

Abstract
Cornelia de Lange syndrome (CdLS) is a clinically heterogeneous developmental disorder characterized by facial dysmorphia, upper limb malformations, growth and cognitive retardation. Mutations in the sister chromatid cohesion factor genes NIPBL, SMC1A and SMC3 are present in ∼65% of CdLS patients. In addition to their canonical roles in chromosome segregation, the cohesin proteins are involved in other biological processes such as regulation of gene expression, DNA repair and maintenance of genome stability. To gain insights into the molecular basis of CdLS, we analyzed the affinity of mutated SMC1A and SMC3 hinge domains for DNA. Mutated hinge dimers bind DNA with higher affinity than wild-type proteins. SMC1A- and SMC3-mutated CdLS cell lines display genomic instability and sensitivity to ionizing radiation and interstrand crosslinking agents. We propose that SMC1A and SMC3 CdLS mutations affect the dynamic association between SMC proteins and DNA, providing new clues to the underlying molecular cause of CdLS.
INTRODUCTION
Cornelia de Lange syndrome (CdLS, MIM 122470, 300590, 610759) is a severe, clinically heterogeneous developmental disorder characterized by facial dysmorphia, upper limb malformations, growth and cognitive retardation and gastrointestinal abnormalities. Two reports showed that mutations in sister chromatid cohesion factor NIPBL cause CdLS (1,2). Extensive analysis of a large series of patients clearly showed that single-allele mutations at the NIPBL locus account for ∼60% of CdLS cases. However, the lack of NIPBL mutations in ∼40% of the patients suggested that CdLS was genetically heterogeneous, a hypothesis also supported by the marked variability of the clinical picture (3). Recently, we and others found mutations in the SMC1A and SMC3 genes, which code for subunits of the cohesin complex (4,5).
The essential role of the cohesin complex is to provide cohesion between sister chromatids from their emergence in the process of replication and until their separation in anaphase. The core cohesin complex is formed by four proteins, SMC1A, SMC3, RAD21 and SA. Each SMC protein folds in half so that its N- and C-termini meet and form a globular ATP-binding domain separated from the other globular domain, so called hinge, by an extended stretch of antiparallel coiled coils. SMC1A and SMC3 strongly bind to each other through the hinge domains on one side and terminal domains, which are bound also to RAD21, on the other side. Together, the three proteins likely form a ring-like structure capable of embracing two chromatids and thus provide a durable bond manifested as sister chromatid cohesion (6–8). How chromosomes enter the ring is to be revealed but there is strong experimental evidence suggesting ring opening by disengagement of SMC1A and SMC3 hinge domains (9). SMC protein hinge domains are capable of binding DNA in vitro (10,11) and it has been shown, in a bacterial SMC protein, that DNA binding by hinge domain stimulates ATP hydrolysis at N- and C-terminal head domain (12). Several SMC1A and SMC3 mutations implicated in CdLS are located at the junction of coiled-coil and hinge domains (4).
The mechanism by which mutations in cohesin genes affect fetal development is still unclear but is likely to involve changes in the control of gene expression at the genomic level. It has recently been shown that cohesin affects the transcription of the genes regulated by the insulator protein CTCF (13–16). On the basis of effects of Nipped-B and cohesin on cut gene expression in Drosophila in vivo, it was proposed that cohesin binding to the cut regulatory region hinders enhancer–promoter interactions and that Nipped-B alleviates this effect by dynamic control of cohesin binding (17–19). In Drosophila, cohesin and Nipped-B are bound preferentially to actively transcribed genes (20).
In addition to its roles in sister chromatid cohesion and the regulation of transcription, the cohesin complex also functions in DNA repair and is required for postreplicative double-strand break repair in Saccharomyces cerevisiae (21,22). A mutation in one subunit of the cohesin complex in Schizosaccharomyces pombe, Rad21, renders cells sensitive to DNA damage (23). The SMC1A-SMC3 heterodimer has also been found in a mammalian protein complex, called RC-1, that promotes repair of gaps and deletions through recombination (24–26). SMC1A becomes phosphorylated following exposure of cells to ionizing irradiation or aphidicolin treatment. These phosphorylation events appear to affect both the arrest of DNA replication and cell survival following DNA damage (27–30).
Analysis of metaphase spreads from CdLS patients carrying mutations in the NIPBL gene showed increased precocious sister chromatid separation (PSCS) and chromosomal breakage, suggesting that there may be some predisposition to chromosomal fragility in a subset of CdLS (31). In addition, NIPBL-mutated CdLS cells have a reduced capacity to tolerate DNA damage, presumably as a result of reduced DNA repair through homologous recombination (32). No evidence of increased sensitivity to DNA-damaging agents has been reported for SMC1A- and SMC3-mutated cell lines.
To gain insights into the molecular basis of CdLS, we investigated the affinity of mutated SMC1A and SMC3 proteins for DNA, focusing on dimeric hinge domains where several CdLS mutations cluster. We find that SMC1A and SMC3 mutations mapping to the hinge domains alter their DNA binding in vitro, i.e. mutated SMC hinge proteins bind DNA with higher affinity than wild-type proteins. To study the phenotypes of these and two other mutations on cellular level, we analyzed cell lines derived from seven CdLS patients, with four different mutations coding for amino acid substitutions R496H, R496C or F1122L and a V58_R62 deletion, in the SMC1A protein, and the E488 deletion in the SMC3 protein. All SMC-mutated cell lines had a higher frequency of spontaneous chromosome aberrations than normal control lines. Moreover, we found that cell lines carrying the R496H, E488del and F1122L mutations display increased sensitivity to DNA damage when compared with three normal control cell lines. Altogether, these findings suggest that SMC1A and SMC3 mutations alter the dynamic association of cohesin with DNA and possibly chromatin, providing, for the first time, a molecular basis for CdLS.
RESULTS
Effect of SMC1A mutations on cohesin/DNA binding
We first investigated the affinity for DNA of SMC1A- and SMC3-mutated proteins. As we showed earlier in electrophoresis mobility shift assay (EMSA), bacterially expressed and purified SMC1A/SMC3 hinge domain dimer binds to DNA in vitro (10). Binding to DNA depends on the presence on each terminus of the monomers of approximately 20 amino acids that represent a transition from globular hinge domains into the coiled-coil regions of the SMC proteins. Since some of the CdLS mutations described earlier (4,5), namely SMC1A E493A, SMC1A R496C, SMC1A R496H and SMC3 E488del, are located in this transitional region, we asked whether the DNA-binding properties would be affected by any of the amino acid substitutions in the SMC1A or the deletion in the SMC3 dimerization partners.
