudates, drains and chyle) [61].
Systemic toxicity
A direct systemic effect of smoke inhalation injury is caused by inhalation of toxic gases during the combustion of organic and inorganic substances. With respect to morbidity and mortality, the two most relevant gases are carbon monoxide (CO) and cyanide. Other toxic gases and their symptoms are listed in Table 1.
Table 1.
Selected toxic compounds of smoke: materials, sources and their pathophysiological effects.
| Toxic compound | Material | Source | Pathophysiology |
|---|---|---|---|
| Arolein/propenal | Acrylics | Aircraft windows, textiles, wall coverings | Severe irritation of upper respiratory tract, mucosa necrosis and death within 10 min with concentrations over 50 ppm [22] |
| Cellulose | Cotton, jute, paper, wood | ||
| Polypropylene | Carpeting, upholstery | ||
|
| |||
| Aldehydes | Acrylics | Aircraft windows, textiles, wall coverings | Corrosive, denatures proteins; formaldehyde: denatures ribonucleic acid |
| Cellulose | Cotton, jute, paper, wood | ||
| Polyamine resins | Household, kitchen goods | ||
|
| |||
| Ammonia | Polyamide | Carpeting, clothing | Airway irritant leading to cough, increasing secretions and bronchoconstriction; ammonium hydroxide: tissue necrosis |
| Polyamine resins | Household, kitchen goods | ||
| Polyurethane | Insulation, upholstery | ||
| Silk, wool | Blankets, clothing, fabrics, furniture | ||
|
| |||
| Carbon monoxide | All materials | All combustible products | Tissue hypoxia, organ failure, death within 1 h with concentrations of 80–90% [14] |
|
| |||
| Hydrogen chloride | Polyester | Clothing, fabrics | Mucosa necrosis and acute bronchitis |
| Polyvinyl chloride | Floor, furniture, upholstery, wall, wire/pipe coating | ||
|
| |||
| Hydrogen cyanide | Fire retardants | Polymeric material | Tissue hypoxia, organ failure, death; death possible with concentrations over1 μg/ml [14] |
| Polyacrylonitrile | Appliances, engineering, plastics | ||
| Polyamide | Carpeting, clothing | ||
| Polyamine resins | Household, kitchen goods | ||
| Polyurethane | Insulation, upholstery | ||
| Silk, wool | Blankets, clothing, fabrics, furniture | ||
|
| |||
| Hydrogen sulfide | Rubber | Tires | Airway irritant, corrosive |
| Silk, wool | Blankets, clothing, fabrics, furniture | ||
|
| |||
| Phosgene | Polyvinyl chloride | Floor, furniture, upholstery, wall, wire/pipe coating | Mucosa necrosis, primary in the small airways and alveoli |
|
| |||
| Sulfur dioxide | Rubber | Tires | Strong irritant to eyes and airways, lower airway injury and pulmonary edema |
Carbon monoxide
Carbon monoxide is not only one of the most frequent immediate causes of death following smoke inhalation injury, but also of poisoning deaths in the USA. It accounts for approximately 15,000 emergency room visits and 500 unintentional deaths each year [62]. CO is an odorless, colorless gas with an affinity for hemoglobin more than 200-times higher than that of oxygen [63]. Accordingly, inhalation of only 0.1% CO mixture can result in a life-threatening COHb level of 50% [14]. Oxygen delivery to organs is further decreased by a left shift of the oxygen–hemoglobin dissociation curve, impairing tissue oxygen availability [64]. In addition, CO inhibits hepatic cytochromes, leading to mitochondrial oxidative stress and membrane damage owing to lipid peroxidation [63].
Clinical symptoms vary depending on the concentration of CO, the duration of exposure and pre-existing morbidity of the patient. Generally, a COHb level of more than 90% may lead to immediate cardiac arrest [65]. Clinical symptoms mainly involve neurological and cardiovascular manifestations. Owing to a lack of alternatives, diagnosis of CO poisoning is still based on the measurement of COHb levels. However, the strength of the correlation between COHb levels and the severity of poisoning, the prognosis or the choice of therapy is discussed controversially [66]. Cellular mechanisms, such as caspase-mediated apoptosis, may play an additional role in CO poisoning [67]. Importantly, it has to be taken into account that the usual pulse oximeter can not differentiate between COHb and oxyhemoglobin and that venous blood underestimates the arterial COHb content [68]. However, there are now devices available (Masimo Set® Rad 57™) enabling physicians or paramedics to determine COHb levels noninvasively at the injury scene. In contrast to usual pulse oximeters, Masimo Set Rad 57 measures seven or more instead of two wavelengths.
Cyanide
Hydrogen cyanide (HCN) represents the gaseous form of cyanide (CN), which is a colorless gas with the odor of bitter almonds; however, HCN is difficult to detect at the site of a fire, as most inhalation injuries represent mixed intoxications. Notably, CN is a normal human metabolite as reflected by plasma levels of 0.3 mg/l in nonsmokers and 0.5 mg/l in smokers [69]. All cells, but mostly those in the liver, are able to convert CN into thiocyanide, which is excreted in the urine via the enzyme rhodanase. In case of large amounts of CN, this system can be overcome, especially in burned or traumatized patients who are hypovolemic [22]. Toxicity of CN is based on hypoxic states owing to the reversible inhibition of cytochrome c oxidase, which is the terminal oxidase of the respiratory chain. The definition of fatal blood levels, however, varies from 1–3 to 5 mg/l [70]. The lack of a precise definition may be one reason for controversial discussions regarding the role of CN poisoning within smoke inhalation injury. Advocates cite studies reporting increased CN plasma levels in nonsurvivors as compared with survivors of fire disasters [71,72]. By contrast, opponents argue that CN is likely to be rapidly consumed in fires. Davies and colleagues described CN concentrations in the smoke of 250 ppm, which went down to below 10 ppm at 8 min [73]. Neither the short-term exposure limit (15 ppm), nor the short-term lethal concentration (350 ppm) were exceeded [70]. Nevertheless, CN originates from numerous compounds during combustion (Table 1), resulting in an increased probability of CN intoxication in a fire victim. At the same time, diagnosis of CN poisoning represents a challenge at the injury scene, as generally described symptoms (e.g., increased lactate levels and base deficit, or metabolic acidosis) may also be caused by asphyxiation, under-resuscitation, CO poisoning or associated traumatic injury. However, owing to the high probability of its presence at a fire scene (especially in case of combustion of certain materials such as plastics), in combination with the difficult diagnosis and its lethal potential, CN intoxication should be carefully considered in every patient with smoke inhalation injury.
Management
At the injury scene
The first priority at the injury scene is rescue of the victim from the source of fire to stop the exposure time. This is usually the responsibility of firefighters. In order to reduce COHb levels as soon as possible, a high flow of 100% oxygen should be administered via facemask immediately. The next step includes a short but careful body check to estimate the extent of smoke inhalation and to assess accompanying injuries such as burns and/or trauma. In addition, it is important to determine whether the victim has been exposed to an explosion, since this can cause barotrauma to the lung. If possible, information about comorbidities should be obtained. Indications of inhalation injury are summarized in Box 1. Usual cardiopulmonary monitoring (electrocardiogram, pulse oxymeter and noninvasive blood pressure) should be established.
Box 1. Indications and symptoms of inhalation injury.
Facial and neck burns
Burned lips and vibrissae
Soot-containing airway secretions
Pathological respiration patterns (coughing, stridor and hoarseness)
Dyspnea
Cyanosis
Neurological symptoms (current or past unconsciousness, dizziness, nausea and vomiting)
After these basic measures, a decision needs to be made on how best to secure the airway of the patient. On the one hand, the risk of a rapidly developing airway edema has to be taken into account even if no dyspnea is present. On the other hand, endotracheal intubation itself, especially at the injury scene, is associated with undeniable risks such as esophageal intubation, aspiration, barotrauma or laryngeal trauma. Therefore, in the authors’ opinion, prophylactic endotracheal intubation is not generally advised in every patient and, depending on the technical skills, for every physician or paramedic. Nevertheless, it is important to consider that airway management will only become more difficult over time. Patients with heat and smoke inhalation injury combined with extensive face or neck burns definitely have to be intubated. In the case of oral burn without inhalation injury, an airway secured early represents the safest approach. However, victims with smoke inhalation injury but no facial or neck burns can be carefully observed and later be intubated, if necessary [22].
If the patient is endotracheally intubated, the tube should be carefully secured. Accidental removal of the endotracheal tube is easy and may be lethal. In cases of vocal cord damage, tracheostomy may be necessary to prevent further damage. The patient’s head should be elevated to minimize facial and airway edema. As a matter of course, hemodynamic stability is a prerequisite. Aerolized epinephrine or corticosteroids may be beneficial to reduce upper airway edema [22]. However, there is no conclusive evidence for the efficacy of these treatment strategies. After initial stabilization of the patient, it might be very helpful for further treatment to obtain information about the fire source, combustion products and the estimated duration of exposure. In case of specific intoxications, for example, HCN poisoning, recommended therapies should be started (please see the section entitled ‘Mechanical ventilation strategies’).
In-hospital treatment
After adequate airway management and arrival in the emergency room, the treatment of accompanying burn injuries or trauma usually represents the first, most immediate priority. Inhalation injury, by comparison, often develops with a latency of several hours. Nevertheless, airway management and oxygenation status of the patient, regardless if intubated or not, need to be reevaluated frequently to allow clinicians to react to the dynamic development of smoke inhalation injury [22]. After stabilization of cardiopulmonary hemodynamics and pulmonary gas exchange, the assumed diagnosis of smoke inhalation injury needs to be verified. However, as there are currently no uniform criteria available, diagnosis of smoke inhalation injury is usually a subjective decision based on a combination of history (please see the section entitled ‘At the injury scene’) and physical examination, which are confirmed by diagnostics [12]. The bronchoscopical examination of the airway represents the gold standard to detect a pathognomonic mucosal hyperemia. Chest radiographs may show signs of diffuse atelectases, pulmonary edema or bronchopneumonia. However, during the initial period, the degree of injury is usually underestimated based on the chest x-ray, as the injury is mainly confined to the airways [74].
