

Abstract
The cell surface organelle called the cilium is essential for preventing kidney cyst formation and for establishing left–right asymmetry of the vertebrate body plan. Recent advances suggest that the cilium functions as a sensory organelle in vertebrate cells for multiple signaling pathways such as the hedgehog and the Wnt pathways. Prompted by kidney cyst formation in tuberous sclerosis complex (TSC) patients and rodent models, we investigated the role of the cilium in the TSC–target of rapamycin (TOR) pathway using zebrafish. TSC1 and TSC2 genes are causal for TSC, and their protein products form a complex in the TOR pathway that integrates environmental signals to regulate cell growth, proliferation and survival. Two TSC1 homologs were identified in zebrafish, which we refer to as tsc1a and tsc1b. Morpholino knockdown of tsc1a led to a ciliary phenotype including kidney cyst formation and left–right asymmetry defects. Tsc1a was observed to localize to the Golgi, but morpholinos against it, nonetheless, acted synthetically with ciliary genes in producing kidney cysts. Consistent with a role of the cilium in the same pathway as Tsc genes, the TOR pathway is aberrantly activated in ciliary mutants, resembling the effect of tsc1a knockdown. Moreover, kidney cyst formation in ciliary mutants was blocked by the Tor inhibitor, rapamycin. Surprisingly, we observed elongation of cilia in tsc1a knockdown animals. Together, these data suggest a signaling network between the cilium and the TOR pathway in that ciliary signals can feed into the TOR pathway and that Tsc1a regulates the length of the cilium itself.
INTRODUCTION
Protruding from the cell surface into the environment, the cilium is ideally situated to function as an antenna for the cell. In vertebrate cells, the cilium is almost ubiquitously present and multiple signaling receptors, including PDGFRα and G protein-coupled receptors, have been localized to the cilium (1–3), suggesting a unique role of the cilium in mediating extra-cellular signals to downstream pathways. In agreement with a sensory role, recently, the cilium has been linked to a growing list of signaling pathways such as the hedgehog and the Wnt pathways (4–11).
Intraflagellar transport (IFT) is a process involved in ciliary assembly and maintenance, which delivers and recycles ciliary components into and out of the cilium, respectively (12). IFT was originally observed in the flagella of the green algae, Chlamydomonas (13), and its significance with regard to ciliary formation and function was realized when a Chlamydomonas homolog of the IFT particle polypeptide ift88/polaris was linked to autosomal-dominant polycystic kidney disease (ADPKD) in mice (14,15). These studies demonstrated that defects in cilia formation and function on renal epithelial cells lead to cyst formation. Consistent with an important signaling role of the cilium in vertebrate systems, defects in ciliary formation or function can also lead to early developmental deficiencies, most notably abnormal left–right asymmetry of the body plan (16–18). In addition, defects in IFT in mice have also been implicated in aberrant Hedgehog signaling, resulting in abnormalities in neural tube patterning and limb development (7,8).
Kidney cysts, which are epithelium-lined and liquid-filled sacs, are thought to result from the over-proliferation of renal epithelial cells and are hallmarks of PKD (19). Importantly, the cilium seems to be at the center of PKD. Multiple PKD genes, such as PKD1, PKD2, INVERSIN and Bardet–Biedl syndrome genes, have been linked to the cilium directly or indirectly (reviewed in 20). Conversely, mutations disrupting cilia formation and/or function in the kidney almost inevitably lead to kidney cyst formation. These results led to the cilium model of PKD, which postulates that cilia on renal epithelial cells transduce an anti-proliferative signal into cells (12,21). Ciliary defects can therefore lead to cell over-proliferation and eventually kidney cyst formation. However, the downstream molecular pathways responsible for such cilium-mediated cell proliferation is still not fully understood.
Intriguingly, in addition to PKD, kidney cyst formation is also frequently seen in another disease, tuberous sclerosis complex (TSC), an autosomal-dominant genetic disorder characterized by benign tumors that develop in numerous organs (reviewed in 22). Mutations in a single allele of either the TSC1 or TSC2 gene lead to TSC. However, it is believed that the development of the disease requires somatic inactivation of the second wild-type TSC allele (23). The role of TSC genes in kidney cyst formation is also supported by the Eker rat model, which carries a germline mutation in one allele of Tsc2 and develops early onset PKD (24,25). Furthermore, in humans, large deletions removing both the TSC2 gene and the adjacent PKD1 gene have been found in PKD patients with more severe and early onset symptoms (26).
The Tsc1 (also known as Hamartin) and Tsc2 (also known as Tuberin) proteins play a well-defined role in the target of rapamycin (TOR) pathway: they form a heterodimer that can negatively regulate the TOR kinase and eventually regulate cell growth, proliferation and survival (reviewed in 27).
Given the critical role of the cilium in PKD, the observation of kidney cyst formation in TSC raises an interesting question: whether the cilium plays a role in the TSC–TOR pathway. To date, the role of the Tsc genes during development remains not fully analyzed. Both Tsc1 and Tsc2 are essential genes as mouse knockouts die at embryonic day 10.5 with an open neural tube phenotype (28,29). Such early lethality precludes the analysis of kidney cyst formation in these mouse knockout models, and the status of laterality in TSC mutants is also unclear. However, emerging evidence supports a functional link between the cilium and the TSC/TOR pathway. For example, studies in mouse and tissue culture suggest that PKD1, a causal gene for ADPKD, interacts with the TSC1–mammalian TOR (mTOR) pathway (30,31). Intriguingly, rapamycin, an inhibitor of mTOR, can delay cyst progression in rodent PKD models (30).
Based on the above evidence, we postulated that the cilium may regulate the TSC–TOR pathway. We tested our hypothesis by examining tsc1 loss-of-function phenotypes and the status of the TOR pathway in available ciliary mutants in the zebrafish model system. Multiple ciliary mutants have been isolated in previous genetic screens in zebrafish (17,32). Notably, reduction of multiple IFT genes, which encode components of IFT particles (12,13), leads to kidney cyst formation, laterality defects and ventrally curved body axes (17,18). Here, we show that (i) knockdown of tsc1a in zebrafish leads to phenotypes characteristic of ciliary mutants; (ii) tsc1a genetically interacts with IFT genes; (iii) the TOR pathway is up-regulated in ift morphants (knockdown animals generated by injection of anti-sense morpholino oligo), similar to that observed in tsc1a morphants, and the Tor inhibitor rapamycin can inhibit cyst formation in ift mutants and (iv) cilia are abnormally long in tsc1a morphants. Together, these data suggest the existence of an interactive network between the cilium and the TOR pathway, as ciliary signals can regulate the TOR pathway whereas Tsc1a is involved in cilia length control.
RESULTS
There are two TSC1 homologs in the zebrafish genome
To analyze tsc1 function in zebrafish, we BLAST searched the zebrafish-annotated genome database (VEGA) and the NCBI EST and non-redundant databases for Tsc1 homologs. We identified two Tsc1-like genes, located on chromosomes 5 and 21, which we hereafter refer to as tsc1a and tsc1b. The genes encode predicted proteins of 126 and 137 kDa that are 44% identical with each other. They share 36 and 44% sequence identity, respectively, with the human hamartin (TSC1) protein. BLAST searches of mammalian databases with either protein yielded TSC1 as the top hit, supporting the idea that both are orthologs of TSC1. As there are two orthologs in fish, we will refer to the fish proteins as Tsc1a and Tsc1b, instead of hamartin. This finding raises a possibility that the cellular functions associated with mammalian Tsc1 are partitioned between two Tsc1 homologs in zebrafish.
To assess the conservation of the TOR pathway in zebrafish, we performed blast searches for other components of the TOR pathway using the Zv7 assembly of the zebrafish genome. In addition to the previously known homologs of S6K, rictor, raptor and TOR (33), single homologs for Tsc2 and Rheb were identified (data not shown).
