

Abstract
Huntington’s disease (HD) is caused by an expansion of a CAG trinucleotide sequence that encodes a polyglutamine tract in the huntingtin (Htt) protein. Expansion of the polyglutamine tract above 35 repeats causes disease, with the age of onset inversely related to the degree of expansion above this number. Growing evidence suggests that mitochondrial function is compromised during HD pathogenesis, but how this occurs is not understood. We examined mitochondrial properties of HeLa cells that expressed green fluorescent protein (GFP)- or FLAG-tagged N-terminal portions of the Htt protein containing either, 17, 28, 74 or 138 polyglutamine repeats. Immunofluorescence staining of cells using antibodies against Tom20, a mitochondrion localized protein, revealed that cells expressing Htt proteins with 74 or 138 polyglutamine repeats were more sensitized to oxidative stress-induced mitochondria fragmentation and had reduced ATP levels compared with cells expressing Htt proteins with 17 or 28 polyglutamine repeats. By measuring changes in fluorescence of a photoactivated GFP protein targeted to mitochondria, we found that cells expressing red fluorescent protein (RFP)-tagged Htt protein containing 74 polyglutamine repeats had mitochondria that displayed reduced movement and fusion than cells expressing RFP-Htt protein with 28 polyglutamine repeats. Overexpression of Drp-1K38A, a dominant-negative mitochondria-fission mutant, or Mfn2, a protein that promotes mitochondria fusion, suppressed polyglutamine-induced mitochondria fragmentation, the reduction of ATP levels and cell death. In a Caenorhabditis elegans model of HD, we found that reduction of Drp-1 expression by RNA interference rescued the motility defect associated with the expression of Htt proteins with polyglutamine repeats. These results suggest that the increase in cytotoxicity induced by Htt proteins containing expanded polyglutamine tracts is likely mediated, at least in part, by an alteration in normal mitochondrial dynamics, which results in increased mitochondrial fragmentation. Furthermore, our results suggest that it might be possible to reverse polyglutamine-induced cytotoxicity by preventing mitochondrial fragmentation.
INTRODUCTION
Huntington’s disease (HD) is a devastating neurodegenerative disorder whose clinical characteristics include progressive cognitive impairment, abnormal movements, psychiatric disturbance and dementia. Pathologically, HD is characterized by a predominant loss of neurons in the striatal and cortical areas of the brain and by formation of neuronal inclusions (1). Genetically, HD is caused by abnormal expansion of a CAG trinucleotide repeat found in the first exon of huntingtin (Htt) gene, which becomes translated into long stretch of glutamines in an ∼350 kDa protein of unclear function (1,2). Individuals containing a tract of more than 35 polyglutamine repeats in Htt protein will develop HD, whereas those with fewer polyglutamine repeats tend not to develop the disease (3). In addition to HD, there are at least eight other neurodegenerative disorders that are caused by expansions in polyglutamine tracts in proteins that are otherwise unrelated in sequence (4).
The mechanism(s) by which expanded polyglutamine proteins induce disease is still unclear. However, in the case of HD, there is growing evidence to suggest that pathogenesis might be linked directly, or indirectly, to a perturbation of mitochondria function (5,6). First, nuclear magnetic resonance imaging has revealed that HD-afflicted individuals display increased production of lactate, which is considered an indicator of compromised mitochondrial function, in the cerebral cortex and basal ganglia, compared with unaffected people (7). Second, biochemical measurements indicate that mitochondria isolated from HD patients or from transgenic mouse models of HD have reduced complex II and II–III activity compared with those isolated from normal human subjects or non-HD transgenic mice (8). Furthermore, animals treated with mitochondrial complex II inhibitors, malonate or 3-nitropropionic acid, develop a movement disorder that is associated with loss of medium spiny neurons in the striatum, similar to clinical and pathological features seen in humans afflicted with HD (9,10). Third, mitochondria isolated from a knock-in mouse model of HD contain lower amounts of ATP than those isolated from non-affected animals (10). Finally, mitochondria isolated from HD patients or HD-transgenic mouse brains display a polyglutamine length-dependent perturbation in depolarization of membrane potential and in calcium homeostasis (11). Despite these observations, it remains unclear how mitochondria become injured and dysfunctional in HD.