We tested the ability of the mutated hinge dimers to bind DNA in EMSA using a 200 bp double-stranded rDNA fragment, which was earlier shown to bind the wild-type SMC1A/SMC3 hinge dimer (10). All the mutant dimers were able to bind DNA, retaining most of the DNA fragment at the very top of the gel at higher protein concentrations (Fig. 1F–J), which is indicative of DNA–protein network formation.
Figure 1.
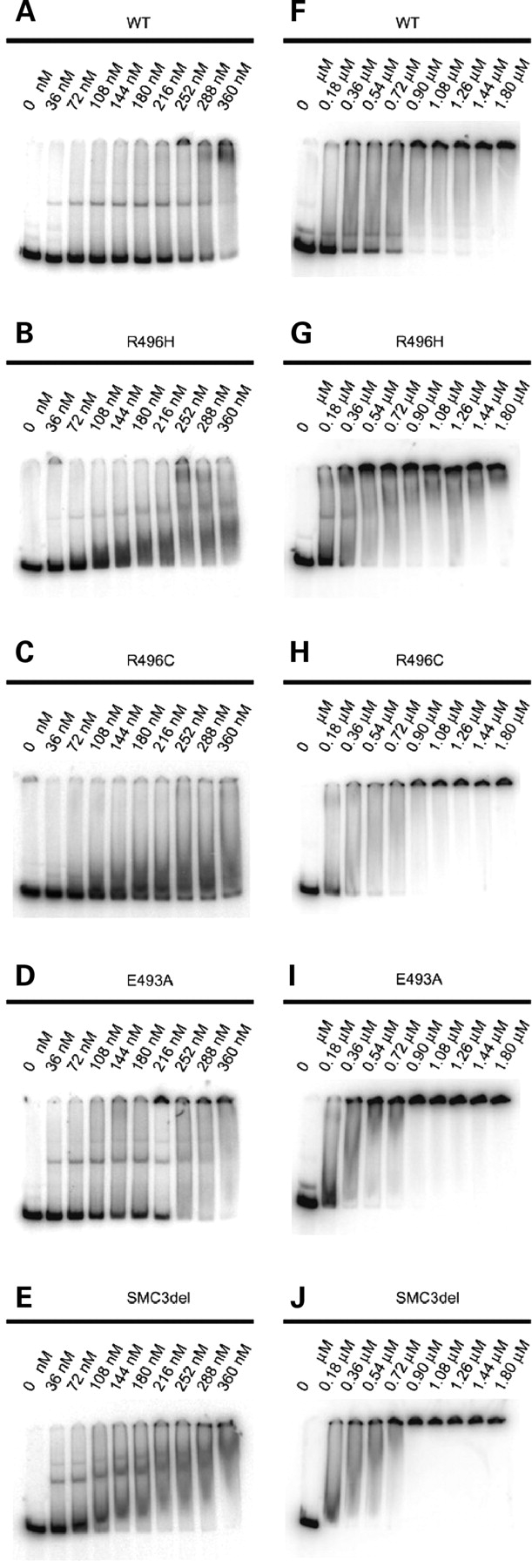
EMSA with SMC1A/SMC3 hinge dimers. Three nanograms of a 200 bp rDNA fragment, 32P 5′-labeled, was incubated with the wild-type SMC1A/SMC3 (A and F), and mutant SMC1A-R496H/SMC3 (B and G), SMC1A-R496C/SMC3 (C and H), SMC1A-E493A (D and I), or SMC1A/SMC3-delE488 (E and J) hinge peptide dimers. The concentrations of the hinge dimers are indicated above.
In order to detect possible subtle differences in the DNA-binding properties, we titrated the mutant dimers into the binding reaction (Fig. 1A–E). Interestingly, in the range of 108–360 nm, the mutant dimers bound DNA more efficiently than the wild-type dimer, leaving less of the free substrate at the bottom of the gel (Fig. 1A–E). Since the DNA-binding efficiency of a given dimer could vary slightly between the individual protein preparations, we repeated the assay for each dimer variant three times with independently purified hinge dimers. We quantified the amount of unbound DNA at the bottom of each lane by PhosphoImager and normalized it to the signal detected in the absence of the hinge protein (Figs 1 and 2). For the SMC1A-R496H/SMC3 and SMC1A-R496C/SMC3 mutants (Fig. 2A and B), the resulting mean values were lower than those of the wild-type. The difference between the wild-type and SMC1A-E493A/SMC3 or SMC1A/SMC3delE488 dimers was more pronounced (Fig. 2C and D). The wild-type hinge protein bound half of the DNA substrate at 250 nm, whereas the SMC1-E493A/SMC3 and SMC1A/SMC3delE488 mutants bound 50% of DNA at or below 100 nm. At 250 nm, both mutants bound >90% of DNA. We conclude that among the mutations mapping in the hinge domain of SMC1A and SMC3, the substitution E493A in SMC1A and the deletion of E488 in SMC3 cause the strongest increase in the affinity of the hinge-coiled-coil transition region to DNA.
Figure 2.

Relative efficiency of DNA binding. Signals from unbound DNA fragments at the bottom of the gel (Fig. 1A–E) were quantified with PhosphoImager and normalized to the signal in the first lane, where no hinge protein was added. The resulting values are plotted against hinge dimer protein concentration (nm). Mean values for the wild-type SMC1A/SMC3 hinge protein are compared with the SMC1A-R496H/SMC3 (A), SMC1A-R496C/SMC3 (B), SMC1A-E493A/SMC3 (C) or SMC1A/SMC3delE488 (D) dimers. The standard errors of the mean are indicated.