Knockdown of tsc1a leads to kidney cyst formation and body curvature in zebrafish
To study the function of tsc1a and tsc1b in zebrafish development, we designed morpholino oligonucleotides against the translational initiation site of either Tsc1a or Tsc1b (AUG morpholino). About 0.25–0.50 pmol of oligo was injected into 1–4-cell stage embryos. Knockdown of Tsc1a produced phenotypes similar to those seen in multiple known cystic kidney mutants, including three ift mutants (17) (Fig. 1A and B). More specifically, the tsc1a morphant embryos exhibited a ventral-directed body curvature (69%) and bilateral cysts (51%, n = 132) located within the tubular/glomerular region of the pronephros. The tsc1b morphant embryos, in contrast, developed curved bodies, but lacked kidney cysts (see Supplementary Material, Fig. S1). We therefore focused our analysis on the tsc1a morphants. To verify the specificity of the tsc1a morpholino, we first tested a control morpholino with five mismatched bases. This control oligo was much less potent in producing the body curvature and kidney cyst phenotype (2% cysts and 9% body curvature). To further test the specificity of the observed phenotypes, we injected a morpholino blocking a splice site in tsc1a and found that it generated a similar cystic kidney and body curvature phenotype (data not shown). RT–PCR confirmed that the Tsc1a transcript was disrupted, whereas tsc1b transcript levels were unaffected (Fig. 1C), verifying that the phenotype observed is a specific consequence of tsc1a knockdown. We further analyzed the cystic kidney phenotype of the tsc1a morphant in detail. In histological cross-sections through the glomerular-tubular region, large cysts are apparent in the tsc1a morphant embryo (Fig. 1D, E and H). In more posterior regions, dilation of the pronephric duct lumens is evident, compared with wild-type embryos (Fig. 1F–H). In addition, electron micrographs of cross-sections taken through the mid-pronephric duct also show luminal expansion in tsc1a morphants (Fig. 1I and J). This defect was accompanied by what appeared to be a decrease in the density of microvilli. To verify this, we counted the number of intact microvilli in EM cross-sections of both wild-type and tsc1a morphant kidney ducts and found an average of 45 and 47, respectively (n = 5 each), with no significant difference (P = 0.73). These results indicate that the number of microvilli does not change significantly, despite the increase in the volume of the duct lumen. The phenotypic similarities between tsc1a morphants and ift mutant embryos, together with the previously appreciated link between IFT and the cilium and Tsc1's regulatory role in growth and proliferation, suggest that there may be a functional connection between the cilium and Tsc1.
Figure 1.
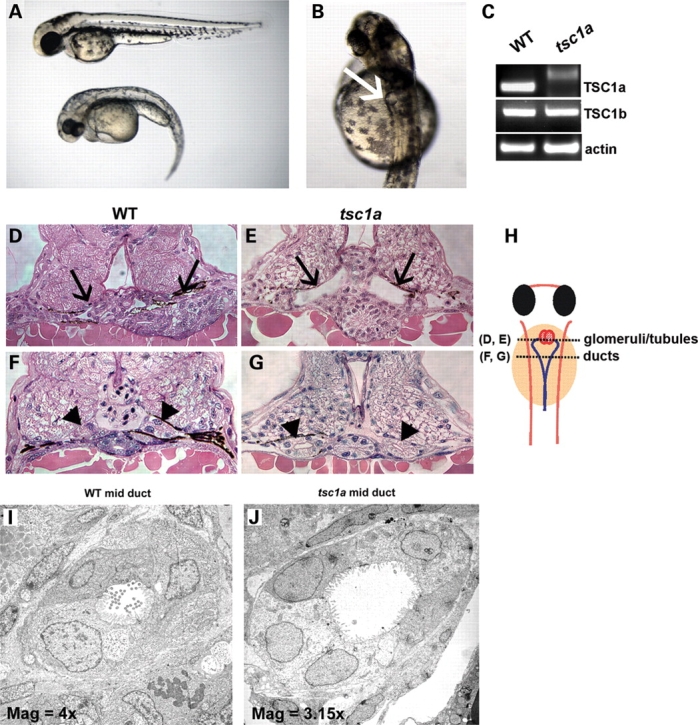
Phenotype of Tsc1a knockdown in zebrafish embryos. (A) One-cell stage wild-type embryos injected with AUG morpolinos to Tsc1a develop a ventral-directed body curvature (bottom embryo) compared with uninjected controls (top embryo). (B) A close up of the morphant in (A) showing a cyst pointed by an arrow. (C) RT–PCR from lysates of wild-type and tsc1a morphants injected with the splicing site morpholino, showing that splicing is blocked in the tsc1a transcript whereas tsc1b mRNA is unaffected. Primers against actin are used as a loading control. (D–J). Sections of tsc1a morphants and wild-type control embryos. Histological cross-sections of day 3 wild-type (D) and tsc1a morphant (E) embryos reveal cysts in the tubular and glomerular regions (black arrow) in tsc1a knock down fish. In more posterior regions, expansion of the pronephric duct lumens (arrow head) is evident in tsc1a embryos (G) compared with uninjected controls (F). (H) Drawing of a zebrafish embryo from dorsal view. Dotted lines represent regions [glomeruli/tubules (D) and (E) and ducts (F) and (G)] from where histological cross-sections are taken. Transmission electron micrographs of mid-duct regions from wild-type (I) and tsc1a (J) embryos also illustrate that knock down of Tsc1a results in duct lumen dilation as well as a decrease in the density of microvilli and pronephric cilia. WT, wild-type; Mag, magnification ×1000.
Tsc1a is required for the establishment of left–right asymmetry of the body plan
Another hallmark of ciliary defects is the abnormal left–right asymmetric body plan, which has been observed in both human patients as well as in mouse and zebrafish models (18,34–39). Accordingly, we next investigated whether reduced levels of Tsc1a would produce laterality defects. To test for this, we performed in situ hybridization for cmlc2 (cardiac myosin light chain 2), which is expressed in atrial and ventricular cardiomyocytes in the developing heart (36). In wild-type control embryos at 26 hours post fertilization (hpf), cmlc2 expression was found on the left side of the embryo 100% of the time (Fig. 2A and Table 1), but only 64% of the tsc1a morphant embryos showed left-sided expression (Fig. 2A and Table 1), with the remaining 36% shifted more centrally, suggesting that heart jogging was disrupted in some of these embryos. To further narrow down the stage at which tsc1a affects laterality, we examined the expression domain of southpaw by in situ hybridization. southpaw encodes a nodal-related protein in zebrafish and is the earliest known marker for asymmetry in zebrafish. In control embryos, southpaw is correctly expressed on the left side of embryos 87% of the time (Fig. 2B), and either right or bilateral 2% of the time each, or absent 9%. In contrast, in tsc1a morphants, southpaw expression was observed on the left 29% of the time, right 20%, bilateral 27% and absent 24% of the time (Fig. 2B, tsc1a and Table 1), suggesting that tsc1a functions upstream from southpaw, in a process closely associated with ciliary functions.
Figure 2.

Tsc1a is required for left–right body patterning. (A) Dorsal view of ∼26 hpf embryos, showing that cardiac myosin light chain 2 (cmlc2) expression is shifted more centrally in ∼36% of the tsc1a embryos compared with 100% left-sided expression in WT fish (summarized in Table 1). (B) southpaw is primarily expressed in the left lateral plate mesoderm in wild-type embryos but is randomly distributed in tsc1a knockdown embryos. Dorsal view showing WT or tsc1a embryos at the 20 somite stage. spaw, southpaw.
Table 1.