Although many observational studies compared outcomes with various grading systems [75–77], there is still no uniform algorithm for assessing inhalation injury. As a result, no reliable indicators of progressive respiratory failure in patients with smoke inhalation injury have been identified so far [78,79]. This failure is largely explained by the extreme heterogeneity of the clinical presentation. In addition, the delay in the manifestation and development of acute lung injury (ALI) as a consequence of SIRS, initiated by accompanying burns or trauma, complicate the evaluation of the isolated effects of smoke inhalation. Useful tools to monitor smoke inhalation injury include frequent blood gas and sputum analyses [22].
Appropriate fluid resuscitation of patients with smoke inhalation injury is still subject to controversial debates. It has been demonstrated that smoke inhalation injury increases fluid requirements in burned patients [6,80]. This, of course, does not inevitably indicate that isolated smoke inhalation injury is associated with increased fluid requirements. By contrast, over-resuscitation may increase pulmonary microvascular pressures and might thereby lead to increased edema formation under the high permeability conditions in early lung injury [57,81]. In a retrospective study of more than 2346 trauma patients, Plurad et al. found an increased incidence of late post-traumatic acute respiratory distress syndrome (ARDS) with rising volumes of administered cristalloids and packed red blood cells [82]. Unfortunately, there is currently no evidence for the specific patient population of isolated smoke inhalation injury. Against the background of no proven benefit and the potential risk of detrimental effects, however, increased amount of fluids should be avoided in patients with isolated smoke inhalation injury. Instead, fluid resuscitation should be guided by urine output and hemodynamic parameters of the individual patient. Thereby, rather dynamic parameters, such as changes in pulse pressure, rather than static parameters, such as central venous or pulmonary artery occlusion pressures, might be helpful.
After acute therapy, treatment is mainly focused on three cornerstones: first, maintenance and restoration of a sufficient gas exchange. Thus, respiratory ventilator settings have to be adjusted frequently to guarantee the most efficient ventilatory support and to minimize ventilation-associated side effects, such as baro- or volutrauma (please see secion entitled ‘Nebulized treatments’). Second, vigorous bronchial toilet should be performed to clear the airway and avoid airway occlusion. Mucociliary action is extensively impaired owing to the extensive structural damage of heat and chemicals in the smoke [22]. In combination with an impaired cough reflex and increased tenacious secretions, this injury bears a high risk of occlusion of smaller airways, leading to atelectasis and infection [83]. Third, therefore, very careful infection surveillance is advised. Impairment of alveolar macrophage function and post-traumatic immunodeficiency also represent important risk factors, but a prophylactic administration of antibiotics is not recommended to avoid the development of resistant organisms [84].
Treatment
The following section provides a brief overview of current therapeutic strategies in respect to smoke inhalation injury. These recommendations are based on limited evidence from clinical and experimental trials as well as expert opinions resulting from clinical experience.
Mechanical ventilation strategies
Several years ago, mechanical ventilation was performed by using high inspiratory oxygen concentrations and tidal volumes of 10–15 ml/kg in a volume-controlled mode. However, “this approach may have violated the injunctive, first to do no harm (Primum non nocere)” [85]. During the last two decades, mechanical ventilation has been investigated extensively. Low tidal volume ventilation with associated permissive hypercapnia has been shown to effectively reduce ventilator-induced lung injury, and currently represents the standard of care [86]. The Assessment of Low Tidal Volume and Elevated End Expiratory Pressure to Obviate Acute Lung Injury (ALVEOLI) trial revealed no effects of higher positive end-expiratory pressure (PEEP) levels in ARDS patients [87]. In addition, prone positioning has been shown to have no beneficial effect on mortality, despite a transient improvement in oxygenation [88].
Although smoke inhalation predisposes patients to ALI and ARDS, especially in combination with burns, it remains to be determined if these results also apply to the specific patient population after smoke inhalation. In the ARDS Network Study concerning low tidal volume ventilation, for example, patients with burns of more than 30% total body surface area (TBSA) have been excluded. A direct transfer of these approaches may be limited in clinical practice owing to the unique clinical and pathophysiological features of smoke inhalation. Ventilation protocols differ not only between different burn centers but also between individual physicians. Apart from conventional pressure-controlled low tidal volume ventilation, multiple strategies for mechanical ventilation are currently used for the treatment of smoke inhalation injury, isolated as well as in combination with burns.
Airway pressure release ventilation (APRV), first described in 1987 by Stock and colleagues [89], is a time-triggered, pressure-limited, time-cycled ventilation mode [90]. It provides two levels of airway pressures during two time periods: a long, high-level and a short, low-level period. In this way, lower peak but higher mean airway pressures are produced compared with conventional ventilation strategies, resulting in lower tidal volumes and more effective alveolar recruitment. In addition, APRV allows spontaneous breathing during the whole inspiration–expiration cycle. Possible side effects include higher intrinsic PEEP, secondary to increased airway resistance and short expiratory times, resulting in hyperinflation of the lungs [90]. Two randomized, controlled trials in mechanically ventilated patients – not patients with smoke inhalation injury – revealed beneficial effects on oxygenation and lower end-inflation pressures [91,92]. However, there was no reduction in mortality. In conclusion, further research is necessary to determine whether APRV represents a beneficial approach for the ventilation of patients suffering from smoke inhalation injury.
The volumetric diffusive ventilator (VDR; Percussionaire Corp., Sandpoint, ID, USA) is a pneumatically powered, pressure-limited ventilator that stacks oscillatory breaths to a selected peak airway pressure by means of a sliding venturi called Phasitron®, resulting in low tidal volumes. Exhalation is passive and a level of continuous positive airway pressure can be selected. In addition, VDR re-establishes physiologic diffusive gas exchange, while standard ventilation modes induce a convective gas exchange [93]. In contrast to APRV, the effects of VDR have been studied in patients with smoke inhalation injury. A prospective clinical analysis revealed an improved gas exchange and a decrease in peak pressures [94]. In addition, a retrospective study in 330 patients with inhalation injury even reported a lower mortality rate [95]. While these studies compared the VDR to high-volume ventilatory strategies, data regarding a comparison with modern low tidal volume ventilation are still lacking. This may represent one reason why VDR is not universally accepted. Another factor might be that the VDR differs from other ventilators, and therefore requires special training. In addition, tidal and minute volumes cannot be monitored and humidified air, as well as nebulized saline, are necessary to prevent airway desiccation [93].
High-frequency oscillatory ventilation (HFOV) uses extremely low tidal volumes of 1–2 ml/kg applied with high frequencies of 3–15 Hz and mean airway pressures of 30–40 cmH2O. This ventilation regime prevents alveolar trauma [96] and leads to improved lung recruitment, thereby improving oxygenation, allowing lower inspiratory oxygen concentrations and consequently limiting oxygen toxicity [97]. However, its use after smoke inhalation injury may be limited by copious secretions and small airway obstruction. In addition, gas trapping and hypercapnia will be hard to control using HFOV [97]. Accordingly, HFOV failed to improve PaO2:FiO2 ratios in patients with combined burn and smoke inhalation injury, while it improved oxygenation in isolated burn patients within 8 h [97,98]. Furthermore, the administration of nebulized adjunctive therapies is not possible with this ventilation strategy.
Taken together, all these ventilation modes, with the HFOV as an extreme, apply low tidal volumes and high mean airway pressures or PEEP, respectively, to improve oxygenation and airway recruitment. Patients with inhalation injury suffer from increased mucous secretions and even airway cast formation to a greater extent than ARDS patients in general. Therefore, clearance of secretions represents an essential factor within the therapeutic regime. However, airway clearance is best supported by high tidal volumes and low PEEP [81], thereby contradicting the ventilation strategies for improved oxygenation. As a consequence, physicians need to balance these competing objectives based on the individual situation of the patient until future studies might provide more evidence.
External arterial venous CO2 removal represents a unique form of CO2 removal, which is driven by endogenous arterial pressure [99]. Several studies in animal models of smoke inhalation injury revealed a reduction in morbidity and mortality [100–102]. However, clinical evidence is lacking.
In conclusion, mechanical ventilation of patients after smoke inhalation injury will further represent a tightrope walk between providing sufficient oxygenation and causing as little collateral harm as possible. While there are several specific ventilation devices and strategies available, large, randomized, controlled multicenter studies are warranted to clarify their potential usefulness in patients with smoke inhalation injury.
Nebulized treatments
Against the background that the lung is the primary injured organ following smoke inhalation injury and that mechanical ventilation is frequently necessary, the administration of therapeutic compounds via nebulization directly to the affected organ seems to be more than reasonable. Based on the complex pathogenesis of smoke inhalation injury, drugs with different mechanisms of action, such as bronchodilatators, anticoagulants, antioxidants and corticosteroids have been investigated. In each case, however, the key to any successful aerosol therapy is the consistent delivery into the lung and to the distal airways.