Mitochondria are the main power-generating organelle of eukaryotic cells, producing ATP, the primary source of energy of cells. Interestingly, mitochondria also harbor several key regulators of cell death including, Bcl-2 family of proteins, cytochrome c and other pro-apoptotic factors. When released into the cytoplasm, these factors can trigger cell death programs. Dysfunction in mitochondrial energy metabolism can lead to a reduction in ATP production, impaired calcium homeostasis and enhanced generation of reactive oxygen species, which, if left unchecked, can eventually lead to the execution of the cell death program (5).
Mitochondria are highly dynamic organelles that constantly change shape and structure in response to different stimuli and metabolic demands of the cell. Their shape is chiefly regulated by cycles of fusion and fission (12,13). During apoptosis, mitochondria undergo extensive fragmentation, which precedes caspase activation, whereas inhibition of the mitochondrial fission machinery blocks or delays cell death (14). The two opposing processes, fusion and fission, are controlled by evolutionarily conserved large GTPases belonging to the dynamin family. In mammals, Opa1, mitofusin 1 (Mfn 1) and mitofusin 2 (Mfn2) regulate fusion, whereas dynamin-related protein 1 (Drp-1) control mitochondrial fission (12,14,15). Recently, it was demonstrated that mitochondrial fission was causal in oxidative stress-mediated neuronal death (16). Overexpression of either Mfn 1, a dominant-negative Drp-1 mutant, Drp-1K38A, or Mfn2 promotes mitochondrial fusion and reduces oxidative stress-induced cell death (16–19). Since mitochondrial dysfunction has been implicated in HD pathogenesis, we investigated whether Htt proteins containing different lengths of polyglutamine repeats induce alterations in mitochondrial fragmentation. We demonstrate that overexpression of Htt proteins containing polyglutamine repeats in the pathological range, but not those in the non-pathological range, increases oxidative stress-induced mitochondrial fragmentation in HeLa cells, which correlates with increased caspase-3 activation and cell death. We further demonstrate that overexpression of proteins that stimulate mitochondria fusion attenuates the toxicity of Htt proteins containing expanded polyglutamine tracts in both cells and animals.
RESULTS
Differential sensitivity of HeLa cell lines expressing Htt-exon-1 proteins containing either a normal or expanded polyglutamine tract to oxidative stress-induced mitochondrial fragmentation
In our previous report, we showed that HeLa cells stably expressing green fluorescent protein (GFP)-tagged Htt exon-1 protein containing 74 polyglutamine repeats (GFP-Htt74Q) are more sensitive to oxidative stress-induced cell death compared with a cell line expressing the Htt-fusion protein with 28 polyglutamine repeats (GFP-Htt28Q) (20). Because of the functional interrelationship between mitochondrial dysfunction and oxidative stress, we examined whether the differential sensitivity of the HeLa cell lines to oxidative stress-induced injury was associated with changes in mitochondria morphology. To examine this possibility, we exposed HeLa cell lines expressing GFP-Htt28Q and GFP-Htt74Q, respectively, to culture conditions that would promote low amounts of oxidative stress, by culturing the cells in either 1.25% fetal bovine serum (FBS) or 25 µm H2O2, and then examining them following immunostaining with antibodies against the outer mitochondrial membrane-localized protein, Tom20 (21,22). As shown in Figure 1A and 1B, many of the cells stably expressing GFP-Htt28Q fusion protein when cultured in medium containing low serum contained mitochondria that were both elongated and dispersed throughout the cytoplasm, similar to the phenotype seen in the parental HeLa cell line from which the stable GFP-Htt28Q clone was derived (data not shown). By contrast, most of the cells expressing GFP-Htt74Q fusion contained mitochondria that appeared to be cluster around the nucleus and which were smaller in length, indicative of mitochondrial fragmentation (Fig. 1A and B). The heightened sensitivity of GFP-Htt74Q-expressing cells to oxidative stress-induced mitochondrial fragmentation after serum deprivation (Fig. 1C) was also seen upon treatment of the cells with H2O2 (Fig. 1D). Interestingly, even when cultured in normal growth conditions (10% FBS), we observed a low, but a noticeable increase in basal mitochondrial fragmentation in the GFP-Htt74Q-expressing cells compared with the GFP-Htt28Q-expressing cells (Fig. 1C).