Expression pattern of cmlc2 and southpaw
| Probe | Sample | n | Left | Right | Center | Absent |
|---|---|---|---|---|---|---|
| Cmlc2 | Uninj. | 37 | 100 | 0 | 0 | 0 |
| tsc1a | 36 | 64 | 0 | 36 | 0 | |
| Spaw | Uninj. | 90 | 87 | 2 | 2 | 9 |
| tsc1a | 155 | 29 | 20 | 27 | 24 |
In southpaw samples, ‘center’ refers to bilateral signal.
tsc1a genetically interacts with IFT genes
The phenotypic similarities between tsc1a morphants and known ciliary mutants in zebrafish suggest that these genes may function in a common cellular process. Partial knockdown of a key component in a cellular pathway often generates a sensitized genetic background, and in such a background, disruption of other components in the same pathway will lead to a synergistic effect. We thus specifically tested whether suboptimal doses of the tsc1a morpholino and an IFT morpholino could synergize with each other. Low doses of morpholinos to tsc1a, ift81 or ift172 were injected into wild-type embryos at concentrations that would result in low frequency, low severity phenotypes. Embryos were then co-injected with similar doses of tsc1a morpholinos in combination with either of the two ift antisense oligonucleotides. At 2 days post fertilization (dpf), embryos were scored for the presence of pronephric cysts. Embryos injected with single, low-dose morpholinos (0.25 pmol) exhibited a mild cystic kidney phenotype, i.e. ∼17% of the tsc1a embryos had cysts [Fig. 3A and C (n = 3, with 151, 58 and 67 embryos), and B and C (n = 3, each with at least 49 embryos), tsc1a]. Similarly, the frequency of cysts in the ift samples was ∼9% [Fig. 3A and C (n = 3, with at least 70 embryos per sample) and B and C (n = 3, with at least 57 embryos per sample), ift]. When the same doses of morpholinos were combined (i.e. tsc1a with ift81 or ift172), a dramatic increase in the frequency of cysts [58 and 57%, respectively, Fig. 3C (n = 3, with at least 72 embryos per sample)] was observed in both groups, suggesting a synergistic rather than an additive effect (Fig. 3A and B, tsc1a/ift; Fig. 3C, both). In addition to the increased frequency of pronephric cysts, the co-injected embryos exhibited a more pronounced body curvature phenotype (Fig. 3A and B, tsc1a/ift). To further rule out the possibility that the synergistic effect was caused by an increased amount of morpholino oligo used in double-injected samples, we co-injected the tsc1a morpholino together with a standard control morpholino. Results showed that the inclusion of the control morpholino did not affect the phenotype significantly. Specifically, 11.4% of the embryos showed kidney cysts when injected with 0.17 pmol of tsc1a morpholino alone, whereas 10.5% showed kidney cyst when injected with tsc1a morpholino in combination with the control morpholino (n = 3, each with at least 24 embryos, P = 0.72). Together, these findings provide genetic evidence that tsc1a may function in a similar pathway as ift genes.
Figure 3.

A synergistic interaction exists between tsc1a and ift genes. Sub-phenotypic doses of AUG morpholinos to tsc1a and either ift81 (A) or ift172 (B) were co-injected into 1-cell, wild-type embryos or injected individually. The coinjected embryos display a dramatic increase in the frequency of kidney cysts compared with those injected with a single morpholino (see bar graph, C) (*P < 0.01, **P < 0.02). mph, morphants.
Tsc1a is localized to the Golgi complex
The close association between the cilium and Tsc1a suggests that Tsc1a may function from the cilium. To determine the subcellular localization pattern of Tsc1a in zebrafish embryos, we developed polyclonal antibodies against Tsc1a. Two regions of Tsc1a corresponding to amino acids 580–736 and 980–1108 were fused to maltose-binding protein (MBP) in two different constructs. Recombinant proteins were generated in bacteria and used to generate polyclonal antisera (Tsc1-1 and Tsc1-2). The antisera were subsequently purified via affinity purification. On immunoblots, both antibodies recognize a ∼127 kDa band in wild-type samples. This signal is greatly diminished in lysates prepared from tsc1a morphant embryos (a western blot using the Tsc1-1 antibody is shown in Fig. 4A). Immuno-staining experiments using the Tsc1-2 antibody revealed that Tsc1a is ubiquitously expressed throughout the embryo (Fig. 4B). The staining signal is greatly reduced in tsc1a morphants (Fig. 4C), verifying the specificity of the antibody.
Figure 4.
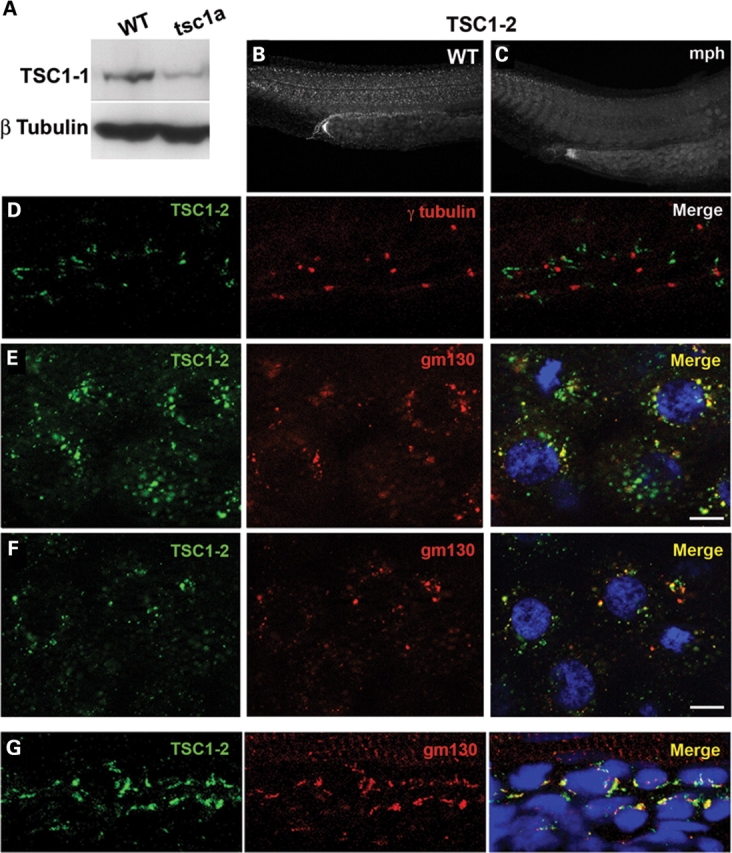
Localization of Tsc1a. (A) Affinity-purified Tsc1-1 antibodies recognize an ∼126 kDa band that is diminished in tsc1a embryo lysates. β-tubulin is used as a loading control. (B) Tsc1a immunolocalization in 1 dpf, whole-mount embryos is ubiquitous. (C) Knockdown of Tsc1a using an AUG morpholino against tsc1a reduces the Tsc1-2 signal. (D) Segment from the pronephric duct of a wild-type, 1 dpf embryo. Immunofluorescence using Tsc1-2 (green) and anti-γ-tubulin (red). The Tsc1-2 signal does not overlap with the centrosomes/basal bodies from pronephric duct epithelial cells. (E) The Tsc1-2 (green) fluorescent signal overlaps with the cis-Golgi marker gm130 (red) in blastula-stage embryos. (F) Both the Tsc1a (green) and gm130 (red) signals disperse following BFA treatment. (D–F) Nuclei stained with DAPI (blue). (G) A segment from the pronephric duct of 2 dpf embryos. Tsc1a (Tsc1-2, green) significantly colocalizes with the cis-Golgi (gm130, red). Nuclei stained with TOTO-3. WT, wild-type; tsc1a, tsc1a morphant.
It has been previously reported that Tsc1 localizes to the centrosome in cell culture (40). To determine whether Tsc1a associates with the centrosome or the cilium, we performed immunofluorescence experiments on day 1 wild-type embryos using the Tsc1-2 antibody and antibodies to γ-tubulin, which labels the centrosome, or acetylated tubulin, which labels the cilium. We found no obvious overlap between γ-tubulin and Tsc1a in the pronephric duct (Fig. 4D). In addition, Tsc1a localization to the cilium is below detection (data not shown).