In order to test whether CdLS mutations affect the binding preference of the SMC1A/SMC3 hinge dimer protein for a 200 bp rDNA fragment (10), which contains palindromic sequences and has high potential of forming secondary structures, we performed competition experiments with a 232 bp pUC19 plasmid DNA fragment that lacks repetitive or palindromic sequences, and with single-stranded or double-stranded circular M13mp18 DNA. Competition assays were done with the amount of hinge protein sufficient to shift ∼98% of the rDNA fragment in the absence of a competitor. The tested concentrations were 0.79 µm for the wild-type dimer, 0.36 µm for SMC1A-R496H/SMC3, 0.54 µm for SMC1A-R496C/SMC3, 0.29 µm for SMC1A-E493A/SMC3 and 0.22 µm for the SMC1A/SMC3delE488 mutant.
For the wild-type dimer, all three additional substrates were relatively weak competitors, with 50% competition at 3 ng of the rDNA fragment and ∼60 ng of a 232 bp pUC19 fragment or between 90 and 120 ng of single-stranded or double-stranded M13 DNA (Supplementary Material, Fig. S1A). Similar to the wild-type dimer, all the mutants demonstrated a strong preference for binding with the rDNA fragment over the pUC19 fragment or M13 DNA (Supplementary Material, Fig. S1B–E). Thus the hinge dimer-binding preference for double-stranded DNA fragments with a high probability to form secondary structures (10) is preserved in the dimers with CdLS mutations.
Spontaneous genomic instability and cellular response to DNA-damaging agents in SMC1A- and SMC3-mutated CdLS cell lines
CdLS cell lines were analyzed for spontaneous chromosome aberrations and the results are reported in Figure 3. The frequency of spontaneous chromosome aberrations in SMC1A- and SMC3-mutated cell lines is significantly higher than in three control cell lines, LCL-N, AG09393 and AG09387. CdL107 with the V58_R62 deletion located in SMC1A N-terminal head domain showed the highest frequency, 0.63 ± 0.093 chromosome aberrations per cell, whereas the CdL060 and CdL203, both carrying the same R496H mutation, showed a very similar frequency, 0.54 ± 0.090 and 0.49 ± 0.065 chromosome aberrations per cell, respectively. It is noteworthy that the cell line derived from the CdLSS patient, carrying the R496C mutation, showed 0.31 ± 0.057, whereas the line derived from his mother, CdLVH, had a frequency of 0.15 ± 0.027 chromosome aberrations per cell. Finally, the CdL074 patient carrying the mutation F1122L showed a 0.59 ± 0.081 frequency, whereas the CdL057 patient, with the E488del mutation in SMC3, showed a 0.6 ± 0.065 frequency. Chromosome aberrations occurred as gaps, breaks and rare fragments (Table 1, Fig. 4A–C).
Figure 3.

Spontaneous chromosome aberrations. Increased number of spontaneous chromosome aberrations in SMC1A- and SMC3-mutated cell lines and three control cell lines (LCL-N, AG09387, AG09393). A hundred metaphase spreads for each subject were analyzed.
Table 1.
Spontaneous aberration types in SMC1A- and SMC3-mutated cell lines
| Patient | Aberration type |
||
|---|---|---|---|
| Gap | Break | Fragment | |
| LCL-N (control) | 8 | 2 | 0 |
| AG09387 (control) | 8 | 3 | 0 |
| AG09393 (control) | 7 | 2 | 0 |
| CdL060 | 36 | 17 | 1 |
| CdL203 | 32 | 17 | 0 |
| CdL107 | 47 | 15 | 1 |
| CdLSS | 19 | 12 | 0 |
| CdLVH | 12 | 3 | 0 |
| CdL074 | 46 | 11 | 2 |
| CdL057 | 43 | 17 | 0 |
Figure 4.

Occurrence of spontaneous genomic instability. Metaphases showing spontaneous chromosome aberrations. These representative metaphases are from lymphoblast cell line CdL074: fragment (A), break (B) and gap (C).
All SMC mutations maintain the frame of the protein. To investigate their effects on SMC protein levels, we analyzed the SMC1A and SMC3 protein expression by western blot. Specific SMC1A and SMC3 bands were detected in all SMC1A- and SMC3-mutated cell lines at an intensity similar to the control cell line (Fig. 5A and B). Next we investigated the progression of the cell cycle in SMC-mutated cell lines. Cells were stained with propidium iodide and analyzed by flow cytometry. Results showed that cell cycle progression in SMC-mutated cell lines was very similar to the control cell line (Supplementary Material, Fig. S3). In summary, these data suggest that SMC mutations do not impair either protein levels or cell cycle progression.
Figure 5.

SMC1A and SMC3 in CdLS cell lines. (A) Western blotting showing SMC1A level in LCL-N (1), CdLSS (2), CdLVH (3), CdL060 (4), CdL074 (5), CdL107 (6), CdL203 (7); (B) SMC3 level in LCL-N (1) and CdL075 (2).
Since CdLS cell lines with mutations in the NIPBL gene showed PSCS (31), metaphases were also scored for PSCS during chromosome aberration analysis. A cell was considered positive for PSCS if all or the majority of sister chromatids in the metaphase spread demonstrated sister chromatid separation. On the basis of this criterion, no difference was found among SMC1A- and SMC3-mutated and control cell lines (Supplementary Material, Table S1).
Previously, we showed that inhibition of the SMC1A gene by RNA interference led to both chromosome aberrations and fragile site expression (27). Therefore, we explored whether spontaneous chromosome aberrations occurred at fragile sites in SMC1A-mutated patients. We found that all chromosome aberrations were random and no fragile site was involved (data not shown). We next investigated how the CdLS cell lines respond to APH, a DNA polymerase inhibitor and a known inducer of fragile site expression. After treatment of SMC1A- or SMC3-mutated cell lines with 0.4 µm APH, they showed an increase of total number of chromosome aberrations as well as a higher frequency of aberrations localized to known fragile sites, compared with the control LCL-N cell line (Fig. 6). In some cases, mitotic catastrophe occurred, preventing chromosome analysis (Supplementary Material, Fig. S4). Again, among cell lines carrying the mutations localized to the hinge domains, CdL057 (SMC3-E488del) showed the highest number of chromosome aberrations and fragile sites in response to inhibition of replication by APH.
Figure 6.

Cellular response to APH treatment. Increased fragile site expression in SMC1A- and SMC3-mutated cell lines. CdLS cell lines were treated with 0.4 µm APH for 26 h and then analyzed for chromosome aberrations and their localization to fragile sites. Black rectangles represent random chromosome aberrations and white rectangles represent chromosome aberrations located at fragile sites. A hundred metaphase spreads for each subject were analyzed.