Figure 1.

Oxidative stress-induced mitochondrial fragmentation in HeLa cells stably expressing GFP-Htt polyglutamine fusion proteins. HeLa cell lines stably expressing either GFP-Htt28Q or GFP-Htt74Q proteins were either treated with 25 µm H2O2 for 30 min or incubated in culture medium containing a lower concentration of FBS (1.25%) for 18 h and then fixed with paraformadehyde and immunostained with a monoclonal anti-Tom20 antibody. (A) Representative microscopy images of GFP fluorescence (left panels) and Tom20 staining (right panels) of the same group of cells viewed with a 40× objective lens in cells stable expressing either GFP-Htt28Q (upper panels) or GFP-Htt74Q (lower panels) fusion proteins. Note that GFP-Htt74Q-expressing cells display increased mitochondria fragmentation. Bar: 10 µm. (B) Same as (A), except imaged with a 100× objective lens. Bar: 10 µm. (C) Quantification of cells containing fragmented mitochondria in the GFP-Htt28Q and GFP-Htt74Q expressing cell lines when cultured under normal growth conditions (10% FBS; control) or in the presence of 1.25% FBS. Approximately 300 GFP-Htt28Q- and 1014 GFP-Htt74Q-expressing cells were counted for these analyses, respectively. Data shown are shown as mean ± SDM (*P < 0.05). (D) Quantification of mitochondrial fragmentation in untransfected HeLa cells and in the GFP-Htt28Q and GFP-Htt74Q-expressing cell lines when cultured in the presence of 25 µm H2O2 for 30 min. Approximately 1000 cells were analyzed for this analysis. GFP-Htt74Q-expressing cells exhibited significantly greater mitochondrial fragmentation compared with untransfected HeLa cells or GFP-Htt28Q-expressing cells. *P < 0.05.
Electron microscopic examination of the GFP-Htt-expressing cell lines treated with 25 µm H2O2 revealed a pronounced difference in the morphology of mitochondria at the ultra structural level in GFP-Htt74Q-expressing cells compared with GFP-Htt28Q-expressing cells (Fig. 2). Notably, ∼88.99 ± 12.17% of the mitochondria in GFP-Htt74Q-expressing cells contained highly disorganized cristae and, furthermore, regions of their mitochondrial matrix were considerably less electron-dense (indicated by arrows, Fig. 2B and C), which is in contrast to only 16.45 ± 12.17% that displayed this phenotype in GFP-Htt28Q-expressing cells (Fig. 2A and C). The presence of disorganized cristae and dilution of the mitochondria matrix are a reflection of mitochondrial damage. Thus, the expression of GFP-Htt74Q not only appears to sensitize cells to an increase in H2O2-induced mitochondrial fragmentation but also leads to defects in the structure of mitochondria. Taken together, these results suggest that the expression of the expanded GFP-Htt74Q protein sensitizes cells to increased damage and fragmentation of mitochondria.
Figure 2.