The subcellular distribution pattern of Tsc1a resembles the Golgi complex. We therefore tested whether it is indeed localized to the Golgi. We used embryos from two different time points: the blastula stage and 48 hpf. We labeled the cis-Golgi using an antibody against one of its structural components, gm130, and found that in embryos at the blastula stage, it largely overlaps with Tsc1a (Fig. 4E). In the pronephric duct at 48 hpf, we found a similar pattern (Fig. 4G). This pattern of overlap is common throughout the embryo (data not shown), suggesting that the Golgi complex may be a primary site for Tsc1a function. To further verify that Tsc1a localizes to the Golgi, wild-type blastula stage embryos were incubated with brefeldin-A (BFA) in 1% dimethyl sulfoxide (DMSO) or 1% DMSO alone for 60 min. BFA blocks vesicle transport from the ER to the Golgi and eventually leads to Golgi dispersal (41). In DMSO-treated embryos, Tsc1a partially overlapped with gm130 in a perinuclear pattern similar to that seen in untreated embryos (Fig. 4E). A 60 min 15 µg/ml BFA treatment, however, caused both the gm130 and Tsc1a signals to disperse throughout the cytoplasm of the cells, often in an overlapping pattern (Fig. 4F), providing further support that the Tsc1a protein is in part associated with the cis-Golgi. Treatment with BFA, however, did not phenocopy the tsc1a morphants because embryos treated at the shield stage with 50 µg/ml BFA all died by day 2, indicating that lack of an intact Golgi has more severe consequences than the absence of Tsc1a alone.
The TOR pathway is up-regulated in zebrafish tsc1a morphants and ift morphants
Tsc1 plays a critical role in growth and proliferation control through suppressing the TOR pathway. As ift mutants show very similar phenotypes as tsc1a morphants, we asked whether the TOR pathway is aberrantly activated in ift mutants. Multiple markers can be used to interrogate the status of the mTOR pathway. For example, elevated mTOR activity leads to phosphorylation and activation of p70S6K, which subsequently phosphorylates S6 and leads to an increase in ribosome biogenesis (42–44), whereas levels of IRS1 drop due to a negative feedback loop induced by elevated mTOR activity (45,46). We compared the protein levels and phosphorylation levels of key TOR components in wild-type, tsc1a and ift172 morphant embryos. As expected, the TOR pathway is activated in tsc1a morphants (Fig. 5A). Excitingly, ift morphants show a similar change. Western blots performed on ∼28 hpf embryo lysates revealed that, in tsc1a and ift172 morpholino-injected samples, phosphorylation of the TOR effector p70S6K (P∼p70S6K), was increased, whereas the level of total p70S6Kwas unchanged (Fig. 5A). Providing further support for an up-regulation of the TOR pathway, levels of IRS1 were reduced in these same samples when compared with uninjected controls (Fig. 5A). Because the reduction in IRS1 levels was moderate, we quantified the changes using AutoQuant software, and used the β-tubulin bands as controls. The results indicated that IRS1 levels dropped 34 and 44% compared with wild-type in tsc1a and ift172 morphants, respectively. Together, these data demonstrate that ciliary defects can lead to aberrant activation of the TOR pathway.
Figure 5.

(A) The TOR pathway is similarly up-regulated in tsc1a and ift172 morphants. Western blots were performed on day 1 deyolked, whole embryo lysates. Levels of activated p70S6K (P∼p70S6K) are elevated and IRS1 levels are reduced in tsc1a and ift172 morpholino-injected embryos compared with uninjected controls (WT). Total p70S6K and β-tubulin are used as loading controls. (B) Rapamycin function is conserved in zebrafish. Western blots against P∼p70S6K and p70S6K as control, on lysates of wild-type embryos treated with 500 nM rapamycin in 1% DMSO or 1% DMSO alone from high to tail bud stages. The P∼p70S6K signal is absent in the rapamycin-treated sample, suggesting that rapamycin suppressed TOR activity. (C) Rapamycin restricts the pronephric cyst size in ift mutant embryos. ift81 or ift57 embryos were incubated with 200 or 300 nM rapamycin, respectively, or 1% DMSO as a control, beginning ∼30 hpf. The DMSO-treated embryos developed large pronephric cysts by day 5; however, cysts were often not visible or were greatly reduced in size in rapamycin-treated embryos.
Rapamycin treatment can partially rescue the phenotypes of ift57 or ift81 mutants
Ectopic activation of the TOR pathway in ift mutants/morphants suggests that one way to reverse their phenotypes is to inhibit TOR activity. Rapamycin can effectively inhibit TOR in the TORC1 complex in mammals. Therefore, we directly tested rapamycin on ift mutants. The conservation of rapamycin activity in zebrafish has been previously demonstrated (33). Consistently, by performing a short incubation of wild-type, high stage embryos, through tail bud stage with 500 nM rapamycin, we were able to see a dramatic drop in P∼p70S6K to virtually undetectable levels compared with DMSO-treated embryos (Fig. 5B). For our cyst rescue experiments, we titrated concentrations and incubation periods with rapamycin and found that concentrations above 300 nM were toxic to embryos on 0 dpf, which suggested that a strong inhibition of p70S6K was severely detrimental. For long-term incubation, though, we treated ift57hi3417 or ift81hi409 mutants daily with 300 or 200 nM rapamycin (or 1% DMSO as a control) beginning from ∼30 hpf. On day 5 of development, we took images of treated embryos (Fig. 5C) and measured the area of cysts and length of fish embryos using Metamorph. We standardized cyst size by dividing cyst area by fish length. Results show that the size of cysts was significantly reduced in embryos treated with rapamycin. The average standardized cyst size for DMSO-treated ift57hi3417 mutant embryos was 5.78, but only 0.66 for drug-treated embryos (Table 2) (P < 0.0005). For ift81hi409, the standardized cyst size was 2.58 in DMSO-treated samples, but only 0.25 in drug-treated embryos (Table 2) (P < 0.04). However, the curvature of the embryos was not affected by rapamycin treatment (Fig. 5C). To rule out the possibility that the curvature phenotype is caused by defects earlier in development, we treated embryos with 200 nM rapamycin from ∼10 hpf, a time point before the lengthening of the trunk. Again, the curvature phenotype was not suppressed by rapamycin treatment (data not shown), suggesting that rapamycin can only partially rescue ift mutants.
Table 2.
Cyst area and embryo length measurements derived from ift57 and ift81 fish using Metamorph
| Strain | Treatment | n | Cyst area | Length | Area/length |
|---|---|---|---|---|---|
| Ift57 | 1% DMSO | 14 | 10047 | 1738 | 5.78* |
| 300 nm rapa | 15 | 1161 | 1756 | 0.66 | |
| Ift81 | 1% DMSO | 11 | 3971 | 1541 | 2.58** |
| 200 nm rapa | 19 | 378 | 1537 | 0.25 |
Cyst sizes were standardized by dividing cyst area by embryo length.
*P < 0.0005.
**P < 0.04.
Ciliary length is increased in tsc1a morphants
As tsc1a morphants show almost identical phenotypes as ift mutants and as the functions of ift genes are thought to be exclusively cilia-related, we analyzed the formation of the cilium in tsc1a morphants. We fixed tsc1a morphants at ∼20 hpf and labeled the basal body with anti-γ tubulin and cilia with an antibody to a ciliary protein, Scorpion/Arl13b (47). We first examined cilia in the pronephric duct. In wild-type embryos, the average length of cilia is 7.4 µm (Fig. 6A and E), consistent with previously published results (18). Intriguingly, cilia in tsc1a morphants can reach 12.4 µm on average (Fig. 6B and E). As cilia in the pronephric duct tend to bundle with each other and thus complicate the measurement of cilia length, we further measured cilia length in otic vesicles. Here, we found that the cilia length in uninjected control embryos measured 1.8 µm (Fig. 6C and E), whereas those in tsc1a embryos averaged 3.1 µm (Fig. 6D and E), suggesting that disrupted cilia length control is systemic in tsc1a morphant embryos. The bar graph in Figure 6E summarizes these results. It has been previously demonstrated that lack of ciliary beating in the zebrafish pronephric duct can lead to the formation of kidney cysts (18). Therefore, we analyzed the motility of cilia in the ducts of tsc1a morphant embryos using Metamorph. In the anterior ducts of wild-type, day 3 embryos, we measured average ciliary beating at 42.5 ± 11.1 beats/s (n = 5), which is in agreement with previous reports (Supplementary Material, Movie S1) (18). We also observed that the anterior pronephric duct cilia found in similar staged tsc1a morphant embryos beat at an average rate of 49 ± 11.8 beats/s (n = 3) (Supplementary Material, Movie S2), with no significant difference (P = 0.40, see bar graph, Fig. 6F). Therefore, knock down of tsc1a does not severely affect frequency of cilia beating.
