

Abstract
Periventricular heterotopia (PH) is a disorder characterized by neuronal nodules, ectopically positioned along the lateral ventricles of the cerebral cortex. Mutations in either of two human genes, Filamin A (FLNA) or ADP-ribosylation factor guanine exchange factor 2 (ARFGEF2), cause PH (Fox et al. in ‘Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia'. Neuron, 21, 1315–1325, 1998; Sheen et al. in ‘Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex'. Nat. Genet., 36, 69–76, 2004). Recent studies have shown that mutations in mitogen-activated protein kinase kinase kinase-4 (Mekk4), an indirect interactor with FlnA, also lead to periventricular nodule formation in mice (Sarkisian et al. in ‘MEKK4 signaling regulates filamin expression and neuronal migration'. Neuron, 52, 789–801, 2006). Here we show that neurons in post-mortem human PH brains migrated appropriately into the cortex, that periventricular nodules were primarily composed of later-born neurons, and that the neuroependyma was disrupted in all PH cases. As studied in the mouse, loss of FlnA or Big2 function in neural precursors impaired neuronal migration from the germinal zone, disrupted cell adhesion and compromised neuroepithelial integrity. Finally, the hydrocephalus with hop gait (hyh) mouse, which harbors a mutation in Napa [encoding N-ethylmaleimide-sensitive factor attachment protein alpha (α-SNAP)], also develops a progressive denudation of the neuroepithelium, leading to periventicular nodule formation. Previous studies have shown that Arfgef2 and Napa direct vesicle trafficking and fusion, whereas FlnA associates dynamically with the Golgi membranes during budding and trafficking of transport vesicles. Our current findings suggest that PH formation arises from a final common pathway involving disruption of vesicle trafficking, leading to impaired cell adhesion and loss of neuroependymal integrity.
INTRODUCTION
Impairments in neuronal migration, particularly in cell motility, are thought to give rise to several genetic malformations of the developing cortex: lissencephaly (smooth brain), subcortical band heterotopia (heterotopic neurons arrested under the normal cerebral cortex) and periventricular heterotopia (PH; nodules of neurons lining the lateral ventricles). In each of these disorders, early post-mitotic neuroblasts fail to migrate (or have impairments in migration) from the ventricular zone (VZ) into the cortical plate. These impairments in neuronal motility are thought to give rise to a thickened cortex with loss of normal cortical lamination, in lissencephaly (1,2); an abnormal layer of neurons below a normal cortex, in subcortical band heterotopia (3–5); and nodules of neurons along the lateral ventricles and beneath a normal-appearing cortex, PH (6,7).
Understanding the genetic mechanisms underlying impaired neuronal motility have aided in elucidating the causes of many human cortical malformations. Neurons harboring a heterozygous LIS1 mutation (the genetic cause of classical lissencephaly) show abnormal dynein localization and reduced cell motility (8–10). Lis1 interactions with dynein and doublecortin support a role of some type for this protein in cell motility (11–15). In subcortical band heterotopia, neurons harboring a mutation in the X-linked gene doublecortin (DCX) also undergo delayed neuronal migration (16,17). One role for DCX in neuronal migration could be in polymerization of microtubules (18). After X-inactivation, hemizygous males with no normal DCX developed lissencephaly, whereas females with a DCX mutation developed subcortical band heterotopia (16,17). In mice, mutations in Dcx and in the related Dclk gene disrupt neuronal migration to the cortex (19,20). In addition, cell-autonomous inhibition of Dcx by RNAi was found to induce abnormal neuronal morphology and was sufficient to impair neuronal migration (19,21). Therefore, the band heterotopia with DCX mutations appeared to reflect cell-autonomous roles for DCX in neuronal migration. Lastly, a recent case of human mosaicism in LIS1 also resulted in subcortical band heterotopia (22), consistent with the view that both DCX and LIS1 are involved in some aspect of cell motility.
As is the case for DCX mutations, PH due to mutations in the X-linked filamin-A (FLNA) gene also is characterized as having a cell motility defect (23–26), but the evidence for this is not definitive. Mutations in FLNA resulted in PH in females and lethality in hemizygous males (27,28). FLNA encodes a 280 kDa actin-binding phosphoprotein that is widely expressed throughout (and outside of) the nervous system (29). FLNA homodimers regulate the actin cytoskeleton through interactions via the protein's multiple receptor-binding regions, with effects on regulating cell stability, protrusion and motility (30). Moreover, FLNA-deficient melanoma cells show impaired cell motility (31). FLNA also promotes actin branching, tethers large actin filaments and holds them in a perpendicular arrangement (30,32–34). The resulting three-dimensional orthogonal network of actin filaments represents a characteristic cortical actin structure at the leading edge of the migrating cell. Thus, FLNA is believed to be essential for mammalian cell locomotion through its stabilization of loose microfilament nets. However, neither null mutations in FlnA, nor cell-autonomous removal of FlnA in neural crest cells, has resulted in cell-autonomous defects in neural migration (35,36), suggesting that some as yet unknown function of FlnA is the essential function in neural development.
Other mechanisms for the formation of periventricular nodules have been proposed in light of the identification of a second cause of PH: mutations in the ADP-ribosylation factor guanine exchange factor 2 (ARFGEF2) gene (37). ARFGEF2 encodes the brefeldin A-inhibited guanine nucleotide-exchange factor 2 (BIG2), a protein that regulates exchange of GDP for GTP, as required by the ADP ribosylation factors (ARFs). Since the ARF proteins are primarily involved in intracellular membrane and vesicular trafficking, it is unknown how FLNA and ARFGEF2, two genes with notably divergent functions, both give rise to the CNS disorder PH. The congenital microcephaly observed in affected individuals with ARFGEF2 mutations would suggest a failure in development within the neural precursor population, rather than a failure in post-mitotic neurons with impaired migration (37,38). Finally, while the clusters of neurons along the lateral ventricles in PH could still be indicative of a failure of neurons to migrate from the VZ, PH with microcephaly is an autosomal recessive disorder, meaning that all progenitors and neurons harbor the mutation. The relative preservation of the overlying cortex implies that the actual motility of neurons is preserved (37). Thus, if FLNA and ARFGEF2 give rise to PH by a common pathway (as radiographic studies of subjects with the two types of the disorder strongly suggest), the cellular and molecular bases of that pathway remain unexplained.
To pursue mechanisms underlying PH formation, we examined post-mortem human brains in four cases of PH, and correlated these observations with findings from both in vitro and in vivo mouse studies in which FlnA and Big2 function was disrupted. We found that many neurons, including those that harbored a mutation in FLNA, appeared to be positioned properly in the human cerebral cortex. Moreover, closer examination of the lateral ventricular lining suggested that the integrity of the neuroependyma was invariably compromised along the ventricular lining, in the four cases. After inhibition of FlnA function in mice, many affected cells were found to be restricted to the ventricular lining or were found to be situated adjacent to a disrupted neuroependymal lining, with loss of cell-to-cell contacts. A similar loss of cell adhesion within the affected cells was seen in vitro. Disruption of Big2 through brefeldin-A (BFA) treatment led to a comparable disruption of the neuroependymal lining, and to periventricular nodule formation in mice. Finally, loss of alpha-SNAP function in the hydrocephalus with hop gait (hyh) mouse, which harbors a mutation in Napa, leads to denudation of the neuroepithelium, and to periventricular nodule formation. Overall, these observations raise the possibility that PH does not merely reflect a problem in neuronal motility; rather, it also entails a disruption of cell morphology and cell–cell integrity along the neuroependymal lining, mediated through vesicle trafficking-dependent pathways.
RESULTS
Heterotopic neurons within human PH
In the current studies, we examined the cerebral cortex and the ventricular lining from four human cases of PH by standard histology and immunostaining. Case 1 was an 83-year-old male with PH (no FLNA mutation analysis could be performed, given the poor DNA quality). Case 2 was a 27-year-old female with PH (no FLNA mutation analysis could be performed, given the poor DNA quality; however, she had glomeruloid vasculopathy consistent with a FLNA mutation). Case 3 was a 2-month-old female with PH (with a heterozygous point mutation (G→A) at bp 5290, resulting in an Ala→Thr substitution at amino acid position 1764, but it remains unclear whether this mutation was pathological). Case 4 was a 37 week gestational age (GA) male with PH [from a previously reported family with FLNA mutations (39)].
Neuronal nodules bilaterally lining the lateral ventricles (the hallmark of PH) were observed in Case 3, the 2-month-old female, by MRI (Fig. 1A, white arrows) and by examination of post-mortem brain tissue (Fig. 1B, black arrow). The cerebral cortex and the basal ganglia otherwise appeared normal. Hemotoxylin–eosin (H–E) staining showed a normal six-layered cerebral cortex with nodules lining the underlying lateral ventricle (Fig. 1C). Occasional regions of dysplasia were observed in the cortex (Fig. 1D), with large pyramidal neurons seen inappropriately in the more superficial layers of the cortex, although the lamination across the vast majority of the cortex appeared normal. Higher magnification of the heterotopia revealed the presence of disorganized cells residing in these nodules (Fig. 1E). Closer examination of the heterotopia, using various neuron-specific markers (neurofilament, NeuN, Tbr1, c-Neu, calbindin and Tle-1), demonstrated the presence of heterogeneous types of post-mitotic neurons in these nodules (Fig. 1E), consistent with prior findings (40).
Figure 1.

Histopathology in human periventricular heterotopia (PH). (A) T2-weighted MR image of the brain from a 2-month-old female with PH (Case 3). Small gray matter nodules of neurons (white arrowheads) can be seen lining the lateral ventricles. (B) Gross specimen of the same case demonstrates a periventricular nodule (black arrow) with otherwise normal appearing cortex and basal ganglia. (C) Hemotoxylin–eosin staining shows a normal six-layered cortex and contiguous nodules (asterisks) along the lateral ventricle (higher magnification to the right). (D) Occasional regions of cortical dysplasia are seen with larger pyramidal neurons ectopically located in the more superficial layers of the cortex. However, the vast majority of the cortical layers appear normal. (E) The nodule is comprised of neurons, expressing a variety of neuronal-specific markers.
Preserved cerebral cortical lamination and interneuron migration in human PH brain
The lamination of the cerebral cortex in individuals with PH has been shown to be grossly normal (41,42). However, those studies did not use layer-specific markers. Neurons that are born early in cerebral cortical development take up final positions in the lower layers of the mature cerebral cortex and are detected in the human cerebral cortex by the neuronal marker FOXP1 (43–45). In the embryonic brain, however, FOXP1 is seen in the more superficial layers II/III of the brain (45). Neurons that are born later during cerebral cortical development migrate past these deep-layer cells and take their final positions in the upper layers of the mature cerebral cortex. These cells are detected with the neuronal marker CUX1 (46). CUX1 staining was only successfully performed on frozen sections from the 2-month-old female (Case 3), given the formalin fixation of the other brain samples.
In the PH cerebral cortex of the 2-month-old female (Case 3), FOXP1 immunoreactivity was noted in the deep cortical layers, whereas CUX1 was observed in the upper superficial layers of the cortex, with no blurring of the cortical layer boundaries (Fig. 2A). Similar findings of largely preserved lamination by Nissl staining were seen in the other documented cases of PH (Cases 1 and 2) in which the clinical and MRI features were consistent with FLNA mutations (Fig. 2C and D). FOXP1 staining in the samples from the 27-year-old woman (Case 2) with PH also demonstrated normal expression of FOXP1 in the deeper cortical layers, consistent with the interpretation that cortical lamination is preserved in PH (Fig. 2D). Preserved lamination was observed even in a male (Case 4; 37-week GA male) with a presumed X-linked FLNA mutation [the family of this child had documented FLNA mutations and PH (39)] in whom all neurons harbored the mutation (Fig. 2E). Examination of the brain from the male case of PH (Case 4) revealed FOXP1 expression in a narrow band across the superficial cortical layers II/III, consistent with other reports of FOXP1 staining in the upper layers of the cerebral cortex in embryonic human brain (45), and once again suggesting appropriate migration during this early stage in development (Fig. 2E). Overall, these findings indicate that cortical lamination and therefore migration were largely preserved in the cerebral cortex of individuals with PH.
