

Abstract
Barrett's esophagus (BE) is the replacement of normal squamous esophageal mucosa with an intestinalized columnar epithelium. The molecular mechanisms underlying its development are not understood. Cdx2 is an intestine-specific transcription factor that is ectopically expressed in BE, but its role in this process is unclear. Herein, we describe a novel cell culture model for BE. Retroviral-mediated Cdx2 expression in immortalized human esophageal keratinocytes [EPC-human telomerase reverse transcriptase (hTERT)] could transiently be established but not maintained and was associated with a reduction in cell proliferation. Coexpression of cyclin D1, but not a dominant-negative p53, rescued proliferation in the Cdx2-expressing cells. Cdx2 expression in the EPC-hTERT.D1 cells decreased cell proliferation but did not induce intestinalization. We investigated for other treatments to enhance intestinalization and found that acidic culture conditions uniformly killed EPC-hTERT.D1.Cdx2 cells. However, treatment with 5-aza-2-deoxycytidine (5-AzaC) to demethylate epigenetically silenced genes did appear to be tolerated. Multiple Cdx2 target genes, markers of intestinal differentiation and markers of BE, were induced by this 5-AzaC treatment. More interestingly, the expression level of several of these genes was enhanced only in the EPC-hTERT.D1-Cdx2 cells treated with 5-AzaC. Two of these, SLC26a3/DRA (downregulated in adenoma) and Na+/H+ exchanger 2 (NHE2), were not previously known to be elevated in BE; however, we confirmed their elevation in BE tissue samples. 5-AzaC treatment also induced cell senescence, even at low doses. We conclude that ectopic proliferation signals, alterations in epigenetic gene regulation and the inhibition of tumor suppressor mechanisms are required for Cdx2-mediated intestinalization of human esophageal keratinocytes in BE.
Introduction
Barrett’s esophagus (BE) occurs at the gastroesophageal junction and is characterized by the replacement of the normal esophageal squamous epithelium with a specialized intestinal columnar epithelium. It is of clinical importance because it is a premalignant condition that considerably increases an individual’s risk for esophageal adenocarcinoma (EAC) (1,2). However, the molecular events that give rise to BE remain to be elucidated.
The intestinal cell phenotype that characterizes BE is normally manifested only in the small intestine and colon. However, intestinal metaplasia precedes the development of cancer in several gastrointestinal tissues (3,4). Typically, expression of the intestine-specific transcription factor Cdx2 is detected early in these preneoplastic lesions (5–9). Cdx2 is a caudal-related homeobox transcription factor. It is well known for its role in promoting intestine-specific gene expression and intestinal cell differentiation (10). A causal link between the ectopic expression of Cdx2 and gastric intestinal metaplasia has been established using a transgenic mouse approach (11,12). However, similar proof for Cdx2 as the causative factor in BE has remained elusive.
One limitation in the study of the genesis of BE has been the lack of suitable experimental cell culture models. The cell culture models used to date have largely been human BE tissue explants and EAC cell lines, with some recent work in primary Barrett’s cell lines. Acid exposure has been suggested to enhance proliferation and promote intestinal differentiation based on studies with these models (13–16). Barrett’s cell lines have recently been developed (17,18). While useful for studies of the progression from BE to EAC, they are less useful for understanding how BE emerges within the normal esophageal epithelium.
We and others have sought to establish whether esophageal squamous keratinocytes can be induced to transdifferentiate in vitro into BE epithelial cells. Chronic acid and bile acid treatment of rat and murine esophageal keratinocytes induces low levels of Cdx2 expression but does not induce an intestinal cell phenotype (19,20). More recently, it was demonstrated that primary human esophageal keratinocytes treated with bone morphogenetic protein 4 began expressing BE-associated cytokeratins and a more intestinalized pattern of gene expression (21). In another study using immortalized human esophageal keratinocytes (HET1A), Cdx2 expression was associated with increased cell proliferation and an intestinal pattern of gene expression (22). One limitation of this study is that HET1A cells are immortalized by ectopic expression of the SV40 T antigen, a viral oncoprotein that inactivates p53 and the retinoblastoma protein (pRb), among other targets (23).
Given these previous reports, we hypothesized that Cdx2 expression in normal human keratinocytes would induce an intestinal pattern of gene expression similar to that seen in human BE. In the present study, we identify several critical parameters required to intestinalize a human esophageal cell line known as EPC-human telomerase reverse transcriptase (hTERT). Expression of Cdx2 alone in these cells did not induce intestinalization but rather severely inhibited cell proliferation. However, agents that promoted chromatin remodeling synergized with Cdx2 to enhance the expression of intestine-specific and BE-associated genes. These agents also uniformly induced cell senescence. We conclude that ectopic proliferation signals, alterations in epigenetic gene regulation and the inhibition of tumor suppressor mechanisms are necessary for Cdx2-mediated intestinalization of normal human esophageal keratinocytes.
Materials and methods
Cell culture and transfections
Immortalized human primary esophageal epithelial cells (EPC-hTERT) were developed and maintained as described previously (24,25). The pBPSTR, pBabe-puro-p53R175H, MIGR1 and MIGR-Cdx2 retroviral vectors were described previously (26–28). The MIGR1 vector includes an internal ribosomal entry site and a 3′ green fluorescent protein (GFP) complementary DNA (cDNA) with a 5′ multicloning site into which a Cdx2 cDNA was cloned. We utilized the pBPSTR retroviral vector to express cyclin D1 under the control of a Tet-Off promoter (28). Retroviral vectors were transfected into Phoenix-Ampho packaging cells (gift of Dr Garry Nolan, Stanford University, Palo Alto, CA) using Lipofectamine™ 2000 reagent (Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions. Infectious retroviral supernatants were isolated at 48 h, purified as described elsewhere (29) and stored at −70°C. Cells infected with the MIGR1-based retrovirus can be identified and sorted by their GFP fluorescence described (26).
EPC-hTERT cells were infected with pBPSTR-D1 virus, subjected to puromycin selection and then infected with MIGR-Cdx2 and MIGR1 retroviral vectors. GFP-expressing cells were then isolated by fluorescence-activated cell sorting. For the 5-aza-2-deoxycytidine (5-AzaC) treatment, cells were plated at a density of 5 × 105 per 10 cm dish. The cells were grown in growth medium without or with the demethylating agent 5-AzaC (1, 5 and 10 μmol/l; Sigma, St Louis, MO) for 120 h. The medium was replaced with fresh medium every other day. The equivalent volume of vehicle (50% dimethyl sulfoxide for 5-AzaC) was applied as control.
Cell proliferation assays
Cell proliferation was determined using two methods: (i) the WST-1 assay and (ii) [H3]thymidine incorporation. Cell proliferation was quantified by colorimetry based on the metabolic cleavage of the tetrazolium salt WST-1 in viable cells as recommended by the manufacturer (Roche Applied Science, Mannheim, Germany). To determine cell proliferation by [H3]thymidine incorporation, the stably infected cells were incubated with [H3]thymidine (1 μCi) (PerkinElmer Life and Analytical Sciences, Shelton, CT) and cells were incubated for 24 h. Cells were harvested for DNA synthesis and cell proliferation analysis was performed using an LS 6500 Multi-Purpose Scintillation Counter (Beckman Coulter, Fullerton, CA). For each cell line, the mean value of six separate experiments was calculated. The assay was repeated at least three times.
Tissue specimens
Fresh tissues were obtained from the Hospital of the Pennsylvania through the Cooperative Human Tissue Network. Fresh specimens were frozen in liquid nitrogen immediately after surgery and stored at −80°C for RNA or protein analysis. Formalin-fixed, paraffin-embedded human sections were used for the immunohistochemical study. Histological analysis was performed by pathologists using standard diagnostic criteria and patient’s informed consent was obtained by the Cooperative Human Tissue Network participating hospitals in accordance with institutional review board standards and guidelines. Six paired human esophageal samples consisting of BE and matched normal or benign esophageal tissues (from the margins of the excised Barrett’s) were obtained. Three normal colon biopsies were obtained from patients undergoing surgical resections.