Figure 6.

Cilia are longer than normal in the otic vesicles and pronephric ducts of tsc1a morphants. Cilia and basal bodies within embryos from 1 dpf (A and B) or 20-somite (C and D) stages were fluorescently labeled using antibodies against Scorpion (Arl13b), a ciliary protein and γ-tubulin. Confocal Z-series were captured using a Zeiss LSM 510 Meta. The LSM Image Browser software was used to measure ciliary length from the basal body to the distal tip [see bar graph in (E) for results]. Cilia from the pronephric ducts of wild-type embryos average 7.4 µm, whereas those in the kidneys of Tsc1a embryos average 12.4 µm long (*P < 0.0003). (B) Average cilia length within the otic vesicles of wild-type embryos was measured at 1.8 µm; however, otic vesicle cilia averaged 3.1 µm (**P < 0.01) after knock down of Tsc1a. (F) Cilia in the pronephric ducts of tsc1a embryos beat at a rate similar to those in WT ducts. Bar graph illustrating the average beats/second for cilia in the pronephric ducts of WT (42.5, n = 5) and tsc1a morphant (49, n = 3) embryos. The difference is not statistically significant (P = 0.4). WT, wild type; tsc1a, tsc1a morphant.
DISCUSSION
The cilium affects the Tor pathway upstream from S6K
The critical role of the cilium in kidney cyst formation, together with the observation of kidney cyst formation in TSC patients and rodent models, prompted us to ask whether there is a functional connection between the cilium and Tsc genes. Literature on this question is limited yet intriguing. In both Tsc1 and Tsc2 heterozygous mice (Tsc1+/−, or Tsc2+/−), the most prominent phenotype is renal and extra-renal tumors (28,29). However, homozygous mutants for either Tsc1 or Tsc2 die around embryonic day 10.5 with an open neural tube (28,29), the same phenotype seen in multiple ciliary mutants in the mouse (8).
In this study, we provided phenotypic, genetic and biochemical evidence to support the existence of such a functional link between the cilium and the TSC/TOR pathway. We observed that knockdown of tsc1a in zebrafish leads to body curvature, kidney cyst formation and defects in establishing left–right asymmetry of the body plan, phenotypes characteristic of ciliary mutants. We additionally observed that partial knockdown of tsc1a and ift genes synergized with each other, supporting common functions for tsc1a and ift genes. Our findings that phosphorylated p70S6K is elevated in tsc1a and ift morphants and that the TOR inhibitor rapamycin can inhibit cyst formation in ift mutants suggest that ciliary signals feed into the TOR pathway upstream of p70S6K.
Our results are also in agreement with previous studies, suggesting a connection between the TOR pathway and PKD. It was shown that the TOR pathway is inappropriately activated in cyst-lining epithelial cells and that rapamycin treatment may be able to reduce the size of polycystic kidneys in mouse models (30).
The molecular nature of the connection between the mTOR pathway and PKD is controversial. For example, it was shown in a tissue culture study that TSC2 is required for the proper membrane localization of PKD1 (31), suggesting that PKD1 functions downstream of TSC2. It was also reported that the cytoplasmic tail of PKD1 binds to TSC2 and mTOR and can retarget TSC2 and mTOR to the Gogli apparatus (30), putting PKD1 upstream of TSC2 and mTOR. Future challenges will be to clarify the molecular connection between the cilium and the TOR pathway, especially to determine how and at which step within the TOR pathway a ciliary signal is incorporated.
TSC genes and trafficking
In this study, we show that Tsc1a is associated with the Golgi complex both at the blastula stage and in differentiated kidney epithelial cells, raising the interesting possibility that one aspect of Tsc1a function is controlling protein trafficking from the Golgi, which in turn mediates the signal from the cilium. Indeed, although the best-known function of the TSC complex is to restrain the activity of mTOR through Rheb, a less well-appreciated area is a potential role of Tsc1 and Tsc2 in protein trafficking. In fission yeast, both TSC1 and TSC2 are required for the proper targeting of an amino acid permease to the plasma membrane (48). In mammalian cells, the TSC complex was reported to be involved in post-Golgi transport of VSVG and caveolin-1 (49). In Tsc2−/− cells, caveolin-1 becomes restricted to cytoplasmic vesicles and treatment with rapamycin can restore its localization to the plasma membrane (49). Interestingly, the trafficking of PKD1 to the cell membrane was reported to require functional TSC2 in cultured cells (31). Various subcellular localization patterns have been reported for TSC1 and TSC2 (49–54). Some of the patterns are consistent with a trafficking role. For example, TSC2 has been shown to be a component of lipid rafts (49). In addition, both proteins were reported to be in vesicular structures in the cytosol (50,51). Furthermore, over-expression of a membrane-tethered N-terminal region of PKD1 was shown to retarget TSC2 from the cytosol to the Golgi (30). The relationship between TSC and IFT may involve trafficking as well because IFT20 is known to shuttle between the Golgi and cilium and is believed to help deliver ciliary membrane proteins such as polycystin-2 to the cilium (55).
Tsc1 and cilia length control
In this study, we show that knockdown of tsc1a in zebrafish leads to phenotypes seen in ift mutants. As IFT genes are essential for cilia biogenesis, tsc1a could be involved in cilia formation. Surprisingly, cilia are elongated in tsc1a morphants. The length of cilia in different tissues is diverse yet stereotypic. For example, cilia in the zebrafish pronephric duct range from 7 to 9 µm long, whereas cilia in the zebrafish spinal canal are around 2 µm long (18). Genetic screens in Chlamydomonas yielded four long-flagella mutants (lf1-4) (56–59). Two of the LF genes encode kinases (57,58), suggesting that a potential signaling cascade is involved in cilia length control. Long cilia have also been found in the jck/Nek8 mouse that develops PKD (60,61). Intriguingly, treatment of Chlamydomonas with the GSK3 inhibitor lithium can induce flagellar elongation (59), pointing to a possible role of the Wnt pathway in ciliary length control. This result is particularly interesting in light of recent findings that genes involved in ciliary functions also regulate Wnt pathways (5,10,62,63). Alternatively, regulation of cilia length by GSK3 may occur independent of the Wnt pathway. For example, it has been previously demonstrated that in the absence of TSC1 or TSC2, p70S6K can directly inhibit GSK3 through phosphorylation (64). Exactly how this inhibition would lead to long cilia is still unclear; however, the defect in ciliary length may represent a global misregulation of microtubule growth as has been previously suggested (59), because TSC2−/− cells grow elongated microtubules that fail to halt at the cell cortex. Interestingly, this defect can be rescued by the reduction of the microtubule tip-binding protein CLIP-170 or by rapamycin treatment (65).
Our result that cilia are abnormally long in tsc1a morphants implicates the TOR pathway in size control of this organelle. It is possible that a feedback mechanism downstream of the cilium plays a role in regulating cilia length. For example, signaling activity by the cilium may serve as an indicator for the cilia assembly/maintenance machinery that a functional cilium has formed and thus downregulates this machinery. In this model, Tsc1a regulates cilia length indirectly by functioning downstream of the cilium. Consistently, a recent study in Caenorhabditis elegans sensory neurons demonstrates that the structure of the cilium is remodeled in response to abnormal levels of sensory activity (66). Currently, although multiple players have been identified for cilia length control, how these players function to achieve this control remains unclear.
MATERIALS AND METHODS
Fish strains and husbandry
Zebrafish were raised and maintained by standard protocols (67). Wild-type embryos were derived from crosses of Tab 5 or Tab 14 adult fish. Mutant embryos were generated from heterozygous crosses.