Figure 2.
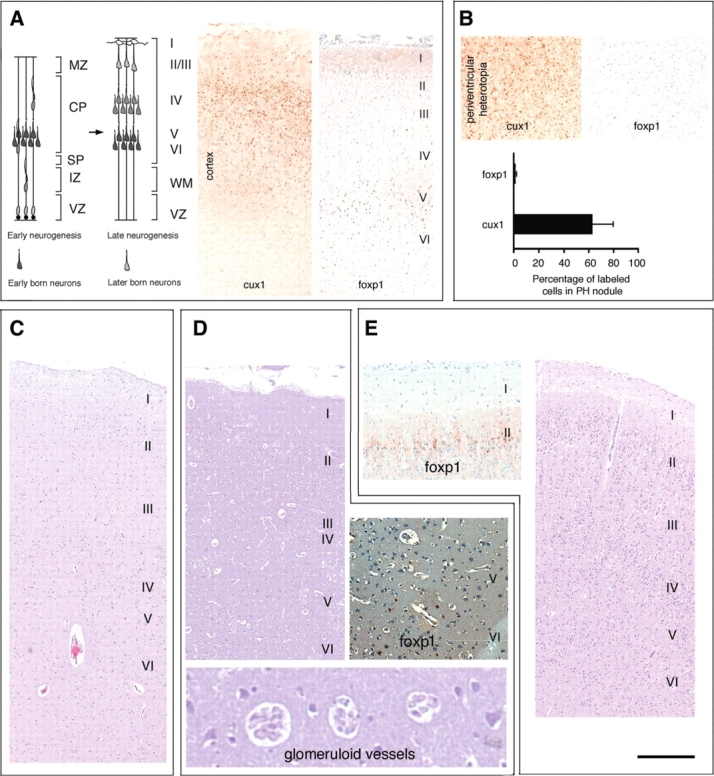
Neurons expressing the mutant FLNA protein migrate appropriately into the cerebral cortex. (A) Photomicrographs of the cerebral cortex and the nodular heterotopia from the 2-month-old female, immunostained with the superficial layer marker (CUX1) and the deep-layer marker (FOXP1), demonstrate a normal cortical lamination with no blurring of the cortical layer boundaries. (B) Photomicrograph of the heterotopic nodules in the 2-month-old female demonstrate increased numbers of CUX1-positive neurons, as compared to FOXP1-positive neurons, suggesting that these nodules are composed of later-born neurons. (C) Bright field photomicrograph of the cortex of an 83-year-old male with PH. The cortical layers appear discrete with no blurring of laminae on Nissl stain. (D) Nissl stained cortical section of a 27-year-old female similarly shows preservation of the layers. Immunostaining for FOXP1, which labels the deep-layer neurons, shows positive and appropriate staining in laminae V/VI. Glomeroid vascular anomalies were also seen in the cortex and have previously been reported in PH cases due to FLNA mutations. (E) Nissl stained cortical section of a 37-week GA male derived from a familial PH pedigree with a previously reported FLNA mutation shows appropriate cortical layering by Nissl stain. Positive immunostaining for FOXP1 which in embryonic human brain labels the neurons of the superficial layers (45).
Several other observations suggest that abnormal neuronal motility is not entirely responsible for periventricular nodule formation. In both the cortex and the nodules lining the lateral ventricles of PH brains, cells expressed the inhibitory neuronal markers GABA and GAD, in addition to the excitatory neuronal marker glutamate (Supplementary Material, Fig. S1), demonstrating that both inhibitory and excitatory neurons are found within the periventricular heterotopic nodules. Since many inhibitory interneurons are derived from the ganglionic eminence, the GABAergic neurons likely migrated long distances to arrive within the nodules. Secondly, CUX1 expression was observed in ∼60% of the neurons forming the nodules lining the lateral ventricles, but there was an almost complete absence of FOXP1 expression (only ∼2%) (Fig. 2B). FOXP1 is largely expressed in the earlier-born, deep-layer neurons of human cortex, while CUX1 is seen in the more superficial, later-born neurons of layers II/III of the cortex. Absence of FOXP1-positive neurons in the nodules suggests that earlier-born neurons were more capable of migrating into the cortex than were later-born neurons. Conversely, the predominance of CUX1-positive neurons in the nodules suggests impairments in neuronal migration later during cortical development. Taken together, these data indicate that nodule formation does not solely reflect a cell-autonomous neuronal motility deficiency, but also additionally a non-cell autonomous defect, such as a disruption of some developmental process along the VZ, that becomes more apparent later in development and that affects later-born neurons more severely.
Disruption of the neuroependyma in human PH brain
Since a mutation in either FLNA or ARFGEF2 results in human PH, we examined whether expression and/or localization of these proteins was disrupted in human PH brain tissue. Our prior studies had shown that FLNA and BIG2 (coded for by ARFGEF2) were co-expressed along the ventricular lining, the region where both of these proteins had their highest expression (47). In the brain of Case 3, the 2-month-old female, neurospheres isolated from the lining of the lateral ventricles demonstrated higher expression levels of FLNA than did the differentiated neurons/glia in the cortex and in the nodules (Supplementary Material, Fig. S2), suggesting that this protein serves a developmental function within the neural stem cell (NSC) population.
Because NSCs typically reside along the germinal matrix of the lateral ventricles, we closely examined the VZ and subventricular zone (SVZ). In our gross histological examination of the PH brain, we noted a marked disruption of the neuroependymal lining next to the periventricular nodules. In control, age-matched brain, FLNA was tightly localized to the apical side of the neuroependyma of the lateral ventricles [Fig. 3A and B (left column)]. However, in the PH brain, FLNA immunoreactivity appeared diffusely localized and often discontinuous, particularly along the ependymal lining of the lateral ventricles and adjacent to the nodules [Fig. 3A and B (right column)]. Similar results were obtained with the neural adhesion markers α-catenin and β-catenin (Fig. 3C and D), the PH-related BIG2 protein (Fig. 3E) and the neuroepithelial markers vimentin and S100B (Fig. 3F and G): disruption of the normal contiguous expression of these markers was seen along the neuroepithelium in PH brains. Disruption of the neuroependymal lining also was observed, as a discontinuity in the staining of FLNA and β-catenin along the neuroependyma in the brain of Case 4 (37-week-old GA male PH brain; Fig. 3H), and in regions immediately below the nodules and in areas with a normal-appearing overlying white matter and cortex without nodules, in both Cases 1 and 2 of PH (Supplementary Material, Fig. S3). The distinct co-localization of FLNA and BIG2 (the proteins responsible for PH) (27,37) to the neuroepithelium, and the apparent disruption of both these and other neuroepithelial markers along the neuroependyma, raised the possibility that PH results from a breakdown of the integrity of the neuroepithelial lining of the lateral ventricle, thereby giving rise to the ectopic neuronal nodules via defects in neuronal migration. Areas of ependymal breakdown without associated nodules could have arisen after the period of neuronal migration. Similarly, areas where the ependyma remained intact suggested that normal radial glial differentiation into the ependyma could occur (Fig. 3G).
Figure 3.
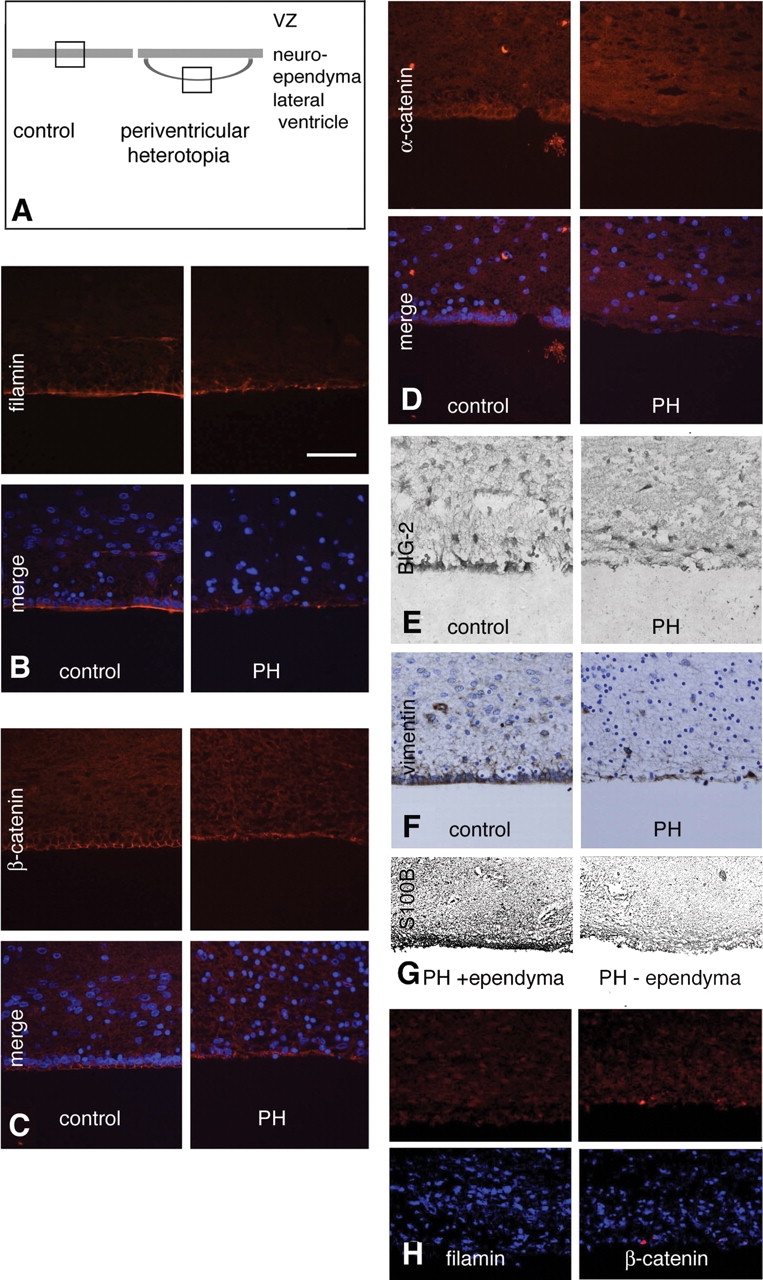
Disruption of the neuroependyma lining along the periventricular nodules in PH brains. (A) Schematic demonstrates the region of impaired neuroepithelial integrity in the neuronal nodules. (B–F) Photomicrographs of tissues from control and PH cases (2-month-old female with PH), following immunostaining for FLNA (B), β-catenin (C), α-catenin (D), BIG-2 (E), vimentin (F) and S100B (G) shows loss of the neuroependymal lining along the heterotopia. Moreover, in areas within PH brains, where the ependyma is intact, there is no disruption of radial glial differentiation as assessed by S100B staining (G). Finally, both FLNA and BIG2 (the protein encoding the gene shown to cause the autosomal recessive form of PH) (37) expression patterns are notably confined to the neuroepithelial lining (B and E). (H) Fluorescent photomicrographs of FLNA and β-catenin staining along the neuroependymal lining of the ventricular zone in a 37-week-old GA male with PH and a FLNA mutation demonstrating the disruption along the ventricular zone.