Bronchodilators, such as β2-agonists, improve respiratory mechanics by decreasing airflow resistance and peak airway pressures. This results in improved dynamic compliance [103]. In addition, β2-agonists provide anti-inflammatory properties, represented by a decrease in inflammatory mediators such as histamine, leukotrienes and TNF-α [104,105]. Finally, β2-agonists are associated with improved airspace fluid clearance and stimulation of epithelial repair [106]. While clinical evidence for ALI/ARDS in general is present [107,108], the literature discussing smoke inhalation is limited. Using an established ovine model of combined burn (40% cutaneous third-degree burn) and smoke inhalation injury (48 breath of cotton smoke, average COHb of 75% injury), our research group was able to show a reduction of pulmonary vascular permeability, pulmonary edema and airway pressures, as well as an improvement of the PaO2:FiO2 ratio by continuous administration of nebulized albuterol at 20 or 40 mg/h compared with placebo treatment [109]. Clinical evidence is restricted to retrospective data in children. Continuous nebulized albuterol (10–40 mg/h) in children with smoke inhalation injury and a PaO2:FiO2 ratio less than 200 mmHg led to improved oxygenation and lung compliance in the first 72 h after treatment. No serious adverse events (e.g., tachycardia or hypokalemia) have been reported [110].
Inhaled NO is thought to improve oxygenation and to reduce increased pulmonary vascular resistance by reversing the ventilation-perfusion mismatch in ARDS owing to selective vasodilation in well-ventilated lung areas. Experimental studies in animals with smoke inhalation injury revealed a consistent reduction in pulmonary hypertension but variable results regarding pulmonary shunting [111,112]. Single-center case studies in patients with combined burn and smoke inhalation injury demonstrated a significant increase in the PaO2:FiO2 ratio, with no additional advantage of doses exceeding 20 ppm [113,114]. Several randomized, controlled trials as well as recent meta-analyses in the general population of ARDS patients revealed only a positive effect on the patient’s oxygenation for 24 h (in some studies until 48 h) [115–117], but no advantages in respect to mortality, ventilator-free days, duration of ventilation or pulmonary hypertension could be verified. On the contrary, there was even a strong trend towards increased mortality in patients receiving NO as compared with placebo treatment [115].
Against the evidence of beneficial effects of NO synthesis inhibition [27,29,118], inhaled administration of NO for the treatment of smoke inhalation injury seems questionable. While a possibly increased production of RNS is only hypothetical at this time point, there are several known, unwanted effects of nebulized NO administration. Long-term treatment might lead to the diffusion of NO into poorly ventilated areas, abolishing the selective pulmonary vasoconstriction and consecutively increasing pulmonary shunt volume. In addition, surfactant inhibition, edematous changes and continuing fibrosis may over-ride any benefit of inhaled NO therapy. Although findings are controversial, renal dysfunction and methemoglobinemia have also been described [115]. Based on the current literature, the standard treatment of smoke inhalation injury with inhaled NO cannot be recommended. However, short-term inhaled administration of NO might be considered as a rescue treatment to improve oxygenation for a short period in patients with acute, life-threatening hypoxemia [116].
Owing to increased procoagulatory activity following smoke inhalation injury [43], the aerolized administration of anticoagulants seems to be more than promising. In an ovine model, Brown et al. first described a reduction of mortality after smoke inhalation induced ARDS by aerolized heparin in 1988 [119]. Interestingly, Murakami and colleagues reported that heparin nebulization had only partial or no beneficial effects in ovine ARDS after combined burn and smoke inhalation injury [120]. This might be explained by the lower levels of antithrombin in bronchoalveolar lavage fluid after combined burn and smoke inhalation injury, as the effects of heparin depend on the binding to antithrombin, thereby enhancing the ability of antithrombin to inhibit fibrin and factor Xa by 2000–6000-fold [114]. In accordance, combined nebulization of heparin and recombinant human antithrombin (rhAT) in the same model significantly improved pulmonary function by reducing airway obstruction [121]. However, heparin binding to antithrombin inhibits the anti-inflammatory effects of antithrombin [43]. In addition, aerolized antithrombin administration did not affect the reduced systemic plasma levels of antithrombin [121]. Recently, our research group was able to demonstrate that the combination of intravenous rhAT and nebulized heparin effectively attenuated pulmonary injury following combined burn and smoke inhalation injury [122]. In children with combined burn and smoke inhalation injury, nebulization of heparin and N-acetylcysteine significantly decreased re-intubation rates, the incidence of atelectasis and mortality [123]. Notably, these children also received transfusions of fresh frozen plasma, a rich source of antithrombin. These promising results, however, need to be verified in randomized, controlled multicenter trials.
As mentioned earlier, following combined burn and smoke inhalation injury, the oxidative–antioxidative balance is disturbed by an increase in ROS and a parallel decrease in antioxidants [124]. Accordingly, the antioxidant Vitamin E or α-tocopherol is markedly reduced in patients with major burns (>50% TBSA) and combined smoke inhalation injury [60]. Our study group reported that prophylactic administration of α-tocopherol (5 mg/kg orally) 24 h prior to injury attenuated the decrease in pulmonary function and the increase in vascular permeability that occurs after combined burn and smoke inhalation injury in sheep [125]. Of special interest are two studies in sheep after combined 40% TBSA burn and smoke inhalation injury, showing the practicability and efficacy of local antioxidant administration to the lung by nebulization [126,127]. Both α- and γ-tocopherol attenuated the progressive decrease in pulmonary function represented by higher PaO2:FiO2 ratios compared with control animals. In addition, nebulized γ-tocopherol decreased lung lymph flow, lung wet-to-dry weight ratio, and markers of oxidative and nitrosative tissue injury more effectively than α-tocopherol. Plasma levels of γ-tocopherol were not increased, suggesting a successful local administration potentially avoiding systemic side effects. The superiority of γ-tocopherol over α-tocopherol might be based on the higher ability of γ-tocopherol to scavenge RNS as well as ROS [128].
Intravenous treatments
While the efficacy of aerolized compounds always depends on consistent delivery into the lung and to the distal airways, the intravenous injection of a compound represents a relatively sure way of administration. Accordingly, the following section discusses some of the intravenous, therapeutic approaches of smoke inhalation injury.
The progressively developing upper airway edema induced by smoke inhalation injury represents one of the major indications for endotracheal intubation. Aerolized epinephrine or corticosteroids as therapeutic approaches have been proposed [22]; however, conclusive evidence for these treatment strategies is still missing. One of the reasons might be the varying efficacy of the aerolized drug delivery, especially in emergency situations. Therefore, intravenous administration of corticosteroids should be the treatment of choice.
Vitamin C has been shown to decrease oxidative damage in several experimental [129,130] and clinical [131,132] trials in different critical diseases. Unfortunately, to the best of our knowledge, there are no studies investigating the effects of Vitamin C administration on isolated smoke inhalation injury. However, Tanaka and colleagues revealed very impressive results in a randomized, controlled trial by adding 66 mg/kg/h Vitamin C to the continuous infusion in patients who had over 30% TBSA burns [132]. Fluid requirements, body weight gain, wound edema and duration of mechanical ventilation were significantly reduced. Notably, 73% of the included patients in this study were diagnosed with inhalation injury after admission to the hospital, with equal distribution between groups. A subgroup analysis revealed improved oxygenation, represented by higher PaO2:FiO2 ratios, beginning at 18 h after injury. In addition, duration of mechanical ventilation was reduced in patients receiving Vitamin C (12 ± 9 days) as compared with the control group (21 ± 16 days). No differences were seen in PEEP level, inspiratory oxygen fraction or the incidence of pneumonia. Nevertheless, it must be explicitly stated that no direct data exist to confirm a beneficial effect of intravenous Vitamin C treatment on patients with smoke inhalation injury.
As mentioned in the section ‘Nebulized treatments’, combined burn and smoke inhalation injury causes an acquired antithrombin deficiency, which represents an independent predictor of length of hospital stay and mortality [133,134]. In addition to its anticoagulatory properties, antithrombin provides anti-inflammatory effects. Antithrombin inhibits the inflammatory signal transduction in and promotes prostacyclin release from endothelial cells. Thereby, anti-thrombin directly attenuates lung inflammation and edema after combined burn and smoke inhalation injury [135]. Murakami et al. reported decreased airway obstruction by casts, higher urine outputs and mean arterial pressures after/or during the continuous administration of 100 μg/kg rhAT over 24 h as compared with control animals in an ovine model of smoke inhalation injury and pneumonia [136]. In a small clinical study of Kowal-Vern and colleagues, patients with 20% or more TBSA burn and smoke inhalation injury who received rhAT had increased PaO2:FiO2 ratios and significantly fewer episodes of pneumonia compared with standard therapy alone [113]. In conclusion, restoring physiological antithrombin levels in patients with smoke inhalation injury has the potential to improve survival and to decrease inflammation. Again, however, clinical evidence regarding isolated smoke inhalation injury is missing.
Specific intoxications
Carbon monoxide
Current treatment recommendations of CO poisoning include cessation of exposure, administration of 100% oxygen and supportive care [67]. Hyperbaric oxygenation therapy (HBO) is sometimes used to encourage rapid displacement of CO from hemoglobin and to reduce the duration of the hypoxic state. Administration of 100% oxygen at 3 atmospheres reduces the CO half-life from 250 min at ambient pressure to 30 min [14]. In spite of this efficiency, the use of HBO remains controversial [67] owing to the questioned correlation between COHb levels and outcomes [66] in combination with limited access to the patient during HBO therapy, which has a severe impact on treatment quality of combined burn injuries.
Key issues
Smoke inhalation injury continues to increase morbidity and mortality in burn patients not only in the third world, but also in industrialized countries.
Smoke inhalation injury can cause thermal and chemical injuries, as well as systemic toxicity.
The ignition source, the size of the particles in the smoke, the duration of exposure and the solubility of the gases all determine the location and the severity of smoke inhalation injury.