Constructs, morpholinos and mRNAs
Morpholinos targeted to translational start sites include the following: standard control morpholino: 5′-CCTCTTACCTCAGTTACAATTTATA-3′; Tsc1a: 5′-CCATAGTTGTGCAGGACAGTGGGCA-3′; 5′-AGAGATCAGTCCTCACCTTCACCAC-3′; Tsc1a mismatch control: 5′-CCATACTTCTGCAGCACACTGGCCAA-3′; Ift81: 5′-CGATAAATTTAAGCTGTTCGCTCAT-3′ and Ift172: 5′-GACTCAGGGCAGTTATAAGAACGTA-3′. Base pairs 1879–2373 (amplified using primers 5′-CAGAATTCCCGAAGGTGGCATCTCTGTTTTT-3′ and 5′-GCTCTAGATTAATGGTGATGGTGATGGTGCTCTCTGTCTTCCTGTATGTGC-3′) or 3079–3496 (using primers 5′-CAGAATTCTCTCTACCGGCTGACCTGA-3′ and 5′-GCTCTAGATTAATGGTGATGGTGATGGTGGTCCTGATGCCTCCTACTCTG-3′) from the Tsc1a-coding region were subcloned into the EcoRI and XbaI sites of pMAL C2 (New England Biolabs, Inc.) to generate MBP fusion proteins with 3′ 6x His tags. Over-produced proteins were purified on amylose columns and used as antigens to generate polyclonal antisera TSC1-1 and TSC1-2, respectively.
Morpholino and mRNA injections
About ∼0.125–0.500 pmol of translational start site blocking morpholinos or 50–100 ng in vitro synthesized mRNAs were injected into embryos at the 1–4-cell stage. Embryos developed at 28.5°C in embryo medium (5 mm NaCl, 0.2 mm KCl, 0.3 mm CaCl2, 0.7 mm MgSO4, 1×10−5% methylene blue).
Preparation of embryo lysates and western blots
Embryo lysates were prepared with ∼24–48 hpf embryos as previously described (68) with minor modifications. Prepared samples were loaded onto 4–20% Tris–HCl mini gradient gels (BioRad), then transferred to nitrocellulose. Immunoblots were carried out using Tris-buffered saline (TBS), pH 7.4, 5% milk, 0.1% Tween 20 for primary and secondary antibodies. After a final wash in TBS, 0.5% Triton X-100, blots were incubated in Western Lightning Chemiluminescence Plus (PerkinElmer LAS, Inc.) and exposed to film.
Antibodies
Antibodies used include TSC1-1 and TSC1-2 polyclonal antibodies (Sigma-Genosys); anti-Scorpion polyclonal antibody (Covance); BD Biosciences: gm130, anti-IRS-1 and anti-p70 S6K; Cell Signaling: anti-Phospho-p70 S6 Kinase (Thr389) and Sigma: anti-β tubulin Clone 2.1, anti-acetylated α-tubulin. Secondary antibodies are from Jackson Immuno Research Labs and Invitrogen/Molecular Probes.
Quantification of Western blots
X-ray films were scanned at 300 dpi and then analyzed using AutoQuant software, version X1.4.1 (Media Cybernetics). Values for each sample were normalized through background subtraction and then divided by the value for the corresponding β-tubulin band.
Statistical analysis
P-values were derived from the Student's t-test using Microsoft Excel.
Electron microscopy
Forty-eight hpf embryos were fixed with Karnovsky fixative for 1 h at 4°C, washed with 0.1 M sodium cacodylate, pH 7.4, then post-fixed with Palade's osmium for 1 h at 4°C, shielded from light. Following a second wash, embryos were stained with Kellenburger's solution for 1 h at RT, washed in double distilled water, then put through an ethanol series, propylene oxide, 50/50 propylene oxide/epon, then two incubations in 100% epon. Embedded embryos were sectioned at 400 nm before staining with 2% uranyl acetate. Micrographs were taken on a Zeiss 910 electron microscope.
Whole-mount immunostaining
Embryos at various time points were fixed in either formalin or Dent's fixative (80% methanol and 20% DMSO). Formalin-fixed samples were permeabilized in ice cold acetone. Embryos were blocked with 10% fetal bovine serum in phosphate-buffered saline, 0.1% Tween-20 (PBT) before incubation with primary antibodies in blocking solution. Before and after secondary antibody incubation, embryos were washed extensively with PBT. Embryos were deyolked and then mounted using Vectashield mounting medium (Vector Laboratories) or Prolong Gold (Invitrogen/Molecular Probes). Images were captured using a Zeiss 510 Meta confocal microscope or a Zeiss Axioplan microscope and processed using Photoshop.
High-speed videomicroscopy
A previously published protocol was followed (18) with minor modifications. Embryos were incubated in 75 µM phenylthiourea in embryo medium starting on day 1. On day 3, embryos were mounted in 6% methylcellulose containing 50 mm 2,3-butanedione monoxime and 0.02% tricaine. Cilia beating was recorded using a 20×/0.4 DIC lens on a Nikon Eclipse TE2000-U microscope equipped with a Nikon Photometrics Coolsnap HQ camera under stream acquisition at 200 frames/s for 1 s, analyzed and converted into an avi format using Metamorph v8.1.2.0. Movies are played at 10 frames/s.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work was supported by grants from the United States Army Medical Research and Material Command (W81XWH-07-10235 to Z.S.), the National Institute of Diabetes and Digestive and Kidney Diseases (ROI DK069528 to Z.S.) and from the Tuberous Sclerosis Alliance to L.M.D.
ACKNOWLEDGEMENTS
We thank Arthur Horwich for his critical review of this manuscript, SueAnn Mentone for her superb skills with histology and EM, members of the Yale Center for PKD Research for their comments and suggestions, and the Reinke and Somlo labs for their technical support and use of their equipment.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Schneider L., Clement C.A., Teilmann S.C., Pazour G.J., Hoffmann E.K., Satir P., Christensen S.T. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr. Biol. 2005;15:1861–1866. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 2.Handel M., Schulz S., Stanarius A., Schreff M., Erdtmann-Vourliotis M., Schmidt H., Wolf G., Hollt V. Selective targeting of somatostatin receptor 3 to neuronal cilia. Neuroscience. 1999;89:909–926. doi: 10.1016/s0306-4522(98)00354-6. [DOI] [PubMed] [Google Scholar]
- 3.Berbari N.F., Lewis J.S., Bishop G.A., Askwith C.C., Mykytyn K. Bardet–Biedl syndrome proteins are required for the localization of G protein-coupled receptors to primary cilia. Proc. Natl Acad. Sci. USA. 2008;105:4242–4246. doi: 10.1073/pnas.0711027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corbit K.C., Aanstad P., Singla V., Norman A.R., Stainier D.Y., Reiter J.F. Vertebrate smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- 5.Corbit K.C., Shyer A.E., Dowdle W.E., Gaulden J., Singla V., Chen M.H., Chuang P.T., Reiter J.F. Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat. Cell Biol. 2008;10:70–76. doi: 10.1038/ncb1670. [DOI] [PubMed] [Google Scholar]
- 6.Gerdes J.M., Liu Y., Zaghloul N.A., Leitch C.C., Lawson S.S., Kato M., Beachy P.A., Beales P.L., DeMartino G.N., Fisher S., et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat. Genet. 2007;39:1350–1360. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- 7.Huangfu D., Anderson K.V. Cilia and Hedgehog responsiveness in the mouse. Proc. Natl Acad. Sci. USA. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huangfu D., Liu A., Rakeman A.S., Murcia N.S., Niswander L., Anderson K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- 9.Kim E., Arnould T., Sellin L.K., Benzing T., Fan M.J., Gruning W., Sokol S.Y., Drummond I., Walz G. The polycystic kidney disease 1 gene product modulates Wnt signaling. J. Biol. Chem. 1999;274:4947–4953. doi: 10.1074/jbc.274.8.4947. [DOI] [PubMed] [Google Scholar]