Loss of FlnA function impairs onset of neural precursor migration and disrupts the neuroependyma
To begin to explore experimentally the molecular mechanisms behind these observations in the human cases of PH, we conducted experiments using the mouse as a model system. Since FLNA has been implicated in neuronal motility, we directly examined the migration of early neuroblasts following in utero electroporation of a FLNA dominant-negative construct (EGFP-ΔABD-FilaminA), to determine whether a neuronal migration defect is the result of a cell-autonomous, or a non-cell-autonomous, defect. Prior studies had shown that loss of FLNA function prevents cells from acquiring consistent directed polarity and reduces motility in the SVZ and the intermediate zone (48). We used a FLNA dominant-negative construct that lacks the actin-binding domain, and therefore is thought to alter FLNA localization and actin-dependent signaling. By 48 h after transfection in embryonic day 16.5 (E16.5) mice, clusters of VZ progenitors expressing the EGFP-ΔABD-FilaminA construct were seen lining the ventricle, whereas progenitors transfected with the control EGFP alone construct were observed migrating toward the cortical plate (Supplementary Material, Fig. S4A). Approximately 80% of progenitors with inhibited FlnA function resided within 50 µm of the neuroependyma (as compared with 60% of control EGFP progenitors, P < 0.05), suggesting that some transfected cells were unable to migrate from the ventricular surface (Supplementary Material, Fig. S4B). Additionally, virtually all of the EGFP-positive cells from both the experimental and control animals stained for nestin and vimentin, suggesting that most of these cells were neural progenitors along the VZ, rather than post-mitotic neurons (Supplementary Material, Fig. S4C). Taken in the context of the prior observations of a normal-appearing cortex in human PH brains, these results suggest that FlnA can disrupt progenitor migration from the ventricular surface, through a cell-autonomous impairment in motility, and/or non-cell- autonomous disruption of the ventricular lining.
Prior studies had shown that loss of FlnA function in mice leads to a disruption of E-cadherin localization along the neuroepithelium (34). To specifically address whether inhibition of FlnA in progenitors along the neuroepithelium contributes to this disruption of the neuroependymal lining, and therefore potentially impairs initial neuronal migration, we blocked FlnA signaling, using the dominant-negative FLNA construct both in vivo and in vitro. Again, we performed in utero electroporations of the developing mouse (E16.5) lateral ventricles and cortex with the dominant-negative EGFP-ΔABD-FilaminA construct (48). Forty-eight hours after electroporation with EGFP- ΔABD-FilaminA, disruptions in the lateral ventricular lining were observed with the neuroependymal markers α- and β-catenin (Fig. 4A and B); effects were not observed in control neuroependyma electroporated with EGFP alone (Fig. 4A and B). These disruptions were most prominent in areas where neuroependymal cells had FlnA signaling disrupted (as assayed by EGFP-ΔABD-FilaminA expression), and where there were specific and localized cell connections to the lateral ventricle (Fig. 4A; higher magnification is lower left insert, for serial sections see Supplementary Material, Fig. S5). Our observations suggested that disruption of FlnA signaling can result in the loss of cell–cell contacts in the polarized neuroependymal cells. To further address this possibility, we transfected the FLNA dominant-negative construct into MDCK cells. When β-catenin was used as a marker for the integrity of cell–cell contacts, we find that impaired FlnA function led to a partial loss of β-catenin expression along the surface of the cell (small arrowheads, Fig. 4C). In addition, we transfected neural progenitors in utero by electroporation of the dominant-negative FLNA plasmid, subsequently dissociated them from the cortex, and cultured them in vitro. Loss of FlnA function in these cells led to impairments in cell spreading and adhesion, consistent with the observed loss of cell–cell contact (Fig. 4D). The loss of cell adhesion and spreading was not a result of increased cell death within the FlnA-inhibited cells (Fig. 4E). Progenitors expressing the dominant-negative FLNA construct exhibited a more rounded appearance, but did not express the cell death marker caspase 3. These findings show that loss of catenin staining is not limited to transfected cells, suggesting that the phenotype is not entirely cell-autonomous. Additionally, the current studies suggest that loss of FlnA function in progenitors along the neuroepithelium is sufficient to reproduce the loss of adhesion seen in FlnA null mice.
Figure 4.

Loss of FlnA function disrupts neuroepithelial cell contacts along the murine ventricular lining. (A) The neuroependymal lining (arrowheads), as assessed by β-catenin staining, appears disrupted 48 h after in utero electroporation of the dominant-negative EGFP-ΔABD-FilaminA construct, by comparison to an EGFP control construct, via confocal microscopy. Electroporation is performed on embryonic day 16.5 (E16.5) embryos and immunostaining is performed in the E18.5 embryo. The number of GFP-positive cells along the lining with co-localized surface expression of β-catenin is quantified relative to the total number of GFP-positive cells, following transfection of either control or EGFP-ΔABD-FilaminA construct. This change in β-catenin distribution and alteration in cell contacts are quantified below (Student's t-test: **P < 0.02; control n = 10, experimental n = 5). (B) The neuroependymal lining, as assessed by α-catenin, is also disrupted 48 h after in utero electroporation of the dominant-negative EGFP-ΔABD-FilaminA construct. Electroporation is performed on E16.5 embryos and immunostaining is performed in the E18.5 embryo. (C) MDCK cell cultures transfected with the dominant-negative EGFP-ΔABD-FilaminA similarly leads to a partial loss of cell–cell contacts (β-catenin, small arrowheads) and alterations in β-catenin distribution as compared to cells transfected with EGFP alone. Cultures are maintained for 48 h after transfection prior to immunostaining with the cell adhesion marker, β-catenin. The number of transfected cells with disrupted cell surface expression of β-catenin was quantified in both control and EGFP-ΔABD-FilaminA positive cells. The alterations in cell contacts are summarized to the right (Student's t-test: **P < 0.02; control n ≥ 3, experimental n ≥ 3 independent experiments). (D) Fluorescent photomicrograph of neural precursors 2 days after transfection either with control EGFP or dominant-negative EGFP-ΔABD-FilaminA constructs, followed by staining for the actin cytoskeleton with phalloidin (rhodamine). Inhibition of FLNA leads to a more rounded appearance in the precursor cells and overall loss in cell membrane spreading, consistent with a disruption in cell adhesion. Cell areas of transfected cells are measured using NIH ImageJ software (Student's t-test: **P < 0.05; control n ≥ 3, experimental n ≥ 3 independent experiments). (E) Fluorescent photomicrograph of neural precursors 2 days after transfection with control EGFP or dominant-negative EGFP-ΔABD-FilaminA constructs, followed by staining for the apoptotic cell death marker caspase3. The rounded appearance of the neural cells following inhibition of FLNA is not due to increased cell death (scale bar = 25 µm).
Loss of Big2 function disrupts the neuroependyma and leads to PH nodule formation
Mutation of ARFGEF2 (which codes for BIG2) causes an autosomal recessive form of PH in humans (37). Big2 has been shown to be expressed at its highest levels along the ventricular lining (47), and both ARFs and GEFs are expressed within the brain during development. Moreover, BFA has been shown to bind the Sec7 domain of Big2 GEF, and to inhibit ARF activity (49). We therefore evaluated in a murine model, whether loss of Big2/ARF activity within neural progenitors in vivo can disrupt the neuroepithelial lining in a manner similar to what occurs with loss of FlnA function. A single injection of BFA (20–40 µm) was performed in E16.5 mice or three serial intraventricular injections were performed in post-natal day P0 mice with subsequent injections on P1 and P2 mice. The inhibition of GEF function led to ventriculomegaly and heterotopic nodule formation along the ventricular lining (Fig. 5A, Supplementary Material, Fig. S6). The ventricular lining appeared to be disrupted, with an aberrant distribution of neurons along the ventricular surface. However, injections in later-born P0 animals led to fewer nodules and fewer mis-sited neurons along the lining, despite multiple BFA injections. This observation likely reflects the end of corticogenesis in the later-age mice. To more closely assess whether these nodules form through a disruption of the ventricular lining, we examined the integrity of the neuroepithelium at time points more closely following BFA injection into embryonic mice. Two hours after Big2/ARF inhibition, the neuroependymal lining of the VZ was not uniformly intact, as indicated by the discontinuous expression of phalloidin and N-cadherin (Fig. 5B), suggestive of an abnormality in the adherens junctions of the neuroependymal cells. By 7 h after Big2/ARF inhibition, the neuroependyma was clearly disrupted, as evidenced by the β-catenin staining pattern (Fig. 5C). Inhibition of Big2/ARF by BFA in vitro suggested a disruption in the vesicle trafficking of adhesion molecules such as β-catenin in MDCK cells (Fig. 5D). In BFA-treated MDCK cells, β-catenin accumulated in the Golgi apparatus, rather than being transported to the cell surface (Fig. 5E). As was seen for altered FlnA function, the loss of Big2 function impaired the targeting of adhesion molecules; this impairment could provide an underlying basis for disruption of the integrity of the neuroepithelium along the VZ.
Figure 5.

Inhibition of Arfgef2/Big2 interrupts the integrity of the neuroependymal lining and leads to periventricular nodule formation in mice. (A) Periventricular heterotopic nodules (PH1 and PH2) and enlarged ventricles (LV) are seen on Cresyl violet stained tissue in a 2-week-old mouse following early post-natal intraventricular injections of 40 µm BFA. Heterotopic nodules (PH1) were seen just below the ventricular surface. Clusters of cells (PH2, arrowheads) also extended beyond the neuroependymal lining to lie directly along the LV. Immunostaining for these ectopically localized cells shows that they express the neuronal marker NeuN (below). To the right, the higher magnification photomicrographs of PH1 and PH2 show neurons ectopically positioned along the ventricle. (B) Two hours after intraventricular injection of BFA into E16.5 mice, phalloidin and N-cadherin staining along the ventricular zone neuroependyma is discontinuous. (C) Within 7 h after intraventricular injection into E16.5 mice, BFA disrupts the continuity of β-catenin staining along the ventricular neuroepithelium. (D) Through inhibition of Big2, BFA impairs transport of adhesion molecules such as β-catenin from the Golgi apparatus to the cell surface in MDCK cells. This impairment in vesicle transport likely contributes to heterotopia formation. (E) After exposure to BFA for 7 h, the trafficking of β-catenin from the Golgi complex to the cell membrane is disrupted.
Loss of alpha-SNAP function leads to denudation of the neuroependyma along the VZ, and induction of periventricular nodules
While FLNA has been implicated in cell motility, this actin-binding protein also associates with the Golgi membrane and regulates endosomal trafficking (50,51). BIG2 directs ARF activation, and ARF proteins are major regulators of vesicle formation in intracellular trafficking (52). Impairments in vesicle transport could disrupt delivery of various adherens junction proteins within both the neuroepithelium/immature ependyma and neural progenitors of the SVZ, thereby compromising the integrity of the neuroependyma. Prior reports have described neuroependymal denudation within the hyh mutant mouse (53–55), which harbors a mutation in the vesicle fusion-related gene Napa (53,56). Prior studies have also shown that polarity markers such as E-cadherin, β-catenin and atypical protein kinase C (aPKC) were abnormally distributed along the neuroepithelial lining (57). We therefore examined whether this mutant mouse develops heterotopic nodules. H-E stained, coronal brain sections from the mutant hyh mouse at various ages showed an impairment of the integrity of the ventricular lining followed a clear temporal and spatial pattern. As early as E12.5, the neuroependyma of the fourth ventricle was disrupted, while that of the lateral ventricles was still unaffected (Fig. 6A and B). At E14.5, a ruffling and disintegration of the neuroependyma lining the ganglionic eminences was seen, leading to a protrusion of cells from the SVZ into the ventricular lumen (Fig. 6C and D). At later ages (E16.5), the disorganization of the VZ progressed rostrally, involving the neuroependyma lining of the septum (Fig. 6E and F). Interestingly, alpha-SNAP immunostaining in wild-type embryos showed this protein to be preferentially expressed in cells along the immature ependyma of the ganglionic eminences and the cerebral cortex (Fig. 6G–J). Moreover, immunostaining for caveolin-1, a membrane-associated protein that is highly expressed in the neuroepithelium (58), revealed disruption of the neuroepithelial architecture (Fig. 7, cf A–B and E–F). We then analyzed the phenotype of the cells protruding into the ventricular lumen and found them to express neuronal markers, likely corresponding to neuronal progenitors or neuroblasts (Fig. 7C and G). Scanning electron microscopy clearly revealed denudation of the neuroepithelium, with progenitor cells and macrophages becoming exposed to the lateral ventricle cavities (Fig. 7D and H). Denudation of neuroepithelium/ependyma and alterations of the SVZ have been shown to occur in human fetuses with a moderate communicating hydrocephalus (58,59).