Electron microscopic examination of mitochondria in HeLa cells stably expressing GFP-Htt polyglutamine fusion proteins. HeLa cell lines stably expressing either GFP-Htt28Q or GFP-Htt74Q proteins were treated with 25 µm H2O2 for 30 min, then fixed and examined by electron microscopy. (A) Representative electron microscopic image of a section taken through two adjacent cells in the GFP-Htt28Q-expressing cell line. Note the variation in size of the mitochondria as well as the close-packed cristae. Bar: 2 µm. (B) Representative electron microscopic image of a section taken through the GFP-Htt74Q expressing cell line. Note that the presence of many small damaged mitochondria that have highly disorganized cristae and whose mitochondrial matrix is considerably less electron-dense (indicated by arrows). Bar: 2 µm. (C) Quantification of the number of abnormal mitochondria (defined as containing both disorganized cristae and a less electron-dense matrix) by electron microscopic examination in the GFP-Htt28Q and GFP-Htt74Q expression lines. For this quantification, 529 and 809 mitochondria seen in 10 and 16 different GFP-Htt28Q- and GFP-Htt74Q cells, respectively, were counted. Data are shown as mean ± SDM (*P < 0.001).
Expanded Htt-polyglutamine proteins induce mitochondrial fragmentation and dysfunction in transiently transfected cells
To confirm our findings that Htt proteins with expanded polyglutamine tracts induce mitochondrial fragmentation, we repeated the analysis this time using transient transfection to express the two GFP-tagged Htt28Q and Htt74Q constructs. As shown in Figure 3A and B, ∼30% of the transiently transfected HeLa cells that expressed GFP-Htt74Q fusion protein contained a preponderance of fragmented mitochondria, when cultured in low serum, compared with only 13% of the cells that expressed GFP-Htt28Q, a result similar to that found with the stable expressing lines. To ensure that these differences were not unique to using the short N-terminal Htt exon-1 fragment (57 amino acids long) or GFP to tag the constructs, we repeated these experiments using a FLAG-tagged N-terminal 548 amino acid fragment of Htt containing either 17 or 138 polyglutamine repeats (23) and again found the construct with the longer polyglutamine tract induced greater mitochondria fragmentation than the construct with the shorter polyglutamine tract (Fig. 3C). To further demonstrate that mitochondrial fragmentation was influenced by the length of the polyglutamine tract and not by the tags used in our constructs, we repeated these experiments using untagged versions of the Htt17Q and Htt138Q constructs. Again, we found that the untagged Htt138Q construct induced greater mitochondrial fragmentation than the untagged Htt17Q construct (Fig. 3D–F).
Figure 3.

Mitochondrial fragmentation and ATP levels of HeLa cells transiently transfected with Htt constructs containing different lengths of polyglutamine repeats. HeLa cells were transfected with plasmid constructs encoding either GFP-, FLAG-tagged or untagged Htt expression constructs containing different lengths of polyglutamine repeats. After transfection, the cells were grown for 18 h in 1.25% FBS and then fixed and immunostained for Tom20 and for the FLAG-expressed protein, where appropriate. Mitochondrial fragmentation in these experiments was quantified by counting between 550 and 1050 cells that stained positive for expression of either the tagged (GFP or FLAG) or untagged Htt-expressed proteins. (A) Representative microscopy images of GFP fluorescence (left panels) and Tom20 staining (right panels) of cells transfected with GFP-Htt28Q (upper panels) or GFP-Htt74Q (lower panels) expression plasmids. Note that the cells expressing GFP-Htt28Q have many tubular mitochondria, whereas those expressing GFP-Htt74Q have numerous small fragmented mitochondria. Bar: 10 µm. (B) Quantification of GFP-expressing cells containing fragmented mitochondria (% of total) following transfection with GFP-Htt28Q and GFP-Htt74Q expression constructs and growth in 1.25% FBS. Data are shown as mean ± SDM (*P < 0.001). (C) Quantification of FLAG-expressing cells containing fragmented mitochondria (% of total) following transfection with FLAG-Htt17Q and FLAG-Htt138Q expression constructs and growth in 1.25% FBS. Data are shown as mean ± SDM (*P < 0.005). (D) HeLa cells were either mock transfected (control) or transfected with untagged Htt17Q or Htt138Q expression constructs. After 24 h, lysates were prepared from the transfected cells and equal amounts of protein were immunoblotted with an anti-huntingtin (upper panel) or anti-actin (lower panel) antibodies. (E) Representative microscopy images of mitochondria visualized by Tom20 staining (right panels) in cells transfected with either Htt17Q (upper panels) or Htt138Q (lower panels) detected with an N-terminal Htt antibody (left panels). Bar: 10 µm. (F) Quantification of the number of transfected cells expressing untagged Htt18Q or Htt138Q proteins cells with fragmented mitochondria grown in conditions similar to (A). Data are shown as mean ± SDM (*P < 0.01). (G) Normalized ATP levels measured in extracts prepared from GFP-Htt28Q or GFP-Htt74Q transfected cells. Data are shown as mean ± SDM (*P < 0.05).