RNA isolation and real-time quantitative polymerase chain reaction analysis
Total RNA was isolated from sorted GFP-expressing cells, human BE tissue and matched normal controls using the RNAeasy kit (Qiagen, Valencia, CA). Five microgram of total RNA was used for cDNA synthesis using the SuperScript™ First-Strand Synthesis Kit (Invitrogen). Reverse transcriptase-negative controls were included. Primer sequences for polymerase chain reaction (PCR) are available in supplementary Table I (available at Carcinogenesis Online). For the reverse transcription (RT)–PCR, cDNA and primers were mixed with SYBR-green PCR Master Mix (Applied Biosystems) and then assayed in an ABI Prism 7000 sequence detection system as directed by the manufacturer. A ribosomal phosphoprotein, 36B4, was used as the normalization control. Fold change in RNA levels was calculated from the Ct values using the formula described previously (26,30). The ΔΔCt values for each gene were averaged across the RNA pools, standard deviations were calculated and statistical comparisons were performed using analysis of variance and Tukey Rank Mean testing. These values were then converted to fold change to graphically report the findings.
Protein extraction and western blot analysis
Whole-cell extract was prepared as described (31). Protein concentration of samples and bovine serum albumin standard was determined using the BCA™ protein assay kit (Pierce Biotechnology, Rockford, IL). Primary antibodies used included: anti-Cdx2 IgG1 (clone CDX2-88) (1:1000; BioGenex, San Ramon, CA), anti-downregulated in adenoma (DRA) [1:1000; Dr Soleimani, University of Cincinnati Medical Center (UCMC) (32)], anti-Na+/H+ exchanger 2 (NHE2) (1:1000, AB3084; Chemicon, Temecula, CA), anti-villin IgG1 (1:500, V34420-150; BD Transduction Laboratories, San Diego, CA), anti-p21 (1:200, sc-397; Santa Cruz Biotechnology, Santa Cruz, CA), anti-p16 (1:5000; gift from Gregory H.Enders, Fox Chase Cancer Center), anti-tubulin (1:4000; Vector Laboratories, Burlingame, CA) and anti-actin (1:1000; Sigma–Aldrich, St. Louis, MO). The secondary antibodies used were all from Sigma–Aldrich and used at 1:3000. Targeted proteins were visualized using an enhanced chemiluminescence detection system (ECL Plus; Amersham Pharmacia Biotech, Buckingham, UK) and exposed to Blue Lite Autorad film (ISC-BioExpress, Kaysville, UT). For quantitative western blotting analysis, the signals were visualized by the STORM840-Scanner phosphoimager (Amersham Biosciences, Piscataway, NJ) following the manufacturer’s instructions, and the appropriate protein bands were quantified using ImageQuant Version 5.2 software (Amersham Biosciences).
Senescence-associated β-galactosidase staining
To determine if the cells had undergone senescence, we used the Senescence β-Galactosidase Staining Kit (Cell Signaling Technology, Beverly, MA) according to the manufacturer’s protocol. Cells stained for senescence-associated β-galactosidase activity were scored by counting five high-power fields.
Immunohistochemistry
Immunostaining assay was performed as described previously (33). In brief, paraffin-embedded sections (6 μm) were pretreated with xylene and then boiled in a microwave oven in 10 mmol/l citric acid buffer. Endogenous peroxidases were quenched using hydrogen peroxide before sections were incubated in avidin D blocking reagent and biotin blocking reagent. Sections were incubated with primary anti-DRA (32) (1:250; gift from Dr Soleimani, UCMC) and anti-NHE2 (diluted 1:250; Chemicon International), followed by incubation with secondary antibodies. Immunohistochemistry was carried out using the Vectastain Elite kit (Vector Laboratories) following the manufacturer’s protocol. The signal was then developed using the 3,3′-diaminobenzidine substrate kit for peroxidases (Vector Laboratories). Stained objects were examined with a Nikon microscope and imaged with a digital camera at indicated magnifications. Normal colon sections were used as a positive control for DRA/SLC26a3 and NHE2. As negative controls, parallel sections were similarly processed without the respective primary antibody for each immunostaining experiment.
Results
We have subcloned a Cdx2 cDNA into an Murine Stem Cell Virus-based retroviral vector to express Cdx2. This vector, called MIGR1, has a GFP cDNA as the selectable marker. The virus we produced, called MIGR-Cdx2, has been successfully used to establish stable Cdx2 expression in colon cancer cells (26,27). This vector expresses both GFP and Cdx2 in a single, bicistronic messenger RNA (mRNA). Retroviral infection and gene expression are established by following GFP protein expression. We therefore used this vector to express Cdx2 in normal human esophageal keratinocytes immortalized by the ectopic expression of human telomerase, called EPC-hTERT cells (24,25). Unlike our experience with colon cancer cells, we found it difficult to establish stable Cdx2 expression in the EPC-hTERT cells. Within 72 h of infection by the retrovirus, we could see abundant GFP-expressing EPC-hTERT cells. However, after 7–10 days in culture, a noticeable decline in GFP expression occurred in those cells in which Cdx2 was coexpressed (Figure 1A). We attempted to sort at 72 h for GFP-expressing cells by flow cytometry to establish a Cdx2-expressing cell line. Although we were able to isolate 25 000 GFP+ cells to start, the population that grew out uniformly had lost GFP and Cdx2 expression. This was not observed when using the empty MIGR1 vector as a control (data not shown).
Fig. 1.
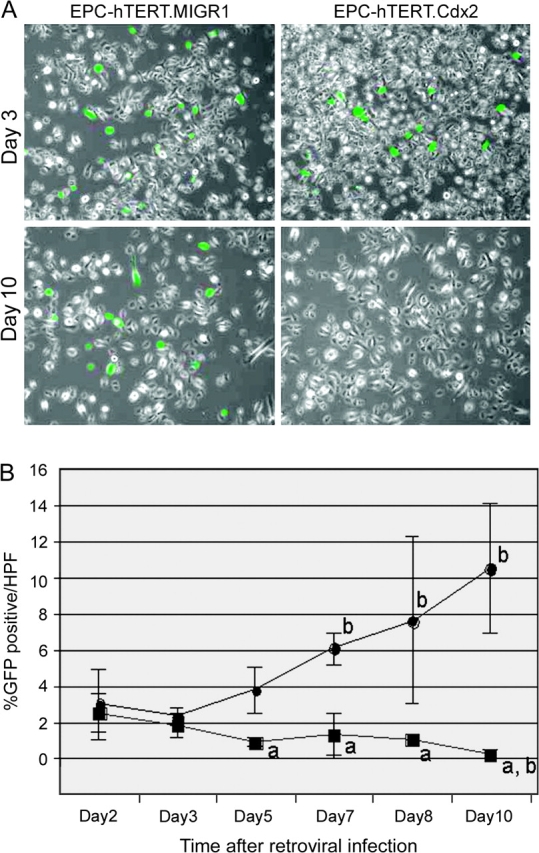
Cdx2 expression is not sustained in normal EPC-hTERT keratinocytes. Human EPC-hTERT keratinocytes were infected with a retroviral vector to induce Cdx2 expression (MIGR-Cdx2) or the control empty viral vector MIGR1. GFP serves as a marker for viral infection and gene expression. (A) Fluorescent image for GFP+ cells overlaid with phase contrast image. At 72 h, nearly equivalent numbers of GFP+ cells with both vectors. After 10 days in culture, repeat images demonstrate highly significant reduction in GFP+ expression in those cells infected with the MIGR-Cdx2 virus. (B) The number of GFP+ cells per total cells in six high power fields (HPFs) was determined on days 2, 3, 5, 7, 8 and 10 post-retroviral infection. Averages and standard deviations were calculated and graphed. Black circle, EPC-hTERT.MIGR. Black square, EPC-hTERT.Cdx2. a, significantly differs from MIGR same day controls using analysis of variance and Tukey Rank Mean testing, P < 0.005. b, significantly differs from day 2 MIGR controls, P < 0.005.
Based on our work in colon cancer cells, where we studied an antiproliferative effect of Cdx2, we suspected that Cdx2 expression was limiting proliferation in our EPC-hTERT cells (10,34,35). To explore this further, we followed GFP expression in EPC-hTERT cells infected by MIGR-Cdx2 virus over 10 days. At 48 h, there was approximately the same number of GFP+ EPC-hTERT cells after infection with the MIGR-Cdx2 virus and the empty vector MIGR1 control. Whereas the percentage of GFP+ cells increased from 3 to 12% in the EPC-hTERT cells receiving the control MIGR1 vector, cells infected with the MIGR-Cdx2 vector gradually lost all GFP expression (Figure 1A). By day 5, there was a statistically significant difference between the Cdx2-expressing and control cells, and virtually all GFP+/Cdx2+ cells were lost by day 10 (Figure 1B). In summary, Cdx2 expression cannot be sustained in a population of normally proliferating esophageal keratinocytes.