Figure 6.

Loss of alpha-SNAP function in hyh mice disrupts the integrity of the neuroepithelium (ventricular zone). Coronal sections, stained with hematoxylin-eosin, through the brain of wild-type (wt) and mutant hyh (hyh) mice, at E12.5, E14.5 and E16.5. A caudal (fourth ventricle) to rostral (lateral ventricles) progressive disruption of the ventricular zone (VZ) lining the ventricular system is shown. (A and B) At E12.5, the neuroepithelium of the ganglionic eminence (GE) appears intact, while neuroepithelium denudation has already started at the fourth ventricle in the hyh mouse (inserts in lower left and right corners). (C and D) At E14.5, loss of neuroepithelium occurs along the caudal GE in the hyh mouse (arrows, compare inset images in C and D). (E and F) By E16.5, a breakdown of neuroepithelium and disorganization of the subventricular zone (SVZ) reaches the rostral horns of lateral ventricles in the hyh mouse (arrows, compare inset images in E and F). In the hyh mouse, denudation of the neuroepithelium leads to protrusion of progenitor cells from the SVZ into the ventricular lumen (top inset imagess in D and F). (G–J) Immunolocalization of alpha-SNAP in the GE in wild-type tissue at E14.5 (G–H) and the cerebral cortex at E16.5 (I–J). (G and I) Alpha-SNAP immunoreactivity is mainly found in the cells of the VZ (VZ, arrows). (H and J) Differential interference contrast (DIC, Nomarski optics) imaging of the same area is shown in G and I, respectively. LV, lateral ventricle. Scale bars: 250 µm (A–F); 500 µm (low magnification insets in A–F); 50 µm (high magnification insets in A–F and G–J).
Figure 7.

Loss of alpha-SNAP function in hyh mice disrupts the progenitor cell population along the neuroependyma. (A–D) Wild-type mice; (E–H) hyh mutant mice. (A) Sagittal, hematoxylin–eosin stained section through the CNS of an E14.5 wild-type mouse. Boxed area is shown in B. (B) Higher magnification image of the area of the ganglionic eminence (GE) shown in A. The integrity of the ventricular zone (VZ)/neuroepithelium and subventricular zone (SVZ) is clearly seen. Inset: caveolin-1, functional marker, also reveals the integrity of the VZ (arrow). (C) Section adjacent to that shown in (B), immunostained for β-III-tubulin. The cells of the VZ are not immunoreactive, while those of the SVZ express the neuronal marker. (D) Scanning electron micrograph of the ventricular surface of the GE of a wild-type E18.5 embryo, showing a mosaic-like arrangement of cells, most of which are monociliated (stem cells/radial glia) (arrows) with few multiciliated cells (immature ependyma). (E) Sagittal hematoxylin–eosin stained section through the CNS of a mutant hyh E14.5 mouse. Boxed area framed is shown in (F). (F) Higher-magnification image of the area of the GE shown in (F). Denudation of the neuroepithelium and exposure of the cells of the SVZ to the ventricular lumen are evident (arrows). A few patches of VZ still remain intact. Inset: Disorganization of the VZ and SVZ (asterisk) can also be shown by immunostaining for caveolin-1. Arrow points to a patch of VZ. (G) Section adjacent to that shown in (F), immunostained for β-III-tubulin. Immunoreactive cells of the SVZ are fully exposed to the ventricular cavity (arrows). VZ indicates an undetached patch of neuroepithelium. (H) Scanning electron micrograph of the ventricular surface of the GE of a mutant hyh (E18.5) embryo, revealing denudation of the neuroepithelium with progenitor cells (PC) lining the VZ with associated macrophages (M). Scale bars: 500 µm (A and E); 50 µm (B, D, F and G); 25 µm (insets in B and F); 10 µm (D and H).
With continued development in the mouse, the multiple areas of neuroepithelium/ependyma denudation give rise to nodules along the lateral ventricles (Fig. 8A–C; Supplementary Material, Fig. S7). Thus, in post-natal mutant hyh mice, heterotopic nodular neurons were frequently observed in the vicinity of the ventricular walls, where the ependymal lining was missing (Fig. 8C–E). In contrast, nodules were never seen below those regions of the ventricular walls that retained the ependymal lining. Because nodules were distinct during the early post-natal period, and frequently found near the SVZ, where a small degree of neurogenesis continues post-natally (Fig. 8C), we studied whether PH neurons originate post-natally from over-proliferation of cells in the SVZ, or whether they correspond to post-mitotic neurons born at embryonic stages and spatially arrested near VZ. We used cumulative BrdU (5-bromo-2′-deoxyuridine) labeling during a 3-day period (P3–P6). Numerous cells of the SVZ were found to be labeled with BrdU, whereas those of the nodules were not, indicating that these cells corresponded to post-mitotic neurons born at embryonic stages (Fig. 8F and G).
Figure 8.

Loss of alpha-SNAP function in hyh mice leads to PH formation. (A) Hematoxylin-eosin (H/E) stained coronal section through the brain of PN14 (P14) hyh mutant mouse, displaying nodular periventricular heterotopias (arrowheads) below the ependymal-denuded ventricular walls. Boxed area is shown in (C). CC, cerebral cortex; HI, hippocampus; LV, lateral ventricle. (B) Adjacent section to that of (A) immunostained for GLUT-1. The ependyma lining the hippocampus (HI) is preserved and GLUT-1 immunoreactive (arrow). The dorsal and lateral walls of the ventricle are denuded (asterisks). (C) Higher-magnification image of the area boxed in (A). Nodular periventricular heterotopias (PH1 and PH2, oval regions delineated by dashed lines) are seen adjacent to the ependymal-denuded ventricular wall (D and E) and the subventricular zone of the lateral wall. (E) Periventricular nodules are formed by cells with large nuclei and prominent nucleoli. The same section shown in (A, C was bleached and used for toluidine staining (E), bleached again and used for immunostaining (D). The two rectangular areas in C are shown in (D) and (E), respectively. (D) Immunostaining with a neuronal marker (β-III-tubulin) shows that the cells forming the periventricular nodules (PH1) in C are indeed neurons (arrows). (E) Similarly, toluidine blue staining demonstrates that periventricular nodules (PH2) in (C) displays a neuronal phenotype. (F) Frontal section through the brain of a hyh mutant mouse injected with BrdU from P4 (PN4) to P6 (PN6) and sacrificed 3 h after the last BrdU injection on P6. PH, periventricular heterotopia; SVZ, subventricular zone; CC, cerebral cortex. (G) Higher-magnification image of a portion of (F) showing that cells within the nodule (PH) do not proliferate post-natally, while those of the SVZ continue to proliferate post-natally and are BrdU-labeled (inset). Scale bars: 200 µm (A, B and F); 50 µm (C and G); 10 µm (D, E, inset in G).
DISCUSSION
A mutation in either of two genes (FLNA or ARFGEF2) has been shown to cause PH in humans (27,28,38). While these two genes appear quite disparate functionally, the similarities in the radiographic characteristics of bilateral heterotopic nodules, between cases of PH arising from FLNA mutation and cases arising from ARFGEF2 mutation, suggest that the two genes have a common final effector, in giving rise to aberrant cortical development. Through examination of post-mortem human PH brains (including a familial male case of PH due to a FLNA mutation), we showed that the vast majority of neurons migrated into the cerebral cortex. Detailed examination of the ependymal lining of the heterotopic nodules revealed a significant disruption in the integrity of the neuroependyma. Experimental disruption of FlnA signaling in mice, both in vivo and in vitro, resulted in a failure to initiate migration, an altered integrity of the ependymal lining and a loss of cell adhesion and cell–cell contacts. Inhibition of Arfgef2/Big2 induced by BFA caused a similar disruption in the neuroepithelial lining and led to formation of periventricular nodules in mice. Finally, we described the formation of nodules in the hyh mouse: this mouse's mutation in the Napa gene disrupts SNARE receptor (SNARE)-mediated vesicle transport and membrane fusion. Because alpha-SNAP is a protein involved in vesicle fusion, it should be widely expressed in the body. However, evidence indicates that it is preferentially expressed in the CNS and at early developmental stages (56); within the CNS, alpha-SNAP is most highly expressed in the VZ and SVZ (present report). This expression pattern would explain why the alpha-SNAP mutation is principally expressed spatially in these zones, and at certain stages of development. Overall, our results suggest that PH, while linked to aberrations in neuronal motility, also can arise from a disruption in the neuroependyma, where loss of FlnA, Arfgef2/Big2 and Napa/alpha-SNAP functions alter vesicle trafficking and the adhesion of neural progenitors.
Genetic studies and clinical observations associated with X-linked PH suggest that the role of FLNA in cell motility is not entirely responsible for periventricular nodule formation. For example, random X-inactivation would imply that ∼50% of the neurons born would not express the normal filamin protein. However, in most reported PH cases and in the post-mortem tissues examined here, the overlying cortex is relatively normal, and only a small proportion of the neurons constitute the nodules (42,60). Moreover, recent genetic analysis has identified FLNA mutations in several male patients, many of whom were not somatic mosaics for FLNA mutations (as was the case for the 37-week GA male included in this study), implying that—despite the fact that all neurons bear the FLNA mutation—the neurons either migrate completely into the cortex or do not migrate at all (but never halfway) (28,39,61). Furthermore, autosomal recessive mutations in ARFGEF2 result in PH with radiographic findings virtually identical to these seen in PH due to a FLNA mutation, except for the presence of the severe microcephaly associated with ARFGEF2 mutation (37). In ARFGEF-related PH, all of the neurons necessarily harbor homozygous mutations in ARFGEF2, but still form heterotopic nodules and a relatively normal-appearing cortex, suggesting that the periventricular nodules are unlikely to be due to motility impairments alone. Finally, the recessive nature of the disorder suggests that PH is not a cell-autonomous process, given that many neurons migrate appropriately even while others do not.
Several aspects of our current findings suggest that PH does not arise solely from a cell-autonomous motility defect. Most neurons actually migrate into the developing cerebral cortex and take up their proper final positions in the appropriate cortical layers, as evidenced by the precise lamination seen with FOXP1 and CUX1 immunostaining. The lack of FOXP1 neuronal expression in the nodules suggests that the earliest-born neurons were able to migrate from the VZ and into the appropriate lower layers of the cerebral cortex (as indicated by the expression of FOXP1 in the deep layers of the cortex). The increased proportion of CUX1-positive neurons in the nodules, however, would argue that the later-born neurons were the ones that were predominantly affected. This preponderance of later-born neurons in the nodules is not necessarily consistent with PH's being a cell-autonomous cell motility problem; if that were the case, the migration of all neuronal populations would be expected to adversely affected. Rather, a loss of ependymal integrity, which occurs later in development, would be a more likely reason why later-born neurons are affected. GABAergic neurons that arise from the ganglionic eminences and undergo long-distance migration to the VZ are found in the nodules suggesting that some migration of this neuronal population occurred appropriately. Finally, recent studies show that the mutant FlnA mouse, while embryonic lethal, does not impair neuronal migration during embryogenesis (35,36). Taken in sum, our data are most consistent with the idea that FLNA mutations cause PH, secondary to impairments in neuronal motility (cell-autonomous), but also PH can arise from a disruption in neural progenitors along the VZ, a disruption that influences the migration of later-born neurons (non-cell-autonomous).