- 10.Oishi I., Kawakami Y., Raya A., Callol-Massot C., Izpisua Belmonte J.C. Regulation of primary cilia formation and left-right patterning in zebrafish by a noncanonical Wnt signaling mediator, duboraya. Nat. Genet. 2006;38:1316–1322. doi: 10.1038/ng1892. [DOI] [PubMed] [Google Scholar]
- 11.Park T.J., Haigo S.L., Wallingford J.B. Ciliogenesis defects in embryos lacking inturned or fuzzy function are associated with failure of planar cell polarity and Hedgehog signaling. Nat. Genet. 2006;38:303–311. doi: 10.1038/ng1753. [DOI] [PubMed] [Google Scholar]
- 12.Rosenbaum J.L., Witman G.B. Intraflagellar transport. Nat. Rev. Mol. Cell Biol. 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- 13.Kozminski K.G., Johnson K.A., Forscher P., Rosenbaum J.L. A motility in the eukaryotic flagellum unrelated to flagellar beating. Proc. Natl Acad. Sci. USA. 1993;90:5519–5523. doi: 10.1073/pnas.90.12.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pazour G.J., Dickert B.L., Vucica Y., Seeley E.S., Rosenbaum J.L., Witman G.B., Cole D.G. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J. Cell Biol. 2000;151:709–718. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoder B.K., Tousson A., Millican L., Wu J.H., Bugg C.E., Jr, Schafer J.A., Balkovetz D.F. Polaris, a protein disrupted in orpk mutant mice, is required for assembly of renal cilium. Am. J. Physiol. Renal. Physiol. 2002;282:F541–F552. doi: 10.1152/ajprenal.00273.2001. [DOI] [PubMed] [Google Scholar]
- 16.Taulman P.D., Haycraft C.J., Balkovetz D.F., Yoder B.K. Polaris, a protein involved in left–right axis patterning, localizes to basal bodies and cilia. Mol. Biol. Cell. 2001;12:589–599. doi: 10.1091/mbc.12.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun Z., Amsterdam A., Pazour G.J., Cole D.G., Miller M.S., Hopkins N. A genetic screen in zebrafish identifies cilia genes as a principal cause of cystic kidney. Development. 2004;131:4085–4093. doi: 10.1242/dev.01240. [DOI] [PubMed] [Google Scholar]
- 18.Kramer-Zucker A.G., Olale F., Haycraft C.J., Yoder B.K., Schier A.F., Drummond I.A. Cilia-driven fluid flow in the zebrafish pronephros, brain and Kupffer's vesicle is required for normal organogenesis. Development. 2005;132:1907–1921. doi: 10.1242/dev.01772. [DOI] [PubMed] [Google Scholar]
- 19.Gabow P.A., Grantham J. Polycystic kidney disease. In: Gottschalk R.S.A.C., editor. Diseases of the Kidney. Boston, MA: Little Brown; 1997. pp. 521–560. [Google Scholar]
- 20.Fliegauf M., Benzing T., Omran H. When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 21.Pazour G.J., Witman G.B. The vertebrate primary cilium is a sensory organelle. Curr. Opin. Cell Biol. 2003;15:105–110. doi: 10.1016/s0955-0674(02)00012-1. [DOI] [PubMed] [Google Scholar]
- 22.Astrinidis A., Henske E.P. Tuberous sclerosis complex: linking growth and energy signaling pathways with human disease. Oncogene. 2005;24:7475–7481. doi: 10.1038/sj.onc.1209090. [DOI] [PubMed] [Google Scholar]
- 23.Green A.J., Smith M., Yates J.R. Loss of heterozygosity on chromosome 16p13.3 in hamartomas from tuberous sclerosis patients. Nat. Genet. 1994;6:193–196. doi: 10.1038/ng0294-193. [DOI] [PubMed] [Google Scholar]
- 24.Yeung R.S., Buetow K.H., Testa J.R., Knudson A.G., Jr Susceptibility to renal carcinoma in the Eker rat involves a tumor suppressor gene on chromosome 10. Proc. Natl Acad. Sci. USA. 1993;90:8038–8042. doi: 10.1073/pnas.90.17.8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cai S., Everitt J.I., Kugo H., Cook J., Kleymenova E., Walker C.L. Polycystic kidney disease as a result of loss of the tuberous sclerosis 2 tumor suppressor gene during development. Am. J. Pathol. 2003;162:457–468. doi: 10.1016/S0002-9440(10)63840-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ong A.C., Harris P.C., Davies D.R., Pritchard L., Rossetti S., Biddolph S., Vaux D.J., Migone N., Ward C.J. Polycystin-1 expression in PKD1, early-onset PKD1, and TSC2/PKD1 cystic tissue. Kidney Int. 1999;56:1324–1333. doi: 10.1046/j.1523-1755.1999.00659.x. [DOI] [PubMed] [Google Scholar]
- 27.Huang J., Manning B.D. The TSC1–TSC2 complex: a molecular switchboard controlling cell growth. Biochem. J. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kobayashi T., Minowa O., Kuno J., Mitani H., Hino O., Noda T. Renal carcinogenesis, hepatic hemangiomatosis, and embryonic lethality caused by a germ-line Tsc2 mutation in mice. Cancer Res. 1999;59:1206–1211. [PubMed] [Google Scholar]
- 29.Kobayashi T., Minowa O., Sugitani Y., Takai S., Mitani H., Kobayashi E., Noda T., Hino O. A germ-line Tsc1 mutation causes tumor development and embryonic lethality that are similar, but not identical to, those caused by Tsc2 mutation in mice. Proc. Natl Acad. Sci. USA. 2001;98:8762–8767. doi: 10.1073/pnas.151033798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shillingford J.M., Murcia N.S., Larson C.H., Low S.H., Hedgepeth R., Brown N., Flask C.A., Novick A.C., Goldfarb D.A., Kramer-Zucker A., et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl Acad. Sci. USA. 2006;103:5466–5471. doi: 10.1073/pnas.0509694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kleymenova E., Ibraghimov-Beskrovnaya O., Kugoh H., Everitt J., Xu H., Kiguchi K., Landes G., Harris P., Walker C. Tuberin-dependent membrane localization of polycystin-1: a functional link between polycystic kidney disease and the TSC2 tumor suppressor gene. Mol. Cell. 2001;7:823–832. doi: 10.1016/s1097-2765(01)00226-x. [DOI] [PubMed] [Google Scholar]
- 32.Drummond I.A., Majumdar A., Hentschel H., Elger M., Solnica-Krezel L., Schier A.F., Neuhauss S.C., Stemple D.L., Zwartkruis F., Rangini Z., et al. Early development of the zebrafish pronephros and analysis of mutations affecting pronephric function. Development. 1998;125:4655–4667. doi: 10.1242/dev.125.23.4655. [DOI] [PubMed] [Google Scholar]
- 33.Makky K., Tekiela J., Mayer A.N. Target of rapamycin (TOR) signaling controls epithelial morphogenesis in the vertebrate intestine. Dev. Biol. 2007;303:501–513. doi: 10.1016/j.ydbio.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Supp D.M., Witte D.P., Potter S.S., Brueckner M. Mutation of an axonemal dynein affects left–right asymmetry in inversus viscerum mice. Nature. 1997;389:963–966. doi: 10.1038/40140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brody S.L., Yan X.H., Wuerffel M.K., Song S.K., Shapiro S.D. Ciliogenesis and left–right axis defects in forkhead factor HFH-4-null mice. Am. J. Respir. Cell Mol. Biol. 2000;23:45–51. doi: 10.1165/ajrcmb.23.1.4070. [DOI] [PubMed] [Google Scholar]
- 36.Chen J.N., van Eeden F.J., Warren K.S., Chin A., Nusslein-Volhard C., Haffter P., Fishman M.C. Left–right pattern of cardiac BMP4 may drive asymmetry of the heart in zebrafish. Development. 1997;124:4373–4382. doi: 10.1242/dev.124.21.4373. [DOI] [PubMed] [Google Scholar]