Ectopic cyclin D1 expression can support Cdx2 in normal human esophageal keratinocytes
There was no evidence for increased cell death or apoptosis in those GFP+/Cdx2+ cells at 72 h after infection (data not shown). Rather, it appeared that the GFP+/Cdx2+ cells were either less proliferative and thereby outcompeted by the other cells or that the retroviral vector was silenced or lost by an unknown mechanism. In colon cancer cells, we had previously observed a reduction in cell proliferation associated with Cdx1 or Cdx2 expression (35). Mechanistically, this was due to a loss in cyclin D1 expression (34). We therefore utilized the pBPSTR retroviral vector to express cyclin D1 under the control of a Tet-Off promoter in the EPC-hTERT parental cells (28). Cyclin D1 levels were increased in the EPC-hTERT cells after infection with the pBPSTR retroviral vector (Figure 2A). The addition of doxycyclin to the cell culture medium resulted in a reduction of cyclin D1 levels to that of the parental EPC-hTERT cells.
Fig. 2.

Cdx2 expression in normal keratinocytes requires exogenous stimulus for proliferation. (A) Western blot analysis demonstrating doxycyclin-regulated cyclin D1 protein expression. Enhanced cyclin D1 protein levels were noted in EPC-hTERT.D1 (EPC.D1) but not control cells (EPC.P) in the absence of doxycyclin. However, doxycyclin (1 μg/ml) in the cell culture medium suppressed this expression. (B) Western blot results illustrating overexpression of Cdx2 in EPC-hTERT.D1-Cdx2 cells but not the EPC-hTERT.D1-MIGR1 control cells. (C) Cdx2 expression in the EPC-hTERT.D1 cells reduces proliferation. [H3]Thymidine incorporation assay in the absence (light gray bars) and presence of doxycyclin (1 μg/ml) (dark gray bars). In the absence of exogenous cyclin D1, Cdx2 expression is associated with a significant reduction in [H3]thymidine incorporation when compared with controls. Averages and standard deviations were calculated (n = 3, P < 0.05). (D) WST-1 cell accumulation studies performed on EPC-hTERT.D1-MIGR1 (EM) and EPC-hTERT.D1-Cdx2 (EX2) cells in the absence or presence of doxycycline (D; 1 μg/ml). There was a significant decrease in the accumulation of EPC-hTERT.D1-Cdx2 cells compared with MIGR1 control cells in the presence or absence of exogenous cyclin D1. Averages and standard deviations from eight separate wells for each treatment group were calculated and graphed. One of three experiments is shown.
EPC-hTERT.D1 cells were subsequently infected with either the MIGR1 or MIGR-Cdx2 retroviruses. GFP+ cells were isolated by fluorescence-activated cell sorting for further study. Unlike our previous attempts, Cdx2 expression was established and maintained over multiple passages (Figure 2B and data not shown). The Cdx2-expressing cells appeared to proliferate slower than the EPC-hTERT.D1.MIGR1 control cells. To explore more fully the effects of Cdx2 expression on EPC-hTERT cell proliferation, we measured [H3]thymidine incorporation in our EPC-hTERT.D1.Cdx2 cells and EPC-hTERT.D1.MIGR1 cells in the absence and presence of doxycyclin. In the absence of doxycyclin, both cell lines readily incorporated the labeled thymidine. However, when cyclin D1 levels were reduced to baseline by the addition of doxycyclin, thymidine incorporation declined considerably (Figure 2C). [H3]Thymidine incorporation by the EPC-hTERT.D1.Cdx2 cells was half that of the controls, suggesting that Cdx2 expression was associated with a significantly reduced rate of cell proliferation when compared with control cells.
We confirmed this finding using the WST-1 assay. EPC-hTERT.D1.Cdx2 cells accumulated more slowly in culture than the control cells, even when cyclin D1 levels were elevated in the absence of doxycyclin (Figure 2D). The addition of doxycyclin further depressed cell numbers in both cell lines. Both of these findings are consistent with our prior observation and probably explain why we were unable to isolate a population of cells stably expressing Cdx2.
We next determined if p53 inactivation would also be permissive for Cdx2 expression in esophageal keratinocytes. We established EPC-hTERT cells expressing a dominant-negative p53 using the pBabe-puro-p53R175H vector (Figure 3A) (28). We then tried again to establish Cdx2 expression using our MIGR-Cdx2 retroviral vector but were unsuccessful. As with the wild-type cells, GFP expression was lost in the dominant-negative p53 cells over 7–10 days after infection, but only in the cells receiving MIGR-Cdx2 and not the control vector MIGR1 (Figure 3B and C). In summary, an enhanced proliferative stimulus, and not p53 inhibition, is required to overcome the antiproliferative effect of Cdx2 on esophageal keratinocytes. Together, these observations suggest that Cdx2 expression cannot be the first event in the transdifferentiation of esophageal keratinocytes into BE epithelial cells.
Fig. 3.
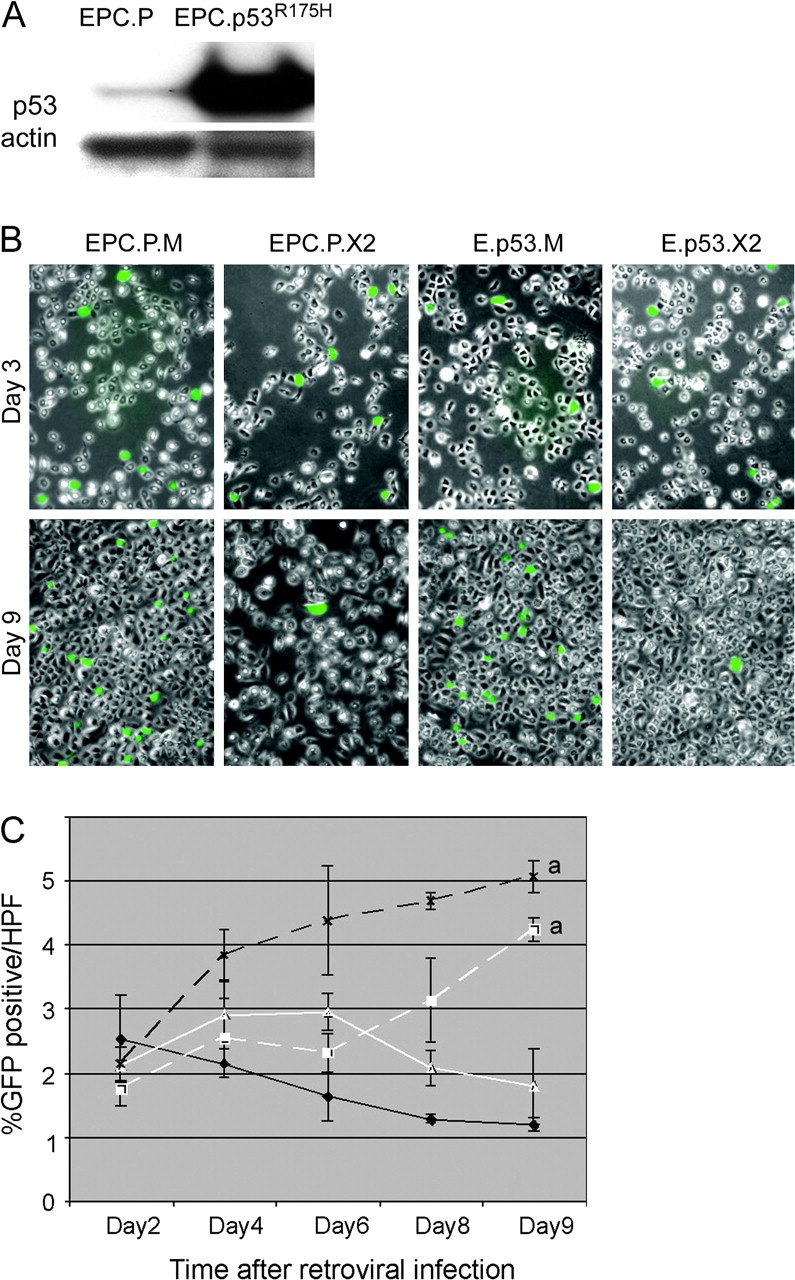
A dominant-negative p53 does not rescue Cdx2 expression in EPC-hTERT keratinocytes. EPC-hTERT keratinocytes expressing a dominant-negative p53 were established using the pBabe-puro-p53R175H vector to direct expression of the p53R175H mutant. (A) Western blot of p53 protein levels in EPC-hTERT cells receiving the empty pBabe control vector (EPC.P) or the pBabe-puro-p53R175H (EPC.p53R175H). The blot was reprobed for actin levels as loading control. (B) As before, fluorescent image for GFP+ cells overlaid with phase contrast image. EPC.P or EPC.p53R175H cells were subsequently infected with the MIGR-Cdx2 virus to induce Cdx2 expression (EPC.P.X2 and E.p53.X2) or the MIGR1 empty control vector (EPC.P.M and E.p53.M). Photodocumentation was obtained at 3 and 9 days post-infection with the MIGR-based vectors. At 3 days, nearly equivalent numbers of GFP+ cells with both vectors. By day 9, there is a highly significant reduction in GFP+ expression in those cells infected with the MIGR-Cdx2 virus. (C) The number of GFP+ cells per total cells in six high-power fields (HPFs) was determined on days 2, 4, 6, 8 and 9 post-retroviral infection. Averages and standard deviations were calculated and graphed. Black X, dashed line: EPC.P.M. White triangle, solid line: EPC.P.X2. White square, dashed line: E.p53.M. Black diamond, solid line: E.p53.X2. a, significantly differs from day 9 MIGR controls using analysis of variance and Tukey Rank Mean testing, P < 0.05.