Previous studies have suggested that PH arises from an impairment of the initial migration of neurons. Inhibition of FlnA signaling by dominant-negative FlnA transfection was shown, in prior work, as well as here, to disrupt the initial migration and the morphology of migratory neuroblasts in the SVZ (48). Furthermore, FilaminA-interacting protein (FILIP) has been suggested to be important, in allowing neurons to exit the VZ (48). Such observations, however, do not necessarily explain the preponderance of later-born neurons in the nodules, unless both FILIP and FLNA are more critical for the decision by later-born migratory neurons to exit the SVZ. Later-born post-mitotic neurons do migrate longer distances and must migrate past earlier-born neurons as they enter the more superficial layers of cortex, suggesting that additional cues are required by the later-born neurons. Our expression data show that both FlnA and Big2 proteins are highly expressed in, and strongly restricted to the neuroependymal lining; they are not highly expressed in the SVZ. Loss of FLNA function has additional effects; it can cause aortic rupture, gut dysmotility and increased skin elasticity (27,62,63), with high FLNA expression restricted to the lining of the blood vessel, intestines and skin, respectively (Supplementary Material, Fig. S8). Defects in the linings of these organs could in part account for the above phenotypes.
Several observations suggest that PH does not result from an over-production of neurons. The brains of individuals with PH that arises from ARFGEF2 mutations are microcephalic, and Big2 inhibition in vitro leads to diminished proliferation of neurons (37). A recent case study of PH suggested that microcephaly in males can be associated with FLNA mutations (64). Neural precursors (neurospheres) and fibroblasts obtained from Case 3 failed to demonstrate any increase in proliferation (data not shown). Similarly, BrdU labeling of proliferating cells in neurospheres obtained from hyh mutant mice showed no difference from wild-type neurospheres (data not shown). The hyh mice showed a progressive loss of proliferative progenitor cells during development (57). Direct inhibition of FlnA signaling in HEK293 cells by dominant-negative over-expression did not produce any increase in cell number (data not shown). Taken together, these observations suggest that nodule formation in the PH brain is not a direct consequence of over-proliferation of neurons along the VZ during cerebral cortical development. However, studies have suggested that endothelial cells can stimulate their own self renewal, and can expand neurogenesis in NSCs (65). Thus, it remains to be determined whether the loss of blood brain barrier integrity along the VZ, or the endothelial vasculature, contributes to an increase in neural progenitor proliferation, and periventricular nodule formation.
Disruption of the neuroepithelial lining, in giving rise to PH, mirrors the loss of structural integrity along the pial surface of the brain that produces cobblestone lissencephaly (66–68). In cobblestone lissencephaly, neurons have no inherent motility defect; rather, they migrate beyond the marginal zone into the leptomeninges and through the external basement membrane during cortical development. The loss of integrity of the molecular layer leads to nodules composed of neurons, on the outermost surface of the brain (thought to resemble cobblestones). Four genes, fukutin, POMGnT1, POMT2 and POMT1, have been associated with cobblestone lissencephaly (69–71); all are implicated in the glycosylation of dystroglycans. Hypoglycosylation of dystroglycan abolishes binding activity for such ligands as laminin, neurexin and agrin, and thereby compromises the integrity of the dystrophin-associated extracellular matrix adhesion complex (72). In a fashion akin to neuronal migration, neural progenitors perform interkinetic nuclear migration in the VZ, with cells in synthesis (S) phase positioned in the upper half of the epithelium away from the ventricular surface (73–75). Cells transitioning from S to growth (G) 2 phase have nuclei that translocate in the cell toward the ventricular surface, and cells in mitosis (M) lie adjacent to the ventricular surface. Following M phase, cells re-entering G1 phase translocate away from the neuroepithelium, while post-mitotic neurons generally migrate toward the pial surface and cortical plate. Loss of the neuroepithelial integrity along the ventricular surface could conceivably disrupt cells exiting M phase, causing them to ‘over-migrate’ or to protrude into the ventricular lumen, giving rise to periventricular nodules. This possibility would be consistent with the observed loss of neuroependymal integrity in the human pathological cases of PH, in the hyh mutant mouse and in the mouse models in which inhibition of FlnA and Big2 occurred. Thus, in some ways, PH reflects a pathological process similar to that seen in cobblestone lissencephaly; the difference is merely that in PH the inner neuroependymal lining is affected, rather than outer surface of the brain.
Several lines of evidence suggest that PH results from a disruption of vesicle trafficking. First, a number of peripheral membrane proteins, including FLNA, associate dynamically with Golgi membranes during the budding and trafficking of transport vesicles in eukaryotic cells (50). FLNA has been shown to regulate furin sorting in the trans-Golgi network/endosomal system (51). Thus, while FLNA regulation of actin may directly alter the cytoskeleton required for migration, an additional role for FLNA, in vesicle trafficking, would not be surprising, given that the actin cytoskeleton is required for vesicle budding and vesicle movement (76–78). Second, other genes implicated in periventricular nodule formation appear to be involved in vesicle trafficking. ARFGEF2 regulates the ARFs, which bind to vesicle coat proteins and adaptors. Similarly, the studies reported in the present paper suggest that mutations in Napa also result in PH in the hyh mouse. Alpha-SNAP is involved in SNARE-mediated vesicle fusion in many cellular contexts (79). Finally, Mekk4, while it has not been directly linked to vesicle trafficking, has been shown to interact with FlnA, and to regulate FlnA expression (86). It remains to be seen whether each of these genes interact directly within the same pathway.
Once we better understand the functions of the genes that regulate the interaction between the actin cytoskeleton and Golgi apparatus (and consequently vesicle transport), during brain development, we will begin to start to form a clearer picture of the causal mechanisms of human PH, and the associated CNS phenotype. Heterotopic nodules are the primary feature seen in this disorder, and disruption in the transport or recycling of adherens junction molecules could explain a breakdown in the neuroepithelial lining. PH, however, is also associated with a thinning of the cortex and in extreme cases, microcephaly. Vesicle transport (via the recycling endosome) is responsible for the delivery of specific lipids and proteins to the cleavage furrow, and is crucial for cell abscission. Disruption of this process would explain the altered rates of symmetric and asymmetric division that lead to changes in neural precursor fate and a thinner cortex in PH. Finally, humans with PH show an elevated incidence of dyslexia and psychiatric disorders (80,81), suggesting problems with synaptic connectivity. Impairments in actin-based vesicle trafficking could also disrupt these contacts; disruption of both FLNA and Big2 has already been shown to disrupt recycling of surface receptors and neurite extension, necessary for dendritic arborization. Further studies are needed to determine whether this common function in vesicle trafficking leads to the multiple observed phenotypes in PH.
MATERIALS AND METHODS
Human tissue, ethical and licensing considerations
The current studies have been approved by the Institutional Review Board (IRB) at the Beth Israel Deaconess Medical Center and Brigham and Women's Hospital. De-identified human discarded tissue was obtained from pathological samples during autopsy. The diagnosis of PH in the 37-week-old GA male (Case 4) and 2-month-old female (Case 3) was made prior to autopsy by prenatal screening through ultrasound and/or confirmed by MR imaging. Confirmation of the diagnosis of PH in the adults cases (Cases 1 and 2) were made at autopsy.
Subjects
Neuropathological examination was performed on four brains with PH: a 37-week GA male (Case 4), a 2-month-old female (Case 3), a 27-year-old female (Case 3) and an 83-year-old male (Case 1).
The 37-week GA male (Case 4) was an archival case (fixed in formalin and stored as a paraffin block); he was born by induction at 36 weeks gestation, and died at 7 days of life, likely from vascular complications. Details have previously been described (39), but the child had PH and came from a family with multiple affected members (including the mother and aunt); he had a documented 8-bp deletion in the intron/exon boundary of exon 25 of the FLNA gene (39). Macroscopically, the brain was small (brain weight 290.5 g; expected 350–400 g by CDC growth chart), and was notable for the presence of multiple nodules of heterotopic gray matter, located bilaterally in the angles of the lateral ventricles. The majority of the cortex showed a normal six-layer pattern. However, microscopic examination did reveal a small area of unlayered polymicrogyria in one insula, as well as abnormal layering in the inferior portion of the temporal lobes. Additional extra-CNS abnormalities, often seen in severe cases of PH arising from FLNA mutations, included atrial and ventricular septal defects with a PDA, thrombocytopenia and a malrotated and shortened gut.
The 2-month-old female (Case 3) with PH was born by Caesarian section after a 40-week gestation. At birth, the infant had an irregular breathing pattern and was placed on a respirator. The infant was noted as having dysmorphic features. Laboratory workup was notable for a mild anemia. The subject had a normal karotype (46,XX). Mitochondrial dysfunction was excluded by the presence of normal activity of the electron transport chain complexes I—IV. She was diagnosed with a ventricular septal defect by an echocardiogram. A brain MRI revealed bilateral subependymal nodular heterotopic gray matter (bilateral PH) and aqueductal stenosis (Fig. 1). During her hospital stay, the infant was observed having at least one tonic-clonic seizure, before being treated with phenobarbital. The subject died at 2 months of age from respiratory failure. Post-mortem brain tissue was obtained immediately following autopsy for histological characterization (frozen tissue), and for generation of both neural progenitors (neurospheres) and skin fibroblasts. No FLNA mutation was detected, upon sequencing of only the coding regions of the gene.
The PH observed in the remaining two adult cases was an incidental finding at autopsy. The 27-year-old female (Case 2) died from acetaminophen overdose. The 83-year-old male (Case 1) also died from causes unrelated to PH. No sequencing for FLNA mutations was performed on these archival samples (fixed in formalin and stored as a paraffin block), due to poor DNA quality.
Mice
The hyh mutant mice (hydrocephalus with hop gait) (B6C3Fe-a/a-hyh/J were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and maintained at Austral de Chile, Valdivia, Chile. Housing, handling, care and processing of animals were carried out following the regulations approved by the council of the American Physiological Society. Mice were fed ad libitum and were maintained under a constant 12 h light/12 h dark photoperiod, and a constant room temperature of 25°C. Heterozygous females were caged together with heterozygous males, and were separated when the presence of a vaginal plug was evident, designated as E0.5. Embryos (E12.5–E18.5) were obtained from anesthetized pregnant dams. Mutant embryos were identified by the absence of the ventral neuroepithelial layer under microscopic analysis, and by genotyping using PCR.
Immunohistochemistry of human and mouse tissues
Immunohistochemistry was performed on tissue sections using standard techniques. In brief, tissue sections were de-paraffinized in serial ethanol washes (if stored in paraffin), placed in blocking solution with phosphate-buffered saline (PBS) containing 10% fetal calf serum, 5% horse serum and 5% goat serum or 10% donkey serum and incubated overnight in the appropriate antibody at 4°C. Antibody staining assessed for neuronal proliferation (Ki-67, courtesy BWH pathology; phospho-histone H3: 1:200 dilution of stock, Upstate Biotechnology), neuronal migration [FOXP1, courtesy A. Banham, using previously described methods (43); Cux-1, courtesy Marta Nieto (46)] and neuronal differentiation (Calbindin, Sigma; c-Neu, Santa Cruz Biotechnology; TLE-1, Abcam; TBR-1, courtesy Dr Hevner; NSE, Zymed; Neu-N, courtesy BWH pathology; neurofilament; Sternberger Monoclonal; GAD, GABA and glutamate, Sigma). To assess the neuroependymal lining, we used neuroepithelial marker antibodies [α- and β-catenin, Transduction laboratories, 1:250 dilution of stock; S100B, Abcam, 1:1000; vimentin, Zymed, stock; FLNA, Novocastra; BIG2, 1:200, using previously described methods (37)]. FLNA staining was also performed on adult paraffin-embedded human sections from the skin, aorta and gut. Tissue sections were processed through standard fluorescent secondary antibodies (Hoechst 33342 dye, Molecular Probes; CY3, Jackson Immunoresearch Laboratories and FITC, Sigma). Cux-1 staining could not be performed on the archival paraffin-embedded tissue samples, since frozen sections are required for such staining.