To determine whether the structural changes of mitochondria are accompanied by changes in mitochondria function, we measured ATP levels in cells transfected with the GFP-tagged Htt constructs. As expected, ATP levels in cells transfected with the GFP-Htt74Q construct were significantly lower (30%) than cells transfected with the GFP-Htt28Q construct (Fig. 3G). These results suggest that expanded polyglutamine proteins not only induce mitochondrial fragmentation but also lead to compromised ATP production.
Expanded polyglutamine proteins interfere with the normal dynamics of mitochondria fusion and fission
Mitochondria are highly dynamic organelles that frequently undergo cycles of fusion and fission (12,14,15). We therefore asked whether the increase in mitochondria fragmentation induced by expanded Htt polyglutamine proteins is associated with an alteration in the normal dynamics of mitochondria fusion and/or fission. To do so, we co-transfected cells with an expression construct encoding a photoactivated GFP protein that is targeted to mitochondria (mito-PAGFP) (24) together with expression constructs encoding either monomeric red fluorescent protein (mRFP) or mRFP-tagged Htt exon 1 containing 28 polyglutamine repeats (mRFP-Htt28Q) or mRFP-Htt74Q. Studies have shown that under normal growth conditions, focal activation of the mito-PAGFP protein in mitochondria dissipates throughout the whole mitochondrial network within the cell over time and that inhibition of mitochondria fusion prevents both the dissipation and decay in fluorescence in mitochondria (24). Using a similar approach, we analyzed changes in GFP fluorescence of mitochondria in the cells that were co-transfected with the PAGFP reporter with each of the different mRFP constructs. An immunoblot analysis of proteins from these transfections confirmed that all three mRFP proteins migrated according to their predicted molecular mass by SDS–PAGE (Fig. 4A). Furthermore, we found that over 50% of mRFP-Htt74Q expressing cells contained fragmented mitochondria compared with only ∼10% in cells expressing either mRFP-Htt28Q or mRFP alone.
Figure 4.
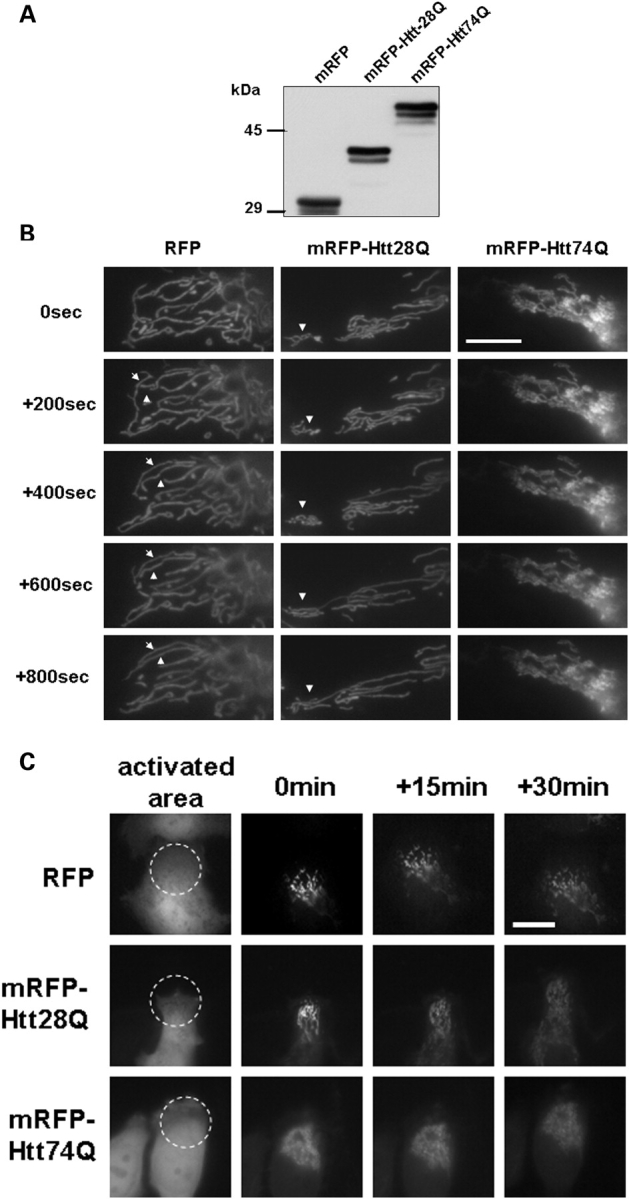

Mitochondrial dynamics in living cells analyzed using GFP photoactivation and time-lapse microscopy. HeLa cells were co-transfected with a mitochondrial matrix targeted photoactivable GFP (mito-PAGFP) and mRFP, or mRFP-Htt28Q or mRFP-Htt74Q expression plasmids. After 24 h transfection, cells were subjected to photoactivation studies, or lysates were prepared from the cells for immunoblot analysis. (A) Expression of mRFP, mRFP-Htt28Q and