Inducing an intestinal pattern of gene expression in human esophageal keratinocytes requires chromatin remodeling in addition to Cdx2 activity
The homeodomain transcription factor Cdx2 is an important regulator of the intestinal cell phenotype including both columnar cell morphology and intestine-specific gene expression (10). After establishing Cdx2 expression in our EPC-hTERT.D1 cells, we evaluated the cells for evidence of intestinalization. By phase contrast and transmission electron microscopy, we observed no significant changes in the morphology of EPC-hTERT.D1.Cdx2 cells when compared with controls (data not shown). We next explored for changes in gene expression patterns that would be consistent with intestinalization. Using quantitative SYBR-green real-time RT–PCR, we tested a panel of genes known to be either Cdx2 transcriptional targets including carbonic anhydrase I (CAI), liver–intestine cadherin (LI-cadherin), lactase-phlorizin hydrolase (LPH), sucrase–isomaltase (SI), claudin-2 and guanylyl cyclase C (GC-C) or genes whose expression is associated with intestinal epithelium [DRA/SLC26A3, Villin, alkaline phosphatase (ALK PHOS), LI-cadherin, SI, LPH, resistin-like molecule β (RELM-β), NHE2, CEACAM6, MUC2 and cytokeratin 20 (KRT20)] for increases in their mRNA levels with Cdx2 expression. In only one gene, CA1, we observed a robust 7-fold increase in mRNA levels associated with Cdx2 (Figure 4A). In the others, there was either no change or no detectable mRNA for that gene (data not shown). This observation stands in contrast to our experience in colon cancer and intestinal cell lines, where Cdx2 expression is associated with robust intestinalization (26,36). It also is quite different from HET1A cells, where Cdx2 expression alone was sufficient to induce a number of intestine-specific and known Cdx2 target genes (22). It suggests that Cdx2 expression alone is insufficient to induce intestinal transdifferentiation of normal esophageal keratinocytes.
Fig. 4.

Cdx2 expression alone has minimal effects on EPC-hTERT intestinal transdifferentiation. (A and B) Quantitative SYBR-green RT–PCR analysis of gene expression in EPC-hTERT.D1-MIGR1 (EPC.D1.M) or EPC-hTERT.D1-Cdx2 (EPC.D1.X2) cells treated for 5 days with 5 μM of 5-azacytidine (5AZA) or diluent control (CTR). Genes tested included Claudin2, CEACAM6, Villin, LPH, CAI, RELM-β, NHE2, KRT20 and DRA/SLC26A3. The PCR control was the phosphoprotein 36B4. ΔCt values were calculated after duplicate PCRs for each sample, then statistical analysis was performed (analysis of variance and Tukey Rank Mean). ΔΔCt values were then calculated and used to determine fold change in expression (n = 6 samples). Black circle = significantly differs from MIGR-diluent control, P < 0.0005; black star = significantly differs from MIGR-diluent control, P < 0.005. (C) Quantitative SYBR-green RT–PCR analysis of CDX1 and CDX2 gene expression in EPC-hTERT.D1.MIGR1 (EPC.D1.M) or EPC-hTERT.D1.Cdx2 (EPC.D1.X2) cells treated with 5 μM 5-azacytidine (5AzaC) or diluent control. PCR controls and the calculation of average ΔΔCt values (and standard deviations) were carried out as before (n = 3). (D) Western blot for CDX1, CDX2, DRA/SLC26a3, villin and NHE2 protein levels in EPC-hTERT.D1.MIGR (MIGR1) or EPC-hTERT.D1.Cdx2 cells (CDX2). Cells were treated with 5 μM 5-azacytidine (5AzaC) or diluent control. The blots were routinely stripped and reprobed for actin levels as loading control. One of three experiments is shown. (E) Quantitative densitometry measurements of selected western band densities. Values were normalized to actin bands, then expressed as fold change compared with EPC-hTERT.D1.MIGR cells treated with diluent (white bars). Black bars, EPC-hTERT.D1.MIGR cells treated with 5 μM 5-azacytidine. Light gray bars, EPC-hTERT.D1.Cdx2 cells with diluent. Dark gray bars, EPC-hTERT.D1.Cdx2 cells with 5 μM 5-azacytidine.
We next sought to determine if other treatments could be applied to the EPC-hTERT.D1.Cdx2 cells to foster intestinalization. Acid exposure of normal murine esophageal keratinocytes, as well as colon and esophageal cancer cells, has modulated cell differentiation and proliferation (13,14,19,20,37). We attempted to replicate this work with our human EPC-hTERT.D1 cells, with and without Cdx2 expression. However, all acid treatments (pH 3.5 or 5.0 for 15 min or 1 h, respectively) uniformly killed the cells (data not shown).
We then considered the possibility that epigenetic processes might be involved in the intestinal transdifferentiation (38–43). Treatment of cells with agents such as 5-AzaC is known to induce chromatin remodeling and can foster cell transdifferentiation (44–48). We treated our EPC-hTERT.D1.Cdx2 and EPC-hTERT.D1.MIGR1 control cells with increasing doses of 5-AzaC for different lengths of time. We isolated total RNA and performed quantitative RT–PCR for our marker genes.
The 5-AzaC treatment enhanced the expression of several previously unexpressed (DRA/SLC26A3, LPH, KRT20 and CEACAM6) and poorly expressed genes (NHE2 and Villin) (Figure 4A). We further determined that 1 μM of 5-AzaC for 10 days or 5 μM for 5 days both yield highly reproducible responses (data not shown). Of the genes tested, there were four patterns of response. One group did not respond to the Cdx2 ± 5-AzaC treatment (LI-cadherin, ALK PHOS, SI, MUC2 and GC-C) (data not shown). Four genes, Claudin-2, CEACAM6, Villin and LPH, were all significantly increased by the 5-AzaC treatment, but Cdx2 expression had no additional effect on their levels (Figure 4A).
In the remaining five genes studied, there was clear evidence of a synergistic interaction between Cdx2 and 5-AzaC. mRNA levels for CAI, which were increased 7-fold with Cdx2 expression alone and only 2-fold at best when treated with 5-AzaC, increased by 35-fold above controls when both treatments were applied. Most interesting of all, four genes responded only to the combination of Cdx2 expression and 5-AzaC treatment (RELM-β, NHE2, KRT20 and DRA/SLC26A3). One of these genes, KRT20, is utilized as a marker for BE. Its induction is therefore of great interest.
Cdx1 and Cdx2 expression in the esophagus has been reportedly induced in esophageal keratinocytes by chromatin remodeling agents (22). We confirmed these findings in our EPC-hTERT.D1.MIGR1 cells, as 5-AzaC treatment induced CDX1 mRNA >20-fold and CDX2 mRNA by nearly 200-fold (Figure 4C). However, despite this significant increase in mRNA levels, it did not necessarily translate into increased protein levels. CDX1 and CDX2 protein remained undetectable in the EPC-hTERT.D1.MIGR1 after 5-AzaC treatment (Figure 4D). Surprisingly, CDX1 mRNA and protein were detected in the EPC-hTERT.D1.CDX2 cells before the addition of 5-AzaC. This was unexpected; however, the CDX transcription factors are known in the intestine to have auto- and cross-regulatory activity.
Protein levels for other genes of interest also did not typically match the measured mRNA responses. CDX2 expression had no apparent effect on SLC26a3/DRA protein levels. Both SLC26a3/DRA and villin protein levels increased only with 5-AzaC treatment (Figure 4D and E). NHE2 protein levels did appear to increase modestly when EPC-hTERT.D1.Cdx2 cells were treated with 5-AzaC compared with controls; however, these levels did not reach the 6-fold induction noted in the mRNA levels. In summary, treatments that alter epigenetic gene regulation in esophageal keratinocytes can augment Cdx2’s effects and lead to stable mRNA expression of many intestine-specific genes. However, protein levels do not correlate with mRNA levels in EPC-hTERT cells treated with 5-AzaC.