For the mouse studies, embryos were sacrificed at various ages and fixed by immersion in 4% paraformaldehyde in PBS at 4°C or Bouin's fixative solution at room temperature. Adult tissue was collected after intracardiac perfusion with PBS followed by 4% paraformaldehyde in PBS or Bouin's fixative solution. Tissues were processed for paraffin embedding, for vibratome sectioning or for cryostat sectioning of frozen sections. Immunohistochemistry was performed on sections as described earlier. Additional antibodies used included alpha-SNAP-specific antibody (clone 4E4) (1:50) (Exalpha Biologicals); neuronal marker beta-III-tubulin (G7121) (1:6000) (Promega); caveolin-1 (N-20) (1:250) (Santa Cruz Biotechnology) and GLUT-1 (kindly provided by Coralia I. Ribas, Memorial Sloan-Kettering Cancer Center, NY, USA). To detect alpha-SNAP signal, we incubated samples sequentially with a biotinylated secondary antibody (Vector Laboratories) and streptavidin Alexa Flour-488 (Molecular Probes) for 30 min each; binding was evaluated on an Olympus Fluoview FV1000 laser scanning confocal microscope.
Bromodeoxyuridine labeling
BrdU (Sigma) was used to label cells undergoing proliferation, around the end of the first post-natal week. Wild-type and hyh mutant mice were injected i.p. with three daily doses of BrdU (100 mg/kg) for three consecutive days (P4–P6) and were sacrificed on P6, 3 h after the last injection. Under anesthesia, the mice were transcardially perfused with heparinized buffered saline, followed by Bouin's fixative. The brains were removed and post-fixed for 48 h in the same fixative. Samples were embedded in paraffin and processed for BrdU immunohistochemistry. Briefly, sections were sequentially incubated with: (i) a monoclonal antibody against BrdU (G3G4; Developmental Studies Hybridoma Bank) at 1:50 for 18 h; (ii) a biotinylated secondary antibody (Vector) for 30 min and (iii) Vectastain Elite ABC reagent (avidin DH: biotinylated horseradish peroxidase H; Vector) for 30 min. The reaction product was detected with 3.3′-diaminobenzidine tetrahydrochloride (Sigma).
Scanning electron microscopy
Wild-type and hyh mutant mice at E18.5 were anesthetized, and their brains were removed and fixed by immersion in 4% paraformaldehyde, 2% glutaraldehyde, 2% acrolein in 0.2 m phosphate buffer, pH 7.4. The blocks of tissue were dissected into small pieces containing different ventricular regions and were post-fixed in the same fixative overnight, at 4°C. After dehydration and critical point drying, the blocks of tissues were coated with gold and visualized in a scanning electron microscope, using a secondary electron detector.
Cellular quantification
The total number of cells labeled by CUX1 or FOXP1 was quantified within random fields represented within the heterotopic nodules. The percentage of cells stained for each marker was calculated by dividing the number of positive labeled cells by the total number of cells. Four fields were quantified within the heterotopic nodules.
In utero electroporations and intraventricular injections
Electroporation procedures using the FLNA dominant-negative construct (EGFP-ΔABD-FilaminA) followed previously published methods (48). In brief, E16.5-pregnant dams were deeply anesthetized with Avertin. The skin overlying the uterus was shaved, sterilized and draped, and incised to reveal the embryos. A 2-μl DNA/EGFP mixture (1:3 µg/µl DNA:GFP ratio) was injected into the ventricle with a Hamilton syringe. A 50 mV, 100-ms duration pulse was applied, using electroporation prongs, across the surface of the embryo's head. For intraventricular injections of BFA (20–40 µm), 1–2 µl injections into E16.5 mice were performed using a pulled glass micropipette. A 1–2 µl intraventricular injection of BFA was performed in P0 mice consecutively over 3 days. Following electroporation or intraventricular injections, the maternal wound was sutured, and the pregnant dam was allowed to recover, and was returned to normal care.
Histological analysis of neuroependymal lining
After survival times of 2–3 days, pregnant mice were deeply anesthetized with sodium pentobarbital and cervically dislocated. The embryos were harvested, decapitated and drop-fixed in 4% paraformaldehyde in 0.1 m PBS overnight. The brains were blocked for coronal sectioning through regions of positive GFP expression, and serial sections 50 µm thick were cut on a vibrating microtome. Alternate sections were processed and mounted for fluorescence and cresyl violet staining, for assessment of cellular morphology, migration of GFP-positive cells from the ventricle and integrity of the neuroependymal lining. Fluorescence sections were air-dried on gelatin-coated slides, and were subsequently either dipped briefly in Histoclear (National Diagnostics) and permanently mounted in Fluoromount (Gurr), or else directly mounted in mineral oil on slides. For the BFA injections, mice were allowed to survive for upwards of 4 weeks prior to sacrifice and histological analyses. Histological analysis was performed using a microscope with epifluorescence and high numerical aperture optics (Zeiss or Olympus microscope), or by confocal microscopy (Harvard Medical School Cell Biology Microscopy Core).
MDCK transfection
MDCK cultures were maintained according to previously published methods (37). For transfection using the dominant-negative FLNA (48), FuGene (Roche) was allowed to equilibrate to room temperature and then 3 µl of FuGene was added to 100 µl of Dulbecco's modified eagle medium (DMEM) without serum. One microgram of the DNA construct was added to each well, allowed to incubate for 15 min and washed. After 6–12 h, medium was removed, and was replaced with DMEM culture medium containing 10% fetal calf serum and 1% penicillin–streptomycin. Twelve to 24 h after transfection, the cultures were fixed, immunostained for the desired markers and examined by confocal microscopy or routine fluorescence (Harvard Medical School Cell Biology Microscopy Core).
Western blot analysis
Protein was extracted from the heterotopic nodules and the cortex by slight modifications of previously described methods, or from brains of mice from various developmental stages (82), solubilized in lysis buffer, separated on a 7.5% SDS–PAGE gel and transferred onto PVDF membrane. Protein from the human brain was probed with anti-filamin (Novacastra) antibodies, and protein from the mouse brain was probed with anti-pan-Arf (Affinity Bioreagents) or anti-Big2 antibodies (37), and detected by enhanced chemiluminescence.
Tissue dissociation and isolation
Methods for VZ dissection and dissociation follow general guidelines used previously in murine cortical cell dissociations (83,84) and in human neurosphere isolation (85). In brief, the brains were cut into coronal sections, and samples were dissected to obtain tissue along the periventricular zone within the frontal cortex; tissue was then minced and washed in cold Hanks-buffered saline solution (HBSS), and placed in trypsin solution at 37°C for 30 min. The sample was then passed through a cell strainer, to isolate single cells, and was washed in (DMEM) with 10% fetal calf serum, to inactivate the trypsin. The dissociated cells were spun down, the medium was aspirated and the cells were placed in neurosphere medium (Cambrex bullet kit) containing EGF, FGF, neural cell survival factor, gentamicin and amphotericin B, for expansion. The cultures were maintained in a 37°C/5% CO2 incubator, and medium was aspirated and renewed on a weekly basis. Individual neurospheres could be dissociated and re-expanded to ensure a clonal population.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work was supported by grants to R.J.F. from the NIMH (1K01MH71801) and a Young Investigator Award from Cure Autism Now; to V.L.S. from Julian and Carol Cohen, Alzheimer's Association, Harvard Center for Neurodegeneration, and 1R21HD054347; to E.M.R. from Fondecyt, Chile (Fondecyt 1070241) and to L.F.B. from DID-UACh (DID-2005-12) and CONICYT, Chile. V.L.S. is a Beckman Young Investigator and Doris Duke Clinical Scientist Development Award Recipient. C.A.W. is an Investigator of the Howard Hughes Medical Institute. A.H.B. and P.J.B. are supported by the Leukaemia Research Fund. We are very grateful to Genaro Alvial and Ricardo Silva (Univerisdad Austral de Chile) for their technical assistance.
ACKNOWLEDGEMENTS
We wish to thank Dr Sato for kindly providing the dominant-negative FLNA construct. The monoclonal antibody against BrdU was obtained from the Developmental Studies Hybridoma Bank at the University of Iowa, Department of Biological Sciences, Iowa City, IA 52242. The authors wish to thank Dr Adriana Verschoor (Wadsworth Center) for critical reading and input on our manuscript.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Barkovich A.J., Koch T.K., Carrol C.L. The spectrum of lissencephaly: report of ten patients analyzed by magnetic resonance imaging. Ann. Neurol. 1991;30:139–146. doi: 10.1002/ana.410300204. [DOI] [PubMed] [Google Scholar]
- 2.Dobyns W.B., Reiner O., Carrozzo R., Ledbetter D.H. Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA. 1993;270:2838–2842. doi: 10.1001/jama.270.23.2838. [DOI] [PubMed] [Google Scholar]
- 3.Dobyns W.B., Andermann E., Andermann F., Czapansky-Beilman D., Dubeau F., Dulac O., Guerrini R., Hirsch B., Ledbetter D.H., Lee N.S., et al. X-linked malformations of neuronal migration. Neurology. 1996;47:331–339. doi: 10.1212/wnl.47.2.331. [DOI] [PubMed] [Google Scholar]
- 4.Hashimoto R., Seki T., Takuma Y., Suzuki N. The ‘double cortex’ syndrome on MRI. Brain Dev. 1993;15:57–59. doi: 10.1016/0387-7604(93)90007-u. discussion: 83–84. [DOI] [PubMed] [Google Scholar]
- 5.Ricci S., Cusmai R., Fariello G., Fusco L., Vigevano F. Double cortex. A neuronal migration anomaly as a possible cause of Lennox–Gastaut syndrome. Arch. Neurol. 1992;49:61–64. doi: 10.1001/archneur.1992.00530250065017. [DOI] [PubMed] [Google Scholar]
- 6.Kamuro K., Tenokuchi Y. Familial periventricular nodular heterotopia. Brain Dev. 1993;15:237–241. doi: 10.1016/0387-7604(93)90073-h. [DOI] [PubMed] [Google Scholar]
- 7.Smith A.S., Weinstein M.A., Quencer R.M., Muroff L.R., Stonesifer K.J., Li F.C., Wener L., Soloman M.A., Cruse R.P., Rosenberg L.H., et al. Association of heterotopic gray matter with seizures: MR imaging. Work in progress. Radiology. 1988;168:195–198. doi: 10.1148/radiology.168.1.3132731. [DOI] [PubMed] [Google Scholar]