mRFP-Htt74Q proteins detected by immunnoblotting proteins lysates from the transfected cells with a rabbit polyclonal anti-RFP antibody. (B) Mitochondria in cells expressing mRFP or mRFP-Htt28Q proteins display more dynamic movement and fusion compared with cells expressing mRFP-Htt74Q protein. HeLa cells that had been co-transfected with mito-PAGFP and mRFP, or mito-PAGFP and mRFP-Htt28Q, or mito-PAGFP and mRFP-Htt74Q were imaged under a fluorescent microscope using a 100× objective lens and those displaying RFP fluorescence were targeted for photoactivation of the co-transfected mito-PAGFP protein by illumination with 405 nm light, and then GFP fluorescence images were captured over the indicated time intervals. Arrowheads point to the mitochondria that fused with one another in the time period shown. Please note that in the GFP-Htt74Q transfected cell, mitochondria were smaller, clustered, lacked dynamic movement and fusion events were very infrequent. (C) Same as in (B), except that the images shown were captured using a 40× objective. The left-hand panels show the mRFP fluorescence seen in a group of cells and the region (indicated by the circle) that was illuminated with 405 nm light to photoactivate the co-expressed PAGFP-mito protein in the various transfected cells. The subsequent GFP fluorescence images captured at 0, 15 and 30 min after GFP photoactivation are shown on the right of the RFP fluorescence image captured for each construct. (D) Quantification of the changes in GFP fluorescence intensity over time in cells transfected with the different mRFP-tagged expression constructs. GFP fluorescence was measured at 0, 15 and 30 min after photoactivation in the photoactivated (a, b and c) and non-activated regions (e, f and g, respectively) in cells transfected with mRFP or mRFP-Htt28Q or mRFP-Htt74Q expression constructs, respectively. The plots depict the results obtained in 10 independent experiments (each shown with a different color). Note that GFP fluorescence in the photoactivated regions, in general, decreases faster in the cells expressing either mRFP, or mRFP-Htt28Q proteins, compared with those expressing mRFP-Htt74Q protein. These changes were accompanied by a gradual increase in GFP fluorescent intensity in the non-activated regions of cells expressing either mRFP or mRFP-Htt28Q proteins, but remained relatively constant in cells expressing mRFP-Htt74Q. These results are consistent with the idea that mitochondria in cells expressing either the mRFP or the mRFP-HttQ28 proteins exhibit greater mitochondria fusion than cells expressing the mRFP-HttQ74 protein.