Gene expression patterns of 5-AzaC-treated EPC-hTERT.D1-Cdx2 cells mimic BE
The patterns of genes induced in EPC-hTERT.D1-Cdx2 cells after 5-AzaC treatment did resemble those reported previously for human BE. The induction of mRNA expression for genes such as KRT20, LPH, CAI and Villin is consistent with BE, based on prior reports (49–51). However, there were no reports for BE-associated induction of NHE2 or DRA/SLC26a3, though both are highly expressed in intestinal epithelium. We therefore investigated for their possible induction in human BE tissue specimens.
Total RNA was isolated from five paired biopsies from BE tissue and adjacent normal squamous epithelium. Quantitative real-time SYBR-green RT–PCR was then performed for two well-described markers for BE (CAI and KRT20) as well as NHE2 and DRA/SLC26a3. In each case, there was a significant increase in the mRNA levels for these four genes in the BE samples when compared with the control normal adjacent esophageal epithelium (Figure 5A). In four of five specimens, there was a 50- to 100-fold increase in NHE2 mRNA levels in BE tissues. DRA/SLC26a3 was increased several 1000-fold in BE tissues compared with normal controls. Similarly, we observed several 100- to several 1000-fold induction of KRT20 and CAI mRNA levels in the BE samples when compared with controls.
Fig. 5.
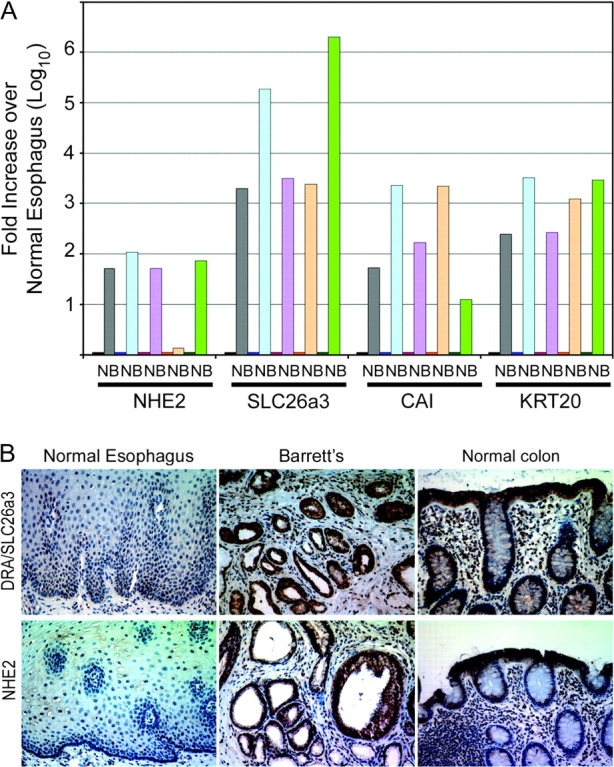
Intestine-associated genes NHE2 and SLC26a3 are induced in human BE. (A) Quantitative real-time PCR measurement of NHE2, DRA/SLC2A3, CAI and KRT20 mRNA levels in biopsies from human BE and adjacent normal esophageal mucosa. (B) Representative immunohistochemical staining for DRA/SLC26A3 and NHE2 protein in BE biopsy specimen. Normal colon and normal esophagus are presented as controls. One of several Barrett’s biopsy specimens is shown.
To confirm that these increased mRNA levels yield significant increases in protein expression, we stained two BE biopsy specimens for DRA/SL26a3 and NHE2 by immunohistochemistry. Robust DRA/SLC26a3 and NHE2 protein expression is detected in both biopsy specimens and the normal colon tissues, but not in the normal esophagus control (Figure 5B). Taken together, these findings suggest that both NHE2 and DRA/SLC26a3 genes are induced in BE. More importantly, it suggests that the intestinalization induced in EPC-hTERT.D1-Cdx2 cells treated with 5-AzaC may mimic BE transdifferentiation, as two novel BE-associated markers were identified using this approach.
Senescence is activated by the chromatin remodeling agent 5-AzaC in EPC-hTERT.D1-Cdx2 and control esophageal keratinocytes
After 5-AzaC treatment, EPC2.hTERT.D1-Cdx2 and MIGR1 control cells exhibit altered cell morphology marked by increased cell volume and subconfluent growth arrest in the normal growth medium. The cells no longer increased in number, despite several days of fresh growth medium. Even low doses of 5-AzaC (1 mM for 72 h) appeared to lead to growth arrest. Additionally, BrdU labeling and WST-1 assays for cell proliferation both suggested that proliferation was absent (data not shown). These features are all suggestive of cell senescence (52).
In fact, 5-AzaC treatment has been associated with cell senescence in other cell systems (52). Therefore, to determine whether 5-AzaC-treated cells were undergoing senescence, we evaluated for the induction of senescence-associated β-galactosidase as well as the expression of p21(Waf1/Cip1) and p16(INK4a), all of which are genetic features of senescence. 5-AzaC treatment induced a flattened and enlarged cell morphology in both EPC-hTERT.D1-Cdx2 and EPC-hTERT.D1-MIGR1 cells. Moreover, senescence-associated β-galactosidase activity was markedly increased (Figure 6A). p21 and p16INK4a protein levels were greatly increased with 5-AzaC treatment when compared with untreated controls (Figure 6B). This increase was not affected by Cdx2 coexpression and is consistent with the induction of senescence. In summary, 5-AzaC treatment of hTERT-immortalized but not transformed human esophageal keratinocytes induces their senescence and withdrawal from the cell cycle.
Fig. 6.
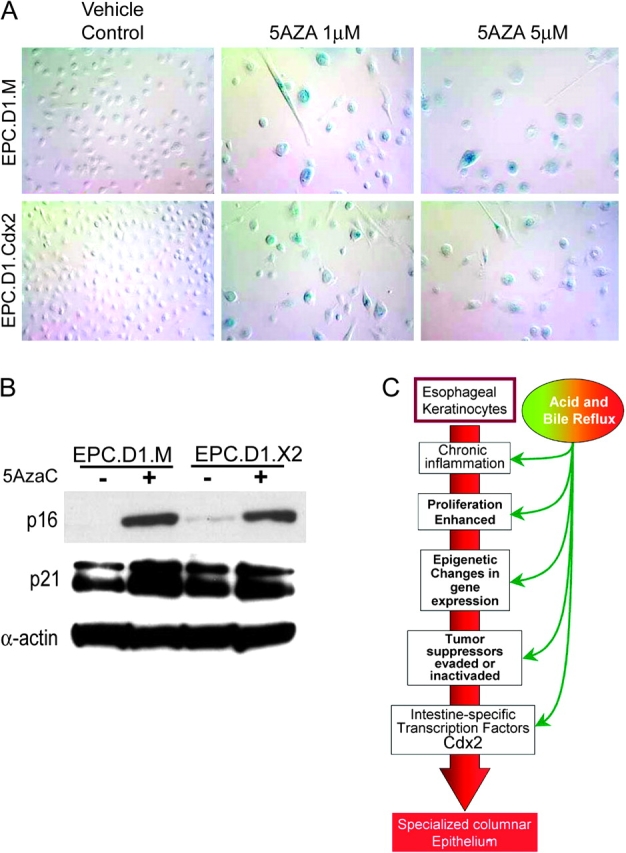
Senescence checkpoint genes are activated by chromatin remodeling agents 5-AzaC in esophageal keratinocytes. After 5-AzaC treatment, EPC-hTERT.D1-Cdx2 and MIGR1 (EPC.D1.X2 and EPC.D1.M, respectively) control cells exhibit characteristics of senescence including altered morphology, increased cell volume and subconfluent growth arrest in the normal medium. (A) Morphology of EPC-hTERT.D1.Cdx2 and control EPC-hTERT.D1.MIGR1 cells after 5AzaC at 1 and 5 μM for 5 days. Induction of senescence-associated β-galactosidase activity was identified by staining (blue). (B) 5-AzaC treatment induces the expression of senescence-associated p16(INK4a) and p21(Waf1/Cip1). Western Blot for p16 and p21 after cells were treated with 1 μM 5-AzaC or control diluent for 5 days. β-Actin served as an internal loading control. (C) Proposed model for the early molecular events preceding the onset of intestinalization in esophageal keratinocytes.