The formation of reactive oxygen and nitrogen species, as well as the procoagulant and antifibrinolytic imbalance of alveolar homeostasis, play a central role in the pathogenesis of smoke inhalation injury.
Massive airway obstruction owing to cast formation, bronchospasm, the increase in bronchial circulation and transvascular fluid flux represent further hallmarks of smoke inhalation injury.
The development of clinical symptoms, such as erythema, dyspnea or massive airway edema, may be delayed, but are ultimately fatal.
Specific intoxications such as cyanide or carbon monoxide need to be considered critically and treated, if necessary.
Clinical management consists preponderantly of supportive care.
Clinical evidence for specific therapeutic approaches is lacking.
Anticoagulants, antioxidants and bronchodilators currently represent the most promising treatment strategies.
Aerolized administration of compounds seems to be feasible, safe and effective.
The optimal ventilation protocol still needs to be defined.
Establishing clear criteria for the diagnosis and grading of smoke inhalation injury is essential for the design and success of randomized, controlled multicenter trials.
A second potential argument for HBO is that CO binds not only to hemoglobin, but also to cytochrome oxidase, and thus delayed neurological sequelae may be prevented by HBO. A Cochrane database review of six randomized, controlled trials, however, did not reveal a beneficial effect of HBO compared with standard treatment with respect to neurological sequelae [137]. These results should be interpreted with care, however, because flaws in design and analyses were evident in all the included trials. In summary, all patients with CO intoxication should be treated with 100% oxygen. HBO might be a useful therapeutic option in patients with severe neurological symptoms and high COHb concentrations (>50%), but without major burns and severe pulmonary injury.
Cyanide
The adequate treatment of HCN poisoning following smoke inhalation injury is currently under discussion. Several antidotes are available: the ‘cyanide antidote kit’ includes amyl nitrite, thiosulfate and sodium nitrite [70], and is based on the classic treatment approach by Chen and Rose [138]. As these substances are methemoglobin generators, which may additionally impair oxygen transport, they should only be used in case of proven diagnosis (increased plasma levels of CN) and under continuous monitoring in an intensive care unit. Methemoglobin chelates CN to form cyanomethemoglobin, which, as it dissociates, allows free CN to be converted to thiocyanite by liver mitochondrial enzymes (rhodanase) using thiosulfate as a substrate. Thiocyanate is then excreted into the urine [138]. n contrast to these antidotes, hydroxycobalamin, a Vitamin B12 derivative, actively binds CN by forming cyanocobalamin, which will be directly excreted via the kidney [139,140]. In case of intoxication with 1 mg CN, 50 mg/kg hydroxycobalamin is recommended [141]. Because it averts methemoglobin production, hydroxycobalamin can even be used in the preclinical setting. Accordingly, hydroxycobalamin represents the active compound of the ‘cyanokit’, which is used in the prehospital management of smoke inhalation injury in Europe with a reported improvement in mortality [140].
Aggressive restoration of cardiopulmonary function augments the hepatic clearance of CN via the enzyme rhodanase [142], and has been reported to be successful in severe CN poisoning (blood levels 5.6–9 mg/l) as well as after ingestion [143–145] or smoke inhalation [146], even without the use of antidotes. Therefore, the standard care of CN poisoning should combine the aggressive supportive therapy with the causal treatment using hydroxycobalamin.
Expert commentary
Against the background of the current literature, there was a remarkable increase in our knowledge regarding the pathogenesis of smoke inhalation injury during the last 10–15 years. There are several promising therapeutic approaches, such as the β2-agonists, antioxidants or anticoagulants, and nebulization of the use of different ventilation modes. In addition, the trend goes to a combination of different compounds rather than a single ‘magic bullet’. Bearing in mind the complex pathophysiology and different clinical presentations of smoke inhalation injury, this is more than justifiable.
At the same time, however, we cannot be satisfied with the clinical progress made. The fact that mortality rates of smoke inhalation injury did not change is a more than obvious indicator for the lacking success of our current efforts. The future challenge, then, will exist largely in transferring our expanding theoretical knowledge adequately into daily clinical practice. Owing to the relatively small patient population, however, no single-center study will be able to recruit enough patients for a randomized, controlled trial with enough power to prove or disprove the efficiency of a therapeutic approach in an adequate time period. To achieve this goal, the cooperation and communication between burn centers should be intensified.
Five-year view
During the recent Consensus Conference for Inhalation Injury (Couer d’Alene, ID, USA), invited experts in the field of smoke inhalation injury met to “define what basic research elements are lacking and what additional information/definitions/technology is needed to advance inhalation injury research” [147]. Definition of criteria for the diagnosis and grading of inhalation injury received the highest ranked priority. These would represent the basis for uniform data acquisition, enabling the evaluation of short- and long-term efficiency of current as well as new therapeutic approaches. Therefore, the authors fully agree and hope that this goal will be achieved within the next 5 years.
Acknowledgments
Financial & competing interests disclosure
Daniel L Traber has received NIH grant NIH-P012 GM066312 – Pathophysiology of lung injury by smoke inhalation. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Contributor Information
Sebastian Rehberg, Department of Anesthesiology, The University of Texas Medical Branch, 301 University Boulevard, Galveston, TX 77555, USA, Tel.: +1 409 772 6405, Fax: +1 409 772 6409, serehber@utmb.edu.
Marc O Maybauer, Department of Anesthesiology, The University of Texas Medical Branch, 301 University Boulevard, Galveston, TX 77555, USA.
Perenlei Enkhbaatar, Department of Anesthesiology, The University of Texas Medical Branch, 301 University Boulevard, Galveston, TX 77555, USA.
Dirk M Maybauer, Department of Anesthesiology, The University of Texas Medical Branch, 301 University Boulevard, Galveston, TX 77555, USA.
Yusuke Yamamoto, Department of Anesthesiology, The University of Texas Medical Branch, 301 University Boulevard, Galveston, TX 77555, USA.
Daniel L Traber, Department of Anesthesiology, The University of Texas Medical Branch, 301 University Boulevard, Galveston, TX 77555, USA.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Alcorta R. Smoke inhalation & acute cyanide poisoning. Hydrogen cyanide poisoning proves increasingly common in smoke-inhalation victims. JEMS. 2004;29(8):S6–S15. [PubMed] [Google Scholar]
- 2.Harrington DT, Biffl WL, Cioffi WG. The Station nightclub fire. J Burn Care Rehabil. 2005;26(2):141–143. doi: 10.1097/01.bcr.0000155537.60909.fc. [DOI] [PubMed] [Google Scholar]
- 3.Yurt RW, Bessey PQ, Bauer GJ, et al. A regional burn center’s response to a disaster: September 11, 2001, and the days beyond. J Burn Care Rehabil. 2005;26(2):117–124. doi: 10.1097/01.bcr.0000155543.46107.e6. [DOI] [PubMed] [Google Scholar]
- 4.CDC. Rapid assessment of injuries among survivors of the terrorist attack on the World Trade Center – New York City, September 2001. JAMA. 2002;287(7):835–838. [PubMed] [Google Scholar]
- 5.Jordan MH, Hollowed KA, Turner DG, Wang DS, Jeng JC. The Pentagon attack of September 11, 2001: a burn center’s experience. J Burn Care Rehabil. 2005;26(2):109–116. doi: 10.1097/01.bcr.0000155539.82870.64. [DOI] [PubMed] [Google Scholar]
- 6.Dai NT, Chen TM, Cheng TY, et al. The comparison of early fluid therapy in extensive flame burns between inhalation and noninhalation injuries. Burns. 1998;24(7):671–675. doi: 10.1016/s0305-4179(98)00092-8. [DOI] [PubMed] [Google Scholar]