- 8.Lo Nigro C., Chong C.S., Smith A.C., Dobyns W.B., Carrozzo R., Ledbetter D.H. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller–Dieker syndrome. Hum. Mol. Genet. 1997;6:157–164. doi: 10.1093/hmg/6.2.157. [DOI] [PubMed] [Google Scholar]
- 9.Mizuguchi M., Takashima S., Kakita A., Yamada M., Ikeda K. Lissencephaly gene product. Localization in the central nervous system and loss of immunoreactivity in Miller–Dieker syndrome. Am. J. Pathol. 1995;147:1142–1151. [PMC free article] [PubMed] [Google Scholar]
- 10.Reiner O., Carrozzo R., Shen Y., Wehnert M., Faustinella F., Dobyns W.B., Caskey C.T., Ledbetter D.H. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- 11.Faulkner N.E., Dujardin D.L., Tai C.Y., Vaughan K.T., O'Connell C.B., Wang Y., Vallee R.B. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat. Cell Biol. 2000;2:784–791. doi: 10.1038/35041020. [DOI] [PubMed] [Google Scholar]
- 12.Niethammer M., Smith D.S., Ayala R., Peng J., Ko J., Lee M.S., Morabito M., Tsai L.H. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 2000;28:697–711. doi: 10.1016/s0896-6273(00)00147-1. [DOI] [PubMed] [Google Scholar]
- 13.Sasaki S., Shionoya A., Ishida M., Gambello M.J., Yingling J., Wynshaw-Boris A., Hirotsune S. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron. 2000;28:681–696. doi: 10.1016/s0896-6273(00)00146-x. [DOI] [PubMed] [Google Scholar]
- 14.Smith D.S., Niethammer M., Ayala R., Zhou Y., Gambello M.J., Wynshaw-Boris A., Tsai L.H. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat. Cell Biol. 2000;2:767–775. doi: 10.1038/35041000. [DOI] [PubMed] [Google Scholar]
- 15.Tromans A. Neurobiology. Convoluted communications. Nature. 2000;407:953. doi: 10.1038/35039654. [DOI] [PubMed] [Google Scholar]
- 16.des Portes V., Pinard J.M., Billuart P., Vinet M.C., Koulakoff A., Carrie A., Gelot A., Dupuis E., Motte J., Berwald-Netter Y., et al. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- 17.Gleeson J.G., Allen K.M., Fox J.W., Lamperti E.D., Berkovic S., Scheffer I., Cooper E.C., Dobyns W.B., Minnerath S.R., Ross M.E., et al. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- 18.Gleeson J.G., Lin P.T., Flanagan L.A., Walsh C.A. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- 19.Deuel T.A., Liu J.S., Corbo J.C., Yoo S.Y., Rorke-Adams L.B., Walsh C.A. Genetic interactions between doublecortin and doublecortin-like kinase in neuronal migration and axon outgrowth. Neuron. 2006;49:41–53. doi: 10.1016/j.neuron.2005.10.038. [DOI] [PubMed] [Google Scholar]
- 20.Koizumi H., Tanaka T., Gleeson J.G. Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron. 2006;49:55–66. doi: 10.1016/j.neuron.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 21.Bai J., Ramos R.L., Ackman J.B., Thomas A.M., Lee R.V., LoTurco J.J. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nat. Neurosci. 2003;6:1277–1283. doi: 10.1038/nn1153. [DOI] [PubMed] [Google Scholar]
- 22.Sicca F., Kelemen A., Genton P., Das S., Mei D., Moro F., Dobyns W.B., Guerrini R. Mosaic mutations of the LIS1 gene cause subcortical band heterotopia. Neurology. 2003;61:1042–1046. doi: 10.1212/wnl.61.8.1042. [DOI] [PubMed] [Google Scholar]
- 23.Couillard-Despres S., Winkler J., Uyanik G., Aigner L. Molecular mechanisms of neuronal migration disorders, quo vadis? Curr. Mol. Med. 2001;1:677–688. doi: 10.2174/1566524013363195. [DOI] [PubMed] [Google Scholar]
- 24.Guerrini R., Filippi T. Neuronal migration disorders, genetics, and epileptogenesis. J. Child Neurol. 2005;20:287–299. doi: 10.1177/08830738050200040401. [DOI] [PubMed] [Google Scholar]
- 25.Pilz D., Stoodley N., Golden J.A. Neuronal migration, cerebral cortical development, and cerebral cortical anomalies. J. Neuropathol. Exp. Neurol. 2002;61:1–11. doi: 10.1093/jnen/61.1.1. [DOI] [PubMed] [Google Scholar]
- 26.Walsh C.A., Goffinet A.M. Potential mechanisms of mutations that affect neuronal migration in man and mouse. Curr. Opin. Genet. Dev. 2000;10:270–274. doi: 10.1016/s0959-437x(00)00076-9. [DOI] [PubMed] [Google Scholar]
- 27.Fox J.W., Lamperti E.D., Eksioglu Y.Z., Hong S.E., Feng Y., Graham D.A., Scheffer I.E., Dobyns W.B., Hirsch B.A., Radtke R.A., et al. Mutations in filamin 1 prevent migration of cerebral cortical neurons in human periventricular heterotopia. Neuron. 1998;21:1315–1325. doi: 10.1016/s0896-6273(00)80651-0. [DOI] [PubMed] [Google Scholar]
- 28.Sheen V.L., Dixon P.H., Fox J.W., Hong S.E., Kinton L., Sisodiya S.M., Duncan J.S., Dubeau F., Scheffer I.E., Schachter S.C., et al. Mutations in the X-linked filamin 1 gene cause periventricular nodular heterotopia in males as well as in females. Hum. Mol. Genet. 2001;10:1775–1783. doi: 10.1093/hmg/10.17.1775. [DOI] [PubMed] [Google Scholar]
- 29.Sheen V.L., Feng Y., Graham D., Takafuta T., Shapiro S.S., Walsh C.A. Filamin A and Filamin B are co-expressed within neurons during periods of neuronal migration and can physically interact. Hum. Mol. Genet. 2002;11:2845–2854. doi: 10.1093/hmg/11.23.2845. [DOI] [PubMed] [Google Scholar]
- 30.Gorlin J.B., Yamin R., Egan S., Stewart M., Stossel T.P., Kwiatkowski D.J., Hartwig J.H. Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): a molecular leaf spring. J. Cell Biol. 1990;111:1089–1105. doi: 10.1083/jcb.111.3.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flanagan L.A., Chou J., Falet H., Neujahr R., Hartwig J.H., Stossel T.P. Filamin A, the Arp2/3 complex, and the morphology and function of cortical actin filaments in human melanoma cells. J. Cell Biol. 2001;155:511–517. doi: 10.1083/jcb.200105148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartwig J.H., Shevlin P. The architecture of actin filaments and the ultrastructural location of actin-binding protein in the periphery of lung macrophages. J. Cell Biol. 1986;103:1007–1020. doi: 10.1083/jcb.103.3.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hartwig J.H., Tyler J., Stossel T.P. Actin-binding protein promotes the bipolar and perpendicular branching of actin filaments. J. Cell Biol. 1980;87:841–848. doi: 10.1083/jcb.87.3.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weihing R.R. Actin-binding and dimerization domains of HeLa cell filamin. Biochemistry. 1988;27:1865–1869. doi: 10.1021/bi00406a011. [DOI] [PubMed] [Google Scholar]
- 35.Feng Y., Chen M.H., Moskowitz I.P., Mendonza A.M., Vidali L., Nakamura F., Kwiatkowski D.J., Walsh C.A. Filamin A (FLNA) is required for cell–cell contact in vascular development and cardiac morphogenesis. Proc. Natl. Acad. Sci. USA. 2006;103:19836–19841. doi: 10.1073/pnas.0609628104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hart A.W., Morgan J.E., Schneider J., West K., McKie L., Bhattacharya S., Jackson I.J., Cross S.H. Cardiac malformations and midline skeletal defects in mice lacking filamin A. Hum. Mol. Genet. 2006;15:2457–2467. doi: 10.1093/hmg/ddl168. [DOI] [PubMed] [Google Scholar]
- 37.Sheen V.L., Ganesh V.S., Topcu M., Sebire G., Bodell A., Hill R.S., Grant P.E., Shugart Y.Y., Imitola J., Khoury S.J., et al. Mutations in ARFGEF2 implicate vesicle trafficking in neural progenitor proliferation and migration in the human cerebral cortex. Nat. Genet. 2004;36:69–76. doi: 10.1038/ng1276. [DOI] [PubMed] [Google Scholar]
- 38.Sheen V.L., Topcu M., Berkovic S., Yalnizoglu D., Blatt I., Bodell A., Hill R.S., Ganesh V.S., Cherry T.J., Shugart Y.Y., et al. Autosomal recessive form of periventricular heterotopia. Neurology. 2003;60:1108–1112. doi: 10.1212/01.wnl.0000055898.00349.02. [DOI] [PubMed] [Google Scholar]
- 39.Guerrini R., Mei D., Sisodiya S., Sicca F., Harding B., Takahashi Y., Dorn T., Yoshida A., Campistol J., Kramer G., et al. Germline and mosaic mutations of FLN1 in men with periventricular heterotopia. Neurology. 2004;63:51–56. doi: 10.1212/01.wnl.0000132818.84827.4d. [DOI] [PubMed] [Google Scholar]
- 40.Santi M.R., Golden J.A. Periventricular heterotopia may result from radial glial fiber disruption. J. Neuropathol. Exp. Neurol. 2001;60:856–862. doi: 10.1093/jnen/60.9.856. [DOI] [PubMed] [Google Scholar]
- 41.Eksioglu Y.Z., Scheffer I.E., Cardenas P., Knoll J., DiMario F., Ramsby G., Berg M., Kamuro K., Berkovic S.F., Duyk G.M., et al. Periventricular heterotopia: an X-linked dominant epilepsy locus causing aberrant cerebral cortical development. Neuron. 1996;16:77–87. doi: 10.1016/s0896-6273(00)80025-2. [DOI] [PubMed] [Google Scholar]
- 42.Kakita A., Hayashi S., Moro F., Guerrini R., Ozawa T., Ono K., Kameyama S., Walsh C.A., Takahashi H. Bilateral periventricular nodular heterotopia due to filamin 1 gene mutation: widespread glomeruloid microvascular anomaly and dysplastic cytoarchitecture in the cerebral cortex. 2002;104:649–657. doi: 10.1007/s00401-002-0594-9. Acta Neuropathol. (Berl.) [DOI] [PubMed] [Google Scholar]
- 43.Banham A.H., Beasley N., Campo E., Fernandez P.L., Fidler C., Gatter K., Jones M., Mason D.Y., Prime J.E., Trougouboff P., et al. The FOXP1 winged helix transcription factor is a novel candidate tumor suppressor gene on chromosome 3p. Cancer Res. 2001;61:8820–8829. [PubMed] [Google Scholar]
- 44.Ferland R.J., Cherry T.J., Preware P.O., Morrisey E.E., Walsh C.A. Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J. Comp. Neurol. 2003;460:266–279. doi: 10.1002/cne.10654. [DOI] [PubMed] [Google Scholar]
- 45.Teramitsu I., Kudo L.C., London S.E., Geschwind D.H., White S.A. Parallel FoxP1 and FoxP2 expression in songbird and human brain predicts functional interaction. J. Neurosci. 2004;24:3152–3163. doi: 10.1523/JNEUROSCI.5589-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nieto M., Monuki E.S., Tang H., Imitola J., Haubst N., Khoury S.J., Cunningham J., Gotz M., Walsh C.A. Expression of Cux-1 and Cux-2 in the subventricular zone and upper layers II-IV of the cerebral cortex. J. Comp. Neurol. 2004;479:168–180. doi: 10.1002/cne.20322. [DOI] [PubMed] [Google Scholar]
- 47.Lu J., Tiao G., Folkerth R., Hecht J., Walsh C., Sheen V. Overlapping expression of ARFGEF2 and Filamin A in the neuroependymal lining of the lateral ventricles: insights into the cause of periventricular heterotopia. J. Comp. Neurol. 2006;494:476–484. doi: 10.1002/cne.20806. [DOI] [PubMed] [Google Scholar]
- 48.Nagano T., Morikubo S., Sato M. Filamin A and FILIP (Filamin A-Interacting Protein) regulate cell polarity and motility in neocortical subventricular and intermediate zones during radial migration. J. Neurosci. 2004;24:9648–9657. doi: 10.1523/JNEUROSCI.2363-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zeghouf M., Guibert B., Zeeh J.C., Cherfils J. Arf, Sec7 and Brefeldin A: a model towards the therapeutic inhibition of guanine nucleotide-exchange factors. Biochem. Soc. Trans. 2005;33:1265–1268. doi: 10.1042/BST0331265. [DOI] [PubMed] [Google Scholar]