Discussion
BE occurs at the gastroesophageal junction and is the replacement of the normal squamous esophageal mucosa with an intestinalized columnar epithelium. It arises in the setting of chronic acid and bile reflux that results in chronic injury and inflammation in the esophageal epithelium. BE has a relatively high progression rate to EAC. This rate has been estimated to be 0.5–1%/year (2). Thus, it is a clinically important condition to understand.
Esophageal epithelium development and BE
Little is known about the molecular mechanisms giving rise to BE from normal squamous esophageal mucosa. The emergence of the specialized columnar epithelium, characteristic of BE, within a stratified squamous epithelium has been difficult to explain. Indeed, even the identity of the progenitor cell that gives rise to BE epithelium remains controversial. Four candidate progenitor cells have been proposed, but there is as yet no consensus around any one of them (4). Our work is based on the hypothesis that BE arises from squamous cell progenitors. We believe there is a great deal of support for this model in the literature.
Studies of human embryology reveal that at 10 weeks of age, the human esophagus is lined by a columnar epithelium. This epithelium is later replaced by a stratified squamous epithelium, which begins in the mid-esophagus and proceeds in both caudad and cephalad directions (53). Interactions between the endodermal and mesodermal layers in part control this transdifferentiation into squamous cells (54–56). In fact, the regional identity of the early endoderm (foregut versus midgut) is very plastic. Co-culture experiments have established that primitive endoderm will differentiate to match the regional identity (foregut or midgut) of the mesoderm with which it is cultured. Thus, it is possible that one or more progenitor cells within the adult esophageal epithelium may retain this early multipotent capability. This is further supported by the observation of hamartomatous polyps with morphologies consistent with squamous, gastric and intestinal epithelium (57,58) that arise in Cdx2 (+/−) heterozygous knockout mice. Theoretically, early progenitor cells with developmental plasticity could differentiate into squamous or glandular epithelium, depending upon local environmental cues and certain disease conditions.
One limitation in the study of BE has been the absence of suitable experimental models. We have approached this limitation by determining if normal esophageal keratinocytes can transdifferentiate into intestinal epithelium using a combination of genetic and biochemical manipulations. This forward genetics approach has been used before with great success in the study of cancerous transformation of normal cells (59). Recently, Liu et al. (22) reported that immortalized human esophageal keratinocytes (HET1A) were intestinalized by expression of the intestine-specific transcription factor Cdx2. That suggested that Cdx2 expression might be associated with increased cell proliferation and an intestinal pattern of gene expression. However, HET1A cells are immortalized by the expression of the SV40 T antigen, which inactivates p53 and pRb function and is associated with altered DNA methylation patterns. These effects may have unintended consequences. For instance, inactivating p53 mutations are acquired late in BE, not with the onset of the intestinal cell phenotype (60). Moreover, normal pRb function is critical for the antiproliferative effect of Cdx1 (61). Thus, its inactivation may alter normal cell responses to Cdx2 expression. Lastly, the SV40 T antigen has been associated with changes in DNA methylation patterns in several cell lines (62). The SV40 T antigen might therefore be masking important early steps in the process of intestinal transdifferentiation. Our findings, reported here, confirm this hypothesis.
Cdx2 expression in human esophageal keratinocytes inhibits proliferation and does not induce an intestinal morphology or pattern of gene expression
We utilized for our studies EPC-hTERT cells, normal human esophageal keratinocytes immortalized by the ectopic expression of human telomerase (hTERT) (24). We demonstrated that Cdx2 expression was not normally maintained in EPC-hTERT cells. Ectopic cyclin D1 expression, but not p53 inhibition, was permissive for long-term Cdx2 expression in EPC-hTERT cells (Figure 2).
We further determined that this was due to a significant antiproliferative effect that Cdx2 expression had upon EPC-hTERT keratinocytes. This finding was unexpected, as Cdx2 expression in HET1A cells is associated with increased cell proliferation (22). We believe this difference is due to the abrogation of the pRb function by the SV40 T antigen in HET1A cells. Studies by us and others suggest that the pRb is critical for Cdx2’s antiproliferative effect (31,35,61). In the absence of normal pRb function, Cdx appears to have a pro-proliferative activity.
We then looked for evidence of intestinalization in our EPC-hTERT.D1.Cdx2 cells. Again, unlike the experience reported with HET1A cells, Cdx2 expression alone in EPC-hTERT cells did not induce an intestinal cell morphology or an intestinal pattern of gene expression. Together, these findings indicate the expression of SV40 T antigen significantly altered keratinocyte response to Cdx2 gene expression.
Changes in DNA methylation and inhibition of cell senescence required for Cdx2-mediated intestinalization of normal human keratinocytes
Since Cdx2 expression alone was insufficient for the induction of intestinalization of EPC-hTERT cells, we tried different culture conditions. Many studies suggest that disordered DNA CpG island methylation contributes to the pathogenesis of BE and its associated dysplasia (38–43), we considered whether modulation of DNA methylation would enhance Cdx2-mediated intestinalization.
CpG methylation is believed to determine whether chromatin is in an open, transcriptionally competent conformation or in a state in which it is compacted and closed-off from transcriptional regulators (63). Agents like 5-AzaC inhibit DNA methyltransferase and globally reduce DNA methylation. Treatment of our EPC-hTERT.D1.Cdx2 cells and controls with 5-AzaC did significantly alter gene expression patterns. More importantly, there were several intestine-specific and BE-associated genes whose expression only increased with the combination of DNA demethtylation and Cdx2 expression. Two of these genes, DRA/SLC26A3 and NHE2, were not previously known to be elevated in BE. Utilizing human BE tissue samples, we confirmed that they were elevated. Thus, our model of esophageal keratinocyte transdifferentiation does appear to mimic the process of intestinal metaplasia seen in BE.
Unexpectedly, we also found that cell senescence was induced in our cells with 5-AzaC treatment. This effect has been reported previously (52). It is the reason our treated cells no longer proliferated and also probably explains the discordance between mRNA and protein levels we observed after treatment (Figure 4D). However, this finding also suggests that important molecular controls exist to prohibit wide-spread epigenetic changes in normal cells. If changes in DNA methylation patterns are essential for initiation of intestinalization in esophageal keratinocytes, then either they must be acquired slowly, so as not to trigger cell senescence, or alternatively the cell senescence mechanism must be inactivated prior to the acquisition of significant changes in DNA methylation. This might be the case, as the tumor suppressor and senescence-associated p16 gene is silenced by promoter hypermethylation early in BE pathogenesis, preceding the onset of dysplasia (64). If cell senescence is indeed inactivated, it argues that a normal mechanism of tumor suppression is dysfunctional in BE cells, and it may explain why the transition to neoplasia occurs more frequently in BE tissues than in the normal esophagus.
Molecular model for BE
Based on our findings, we propose a stepwise model for the earliest molecular events in the progression from normal squamous esophageal epithelium to BE (Figure 6C). Chronic acid and bile reflux induce inflammation, both acute and chronic. Inflammatory mediators induce repair mechanisms that promote cell proliferation of basal cells. Reactive oxygen species liberated by the injury cause oxidative stress and DNA damage, resulting in changes in DNA methylation patterns of all cells including stem cells. These epigenetic changes lead to the induction of Cdx1 and Cdx2 expression and ultimately the activation of an intestinal program for cell differentiation. In theory, intestinalized cells expressing intestinal mucins would better tolerate the acid and bile acid injury caused by gastric reflux and therefore have a competitive advantage, permitting them to replace the squamous epithelium. However, given our unexpected induction of cell senescence with 5-AzaC, we further hypothesize that these pro-proliferative and epigenetic events run the risk of activating tumor suppressor mechanisms like cell senescence in normal epithelial cells. Therefore, these Barrett’s supportive changes are either acquired in a manner that does not trigger tumor suppressors or critical tumor suppressors are inactivated during this process (i.e. p16 silencing by hypermethylation). Additional studies on human Barrett’s samples are needed to clarify the role of tumor suppressor mechanisms in the earliest stages of BE.
In summary, BE can arise from normal esophageal keratinocyte precursors, but requires several molecular events including enhanced cell proliferation, inhibition of cell senescence and epigenetic changes in order for the induction of intestinal metaplasia to occur. Characterizing these early steps in the progression will give us further insights into the molecular pathogenesis of BE and may suggest novel therapeutic and preventative strategies to better treat patients with this condition.