- 7•.Shirani KZ, Pruitt BA, Jr, Mason AD., Jr The influence of inhalation injury and pneumonia on burn mortality. Ann Surg. 1987;205(1):82–87. doi: 10.1097/00000658-198701000-00015. Points out the important role of inhalation injury in patients suffering from burn trauma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki M, Aikawa N, Kobayashi K, Higuchi R. Prognostic implications of inhalation injury in burn patients in Tokyo. Burns. 2005;31(3):331–336. doi: 10.1016/j.burns.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 9.Tredget EE, Shankowsky HA, Taerum TV, Moysa GL, Alton JD. The role of inhalation injury in burn trauma. A Canadian experience. Ann Surg. 1990;212(6):720–727. doi: 10.1097/00000658-199012000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwela DH. Public health and the Air Management Information System (AMIS) Epidemiology. 1999;10(5):647–655. [PubMed] [Google Scholar]
- 11.Schwela D. Cooking smoke: a silent killer. People Planet. 1997;6(3):24–25. [PubMed] [Google Scholar]
- 12.Woodson LC. Diagnosis and grading of inhalation injury. J Burn Care Res. 2009;30(1):143–145. doi: 10.1097/BCR.0b013e3181923b71. [DOI] [PubMed] [Google Scholar]
- 13.Gamsu G, Weintraub RM, Nadel JA. Clearance of tantalum from airways of different caliber in man evaluated by a roentgenographic method. Am Rev Respir Dis. 1973;107(2):214–224. doi: 10.1164/arrd.1973.107.2.214. [DOI] [PubMed] [Google Scholar]
- 14••.Traber DL, Herndon DN, Enkhbaatar P, Maybauer MO, Maybauer DM. The pathophysiology of inhalation injury. In: Herndon DN, editor. Total Burn Care. Saunders Elsevier; PA, USA: 2007. pp. 248–261. The current pathophysiological knowledge about inhalation injury. [Google Scholar]
- 15.Clark WR, Bonaventura M, Myers W. Smoke inhalation and airway management at a regional burn unit: 1974–1983. Part I: diagnosis and consequences of smoke inhalation. J Burn Care Rehabil. 1989;10(1):52–62. doi: 10.1097/00004630-198901000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Friedl HP, Till GO, Trentz O, Ward PA. Roles of histamine, complement and xanthine oxidase in thermal injury of skin. Am J Pathol. 1989;135(1):203–217. [PMC free article] [PubMed] [Google Scholar]
- 17.Granger DN. Role of xanthine oxidase and granulocytes in ischemia–reperfusion injury. Am J Physiol. 1988;255(6 Pt 2):H1269–H1275. doi: 10.1152/ajpheart.1988.255.6.H1269. [DOI] [PubMed] [Google Scholar]
- 18.Maybauer MO, Maybauer DM, Herndon DN, Traber DL. The role of superoxide dismutase in systemic inflammation. Shock. 2006;25(2):206–207. doi: 10.1097/01.shk.0000192121.53048.29. [DOI] [PubMed] [Google Scholar]
- 19.Herndon DN, Abston S, Stein MD. Increased thromboxane B2 levels in the plasma of burned and septic burned patients. Surg Gynecol Obstet. 1984;159(3):210–213. [PubMed] [Google Scholar]
- 20.Vindenes H, Ulvestad E, Bjerknes R. Increased levels of circulating interleukin-8 in patients with large burns: relation to burn size and sepsis. J Trauma. 1995;39(4):635–640. doi: 10.1097/00005373-199510000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Tari C, Baranink J. Upper airway neurological mechanisms. Curr Opin Allergy Clin Immunol. 2002;2:1149. doi: 10.1097/00130832-200202000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Demling RH. Smoke inhalation lung injury: an update. Eplasty. 2008;8:e27. [PMC free article] [PubMed] [Google Scholar]
- 23.Navar PD, Saffle JR, Warden GD. Effect of inhalation injury on fluid resuscitation requirements after thermal injury. Am J Surg. 1985;150(6):716–720. doi: 10.1016/0002-9610(85)90415-5. [DOI] [PubMed] [Google Scholar]
- 24.Cox RA, Burke AS, Soejima K, et al. Airway obstruction in sheep with burn and smoke inhalation injuries. Am J Respir Cell Mol Biol. 2003;29(3 Pt 1):295–302. doi: 10.1165/rcmb.4860. [DOI] [PubMed] [Google Scholar]
- 25.Perez Fontan JJ. On lung nerves and neurogenic injury. Ann Med. 2002;34(4):226–240. doi: 10.1080/078538902320322493. [DOI] [PubMed] [Google Scholar]
- 26.Fontan JJ, Cortright DN, Krause JE, et al. Substance P and neurokinin-1 receptor expression by intrinsic airway neurons in the rat. Am J Physiol Lung Cell Mol Physiol. 2000;278(2):L344–L355. doi: 10.1152/ajplung.2000.278.2.L344. [DOI] [PubMed] [Google Scholar]
- 27.Rehberg S, Maybauer MO, Maybauer DM, et al. The role of nitric oxide and reactive nitrogen species in experimental ARDS. Front Biosci. 2009 doi: 10.2741/s43. (In press) [DOI] [PubMed] [Google Scholar]
- 28.Steudel W, Kirmse M, Weimann J, et al. Exhaled nitric oxide production by nitric oxide synthase-deficient mice. Am J Respir Crit Care Med. 2000;162(4 Pt 1):1262–1267. doi: 10.1164/ajrccm.162.4.9909037. [DOI] [PubMed] [Google Scholar]
- 29.Westphal M, Enkhbaatar P, Schmalstieg FC, et al. Neuronal nitric oxide synthase inhibition attenuates cardiopulmonary dysfunctions after combined burn and smoke inhalation injury in sheep. Crit Care Med. 2008;36(4):1196–1204. doi: 10.1097/CCM.0b013e31816a1a0c. [DOI] [PubMed] [Google Scholar]
- 30.Fink MP. Role of reactive oxygen and nitrogen species in acute respiratory distress syndrome. Curr Opin Crit Care. 2002;8(1):6–11. doi: 10.1097/00075198-200202000-00002. [DOI] [PubMed] [Google Scholar]
- 31.Gero D, Szabo C. Poly(ADP-ribose) polymerase: a new therapeutic target? Curr Opin Anaesthesiol. 2008;21(2):111–121. doi: 10.1097/ACO.0b013e3282f63c15. [DOI] [PubMed] [Google Scholar]
- 32.Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173(1):2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szabo C. DNA strand breakage and activation of poly-ADP ribosyltransferase: a cytotoxic pathway triggered by peroxynitrite. Free Radic Biol Med. 1996;21(6):855–869. doi: 10.1016/0891-5849(96)00170-0. [DOI] [PubMed] [Google Scholar]
- 34.Lobo SM, Orrico SR, Queiroz MM, et al. Pneumonia-induced sepsis and gut injury: effects of a poly-(ADP-ribose) polymerase inhibitor. J Surg Res. 2005;129(2):292–297. doi: 10.1016/j.jss.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 35.Jagtap P, Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005;4(5):421–440. doi: 10.1038/nrd1718. [DOI] [PubMed] [Google Scholar]
- 36.Chiarugi A, Moskowitz MA. Poly(ADP-ribose) polymerase-1 activity promotes NF-κB-driven transcription and microglial activation: implication for neurodegenerative disorders. J Neurochem. 2003;85(2):306–317. doi: 10.1046/j.1471-4159.2003.01684.x. [DOI] [PubMed] [Google Scholar]
- 37.Li LF, Ouyang B, Choukroun G, et al. Stretch-induced IL-8 depends on c-Jun NH2-terminal and nuclear factor-κB-inducing kinases. Am J Physiol Lung Cell Mol Physiol. 2003;285(2):L464–L475. doi: 10.1152/ajplung.00031.2003. [DOI] [PubMed] [Google Scholar]
- 38.Yamaza T, Masuda KF, Tsukiyama Y, et al. NF-κB activation and iNOS expression in the synovial membrane of rat temporomandibular joints after induced synovitis. J Dent Res. 2003;82(3):183–188. doi: 10.1177/154405910308200307. [DOI] [PubMed] [Google Scholar]
- 39.Herndon DN, Traber DL, Niehaus GD, Linares HA, Traber LD. The pathophysiology of smoke inhalation injury in a sheep model. J Trauma. 1984;24(12):1044–1051. doi: 10.1097/00005373-198412000-00007. [DOI] [PubMed] [Google Scholar]
- 40•.Traber DL, Enkhbaatar P. Thermal lung injury and acute smoke inhalation. In: Fishman DA, editor. Fishman’s Pulmonary Diseases and Disorders. McGraw-Hill Medical Publishing Company; NY, USA: 2008. Comprehensive summary of the current knowledge regarding pathophysiology of thermal lung injury and acute smoke inhalation. [Google Scholar]
- 41.Traber DL, Hawkins HK, Enkhbaatar P, et al. The role of the bronchial circulation in the acute lung injury resulting from burn and smoke inhalation. Pulm Pharmacol Ther. 2007;20(2):163–166. doi: 10.1016/j.pupt.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 42.Traber DL, Traber LD. Airway blood flow changes and airway obstruction following lung injury. Arch Physiol Biochem. 2003;111(4):297–300. doi: 10.3109/13813450312331337432. [DOI] [PubMed] [Google Scholar]
- 43.Enkhbaatar P, Herndon DN, Traber DL. Use of nebulized heparin in the treatment of smoke inhalation injury. J Burn Care Res. 2009;30(1):159–162. doi: 10.1097/BCR.0b013e3181923bd3. [DOI] [PubMed] [Google Scholar]
- 44.Abdi S, Herndon D, McGuire J, Traber L, Traber DL. Time course of alterations in lung lymph and bronchial blood flows after inhalation injury. J Burn Care Rehabil. 1990;11(6):510–515. doi: 10.1097/00004630-199011000-00005. [DOI] [PubMed] [Google Scholar]