To study the impact of SMC1A and SMC3 mutations on ionizing radiation (IR) sensitivity, we compared the radiosensitivity of three normal lymphoblastoid cell lines (LCL-N, AG09387 and AG09393) with six CdLS cell lines using a colony survival assay. SMC1A- and SMC3-mutated cell lines, CdL060 (SMC1A-R496H), CdL074 (SMC1A-F1122L), CdLSS (SMC1A-R496C) and CdL057 (SMC3-E488del), showed enhanced IR sensitivity compared with control cells, CdL107 (SMC1A V58_R62del) was moderately sensitive and CdLVH (SMC1A-R496C) was similar to the normal cell line, whereas the positive control, an ATM-deficient cell line (252RM), showed strong hypersensitivity (Fig. 7A). Although individual SMC-mutated cell lines showed variable degrees of sensitivity, most of them showed a sensitivity intermediate between that of the wild type and that of the ATM cell line.
Figure 7.

Cellular response to DNA damage-inducing agents. Clonogenic survival assay of CdLS cell lines treated with 1, 2, 3 Gy of IR (A) and 1, 3, 5 µm of MMC (B). For every treatment, data presented here are the mean of three independent experiments.
Next, we investigated the cellular response to interstrand crosslinking agent mitomycin C (MMC). Following the treatment with MMC, three CdLS cell lines, CdL074 (SMC1A-F1122L), CdL060 (SMC1A-R496H) and CdL057 (SMC3-E488del) showed an increased sensitivity to MMC when compared with the control (Fig. 7B), although they did not reach the high sensitivity level of the Fanconi anemia cell line (HSCFA). Altogether, these findings suggest that SMC-mutated CdLS cell lines display spontaneous genomic instability. Furthermore, R496H and E488del mutations mapping to the hinge domain and affecting its affinity to DNA, or the head domain F1122L mutation, make cells sensitive to DNA-damaging agents.
DISCUSSION
CdLS is a genetic multisystem development disorder. Mutations in NIPBL account for ∼60% of CdLS cases and have been shown to cause both mild and severe forms. Recently, we showed that mutations in SMC1A and SMC3 genes contribute to ∼5% of CdLS cases, resulting in a consistently mild phenotype (4,5). To date, there is no clear explanation why SMC1A and SMC3 mutations are associated with a milder phenotype compared with NIPBL mutations. All SMC1A and SMC3 mutations are missense or in- frame deletions and it is likely that more severe mutations, like those occurring in NIPBL, would not be compatible with life since SMC1A and SMC3 are core structural subunits of the cohesion complex. Notwithstanding the impressive progress in identifying genes linked to CdLS during the last few years, information on the effects of CdLS mutations on molecular and cellular features of individual human cohesin proteins remains elusive. To gain insights into the molecular basis of CdLS, we analyzed both the effects of SMC1A and SMC3 mutations on the binding of SMC hinge dimers to DNA and the cellular response of seven CdLS cell lines, six of them SMC1A-mutated and the last one SMC3-mutated, to DNA damaging agents. We show here, for the first time, that several mutations in SMC1A and SMC3 genes causing CdLS affect the affinity of SMC hinge dimers for DNA. Although the biochemical consequences of this effect are not yet known, we demonstrate that CdLS cell lines are impaired in DNA repair, revealing spontaneous genomic instability and increased sensitivity to irradiation and MMC treatments. Since the mutations tested by EMSA were originally found in patients reaching adulthood and therefore are compatible with both viability and the essential functions of cohesin, we did not expect them to cause drastic changes in the structure and properties of the hinge domains in vitro. Nevertheless, all mutations cause stronger affinity of hinge region to DNA (Figs 1 and 2). The SMC3 E488del dimer binds DNA stronger than the SMC1A E493A dimer, and the latter binds stronger than the SMC1A R496H or R496C. In fact, all these mutations map to the N-terminal coiled-coil/hinge junction and the mutated residues are evolutionary highly conserved (4). When short N- and C-terminal stretches of amino acids, which have high probability to form coiled-coils and represent the transition regions from coiled-coil into hinge domain, are removed from hinge peptides, the SMC1A/SMC3 hinge dimer loses its ability to bind DNA. On the other hand, SMC1A or SMC3 hinge monomers do not bind DNA even in the presence of the outgoing transition region (10). Thus both the presence of the transition regions and the dimerization partner are necessary to form a structure with DNA-binding capacity. Deletion or substitution of positively charged Glu residues makes pI values of N-terminal transition regions more basic. In case of SMC1 E493A, the theoretical pI value of 23 N-terminal amino acids of the hinge peptide changes from 6.04 to 8.49, and for SMC3 E488del, the pI value changes from 9.99 to 10.45, which may slightly increase the affinity to DNA. Similarly, replacement of positively charged Arg residue in the case of R496C or R496H should decrease affinity to DNA, but this is not the case. It is possible that mutations in these positions affect overall folding of the hinge domains, causing subtle impairment of cohesin functions.
Recently it has been shown that if three cohesin subunits, SMC1, SMC3 and Scc1 (yeast ortholog of Rad21), are covalently bound to each other at defined positions by chemical crosslinks, the resulting tripartite molecule can concatenate circular minichromosomes even under denaturing conditions that exclude protein–DNA interactions and protein–protein interactions. Thus cohesin can hold together two DNA molecules by trapping them topologically inside a single ring. In mammalian cells, cohesin bound to chromatin forms two pools, one is labile and present through the most part of the cell cycle and the other is stably bound to chromatin and exists from S-phase until metaphase–anaphase transition (33). The stably bound cohesin most probably represents the complexes that trap sister chromatids inside the ring as a result of sister chromatid cohesion establishment during chromosome replication. It is still unclear how the entrapment of sister chromatids is performed, but one of the models supported by experimental evidence postulates that it involves opening of the ring at the interaction surfaces between hinge domains (9). It is possible that E488del, E493A, R496H and R496C mutations by increasing the affinity of the hinge domains to DNA interfere with the dynamics of cohesion establishment and/or of labile cohesin pool interactions with chromatin.
In yeast, a mutation in the hinge domain, which does not disrupt cohesin complex formation or chromosome binding, was shown to negatively affect proper cohesin localization to specific chromatin regions normally enriched with cohesin and abolish cohesion establishment (34).