Supplementary material
Supplementary Table I can be found at http://carcin.oxfordjournals.org/
Funding
Investigator Sponsored Study Program of AstraZeneca (#IRUSESOM0394 to J.P.L.); National Cancer Institute Program Project (PO1 CA098101) to A.K.R.; Morphology, Cell Culture and Molecular Biology Core Facilities of the Center for Molecular Studies in Digestive and Liver Disease at the University of Pennsylvania (P30-DK50306, PO1 CA098101).
Acknowledgments
We thank Dr Gary Swain for his immunohistochemical and microscopy expertise.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- 5-AzaC
5-aza-2-deoxycytidine
- BE
Barrett’s esophagus
- cDNA
complementary DNA
- DRA
downregulated in adenoma
- EAC
esophageal adenocarcinoma
- GFP
green fluorescent protein
- hTERT
human telomerase reverse transcriptase
- mRNA
messenger RNA
- NHE2
Na+/H+ exchanger 2
- PCR
polymerase chain reaction
- pRb
retinoblastoma protein
- RT
reverse transcription
References
- 1.Morales CP, et al. Hallmarks of cancer progression in Barrett's oesophagus. Lancet. 2002;360:1587–1589. doi: 10.1016/S0140-6736(02)11569-8. [DOI] [PubMed] [Google Scholar]
- 2.Paulson TG, et al. Focus on Barrett's esophagus and esophageal adenocarcinoma. Cancer Cell. 2004;6:11–16. doi: 10.1016/j.ccr.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 3.Yuasa Y. Control of gut differentiation and intestinal-type gastric carcinogenesis. Nat. Rev. Cancer. 2003;3:592–600. doi: 10.1038/nrc1141. [DOI] [PubMed] [Google Scholar]
- 4.Guillem PG. How to make a Barrett esophagus: pathophysiology of columnar metaplasia of the esophagus. Dig. Dis. Sci. 2005;50:415–424. doi: 10.1007/s10620-005-2451-x. [DOI] [PubMed] [Google Scholar]
- 5.Bai YQ, et al. Ectopic expression of homeodomain protein CDX2 in intestinal metaplasia and carcinomas of the stomach. Cancer Lett. 2002;176:47–55. doi: 10.1016/s0304-3835(01)00753-4. [DOI] [PubMed] [Google Scholar]
- 6.Silberg DG, et al. CDX1 protein expression in normal, metaplastic, and neoplastic human alimentary tract epithelium. Gastroenterology. 1997;113:478–486. doi: 10.1053/gast.1997.v113.pm9247467. [DOI] [PubMed] [Google Scholar]
- 7.Satoh K, et al. Aberrant expression of CDX2 in the gastric mucosa with and without intestinal metaplasia: effect of eradication of Helicobacter pylori. Helicobacter. 2002;7:192–198. doi: 10.1046/j.1523-5378.2002.00080.x. [DOI] [PubMed] [Google Scholar]
- 8.Phillips RW, et al. Cdx2 as a marker of epithelial intestinal differentiation in the esophagus. Am. J. Surg. Pathol. 2003;27:1442–1447. doi: 10.1097/00000478-200311000-00006. [DOI] [PubMed] [Google Scholar]
- 9.Eda A, et al. Aberrant expression of CDX2 in Barrett's epithelium and inflammatory esophageal mucosa. J. Gastroenterol. 2003;38:14–22. doi: 10.1007/s005350300001. [DOI] [PubMed] [Google Scholar]
- 10.Guo RJ, et al. The role of Cdx proteins in intestinal development and cancer. Cancer Biol. Ther. 2004;3:593–601. doi: 10.4161/cbt.3.7.913. [DOI] [PubMed] [Google Scholar]
- 11.Silberg DG, et al. Cdx2 ectopic expression induces gastric intestinal metaplasia in transgenic mice. Gastroenterology. 2002;122:689–696. doi: 10.1053/gast.2002.31902. [DOI] [PubMed] [Google Scholar]
- 12.Mutoh H, et al. Conversion of gastric mucosa to intestinal metaplasia in Cdx2-expressing transgenic mice. Biochem. Biophys. Res. Commun. 2002;294:470–479. doi: 10.1016/S0006-291X(02)00480-1. [DOI] [PubMed] [Google Scholar]
- 13.Fitzgerald RC, et al. Dynamic effects of acid on Barrett's esophagus. An ex vivo proliferation and differentiation model. J. Clin. Invest. 1996;98:2120–2128. doi: 10.1172/JCI119018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fitzgerald RC, et al. Acid modulation of HT29 cell growth and differentiation. An in vitro model for Barrett's esophagus. J. Cell Sci. 1997;110:663–671. doi: 10.1242/jcs.110.5.663. [DOI] [PubMed] [Google Scholar]
- 15.Souza RF, et al. Acid exposure activates the mitogen-activated protein kinase pathways in Barrett's esophagus. Gastroenterology. 2002;122:299–307. doi: 10.1053/gast.2002.30993. [DOI] [PubMed] [Google Scholar]
- 16.Kaur BS, et al. Bile salts induce or blunt cell proliferation in Barrett's esophagus in an acid-dependent fashion. Am. J. Physiol. Gastrointest. Liver Physiol. 2000;278:G1000–G1009. doi: 10.1152/ajpgi.2000.278.6.G1000. [DOI] [PubMed] [Google Scholar]
- 17.Palanca-Wessels MC, et al. Extended lifespan of Barrett's esophagus epithelium transduced with the human telomerase catalytic subunit: a useful in vitro model. Carcinogenesis. 2003;24:1183–1190. doi: 10.1093/carcin/bgg076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaiswal K, et al. Bile salt exposure increases proliferation through p38 and ERK MAPK pathways in a non-neoplastic Barrett's cell line. Am. J. Physiol. Gastrointest. Liver Physiol. 2006;290:G335–G342. doi: 10.1152/ajpgi.00167.2005. [DOI] [PubMed] [Google Scholar]
- 19.Marchetti M, et al. Chronic acid exposure leads to activation of the cdx2 intestinal homeobox gene in a long-term culture of mouse esophageal keratinocytes. J. Cell Sci. 2003;116:1429–1436. doi: 10.1242/jcs.00338. [DOI] [PubMed] [Google Scholar]
- 20.Kazumori H, et al. Bile acids directly augment caudal-related homeobox gene Cdx2 expression in esophageal keratinocytes in Barrett's epithelium. Gut. 2005;55:16–25. doi: 10.1136/gut.2005.066209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milano F, et al. Bone morphogenetic protein 4 expressed in esophagitis induces a columnar phenotype in esophageal squamous cells. Gastroenterology. 2007;132:2412–2421. doi: 10.1053/j.gastro.2007.03.026. [DOI] [PubMed] [Google Scholar]
- 22.Liu T, et al. Regulation of Cdx2 expression by promoter methylation, and effects of Cdx2 transfection on morphology and gene expression of human esophageal epithelial cells. Carcinogenesis. 2006;28:488–496. doi: 10.1093/carcin/bgl176. [DOI] [PubMed] [Google Scholar]
- 23.Ahuja D, et al. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene. 2005;24:7729–7745. doi: 10.1038/sj.onc.1209046. [DOI] [PubMed] [Google Scholar]
- 24.Harada H, et al. Telomerase induces immortalization of human esophageal keratinocytes without p16INK4a inactivation. Mol. Cancer Res. 2003;1:729–738. [PubMed] [Google Scholar]
- 25.Okawa T, et al. The functional interplay between EGFR overexpression, hTERT activation, and p53 mutation in esophageal epithelial cells with activation of stromal fibroblasts induces tumor development, invasion, and differentiation. Genes Dev. 2007;21:2788–2803. doi: 10.1101/gad.1544507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keller MS, et al. Cdx1 or Cdx2 expression activates E-cadherin-mediated cell-cell adhesion and compaction in human COLO 205 cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2004;287:G104–G114. doi: 10.1152/ajpgi.00484.2003. [DOI] [PubMed] [Google Scholar]
- 27.Ezaki T, et al. The homeodomain transcription factors Cdx1 and Cdx2 induce E-cadherin adhesion activity by reducing beta- and p120-catenin tyrosine phosphorylation. Am. J. Physiol. Gastrointest. Liver Physiol. 2007;293:G54–G65. doi: 10.1152/ajpgi.00533.2006. [DOI] [PubMed] [Google Scholar]
- 28.Opitz OG, et al. Cyclin D1 overexpression and p53 inactivation immortalize primary oral keratinocytes by a telomerase-independent mechanism. J. Clin. Invest. 2001;108:725–732. doi: 10.1172/JCI11909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pear WS, et al. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl Acad. Sci. USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winer J, et al. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Anal. Biochem. 1999;270:41–49. doi: 10.1006/abio.1999.4085. [DOI] [PubMed] [Google Scholar]