- 45.Seeger W, Stohr G, Wolf HR, Neuhof H. Alteration of surfactant function due to protein leakage: special interaction with fibrin monomer. J Appl Physiol. 1985;58(2):326–338. doi: 10.1152/jappl.1985.58.2.326. [DOI] [PubMed] [Google Scholar]
- 46.Ciano PS, Colvin RB, Dvorak AM, McDonagh J, Dvorak HF. Macrophage migration in fibrin gel matrices. Lab Invest. 1986;54(1):62–70. [PubMed] [Google Scholar]
- 47.Sakurai H, Schmalstieg FC, Traber LD, Hawkins HK, Traber DL. Role of L-selectin in physiological manifestations after burn and smoke inhalation injury in sheep. J Appl Physiol. 1999;86(4):1151–1159. doi: 10.1152/jappl.1999.86.4.1151. [DOI] [PubMed] [Google Scholar]
- 48.Abdi S, Herndon DN, Traber LD, et al. Lung edema formation following inhalation injury: role of the bronchial blood flow. J Appl Physiol. 1991;71(2):727–734. doi: 10.1152/jappl.1991.71.2.727. [DOI] [PubMed] [Google Scholar]
- 49.Efimova O, Volokhov AB, Iliaifar S, Hales CA. Ligation of the bronchial artery in sheep attenuates early pulmonary changes following exposure to smoke. J Appl Physiol. 2000;88(3):888–893. doi: 10.1152/jappl.2000.88.3.888. [DOI] [PubMed] [Google Scholar]
- 50.Hales CA, Barkin P, Jung W, et al. Bronchial artery ligation modifies pulmonary edema after exposure to smoke with acrolein. J Appl Physiol. 1989;67(3):1001–1006. doi: 10.1152/jappl.1989.67.3.1001. [DOI] [PubMed] [Google Scholar]
- 51.Sakurai H, Johnigan R, Kikuchi Y, et al. Effect of reduced bronchial circulation on lung fluid flux after smoke inhalation in sheep. J Appl Physiol. 1998;84(3):980–986. doi: 10.1152/jappl.1998.84.3.980. [DOI] [PubMed] [Google Scholar]
- 52.Sakurai H, Soejima K, Nozaki M, Traber LD, Traber DL. Effect of ablated airway blood flow on systemic and pulmonary microvascular permeability after smoke inhalation in sheep. Burns. 2007;33(7):885–891. doi: 10.1016/j.burns.2006.10.394. [DOI] [PubMed] [Google Scholar]
- 53.Basadre JO, Sugi K, Traber DL, et al. The effect of leukocyte depletion on smoke inhalation injury in sheep. Surgery. 1988;104(2):208–215. [PubMed] [Google Scholar]
- 54.Abdi S, Traber LD, Herndon DN, Rogers CS, Traber DL. Effects of ibuprofen on airway vascular response to cotton smoke injury. Eur J Pharmacol. 1995;293(4):475–481. doi: 10.1016/0926-6917(95)90068-3. [DOI] [PubMed] [Google Scholar]
- 55.Sakurai H, Traber LD, Traber DL. Altered systemic organ blood flow after combined injury with burn and smoke inhalation. Shock. 1998;9(5):369–374. doi: 10.1097/00024382-199805000-00010. [DOI] [PubMed] [Google Scholar]
- 56.Demling RH, Knox J, Youn YK, LaLonde C. Oxygen consumption early postburn becomes oxygen delivery dependent with the addition of smoke inhalation injury. J Trauma. 1992;32(5):593–598. doi: 10.1097/00005373-199205000-00010. discussion 599. [DOI] [PubMed] [Google Scholar]
- 57.Soejima K, Schmalstieg FC, Sakurai H, Traber LD, Traber DL. Pathophysiological analysis of combined burn and smoke inhalation injuries in sheep. Am J Physiol Lung Cell Mol Physiol. 2001;280(6):L1233–L1241. doi: 10.1152/ajplung.2001.280.6.L1233. [DOI] [PubMed] [Google Scholar]
- 58.Rehberg S, Maybauer MO, Maybauer DM, et al. The antioxidants Vitamin E and Vitamin C for nutritional support in critical ill patients: beneficial or harmful? Adv Anaesthesiol Crit Care. 2009;1(1):11–19. [Google Scholar]
- 59.Horton JW. Free radicals and lipid peroxidation mediated injury in burn trauma: the role of antioxidant therapy. Toxicology. 2003;189(1–2):75–88. doi: 10.1016/s0300-483x(03)00154-9. [DOI] [PubMed] [Google Scholar]
- 60.Nguyen TT, Cox CS, Traber DL, et al. Free radical activity and loss of plasma antioxidants, vitamin E, and sulfhydryl groups in patients with burns: the 1993 Moyer Award. J Burn Care Rehabil. 1993;14(6):602–609. doi: 10.1097/00004630-199311000-00004. [DOI] [PubMed] [Google Scholar]
- 61.Berger MM, Shenkin A. Update on clinical micronutrient supplementation studies in the critically ill. Curr Opin Clin Nutr Metab Care. 2006;9(6):711–716. doi: 10.1097/01.mco.0000247466.41661.ba. [DOI] [PubMed] [Google Scholar]
- 62.Carbon monoxide-related deaths – United States, 1999–2004. Morb Mortal Wkly Rep. 2007;56(50):1309–1312. [PubMed] [Google Scholar]
- 63.Weaver LK. Carbon monoxide poisoning. Crit Care Clin. 1999;15(2):297–317. viii. doi: 10.1016/s0749-0704(05)70056-7. [DOI] [PubMed] [Google Scholar]
- 64.Prien T, Traber DL. Toxic smoke compounds and inhalation injury – a review. Burns Incl Therm Inj. 1988;14(6):451–460. doi: 10.1016/s0305-4179(88)80005-6. [DOI] [PubMed] [Google Scholar]
- 65.Traber DL, Maybauer MO, Maybauer DM, Westphal M, Traber LD. Inhalational and acute lung injury. Shock. 2005;24(Suppl 1):82–87. doi: 10.1097/01.shk.0000191338.39154.73. [DOI] [PubMed] [Google Scholar]
- 66.Hardy KR, Thom SR. Pathophysiology and treatment of carbon monoxide poisoning. J Toxicol Clin Toxicol. 1994;32(6):613–629. doi: 10.3109/15563659409017973. [DOI] [PubMed] [Google Scholar]
- 67.Kealey GP. Carbon monoxide toxicity. J Burn Care Res. 2009;30(1):146–147. doi: 10.1097/BCR.0b013e3181923b81. [DOI] [PubMed] [Google Scholar]
- 68.Westphal M, Morita N, Enkhbaatar P, et al. Carboxyhemoglobin formation following smoke inhalation injury in sheep is interrelated with pulmonary shunt fraction. Biochem Biophys Res Commun. 2003;311(3):754–758. doi: 10.1016/j.bbrc.2003.10.063. [DOI] [PubMed] [Google Scholar]
- 69.Hall AH, Rumack BH. Clinical toxicology of cyanide. Ann Emerg Med. 1986;15(9):1067–1074. doi: 10.1016/s0196-0644(86)80131-7. [DOI] [PubMed] [Google Scholar]
- 70.Barillo DJ. Diagnosis and treatment of cyanide toxicity. J Burn Care Res. 2009;30(1):148–152. doi: 10.1097/BCR.0b013e3181923b91. [DOI] [PubMed] [Google Scholar]
- 71.Baud FJ, Barriot P, Toffis V, et al. Elevated blood cyanide concentrations in victims of smoke inhalation. N Engl J Med. 1991;325(25):1761–1766. doi: 10.1056/NEJM199112193252502. [DOI] [PubMed] [Google Scholar]
- 72.Silverman SH, Purdue GF, Hunt JL, Bost RO. Cyanide toxicity in burned patients. J Trauma. 1988;28(2):171–176. doi: 10.1097/00005373-198802000-00007. [DOI] [PubMed] [Google Scholar]
- 73.Davies JW. Toxic chemicals versus lung tissue – an aspect of inhalation injury revisited. The Everett Idris Evans memorial lecture – 1986. J Burn Care Rehabil. 1986;7(3):213–222. doi: 10.1097/00004630-198605000-00004. [DOI] [PubMed] [Google Scholar]
- 74.Lee MJ, O’Connell DJ. The plain chest radiograph after acute smoke inhalation. Clin Radiol. 1988;39(1):33–37. doi: 10.1016/s0009-9260(88)80334-9. [DOI] [PubMed] [Google Scholar]
- 75.Endorf FW, Gamelli RL. Inhalation injury, pulmonary perturbations, and fluid resuscitation. J Burn Care Res. 2007;28(1):80–83. doi: 10.1097/BCR.0B013E31802C889F. [DOI] [PubMed] [Google Scholar]
- 76.Marek K, Piotr W, Stanislaw S, et al. Fibreoptic bronchoscopy in routine clinical practice in confirming the diagnosis and treatment of inhalation burns. Burns. 2007;33(5):554–560. doi: 10.1016/j.burns.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 77.Brown DL, Archer SB, Greenhalgh DG, et al. Inhalation injury severity scoring system: a quantitative method. J Burn Care Rehabil. 1996;17(6 Pt 1):552–557. doi: 10.1097/00004630-199611000-00013. [DOI] [PubMed] [Google Scholar]
- 78.Edelman DA, White MT, Tyburski JG, Wilson RF. Factors affecting prognosis of inhalation injury. J Burn Care Res. 2006;27(6):848–853. doi: 10.1097/01.BCR.0000245493.26814.CE. [DOI] [PubMed] [Google Scholar]
- 79.Sellers BJ, Davis BL, Larkin PW, Morris SE, Saffle JR. Early prediction of prolonged ventilator dependence in thermally injured patients. J Trauma. 1997;43(6):899–903. doi: 10.1097/00005373-199712000-00005. [DOI] [PubMed] [Google Scholar]
- 80.Cancio LC, Chavez S, Alvarado-Ortega M, et al. Predicting increased fluid requirements during the resuscitation of thermally injured patients. J Trauma. 2004;56(2):404–413. doi: 10.1097/01.TA.0000075341.43956.E4. discussion 413–404. [DOI] [PubMed] [Google Scholar]
- 81.Dries DJ. Key questions in ventilator management of the burn-injured patient (second of two parts) J Burn Care Res. 2009;30(2):211–220. doi: 10.1097/BCR.0b013e318198a33f. [DOI] [PubMed] [Google Scholar]