Sister chromatid cohesion not only serves the purpose of faithful chromosome segregation but is also required for postreplicative DNA repair. As shown in yeast, in response to double-strand breaks in G2/M, cohesin is recruited to chromatin flanking the site of damage and cohesion is generated de novo on both broken and undamaged chromosomes (35–38). In mammalian cells, cohesins are important components of DNA repair, as ATM- and ATR-dependent phosphorylation of SMC1 and SMC3 are critical for DNA damage response (30,39,40). CdLS mutations clearly affect the function of cohesin in coping with DNA damage since all SMC-mutated cell lines we studied display spontaneous genomic instability, although at different levels, and some display moderate sensitivity to IR and MMC. Increased genome instability seems to be a common feature of CdLS. In fact, chromosome breakages and a reduced capacity to tolerate DNA damage have been detected in NIPBL-mutated patients (31,32). These findings suggest that genome instability and both irradiation and crosslinking treatments can be useful diagnostic assays before the screening of known CdLS genes. The mean of spontaneous aberrations per cell ranges from 0.15 ± 0.027 to 0.63 ± 0.093 in CdLVH and CdL107, respectively. It is worthy of note that CdL107 and CdL074 have the highest frequency of chromosome aberrations, 0.63 and 0.59, respectively. These patients carry the mutations V58_R62del and F1122L mapping to the head domain near the Walker A motif and the signature motif of the ABC family of ATPases, respectively. It has been hypothesized that the energy necessary to open the hinge is generated by ATP binding or hydrolysis at the head domains (9). Direct interaction of hinge and head domains was suggested by atomic force microscopy images (41) and demonstrated by immunoprecipitation of overexpressed domains (42). It is conceivable that the mutations V58_R62del and F1122L could affect hinge opening or dimerization and thus cohesion establishment.
Most analyzed patients are females and it is expected that they express a wild-type protein besides the mutant allele, because SMC1A escapes X inactivation (43). The finding that the respective cell lines show spontaneous genomic instability suggests that the mechanism in affected females is due to a dominant-negative effect of the altered protein. Alternatively, it is possible that the genomic instability is due to decreased protein levels. This is unlikely, however, because we showed that levels of SMC1A and SMC3 proteins are normal in the mutant cell lines (Fig. 5).
Recently, we showed that the inhibition of SMC1A by iRNA induces fragile site expression (27). We investigated whether SMC1A-mutated cell lines express fragile sites under normal conditions but found no increase in fragile sites. Interestingly, APH treatment induced a higher frequency of fragile sites in CdLS cells than in normal cells. This suggests that SMC1A mutations interfere with the repair of stalled forks, which are generated by APH treatment. Treatment of cells with 0.4 µm APH causes DNA fragmentation and activation of ATM and ATR pathways (27,44) and thus it is highly probable that the increase in fragile site expression in CdLS lines is a consequence of impaired double-strand break repair due to the presence of mutated SMC1A.
It appears likely that DNA damage responses mediated by cohesin proteins involve specific protein–DNA interactions. Altered DNA interactions such as those found in CdLS cells may contribute to their DNA damage hypersensitivity. Whether this contribution is direct through altering the DNA repair function of cohesin proteins, or indirect, i.e. through affecting transcription of DNA repair genes, remains to be determined.
Very recently, four groups mapped cohesin-binding sites on mammalian chromosomes and reported co-localization of cohesin and the CCCTC-binding factor (CTCF), a zinc finger DNA-binding protein (13–16). CTCF is best known for its role as transcriptional insulators. It has been proposed that insulators could interfere with gene transcription through loop formation. Altered DNA binding of CdLS-mutated SMC1A or SMC3 may affect gene expression through impaired spatial organization of genome. This is somewhat reminiscent of the demonstrated role of meiotic SMC1B in chromatin loop formation (45,46) and illustrates a potential role of SMC proteins in remodeling chromatin architecture. It is worthy to note that NIPBL missense mutations identified in CdLS patients affect the interaction of NIPBL with histone deacetylase (47), further supporting a chromatin-remodeling model of CdLS. In conclusion, our data suggest that SMC1A and SMC3 mutations affect the dynamic association of cohesin with DNA, providing a molecular basis for CdLS pathogenesis.
MATERIALS AND METHODS
Cell lines
We analyzed five unrelated and two related CdLS lymphoblastoid cell lines, transformed with Epstein–Barr virus. Phenotypic features of all patients were described previously (4). Briefly, the female patients CdL060 (patient 9P in 4), CdL203 (patient 8P in 4), CdL107 (patient 3P in 4), CdL074 (patient 12P in 4), CdLVH and the male patient CdLSS (patient 6P in 4) carried the following SMC1A mutations: +/R496H, +/R496H, +/V58_R62del, +/F1122L, +/R496C and R496C, respectively. In addition, the male patient CdL057 (patient 2P in 4) carried the +/E488del mutation in the SMC3 gene. Studies were performed after informed consent was obtained from the families, according to the procedures established by the local Ethical Committees. AG09393 and AG09387, purchased from Coriell Cell Repositories, LCL-N, normal human lymphoblastoid cell lines, HSCFA, a Fanconi anemia lymphoblastoid cell line, and 252RM, an ataxia-telangiectasia lymphoblastoid cell line, were used as negative and positive controls, respectively. All the cell lines were cultured in RPMI containing 10% fetal calf serum and antibiotics.
Sensitivity of SMC1A- and SMC3-mutated cell lines to genotoxic agents
For IR-sensitivity studies, SMC1A- and SMC3-mutated cell lines were exposed to 1–3 Gy by a linear accelerator (Philips 75-5) with a 6 MV photon energy source, plated at various dilutions and then cultured for 21 days, as described previously (48). Thereafter, surviving colonies were stained and counted as described previously (49). For MMC sensitivity, cell lines were plated 24 h before treatment with 1, 3, 5 µm MMC for 1 h, then washed and allowed to recover for 6 days (48). For every treatment, data presented here are the mean of three independent experiments.