- 31.Lynch J, et al. The caudal-related homeodomain protein Cdx1 inhibits proliferation of intestinal epithelial cells by down-regulation of D-type cyclins. J. Biol. Chem. 2000;275:4499–4506. doi: 10.1074/jbc.275.6.4499. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z, et al. Identification of an apical Cl(-)/HCO3(-) exchanger in the small intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2002;282:G573–G579. doi: 10.1152/ajpgi.00338.2001. [DOI] [PubMed] [Google Scholar]
- 33.Andl CD, et al. Epidermal growth factor receptor mediates increased cell proliferation, migration, and aggregation in esophageal keratinocytes in vitro and in vivo. J. Biol. Chem. 2003;278:1824–1830. doi: 10.1074/jbc.M209148200. [DOI] [PubMed] [Google Scholar]
- 34.Guo RJ, et al. Cdx1 inhibits human colon cancer cell proliferation by reducing β-catenin/TCF transcriptional activity. J. Biol. Chem. 2004;279:36865–36875. doi: 10.1074/jbc.M405213200. [DOI] [PubMed] [Google Scholar]
- 35.Lynch J, et al. Cdx1 inhibits the proliferation of human colon cancer cells by reducing cyclin D1 gene expression. Oncogene. 2003;22:6395–6407. doi: 10.1038/sj.onc.1206770. [DOI] [PubMed] [Google Scholar]
- 36.Suh E, et al. An intestine-specific homeobox gene regulates proliferation and differentiation. Mol. Cell. Biol. 1996;16:619–625. doi: 10.1128/mcb.16.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ouatu-Lascar R, et al. Differentiation and proliferation in Barrett's esophagus and the effects of acid suppression. Gastroenterology. 1999;117:327–335. doi: 10.1053/gast.1999.0029900327. [DOI] [PubMed] [Google Scholar]
- 38.Jenkins GJ, et al. Genetic pathways involved in the progression of Barrett's metaplasia to adenocarcinoma. Br. J. Surg. 2002;89:824–837. doi: 10.1046/j.1365-2168.2002.02107.x. [DOI] [PubMed] [Google Scholar]
- 39.Clement G, et al. Alterations of the Wnt signaling pathway during the neoplastic progression of Barrett's esophagus. Oncogene. 2006;25:3084–3092. doi: 10.1038/sj.onc.1209338. [DOI] [PubMed] [Google Scholar]
- 40.Zou H, et al. Aberrant methylation of secreted frizzled-related protein genes in esophageal adenocarcinoma and Barrett's esophagus. Int. J. Cancer. 2005;116:584–591. doi: 10.1002/ijc.21045. [DOI] [PubMed] [Google Scholar]
- 41.Clement G, et al. Methylation of APC, TIMP3, and TERT: a new predictive marker to distinguish Barrett's oesophagus patients at risk for malignant transformation. J. Pathol. 2006;208:100–107. doi: 10.1002/path.1884. [DOI] [PubMed] [Google Scholar]
- 42.Eads CA, et al. Fields of aberrant CpG island hypermethylation in Barrett's esophagus and associated adenocarcinoma. Cancer Res. 2000;60:5021–5026. [PubMed] [Google Scholar]
- 43.Eads CA, et al. Epigenetic patterns in the progression of esophageal adenocarcinoma. Cancer Res. 2001;61:3410–3418. [PubMed] [Google Scholar]
- 44.Boukamp P, et al. Progressive stages of “transdifferentiation” from epidermal to mesenchymal phenotype induced by MyoD1 transfection, 5-aza-2′-deoxycytidine treatment, and selection for reduced cell attachment in the human keratinocyte line HaCaT. J. Cell Biol. 1992;116:1257–1271. doi: 10.1083/jcb.116.5.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zelivianski S, et al. Multipathways for transdifferentiation of human prostate cancer cells into neuroendocrine-like phenotype. Biochim. Biophys. Acta. 2001;1539:28–43. doi: 10.1016/s0167-4889(01)00087-8. [DOI] [PubMed] [Google Scholar]
- 46.Kohyama J, et al. Brain from bone: efficient “meta-differentiation” of marrow stroma-derived mature osteoblasts to neurons with Noggin or a demethylating agent. Differentiation. 2001;68:235–244. doi: 10.1046/j.1432-0436.2001.680411.x. [DOI] [PubMed] [Google Scholar]
- 47.Karpf AR, et al. Limited gene activation in tumor and normal epithelial cells treated with the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine. Mol. Pharmacol. 2004;65:18–27. doi: 10.1124/mol.65.1.18. [DOI] [PubMed] [Google Scholar]
- 48.Moscoso I, et al. Differentiation “in vitro” of primary and immortalized porcine mesenchymal stem cells into cardiomyocytes for cell transplantation. Transplant. Proc. 2005;37:481–482. doi: 10.1016/j.transproceed.2004.12.247. [DOI] [PubMed] [Google Scholar]
- 49.Regalado SP, et al. Abundant expression of the intestinal protein villin in Barrett's metaplasia and esophageal adenocarcinomas. Mol. Carcinog. 1998;22:182–189. [PubMed] [Google Scholar]
- 50.Iwata T, et al. Usefulness of cytokeratin immunoreactivity pattern for distinction of Barrett's esophagus from intestinal metaplasia of the stomach. Hepatogastroenterology. 2007;54:1710–1712. [PubMed] [Google Scholar]
- 51.Tanaka H, et al. Expression of small intestinal and colonic phenotypes in complete intestinal metaplasia of the human stomach. Virchows Arch. 2005;447:806–815. doi: 10.1007/s00428-005-0040-1. [DOI] [PubMed] [Google Scholar]
- 52.Tominaga K, et al. Genetics of cellular senescence. Mech. Ageing Dev. 2002;123:927–936. doi: 10.1016/s0047-6374(02)00030-1. [DOI] [PubMed] [Google Scholar]
- 53.Montgomery RK, et al. Development of the human gastrointestinal tract: twenty years of progress. Gastroenterology. 1999;116:702–731. doi: 10.1016/s0016-5085(99)70193-9. [DOI] [PubMed] [Google Scholar]
- 54.Yasugi S. Role of epithelial-mesenchymal interactions in differentiation of epithelium of vertebrate digestive organs. Dev. Growth Differ. 1993;35:1–9. doi: 10.1111/j.1440-169X.1993.00001.x. [DOI] [PubMed] [Google Scholar]
- 55.Horb ME. Patterning the endoderm: the importance of neighbours. Bioessays. 2000;22:599–602. doi: 10.1002/1521-1878(200007)22:7<599::AID-BIES2>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 56.Roberts DJ. Molecular mechanisms of development of the gastrointestinal tract. Dev. Dyn. 2000;219:109–120. doi: 10.1002/1097-0177(2000)9999:9999<::aid-dvdy1047>3.3.co;2-y. [DOI] [PubMed] [Google Scholar]
- 57.Beck F, et al. Reprogramming of intestinal differentiation and intercalary regeneration in Cdx2 mutant mice. Proc. Natl Acad. Sci. USA. 1999;96:7318–7323. doi: 10.1073/pnas.96.13.7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tamai Y, et al. Colonic hamartoma development by anomalous duplication in Cdx2 knockout mice. Cancer Res. 1999;59:2965–2970. [PubMed] [Google Scholar]
- 59.Hahn WC, et al. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 60.Tselepis C, et al. Barrett's esophagus: disregulation of cell cycling and intercellular adhesion in the metaplasia-dysplasia-carcinoma sequence. Digestion. 2000;61:1–5. doi: 10.1159/000007729. [DOI] [PubMed] [Google Scholar]
- 61.Haigis K, et al. The related retinoblastoma (pRb) and p130 proteins cooperate to regulate homeostasis in the intestinal epithelium. J. Biol. Chem. 2006;281:638–647. doi: 10.1074/jbc.M509053200. [DOI] [PubMed] [Google Scholar]
- 62.Li HP, et al. Epigenetic changes in virus-associated human cancers. Cell Res. 2005;15:262–271. doi: 10.1038/sj.cr.7290295. [DOI] [PubMed] [Google Scholar]
- 63.Lund AH, et al. Epigenetics and cancer. Genes Dev. 2004;18:2315–2335. doi: 10.1101/gad.1232504. [DOI] [PubMed] [Google Scholar]
- 64.Maley CC, et al. Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett's esophagus. Cancer Res. 2004;64:3414–3427. doi: 10.1158/0008-5472.CAN-03-3249. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.