- 82.Plurad D, Martin M, Green D, et al. The decreasing incidence of late posttraumatic acute respiratory distress syndrome: the potential role of lung protective ventilation and conservative transfusion practice. J Trauma. 2007;63(1):1–7. doi: 10.1097/TA.0b013e318068b1ed. discussion 8. [DOI] [PubMed] [Google Scholar]
- 83.Mlcak R, Herndon DN. Respiratory care. In: Herndon DN, editor. Total Burn Care. WB Saunders; PA, USA: 2002. pp. 242–267. [Google Scholar]
- 84.Fabian TC. Empiric therapy for pneumonia in the surgical intensive care unit. Am J Surg. 2000;179(2 Suppl 1):18–23. [PubMed] [Google Scholar]
- 85.Peck MD, Koppelman T. Low-tidal-volume ventilation as a strategy to reduce ventilator-associated injury in ALI and ARDS. J Burn Care Res. 2009;30(1):172–175. doi: 10.1097/BCR.0b013e3181923c32. [DOI] [PubMed] [Google Scholar]
- 86••.The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1301–1308. doi: 10.1056/NEJM200005043421801. Represents the standard reference for currently applied ventilation protocols. [DOI] [PubMed] [Google Scholar]
- 87.Brower RG, Lanken PN, MacIntyre N, et al. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. N Engl J Med. 2004;351(4):327–336. doi: 10.1056/NEJMoa032193. [DOI] [PubMed] [Google Scholar]
- 88.Gattinoni L, Tognoni G, Pesenti A, et al. Effect of prone positioning on the survival of patients with acute respiratory failure. N Engl J Med. 2001;345(8):568–573. doi: 10.1056/NEJMoa010043. [DOI] [PubMed] [Google Scholar]
- 89.Stock MC, Downs JB, Frolicher DA. Airway pressure release ventilation. Crit Care Med. 1987;15(5):462–466. doi: 10.1097/00003246-198705000-00002. [DOI] [PubMed] [Google Scholar]
- 90.Mlcak RP. Airway pressure release ventilation. J Burn Care Res. 2009;30(1):176–177. doi: 10.1097/BCR.0b013e3181923c58. [DOI] [PubMed] [Google Scholar]
- 91.Putensen C, Zech S, Wrigge H, et al. Long-term effects of spontaneous breathing during ventilatory support in patients with acute lung injury. Am J Respir Crit Care Med. 2001;164(1):43–49. doi: 10.1164/ajrccm.164.1.2001078. [DOI] [PubMed] [Google Scholar]
- 92.Varpula T, Jousela I, Niemi R, Takkunen O, Pettila V. Combined effects of prone positioning and airway pressure release ventilation on gas exchange in patients with acute lung injury. Acta Anaesthesiol Scand. 2003;47(5):516–524. doi: 10.1034/j.1399-6576.2003.00109.x. [DOI] [PubMed] [Google Scholar]
- 93.Harrington D. Volumetric diffusive ventilator. J Burn Care Res. 2009;30(1):175–176. doi: 10.1097/BCR.0b013e3181923c44. [DOI] [PubMed] [Google Scholar]
- 94.Carman B, Cahill T, Warden G, McCall J. A prospective, randomized comparison of the volume diffusive respirator vs conventional ventilation for ventilation of burned children. 2001 ABA paper. J Burn Care Rehabil. 2002;23(6):444–448. doi: 10.1097/00004630-200211000-00011. [DOI] [PubMed] [Google Scholar]
- 95.Rue LW, 3rd, Cioffi WG, Mason AD, McManus WF, Pruitt BA., Jr Improved survival of burned patients with inhalation injury. Arch Surg. 1993;128(7):772–778. doi: 10.1001/archsurg.1993.01420190066009. discussion 778–780. [DOI] [PubMed] [Google Scholar]
- 96.Froese AB. High-frequency oscillatory ventilation for adult respiratory distress syndrome: let’s get it right this time! Crit Care Med. 1997;25(6):906–908. doi: 10.1097/00003246-199706000-00004. [DOI] [PubMed] [Google Scholar]
- 97.Cartotto R. Use of high frequency oscillatory ventilation in inhalation injury. J Burn Care Res. 2009;30(1):178–181. doi: 10.1097/BCR.0b013e3181923c6a. [DOI] [PubMed] [Google Scholar]
- 98.Cartotto R, Ellis S, Gomez M, Cooper A, Smith T. High frequency oscillatory ventilation in burn patients with the acute respiratory distress syndrome. Burns. 2004;30(5):453–463. doi: 10.1016/j.burns.2004.01.015. [DOI] [PubMed] [Google Scholar]
- 99.Alpard SK, Zwischenberger JB, Tao W, Deyo DJ, Bidani A. Reduced ventilator pressure and improved P/F ratio during percutaneous arteriovenous carbon dioxide removal for severe respiratory failure. Ann Surg. 1999;230(2):215–224. doi: 10.1097/00000658-199908000-00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zwischenberger JB, Wang D, Lick SD, et al. The paracorporeal artificial lung improves 5-day outcomes from lethal smoke/burn-induced acute respiratory distress syndrome in sheep. Ann Thorac Surg. 2002;74(4):1011–1016. doi: 10.1016/s0003-4975(02)03896-1. discussion 1017–1018. [DOI] [PubMed] [Google Scholar]
- 101.Zwischenberger JB, Savage C, Witt SA, et al. Arterio-venous CO2 removal (AVCO2R) perioperative management: rapid recovery and enhanced survival. J Invest Surg. 2002;15(1):15–21. doi: 10.1080/08941930252807741. [DOI] [PubMed] [Google Scholar]
- 102.Schmalstieg FC, Keeney SE, Rudloff HE, et al. Arteriovenous CO2 removal improves survival compared to high frequency percussive and low tidal volume ventilation in a smoke/burn sheep acute respiratory distress syndrome model. Ann Surg. 2007;246(3):512–521. doi: 10.1097/SLA.0b013e318148c6e6. discussion 521–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Morina P, Herrera M, Venegas J, et al. Effects of nebulized salbutamol on respiratory mechanics in adult respiratory distress syndrome. Intensive Care Med. 1997;23(1):58–64. doi: 10.1007/s001340050291. [DOI] [PubMed] [Google Scholar]
- 104.Zhang H, Kim YK, Govindarajan A, et al. Effect of adrenoreceptors on endotoxin-induced cytokines and lipid peroxidation in lung explants. Am J Respir Crit Care Med. 1999;160(5 Pt 1):1703–1710. doi: 10.1164/ajrccm.160.5.9903068. [DOI] [PubMed] [Google Scholar]
- 105.van der Poll T, Coyle SM, Barbosa K, Braxton CC, Lowry SF. Epinephrine inhibits tumor necrosis factor-α and potentiates interleukin 10 production during human endotoxemia. J Clin Invest. 1996;97(3):713–719. doi: 10.1172/JCI118469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McAuley DF, Frank JA, Fang X, Matthay MA. Clinically relevant concentrations of β2-adrenergic agonists stimulate maximal cyclic adenosine monophosphate-dependent airspace fluid clearance and decrease pulmonary edema in experimental acid-induced lung injury. Crit Care Med. 2004;32(7):1470–1476. doi: 10.1097/01.ccm.0000129489.34416.0e. [DOI] [PubMed] [Google Scholar]
- 107.Sartori C, Allemann Y, Duplain H, et al. Salmeterol for the prevention of high-altitude pulmonary edema. N Engl J Med. 2002;346(21):1631–1636. doi: 10.1056/NEJMoa013183. [DOI] [PubMed] [Google Scholar]
- 108.Perkins GD, Gao F, Thickett DR. In vivo and in vitro effects of salbutamol on alveolar epithelial repair in acute lung injury. Thorax. 2008;63(3):215–220. doi: 10.1136/thx.2007.080382. [DOI] [PubMed] [Google Scholar]
- 109.Palmieri TL, Enkhbaatar P, Sheridan R, Traber DL, Greenhalgh DG. Studies of inhaled agents in inhalation injury. J Burn Care Res. 2009;30(1):169–171. doi: 10.1097/BCR.0b013e3181923c1d. [DOI] [PubMed] [Google Scholar]
- 110.Palmieri TL. Use of β-agonists in inhalation injury. J Burn Care Res. 2009;30(1):156–159. doi: 10.1097/BCR.0b013e3181923bc3. [DOI] [PubMed] [Google Scholar]
- 111.Enkhbaatar P, Kikuchi Y, Traber LD, et al. Effect of inhaled nitric oxide on pulmonary vascular hyperpermeability in sheep following smoke inhalation. Burns. 2005;31(8):1013–1019. doi: 10.1016/j.burns.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 112.Booke M, Bradford DW, Hinder F, et al. Inhaled nitric oxide selectively reduces pulmonary hypertension after ovine smoke inhalation but does not improve oxygenation. J Burn Care Rehabil. 1997;18(1 Pt 1):27–33. doi: 10.1097/00004630-199701000-00005. [DOI] [PubMed] [Google Scholar]
- 113.Kowal-Vern A, Walenga JM, McGill V, Gamelli RL. The impact of antithrombin (H) concentrate infusions on pulmonary function in the acute phase of thermal injury. Burns. 2001;27(1):52–60. doi: 10.1016/s0305-4179(00)00057-7. [DOI] [PubMed] [Google Scholar]
- 114.Olson ST, Bjork I, Sheffer R, et al. Role of the antithrombin-binding pentasaccharide in heparin acceleration of antithrombin–proteinase reactions. Resolution of the antithrombin conformational change contribution to heparin rate enhancement. J Biol Chem. 1992;267(18):12528–12538. [PubMed] [Google Scholar]