Genomic instability assay
Spontaneous genomic instability of SMC1A- and SMC3-mutated cell lines was evaluated by standard procedures. Briefly, colcemid was added to the cultures for 90 min followed by a 20 min incubation in 0.075 M KCl at 37°C and multiple changes of Carnoy’s fixative (3:1 methanol:acetic acid). Cells were dropped onto cleaned and wet slides. A hundred metaphases for each patient were analyzed. Chromosomal aberrations, gaps and breaks were visualized by staining slides in Giemsa stain and detected by direct microscope visualization. For fragile sites expression, cells were treated with aphidicolin (APH, 0.4 µm) for 26 h and a hundred metaphases for each subject were analyzed as described previously (27).
Flow cytometry
Samples in suspension were fixed in 70% ethanol at 4°C, then treated with 1 µg/ml RNaseA (Sigma) at 37°C for 20 min and stained with 5 µg/ml propidium iodide (Sigma) and analyzed for DNA content (25 000 cells/sample) with a FACScalibur flow cytometer (Becton Dickinson).
Western blotting
Western blotting was performed as described previously (27). For immunoblot analysis, we used rabbit polyclonal anti-SMC1A and anti-SMC3 antibody (Bethyl). Actin antibody (Santa Cruz Biotechnology) was used as an internal control.
Electrophoresis mobility shift assay
The SMC1A and SMC3 hinge peptides were co-expressed in Escherichia coli and formed heterodimers that were purified using the histidine tag present on the SMC3 peptide as described (10). The site-directed mutagenesis was performed with QuikChange Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer’s instructions. All mutations were confirmed by sequencing.
We used the wild-type SMC1A hinge domain containing amino acids 481–674, the wild-type SMC3 hinge domain containing amino acids 484–690 preceded by a methionine and followed by a glycine–serine linker and a histidine tag, the mutant SMC1A hinge domains carrying amino acid substitutions E493A, R496C or R496H and the SMC3 deletion variant SMC3del, identical to the wild-type SMC3 hinge domain but lacking the amino acid E488. Each mutant domain was paired with a corresponding wild-type dimerization partner. All the mutants were able to form heterodimers, which were purified under the same conditions as wild-type dimers.
The EMSA assays were performed as described previously (10,50). Thirty microliter reaction mixtures contained 3 ng 32P 5′-labeled DNA in 20 mm HEPES (pH 7.5), 1 mm DTT, 100 µg/ml BSA and hinge protein as indicated. After 20 min incubation at room temperature, DNA–protein complexes were resolved by electrophoresis at 4°C in nondenaturing polyacrylamide gels in 20 mm HEPES (pH 7.5), 0.1 mm EDTA. All gels were fixed (60 min in 10% acetic acid, 10% ethanol), dried and exposed for autoradiography and quantification using a PhosphoImager (Molecular Dynamics). Radioactively labeled DNA substrate was a 200 bp EcoRI 5S rDNA fragment excised from an array of 11 head-to-tail repeats (51). This fragment bears palindromic sequences and was found earlier to be efficiently bound by SMC protein domains.
For competition experiments, the following DNA substrates were used: a 232 bp AvaII fragment from pUC19 (bp 1837–2059) and full-length single-stranded or double-stranded (RF I) circular M13mp18 DNA (New England Biolabs). Then 0.3 ng of a 200 bp rDNA fragment, radioactively labeled, was mixed with different amounts of a competitor before adding the hinge dimers. An amount of hinge protein sufficient to shift, on average, 98% of a 200 bp rDNA fragment (in the absence of a competitor) was used.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work is partially funded by grants from Istituto Toscano Tumori to A.M. This work is also funded by Fondazione Cassa di Risparmio delle Province Lombarde (CARIPLO, project number 2006/0780), Istituto Superiore di Sanità, the N.O.B.E.L. project of Fondazione Cariplo to P.V., by NIH R01 GM62517 to R.J. and by PO1 HD052860 NICHD to I.K.
ACKNOWLEDGEMENTS
We thank Alessandra Tonelli (E. Medea Scientific Institute) for her skillful technical assistance and Miria Stefanini (IGM-CNR, Pavia) and Luciana Chessa (Sapienza University, Rome) for providing us with Fanconi anemia and ataxia telangiectasia human lymphoblastoid cell lines, respectively.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Krantz I.D., McCallum J., DeScipio C., Kaur M., Gillis L.A., Yaeger D., Jukofsky L., Wasserman N., Bottani A., Morris C.A., et al. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat. Genet. 2004;36:631–635. doi: 10.1038/ng1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tonkin E.T., Wang T.J., Lisgo S., Bamshad M., Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesin proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat. Genet. 2004;36:636–341. doi: 10.1038/ng1363. [DOI] [PubMed] [Google Scholar]
- 3.Gillis L.A., McCallum J., Kaur M., DeScipio C., Yaeger D., Mariani A., Kline A.D., Li H.H., Devoto M., Jackson L.G., et al. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype–phenotype correlations. Am. J. Hum. Genet. 2004;75:610–623. doi: 10.1086/424698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deardorff M.A., Kaur M., Yaeger D., Rampuria A., Korolev S., Pie J., Gil-Rodriguez C., Arnedo M., Loyes B., Kline A., et al. Mutations in cohesin complex members SMC3 and SMC1A cause a mild variant of Cornelia de Lange syndrome with predominant mental retardation. Am. J. Hum. Genet. 2007;80:485–494. doi: 10.1086/511888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Musio A., Selicorni A., Focarelli M.L., Gervasini C., Milani D., Russo S., Vezzoni P., Larizza L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat. Genet. 2006;38:528–530. doi: 10.1038/ng1779. [DOI] [PubMed] [Google Scholar]