- 50.Ikonen E., de Almeid J.B., Fath K.R., Burgess D.R., Ashman K., Simons K., Stow J.L. Myosin II is associated with Golgi membranes: identification of p200 as nonmuscle myosin II on Golgi-derived vesicles. J. Cell Sci. 1997;110(Pt):2155–2164. doi: 10.1242/jcs.110.18.2155. [DOI] [PubMed] [Google Scholar]
- 51.Liu G., Thomas L., Warren R.A., Enns C.A., Cunningham C.C., Hartwig J.H., Thomas G. Cytoskeletal protein ABP-280 directs the intracellular trafficking of furin and modulates proprotein processing in the endocytic pathway. J. Cell Biol. 1997;139:1719–1733. doi: 10.1083/jcb.139.7.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pasqualato S., Renault L., Cherfils J. Arf, Arl, Arp and Sar proteins: a family of GTP-binding proteins with a structural device for ‘front-back’ communication. EMBO Rep. 2002;3:1035–1041. doi: 10.1093/embo-reports/kvf221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jimenez A.J., Tome M., Paez P., Wagner C., Rodriguez S., Fernandez-Llebrez P., Rodriguez E.M., Perez-Figares J.M. A programmed ependymal denudation precedes congenital hydrocephalus in the hyh mutant mouse. J. Neuropathol. Exp. Neurol. 2001;60:1105–1119. doi: 10.1093/jnen/60.11.1105. [DOI] [PubMed] [Google Scholar]
- 54.Paez P., Batiz L.F., Roales-Bujan R., Rodriguez-Perez L.M., Rodriguez S., Jimenez A.J., Rodriguez E.M., Perez-Figares J.M. Patterned neuropathologic events occurring in hyh congenital hydrocephalic mutant mice. J. Neuropathol. Exp. Neurol. 2007;66:1082–1092. doi: 10.1097/nen.0b013e31815c1952. [DOI] [PubMed] [Google Scholar]
- 55.Wagner C., Batiz L.F., Rodriguez S., Jimenez A.J., Paez P., Tome M., Perez-Figares J.M., Rodriguez E.M. Cellular mechanisms involved in the stenosis and obliteration of the cerebral aqueduct of hyh mutant mice developing congenital hydrocephalus. J. Neuropathol. Exp. Neurol. 2003;62:1019–1040. doi: 10.1093/jnen/62.10.1019. [DOI] [PubMed] [Google Scholar]
- 56.Batiz L.F., Paez P., Jimenez A.J., Rodriguez S., Wagner C., Perez-Figares J.M., Rodriguez E.M. Heterogeneous expression of hydrocephalic phenotype in the hyh mice carrying a point mutation in alpha-SNAP. Neurobiol. Dis. 2006;23:152–168. doi: 10.1016/j.nbd.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 57.Chae T.H., Kim S., Marz K.E., Hanson P.I., Walsh C.A. The hyh mutation uncovers roles for alpha Snap in apical protein localization and control of neural cell fate. Nat. Genet. 2004;36:264–270. doi: 10.1038/ng1302. [DOI] [PubMed] [Google Scholar]
- 58.Dominguez-Pinos M.D., Paez P., Jimenez A.J., Weil B., Arraez M.A., Perez-Figares J.M., Rodriguez E.M. Ependymal denudation and alterations of the subventricular zone occur in human fetuses with a moderate communicating hydrocephalus. J. Neuropathol. Exp. Neurol. 2005;64:595–604. doi: 10.1097/01.jnen.0000171648.86718.bb. [DOI] [PubMed] [Google Scholar]
- 59.de Wit O.A., den Dunnen W.F., Sollie K.M., Munoz R.I., Meiners L.C., Brouwer O.F., Rodriguez E.M., Sival D.A. Pathogenesis of cerebral malformations in human fetuses with meningomyelocele. Cerebrospinal Fluid Res. 2008;5:4. doi: 10.1186/1743-8454-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Poussaint T.Y., Fox J.W., Dobyns W.B., Radtke R., Scheffer I.E., Berkovic S.F., Barnes P.D., Huttenlocher P.R., Walsh C.A. Periventricular nodular heterotopia in patients with filamin-1 gene mutations: neuroimaging findings. Pediatr. Radiol. 2000;30:748–755. doi: 10.1007/s002470000312. [DOI] [PubMed] [Google Scholar]
- 61.Moro F., Carrozzo R., Veggiotti P., Tortorella G., Toniolo D., Volzone A., Guerrini R. Familial periventricular heterotopia: missense and distal truncating mutations of the FLN1 gene. Neurology. 2002;58:916–921. doi: 10.1212/wnl.58.6.916. [DOI] [PubMed] [Google Scholar]
- 62.Gomez-Garre P., Seijo M., Gutierrez-Delicado E., Castro Del Rio M., de la Torre C., Gomez-Abad C., Morales-Corraliza J., Puig M., Serratosa J.M. Ehlers-Danlos syndrome and periventricular nodular heterotopia in a Spanish family with a single FLNA mutation. J. Med. Genet. 2005;43:232–237. doi: 10.1136/jmg.2004.029173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sheen V.L., Jansen A., Chen M.H., Parrini E., Morgan T., Ravenscroft R., Ganesh V., Underwood T., Wiley J., Leventer R., et al. Filamin A mutations cause periventricular heterotopia with Ehlers–Danlos syndrome. Neurology. 2005;64:254–262. doi: 10.1212/01.WNL.0000149512.79621.DF. [DOI] [PubMed] [Google Scholar]
- 64.Gerard-Blanluet M., Sheen V., Machinis K., Neal J., Apse K., Danan C., Sinico M., Brugieres P., Mage K., Ratsimbazafy L., et al. Bilateral periventricular heterotopias in an X-linked dominant transmission in a family with two affected males. Am. J. Med. Genet. A. 2006;140:1041–1046. doi: 10.1002/ajmg.a.31197. [DOI] [PubMed] [Google Scholar]
- 65.Shen Q., Goderie S.K., Jin L., Karanth N., Sun Y., Abramova N., Vincent P., Pumiglia K., Temple S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–1340. doi: 10.1126/science.1095505. [DOI] [PubMed] [Google Scholar]
- 66.Muntoni F., Brockington M., Torelli S., Brown S.C. Defective glycosylation in congenital muscular dystrophies. Curr. Opin. Neurol. 2004;17:205–209. doi: 10.1097/00019052-200404000-00020. [DOI] [PubMed] [Google Scholar]
- 67.van Reeuwijk J., Brunner H.G., van Bokhoven H. Glyc-O-genetics of Walker–Warburg syndrome. Clin. Genet. 2005;67:281–289. doi: 10.1111/j.1399-0004.2004.00368.x. [DOI] [PubMed] [Google Scholar]
- 68.Yamamoto T., Kato Y., Kawaguchi M., Shibata N., Kobayashi M. Expression and localization of fukutin, POMGnT1 and POMT1 in the central nervous system: consideration for functions of fukutin. Med. Electron. Microsc. 2004;37:200–207. doi: 10.1007/s00795-004-0260-5. [DOI] [PubMed] [Google Scholar]
- 69.Kobayashi K., Nakahori Y., Miyake M., Matsumura K., Kondo-Iida E., Nomura Y., Segawa M., Yoshioka M., Saito K., Osawa M., et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388–392. doi: 10.1038/28653. [DOI] [PubMed] [Google Scholar]
- 70.Yoshida A., Kobayashi K., Manya H., Taniguchi K., Kano H., Mizuno M., Inazu T., Mitsuhashi H., Takahashi S., Takeuchi M., et al. Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev. Cell. 2001;1:717–724. doi: 10.1016/s1534-5807(01)00070-3. [DOI] [PubMed] [Google Scholar]
- 71.Beltran-Valero de Bernabe D., Currier S., Steinbrecher A., Celli J., van Beusekom E., van der Zwaag B., Kayserili H., Merlini L., Chitayat D., Dobyns W.B., et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker–Warburg syndrome. Am. J. Hum. Genet. 2002;71:1033–1043. doi: 10.1086/342975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Michele D.E., Barresi R., Kanagawa M., Saito F., Cohn R.D., Satz J.S., Dollar J., Nishino I., Kelley R.I., Somer H., et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- 73.Cai L., Hayes N.L., Nowakowski R.S. Synchrony of clonal cell proliferation and contiguity of clonally related cells: production of mosaicism in the ventricular zone of developing mouse neocortex. J. Neurosci. 1997;17:2088–2100. doi: 10.1523/JNEUROSCI.17-06-02088.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hayes N.L., Nowakowski R.S. Exploiting the dynamics of S-phase tracers in developing brain: interkinetic nuclear migration for cells entering versus leaving the S-phase. Dev. Neurosci. 2000;22:44–55. doi: 10.1159/000017426. [DOI] [PubMed] [Google Scholar]
- 75.Takahashi T., Nowakowski R.S., Caviness V.S., Jr Interkinetic and migratory behavior of a cohort of neocortical neurons arising in the early embryonic murine cerebral wall. J. Neurosci. 1996;16:5762–5776. doi: 10.1523/JNEUROSCI.16-18-05762.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carreno S., Engqvist-Goldstein A.E., Zhang C.X., McDonald K.L., Drubin D.G. Actin dynamics coupled to clathrin-coated vesicle formation at the trans-Golgi network. J. Cell Biol. 2004;165:781–788. doi: 10.1083/jcb.200403120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lanzetti L. Actin in membrane trafficking. Curr. Opin. Cell Biol. 2007;19:453–458. doi: 10.1016/j.ceb.2007.04.017. [DOI] [PubMed] [Google Scholar]
- 78.Smythe E., Ayscough K.R. Actin regulation in endocytosis. J. Cell Sci. 2006;119:4589–4598. doi: 10.1242/jcs.03247. [DOI] [PubMed] [Google Scholar]
- 79.Clary D.O., Griff I.C., Rothman J.E. SNAPs, a family of NSF attachment proteins involved in intracellular membrane fusion in animals and yeast. Cell. 1990;61:709–721. doi: 10.1016/0092-8674(90)90482-t. [DOI] [PubMed] [Google Scholar]
- 80.Chang B.S., Ly J., Appignani B., Bodell A., Apse K.A., Ravenscroft R.S., Sheen V.L., Doherty M.J., Hackney D.B., O'Connor M., et al. Reading impairment in the neuronal migration disorder of periventricular nodular heterotopia. Neurology. 2005;64:799–803. doi: 10.1212/01.WNL.0000152874.57180.AF. [DOI] [PubMed] [Google Scholar]
- 81.Lu J., Sheen V. Periventricular heterotopia. Epilepsy Behav. 2005;7:143–149. doi: 10.1016/j.yebeh.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 82.Sheen V.L., Arnold M.W., Wang Y., Macklis J.D. Neural precursor differentiation following transplantation into neocortex is dependent on intrinsic developmental state and receptor competence. Exp. Neurol. 1999;158:47–62. doi: 10.1006/exnr.1999.7104. [DOI] [PubMed] [Google Scholar]
- 83.Bahn S., Mimmack M., Ryan M., Caldwell M.A., Jauniaux E., Starkey M., Svendsen C.N., Emson P. Neuronal target genes of the neuron-restrictive silencer factor in neurospheres derived from fetuses with Down's syndrome: a gene expression study. Lancet. 2002;359:310–315. doi: 10.1016/S0140-6736(02)07497-4. [DOI] [PubMed] [Google Scholar]
- 84.Sheen V.L., Macklis J.D. Targeted neocortical cell death in adult mice guides migration and differentiation of transplanted embryonic neurons. J. Neurosci. 1995;15:8378–8392. doi: 10.1523/JNEUROSCI.15-12-08378.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sheen V.L., Ferland R.J., Harney M., Hill R.S., Neal J., Banham A.H., Brown P., Chenn A., Corbo J., Hecht J., et al. Impaired proliferation and migration in human Miller-Dieker neural precursors. Ann. Neurol. 2006;60:137–144. doi: 10.1002/ana.20843. [DOI] [PubMed] [Google Scholar]
- 86.Sarkisian M.R., Bartley C.M., Chi H., Nakamura F., Hashimoto-Torii K., Torii M., Flavell R.A., Rakic P. MEKK4 signaling regulates filamin expression and neuronal migration. Neuron. 2006;52:789–801. doi: 10.1016/j.neuron.2006.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.