- 115••.Adhikari NK, Burns KE, Friedrich JO, et al. Effect of nitric oxide on oxygenation and mortality in acute lung injury: systematic review and meta-analysis. BMJ. 2007;334(7597):779. doi: 10.1136/bmj.39139.716794.55. A summary of the present knowledge about the use of nitric oxide (NO) for the treatment of acute respiratory distress syndrome. It concludes that there is currently no evidence for beneficial effects of NO and that it should not be recommended for standard care. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sokol J, Jacobs SE, Bohn D. Inhaled nitric oxide for acute hypoxic respiratory failure in children and adults: a meta-analysis. Anesth Analg. 2003;97(4):989–998. doi: 10.1213/01.ANE.0000078819.48523.26. [DOI] [PubMed] [Google Scholar]
- 117.Taylor RW, Zimmerman JL, Dellinger RP, et al. Low-dose inhaled nitric oxide in patients with acute lung injury: a randomized controlled trial. JAMA. 2004;291(13):1603–1609. doi: 10.1001/jama.291.13.1603. [DOI] [PubMed] [Google Scholar]
- 118.Westphal M, Maybauer DM, Maybauer MO. Who is the bad guy in acute respiratory distress syndrome? Neuronal nitric oxide synthase, inducible nitric oxide synthase, or both? Crit Care Med. 2009;37(1):363–364. doi: 10.1097/CCM.0b013e318192fec3. [DOI] [PubMed] [Google Scholar]
- 119.Brown M, Desai M, Traber LD, Herndon DN, Traber DL. Dimethylsulfoxide with heparin in the treatment of smoke inhalation injury. J Burn Care Rehabil. 1988;9(1):22–25. doi: 10.1097/00004630-198801000-00007. [DOI] [PubMed] [Google Scholar]
- 120.Murakami K, Enkhbaatar P, Morita N, Westphal M, Traber DL. The elevation of airway antithrombin level in smoke inhalation with pneumonia in sheep. Crit Care Med. 2003;31:A28. doi: 10.1097/01.CCM.0000050444.52531.08. [DOI] [PubMed] [Google Scholar]
- 121.Enkhbaatar P, Cox RA, Traber LD, et al. Aerosolized anticoagulants ameliorate acute lung injury in sheep after exposure to burn and smoke inhalation. Crit Care Med. 2007;35(12):2805–2810. doi: 10.1097/01.ccm.0000291647.18329.83. [DOI] [PubMed] [Google Scholar]
- 122••.Enkhbaatar P, Esechie A, Wang J, et al. Combined anticoagulants ameliorate acute lung injury in sheep after burn and smoke inhalation. Clin Sci (Lond) 2008;114(4):321–329. doi: 10.1042/CS20070254. The combination of intravenous recombinant human antithrombin administration and aerolized heparin significantly attenuated lung injury following combined burn and smoke inhalation injury. This therapeutic approach was based on a very carefully conducted serious of studies using different anticoagulants and different ways of administration. [DOI] [PubMed] [Google Scholar]
- 123.Desai MH, Mlcak R, Richardson J, Nichols R, Herndon DN. Reduction in mortality in pediatric patients with inhalation injury with aerosolized heparin/N-acetylcystine [correction of acetylcystine] therapy. J Burn Care Rehabil. 1998;19(3):210–212. doi: 10.1097/00004630-199805000-00004. [DOI] [PubMed] [Google Scholar]
- 124.LaLonde C, Nayak U, Hennigan J, Demling R. Plasma catalase and glutathione levels are decreased in response to inhalation injury. J Burn Care Rehabil. 1997;18(6):515–519. doi: 10.1097/00004630-199711000-00008. [DOI] [PubMed] [Google Scholar]
- 125.Morita N, Shimoda K, Traber MG, et al. Vitamin E attenuates acute lung injury in sheep with burn and smoke inhalation injury. Redox Rep. 2006;11(2):61–70. doi: 10.1179/135100006X101020. [DOI] [PubMed] [Google Scholar]
- 126.Morita N, Traber MG, Enkhbaatar P, et al. Aerosolized α-tocopherol ameliorates acute lung injury following combined burn and smoke inhalation injury in sheep. Shock. 2006;25(3):277–282. doi: 10.1097/01.shk.0000208805.23182.a7. [DOI] [PubMed] [Google Scholar]
- 127.Hamahata A, Enkhbaatar P, Kraft ER, et al. γ-tocopherol nebulization by a lipid aerosolization device improves pulmonary function in sheep with burn and smoke inhalation injury. Free Radic Biol Med. 2008;45(4):425–433. doi: 10.1016/j.freeradbiomed.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Galli F, Piroddi M, Lannone A, et al. A comparison between the antioxidant and peroxynitrite-scavenging functions of the Vitamin E metabolites α- and γ-carboxyethyl-6-hydroxychromans. Int J Vitam Nutr Res. 2004;74(5):362–373. doi: 10.1024/0300-9831.74.5.362. [DOI] [PubMed] [Google Scholar]
- 129.Tyml K, Li F, Wilson JX. Septic impairment of capillary blood flow requires nicotinamide adenine dinucleotide phosphate oxidase but not nitric oxide synthase and is rapidly reversed by ascorbate through an endothelial nitric oxide synthase-dependent mechanism. Crit Care Med. 2008;36(8):2355–2362. doi: 10.1097/CCM.0b013e31818024f6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tanaka H, Matsuda H, Shimazaki S, Hanumadass M, Matsuda T. Reduced resuscitation fluid volume for second-degree burns with delayed initiation of ascorbic acid therapy. Arch Surg. 1997;132(2):158–161. doi: 10.1001/archsurg.1997.01430260056011. [DOI] [PubMed] [Google Scholar]
- 131.Galley HF, Davies MJ, Webster NR. Ascorbyl radical formation in patients with sepsis: effect of ascorbate loading. Free Radic Biol Med. 1996;20(1):139–143. doi: 10.1016/0891-5849(95)02022-5. [DOI] [PubMed] [Google Scholar]
- 132•.Tanaka H, Matsuda T, Miyagantani Y, et al. Reduction of resuscitation fluid volumes in severely burned patients using ascorbic acid administration: a randomized, prospective study. Arch Surg. 2000;135(3):326–331. doi: 10.1001/archsurg.135.3.326. The only randomized, controlled study to date investigating the effects of Vitamin C administration in patients with combined burn and smoke inhalation injury. [DOI] [PubMed] [Google Scholar]
- 133.Enkhbaatar P, Traber DL. Pathophysiology of acute lung injury in combined burn and smoke inhalation injury. Clin Sci (Lond) 2004;107(2):137–143. doi: 10.1042/CS20040135. [DOI] [PubMed] [Google Scholar]
- 134.Niedermayr M, Schramm W, Kamolz L, et al. Antithrombin deficiency and its relationship to severe burns. Burns. 2007;33(2):173–178. doi: 10.1016/j.burns.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 135.Latenser BA. Use of antithrombin III in inhalation injury. J Burn Care Res. 2009;30(1):186–188. doi: 10.1097/BCR.0b013e3181923f08. [DOI] [PubMed] [Google Scholar]
- 136.Murakami K, McGuire R, Cox RA, et al. Recombinant antithrombin attenuates pulmonary inflammation following smoke inhalation and pneumonia in sheep. Crit Care Med. 2003;31(2):577–583. doi: 10.1097/01.CCM.0000050444.52531.08. [DOI] [PubMed] [Google Scholar]
- 137.Juurlink DN, Buckley NA, Stanbrook MB, et al. Hyperbaric oxygen for carbon monoxide poisoning. Cochrane Database Syst Rev. 2005;1:CD002041. doi: 10.1002/14651858.CD002041.pub2. [DOI] [PubMed] [Google Scholar]
- 138.Chen KK, Rose CL, Clorves GHA. Comparative values of several antidotes in cyanide poisoning. Am J Med Sci. 1934;188:767–781. [Google Scholar]
- 139.Maybauer DM, Traber DL, Radermacher P, Herndon DN, Maybauer MO. Treatment strategies for acute smoke inhalation injury. Anaesthesist. 2006;55(9):980–982. 984–988. doi: 10.1007/s00101-006-1050-3. [DOI] [PubMed] [Google Scholar]
- 140.Fortin JL, Giocanti JP, Ruttimann M, Kowalski JJ. Prehospital administration of hydroxocobalamin for smoke inhalation-associated cyanide poisoning: 8 years of experience in the Paris Fire Brigade. Clin Toxicol (Phila) 2006;44(Suppl 1):37–44. doi: 10.1080/15563650600811870. [DOI] [PubMed] [Google Scholar]
- 141.Kafka G, Maybauer DM, Traber DL, Maybauer MO. Treatment of inhalation injury in preclinical emergency medicine. Notfall Rettungsmed. 2007;10:529–540. [Google Scholar]
- 142.Cohen MA, Guzzardi LJ. Inhalation of products of combustion. Ann Emerg Med. 1983;12(10):628–632. doi: 10.1016/s0196-0644(83)80209-1. [DOI] [PubMed] [Google Scholar]
- 143.Brivet F, Delfraissy JF, Duche M, Bertrand P, Dormont J. Acute cyanide poisoning: recovery with non-specific supportive therapy. Intensive Care Med. 1983;9(1):33–35. doi: 10.1007/BF01693704. [DOI] [PubMed] [Google Scholar]
- 144.Caravati EM, Litovitz TL. Pediatric cyanide intoxication and death from an acetonitrile-containing cosmetic. JAMA. 1988;260(23):3470–3473. [PubMed] [Google Scholar]
- 145.Graham DL, Laman D, Theodore J, Robin ED. Acute cyanide poisoning complicated by lactic acidosis and pulmonary edema. Arch Intern Med. 1977;137(8):1051–1055. [PubMed] [Google Scholar]
- 146.Clark CJ, Campbell D, Reid WH. Blood carboxyhaemoglobin and cyanide levels in fire survivors. Lancet. 1981;1(8234):1332–1335. doi: 10.1016/s0140-6736(81)92516-2. [DOI] [PubMed] [Google Scholar]
- 147.Palmieri TL. Inhalation injury consensus conference: conclusions. J Burn Care Res. 2009;30(1):209–210. doi: 10.1097/BCR.0b013e3181923f5c. [DOI] [PubMed] [Google Scholar]