- 6.Haering C.H., Farcas A.M., Arumugam P., Metson J., Nasmyth K. The cohesin ring concatenates sister DNA molecules. Nature. 2008;454:297–301. doi: 10.1038/nature07098. [DOI] [PubMed] [Google Scholar]
- 7.Nasmyth K., Haering C.H. The structure and function of SMC and kleisin complexes. Annu. Rev. Biochem. 2005;74:595–648. doi: 10.1146/annurev.biochem.74.082803.133219. [DOI] [PubMed] [Google Scholar]
- 8.Hirano T. SMC proteins and chromosome mechanics: from bacteria to humans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005;360:507–514. doi: 10.1098/rstb.2004.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gruber S., Arumugam P., Katou Y., Kuglitsch D., Helmhart W., Shirahige K., Nasmyth K. Evidence that loading of cohesin onto chromosomes involves opening of its SMC hinge. Cell. 2006;127:523–537. doi: 10.1016/j.cell.2006.08.048. [DOI] [PubMed] [Google Scholar]
- 10.Chiu A., Revenkova E., Jessberger R. DNA interaction and dimerization of eukaryotic SMC hinge domains. J. Biol. Chem. 2004;279:26233–26242. doi: 10.1074/jbc.M402439200. [DOI] [PubMed] [Google Scholar]
- 11.Hirano M., Hirano T. Hinge-mediated dimerization of SMC protein is essential for its dynamic interaction with DNA. EMBO J. 2002;21:5733–5744. doi: 10.1093/emboj/cdf575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirano M., Hirano T. Opening closed arms: long-distance activation of SMC ATPase by hinge–DNA interactions. Mol. Cell. 2006;21:175–186. doi: 10.1016/j.molcel.2005.11.026. [DOI] [PubMed] [Google Scholar]
- 13.Parelho V., Hadjur S., Spivakov M., Leleu M., Sauer S., Gregson H.C., Jarmuz A., Canzonetta C., Webster Z., Nesterova T., et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008;132:422–433. doi: 10.1016/j.cell.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 14.Rubio E.D., Reiss D.J., Welcsh P.L., Disteche C.M., Filippova G.N., Baliga N.S., Aebersold R., Ranish J.A., Krumm A. CTCF physically links cohesin to chromatin. Proc. Natl Acad. Sci. USA. 2008;105:8309–8314. doi: 10.1073/pnas.0801273105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stedman W., Kang H., Lin S., Kissil J.L., Bartolomei M.S., Lieberman P.M. Cohesins localize with CTCF at the KSHV latency control region and at cellular cmyc and H19/Igf2 insulators. EMBO J. 2008;27:654–666. doi: 10.1038/emboj.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wendt K.S., Yoshida K., Itoh T., Bando M., Koch B., Schirghuber E., Tsutsumi S., Nagae G., Ishihara K., Mishiro T., et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- 17.Dorsett D., Eissenberg J.C., Misulovin Z., Martens A., Redding B., McKim K. Effects of sister chromatid cohesion proteins on cut gene expression during wing development in Drosophila. Development. 2005;132:4743–4753. doi: 10.1242/dev.02064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rollins R.A., Korom M., Aulner N., Martens A., Dorsett D. Drosophila nipped-B protein supports sister chromatid cohesion and opposes the stromalin/Scc3 cohesion factor to facilitate long-range activation of the cut gene. Mol. Cell Biol. 2004;24:3100–3111. doi: 10.1128/MCB.24.8.3100-3111.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dorsett D. Adherin: key to the cohesin ring and Cornelia de Lange syndrome. Curr. Biol. 2004;14:R834–R836. doi: 10.1016/j.cub.2004.09.035. [DOI] [PubMed] [Google Scholar]
- 20.Misulovin Z., Schwartz Y.B., Li X.Y., Kahn T.G., Gause M., Macarthur S., Fay J.C., Eisen M.B., Pirrotta V., Biggin M.D., et al. Association of cohesin and nipped-B with transcriptionally active regions of the Drosophila melanogaster genome. Chromosoma. 2007;117:89–102. doi: 10.1007/s00412-007-0129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schär P., Fäsi M., Jessberger R. SMC1 coordinates DNA double-strand break repair pathways. Nucleic Acids Res. 2004;32:3921–3929. doi: 10.1093/nar/gkh716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sjogren C., Nasmyth K. Sister chromatid cohesion is required for postreplicative double-strand break repair in Saccharomyces cerevisiae. Curr. Biol. 2001;11:991–995. doi: 10.1016/s0960-9822(01)00271-8. [DOI] [PubMed] [Google Scholar]
- 23.Birkenbihl R.P., Subramani S. Cloning and characterization of rad21 an essential gene of Schizosaccharomyces pombe involved in DNA double-strand-break repair. Nucleic Acids Res. 1992;20:6605–6611. doi: 10.1093/nar/20.24.6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stursberg S., Riwar B., Jessberger R. Cloning and characterization of mammalian SMC1 and SMC3 genes and proteins, components of the DNA recombination complexes RC-1. Gene. 1999;228:1–12. doi: 10.1016/s0378-1119(99)00021-9. [DOI] [PubMed] [Google Scholar]
- 25.Jessberger R., Riwar B., Baechtold H., Akhmedov A.T. SMC proteins constitute two subunits of the mammalian recombination complex RC-1. EMBO J. 1996;15:4061–4068. [PMC free article] [PubMed] [Google Scholar]
- 26.Jessberger R., Podust V., Hubscher U., Berg P. A mammalian protein complex that repairs double-strand breaks and deletion by recombination. J. Biol. Chem. 1993;268:15070–15079. [PubMed] [Google Scholar]
- 27.Musio A., Montagna C., Mariani T., Tilenni M., Focarelli M.L., Brait L., Indino E., Benedetti P.A., Chessa L., Albertini A., et al. Smc1 involvement in fragile site expression. Hum. Mol. Genet. 2005;14:525–533. doi: 10.1093/hmg/ddi049. [DOI] [PubMed] [Google Scholar]