- 37.Marszalek J.R., Ruiz-Lozano P., Roberts E., Chien K.R., Goldstein L.S. Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc. Natl Acad. Sci. USA. 1999;96:5043–5048. doi: 10.1073/pnas.96.9.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nonaka S., Tanaka Y., Okada Y., Takeda S., Harada A., Kanai Y., Kido M., Hirokawa N. Randomization of left–right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- 39.Zhao C., Malicki J. Genetic defects of pronephric cilia in zebrafish. Mech. Dev. 2007;124:605–616. doi: 10.1016/j.mod.2007.04.004. [DOI] [PubMed] [Google Scholar]
- 40.Astrinidis A., Senapedis W., Henske E.P. Hamartin, the tuberous sclerosis complex 1 gene product, interacts with polo-like kinase 1 in a phosphorylation-dependent manner. Hum. Mol. Genet. 2006;15:287–297. doi: 10.1093/hmg/ddi444. [DOI] [PubMed] [Google Scholar]
- 41.Klausner R.D., Donaldson J.G., Lippincott-Schwartz J. Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kwiatkowski D.J., Manning B.D. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum. Mol. Genet. 2005;14(Spec No. 2):R251–R258. doi: 10.1093/hmg/ddi260. [DOI] [PubMed] [Google Scholar]
- 43.Harris T.E., Lawrence J.C., Jr TOR signaling. Sci. STKE. 2003;2003 doi: 10.1126/stke.2122003re15. re15. [DOI] [PubMed] [Google Scholar]
- 44.Arsham A.M., Neufeld T.P. Thinking globally and acting locally with TOR. Curr. Opin. Cell Biol. 2006;18:589–597. doi: 10.1016/j.ceb.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 45.Harrington L.S., Findlay G.M., Gray A., Tolkacheva T., Wigfield S., Rebholz H., Barnett J., Leslie N.R., Cheng S., Shepherd P.R., et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 2004;166:213–223. doi: 10.1083/jcb.200403069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shah O.J., Wang Z., Hunter T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 2004;14:1650–1656. doi: 10.1016/j.cub.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 47.Caspary T., Larkins C.E., Anderson K.V. The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell. 2007;12:767–778. doi: 10.1016/j.devcel.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 48.Matsumoto S., Bandyopadhyay A., Kwiatkowski D.J., Maitra U., Matsumoto T. Role of the Tsc1–Tsc2 complex in signaling and transport across the cell membrane in the fission yeast Schizosaccharomyces pombe. Genetics. 2002;161:1053–1063. doi: 10.1093/genetics/161.3.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jones K.A., Jiang X., Yamamoto Y., Yeung R.S. Tuberin is a component of lipid rafts and mediates caveolin-1 localization: role of TSC2 in post-Golgi transport. Exp. Cell Res. 2004;295:512–524. doi: 10.1016/j.yexcr.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 50.Wienecke R., Maize J.C., Jr, Shoarinejad F., Vass W.C., Reed J., Bonifacino J.S., Resau J.H., de Gunzburg J., Yeung R.S., DeClue J.E. Co-localization of the TSC2 product tuberin with its target Rap1 in the Golgi apparatus. Oncogene. 1996;13:913–923. [PubMed] [Google Scholar]
- 51.Plank T.L., Yeung R.S., Henske E.P. Hamartin, the product of the tuberous sclerosis 1 (TSC1) gene, interacts with tuberin and appears to be localized to cytoplasmic vesicles. Cancer Res. 1998;58:4766–4770. [PubMed] [Google Scholar]
- 52.Rosner M., Freilinger A., Hengstschlager M. Akt regulates nuclear/cytoplasmic localization of tuberin. Oncogene. 2006;26:521–531. doi: 10.1038/sj.onc.1209812. [DOI] [PubMed] [Google Scholar]
- 53.Rosner M., Hengstschlager M. Cytoplasmic/nuclear localization of tuberin in different cell lines. Amino Acids. 2007;33:575–579. doi: 10.1007/s00726-007-0541-0. [DOI] [PubMed] [Google Scholar]
- 54.Clements D., Mayer R.J., Johnson S.R. Subcellular distribution of the TSC2 gene product tuberin in human airway smooth muscle cells is driven by multiple localization sequences and is cell-cycle dependent. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007;292:L258–L266. doi: 10.1152/ajplung.00354.2005. [DOI] [PubMed] [Google Scholar]
- 55.Follit J.A., Tuft R.A., Fogarty K.E., Pazour G.J. The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol. Biol. Cell. 2006;17:3781–3792. doi: 10.1091/mbc.E06-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nguyen R.L., Tam L.W., Lefebvre P.A. The LF1 gene of Chlamydomonas reinhardtii encodes a novel protein required for flagellar length control. Genetics. 2005;169:1415–1424. doi: 10.1534/genetics.104.027615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tam L.W., Wilson N.F., Lefebvre P.A. A CDK-related kinase regulates the length and assembly of flagella in Chlamydomonas. J. Cell Biol. 2007;176:819–829. doi: 10.1083/jcb.200610022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Berman S.A., Wilson N.F., Haas N.A., Lefebvre P.A. A novel MAP kinase regulates flagellar length in Chlamydomonas. Curr. Biol. 2003;13:1145–1149. doi: 10.1016/s0960-9822(03)00415-9. [DOI] [PubMed] [Google Scholar]
- 59.Wilson N.F., Lefebvre P.A. Regulation of flagellar assembly by glycogen synthase kinase 3 in Chlamydomonas reinhardtii. Eukaryot. Cell. 2004;3:1307–1319. doi: 10.1128/EC.3.5.1307-1319.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sohara E., Luo Y., Zhang J., Manning D.K., Beier D.R., Zhou J. Nek8 regulates the expression and localization of polycystin-1 and polycystin-2. J. Am. Soc. Nephrol. 2008;19:469–476. doi: 10.1681/ASN.2006090985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith L.A., Bukanov N.O., Husson H., Russo R.J., Barry T.C., Taylor A.L., Beier D.R., Ibraghimov-Beskrovnaya O. Development of polycystic kidney disease in juvenile cystic kidney mice: insights into pathogenesis, ciliary abnormalities, and common features with human disease. J. Am. Soc. Nephrol. 2006;17:2821–2831. doi: 10.1681/ASN.2006020136. [DOI] [PubMed] [Google Scholar]
- 62.Kishimoto N., Cao Y., Park A., Sun Z. Cystic kidney gene seahorse regulates cilia-mediated processes and Wnt pathways. Dev. Cell. 2008;14:954–961. doi: 10.1016/j.devcel.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 63.Ross A.J., May-Simera H., Eichers E.R., Kai M., Hill J., Jagger D.J., Leitch C.C., Chapple J.P., Munro P.M., Fisher S., et al. Disruption of Bardet–Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet. 2005;37:1135–1140. doi: 10.1038/ng1644. [DOI] [PubMed] [Google Scholar]
- 64.Zhang H.H., Lipovsky A.I., Dibble C.C., Sahin M., Manning B.D. S6K1 regulates GSK3 under conditions of mTOR-dependent feedback inhibition of Akt. Mol. Cell. 2006;24:185–197. doi: 10.1016/j.molcel.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang X., Yeung R.S. Regulation of microtubule-dependent protein transport by the TSC2/mammalian target of rapamycin pathway. Cancer Res. 2006;66:5258–5269. doi: 10.1158/0008-5472.CAN-05-4510. [DOI] [PubMed] [Google Scholar]
- 66.Mukhopadhyay S., Lu Y., Shaham S., Sengupta P. Sensory signaling-dependent remodeling of olfactory cilia architecture in C. elegans. Dev. Cell. 2008;14:762–774. doi: 10.1016/j.devcel.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Westerfield M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish. 4th edn. Eugene, OR: University of Oregon Press; 2000. [Google Scholar]
- 68.Link V., Shevchenko A., Heisenberg C.P. Proteomics of early zebrafish embryos. BMC Dev. Biol. 2006;6:1. doi: 10.1186/1471-213X-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.