- 28.Yazdi P.T., Wang Y.I., Zhao S., Patel N., Lee E.Y., Qin J. SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev. 2002;16:571–582. doi: 10.1101/gad.970702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S.T., Xu B., Kastan M.B. Involvement of the cohesin protein, Smc1, in Atm-dependent and independent responses to DNA damage. Genes Dev. 2002;16:560–570. doi: 10.1101/gad.970602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kitagawa R., Bakkenist C.J., McKinnon P.J., Kastan M.B. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004;18:1423–1438. doi: 10.1101/gad.1200304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaur M., DeScipio C., McCallum J., Yaeger D., Devoto M., Jackson L.G., Spinner N.B., Krantz I.D. Precocious sister chromatid separation (PSCS) in Cornelia de Lange syndrome. Am. J. Med. Genet. 2005;138:27–31. doi: 10.1002/ajmg.a.30919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vrouwe M.G., Elghalbzouri-Maghrani E., Meijers M., Schouten P., Godthelp B.C., Bhuiyan Z.A., Redeker E.J., Mannens M.M., Mullenders L.H., Pastink A., et al. Increased DNA damage sensitivity of Cornelia de Lange syndrome cells: evidence for impaired recombinational repair. Hum. Mol. Genet. 2007;16:1478–1487. doi: 10.1093/hmg/ddm098. [DOI] [PubMed] [Google Scholar]
- 33.Gerlich D., Koch B., Dupeux F., Peters J.M., Ellenberg J. Live-cell imaging reveals a stable cohesin–chromatin interaction after but not before DNA replication. Curr. Biol. 2006;16:1571–1578. doi: 10.1016/j.cub.2006.06.068. [DOI] [PubMed] [Google Scholar]
- 34.Milutinovich M., Unal E., Ward C., Skibbens R.V., Koshland D. A multi-step pathway for the establishment of sister chromatid cohesion. PLoS Genet. 2007;3:e12. doi: 10.1371/journal.pgen.0030012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ström L., Karlsson C., Lindroos H.B., Wedahl S., Katou Y., Shirahige K., Sjögren C. Postreplicative formation of cohesion is required for repair and induced by a single DNA break. Science. 2007;317:242–245. doi: 10.1126/science.1140649. [DOI] [PubMed] [Google Scholar]
- 36.Ünal E., Heidinger-Pauli J.M., Koshland D. DNA double-strand breaks trigger genome-wide sister-chromatid cohesion through Eco1 (Ctf7) Science. 2007;317:245–248. doi: 10.1126/science.1140637. [DOI] [PubMed] [Google Scholar]
- 37.Ström L., Lindroos H.B., Shirahige K., Sjögren C. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol. Cell. 2004;16:1003–1015. doi: 10.1016/j.molcel.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 38.Ünal E., Arbel-Eden A., Sattler U., Shroff R., Lichten M., Haber J.E., Koshland D. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol. Cell. 2004;16:991–1002. doi: 10.1016/j.molcel.2004.11.027. [DOI] [PubMed] [Google Scholar]
- 39.Luo H., Li Y., Mu J.J., Zhang J., Tonaka T., Hamamori Y., Jung S.Y., Wang Y., Qin J. Regulation of intra-S phase checkpoint by ionizing radiation (IR)-dependent and IR-independent phosphorylation of SMC3. J. Biol. Chem. 2008;28:19176–19183. doi: 10.1074/jbc.M802299200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garg R., Callens S., Lim D.S., Canman C.E., Kastan M.B., Xu B. Chromatin association of rad17 is required for an ataxia telangiectasia and rad-related kinase-mediated S-phase checkpoint in response to low-dose ultraviolet radiation. Mol. Cancer Res. 2004;2:362–369. [PubMed] [Google Scholar]
- 41.Sakai A., Hizume K., Sutani T., Takeyasu K., Yanagida M. Condensin but not cohesin SMC heterodimer induces DNA reannealing through protein–protein assembly. EMBO J. 2003;22:2764–2775. doi: 10.1093/emboj/cdg247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McIntyre J., Muller E.G., Weitzer S., Snydsman B.E., Davis T.N., Uhlmann F. In vivo analysis of cohesin architecture using FRET in the budding yeast Saccharomyces cerevisiae. EMBO J. 2007;26:3783–3793. doi: 10.1038/sj.emboj.7601793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown C.J., Miller A.P., Carrel L., Rupert J.L., Davies K.E., Willard H.F. The DXS423E gene in Xp11.21 escapes X chromosome inactivation. Hum. Mol. Genet. 1995;4:251–255. doi: 10.1093/hmg/4.2.251. [DOI] [PubMed] [Google Scholar]
- 44.Ozeri-Galai E., Schwartz M., Rahat A., Kerem B. Interplay between ATM and ATR in the regulation of common fragile site stability. Oncogene. 2008;27:2109–2117. doi: 10.1038/sj.onc.1210849. [DOI] [PubMed] [Google Scholar]
- 45.Revenkova E., Eijpe M., Heytingm C., Hodges C.A., Hunt P.A., Liebe B., Scherthan H., Jessberger R. Cohesin SMC1 beta is required for meiotic chromosome dynamics, sister chromatid cohesion and DNA recombination. Nat. Cell Biol. 2004;6:555–562. doi: 10.1038/ncb1135. [DOI] [PubMed] [Google Scholar]
- 46.Novak I., Wang H., Revenkova E., Jessberger R., Scherthan H., Höög C. Cohesin Smc1beta determines meiotic chromatin axis loop organization. J. Cell Biol. 2008;180:83–90. doi: 10.1083/jcb.200706136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jahnke P., Xu W., Wülling M., Albrecht M., Gabriel H., Gillessen-Kaesbach G., Kaiser F.J. The cohesin loading factor NIPBL recruits histone deacetylase to mediate local chromatin modification. Nucleic Acid Res. 2008 doi: 10.1093/nar/gkn688. doi:10.1093/nar/gkn688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Musio A., Marrella V., Sobacchi C., Rucci F., Fariselli L., Giliani S., Lanzi G., Notarangelo L.D., Delia D., Colombo R., et al. Damaging-agent sensitivity of Artemis-deficient cell lines. Eur. J. Immun. 2005;35:1250–1256. doi: 10.1002/eji.200425555. [DOI] [PubMed] [Google Scholar]
- 49.Sun X., Becker-Catania S.G., Chun H.H., Hwang M.J., Huo Y., Wang Z., Mitui M., Sanal O., Chessa L., Crandall B., et al. Early diagnosis of ataxia-telangiectasia using radiosensitivity testing. J. Pediatr. 2002;140:355–361. doi: 10.1067/mpd.2002.123879. [DOI] [PubMed] [Google Scholar]
- 50.Akhmedov A.T., Frei C., Tsai-Pflugfelder M., Kemper B., Gasser S.M., Jessberger R. Structural maintenance of chromosomes protein C-terminal domains bind preferentially to DNA with secondary structure. J. Biol. Chem. 1998;273:24088–24094. doi: 10.1074/jbc.273.37.24088. [DOI] [PubMed] [Google Scholar]
- 51.Logie C., Peterson C.L. Catalytic activity of the yeast SWI/SNF complex on reconstituted nucleosome arrays. EMBO J. 1997;16:6772–6782. doi: 10.1093/emboj/16.22.6772. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.