We next used time-lapse microscopy to follow changes in the distribution and intensity of GFP fluorescence contained in photoactivated regions of interest (ROIs) in cells co-expressing the mito-PAGFP with each of the different mRFP proteins. As shown in Figure 4B and C, mitochondria in the cell transfected with the mRFP and mRFP-Htt28Q constructs underwent relatively rapid remodeling, some of which fused to form long tubular mitochondria at the indicated time points (see additional Supplementary Material, Movies S1 mRFP-Htt28Q and S2 mRFP). By contrast, mitochondria in cells transfected with the mRFP-Htt74Q construct lacked such dynamic behavior. Instead, they tended to remain static and appeared to fragment and roundup over time (see Supplementary Material, Movie S3 mRFP-Htt74Q). We quantified how the intensity of GFP fluorescence in the photoactivated ROIs changed in cells expressing the different mRFP proteins over time. In mRFP and mRFP-Htt28Q-transfected cells, GFP fluorescence decreased rapidly in the 15 min period after its photoactivation (Fig. 4C, 4D, a and b). In accordance with this result, we also observed a concomitant increase in the intensity of GFP fluorescence in the surrounding non-photoactivated regions of the cells, which we hypothesized to arise from rapid segregation of the photoactivated mito-GFP protein into the neighboring mitochondrial network by fusion (Fig. 4D, d and e; Supplementary Material, Movies S1 and S2). In contrast, the intensity of GFP fluorescence in the photoactivated areas of mRFP-Htt74Q-transfected cells reduced more gradually than in the mRFP or mRFP-Htt28Q-transfected cells (Fig. 4C and Dc; Supplementary Material, Movie S3). Moreover, the intensity of GFP fluorescence in the non-activated areas of mRFP-Htt74Q-transfected cells remained relatively constant (Fig. 4Df), which is expected in cells that have reduced mitochondrial fusion.
Overexpression of Drp-1K38A reduces polyglutamine-induced mitochondrial fragmentation, diminution of ATP levels in cells and propagation of cell death
Fission and fusion of mitochondria are regulated by a number of key proteins including dynamin-related protein 1 (Drp1) and mitofusin-2 (Mfn2). Recruitment of Drp1 into mitochondria promotes mitochondrial fission, whereas expression of a dominant-negative mutant, Drp-1K38A, inhibits mitochondrial fission and promotes mitochondrial fusion (15). To determine whether overexpression of the dominant-negative Drp-1K38A mutant reduces mitochondrial fragmentation in cells expressing expanded polyglutamine proteins, we co-transfected HeLa cells with either GFP-Htt28Q or GFP-Htt74Q expression plasmids together with or without the Drp-1K38A expression construct and examined the cells for mitochondria staining using the anti-Tom20 antibody. Considerably fewer cells co-transfected with the Drp-1K38A mutant and GFP-Htt74Q expression constructs contained fragmented mitochondria than cells transfected with the GFP-Htt74Q construct alone (Fig. 5A). This abrogation of mitochondria fragmentation by the Drp-1K38A mutant was recapitulated in additional experiments in which we transfected HeLa cells with the FLAG-tagged Htt17Q and FLAG-Htt138Q expression constructs with or without the Drp-1K38A mutant (Fig. 5B). Because overexpression of Drp-1K38A reduced mitochondria fragmentation in GFP-Htt74Q expressing cells, we measured ATP levels in protein lysates prepared from the transfected cells to see if it abrogates the reduction in ATP levels induced by expression of GFP-Htt74Q fusion protein. As shown in Figure 4C, cells co-transfected with the Drp-1K38A mutant and GFP-Htt74Q contained ATP levels comparable to untransfected cells, or cells transfected with GFP-Htt28Q, which is in sharp contrast to the reduced levels of ATP seen in cells transfected with the GFP-Htt74Q construct alone. Furthermore, cytotoxicity assays revealed that expression of the Drp-1K38A mutant significantly reduced cell death induced by expression of the GFP-Htt74Q construct, as reflected by both decreased nuclear fragmentation (Fig. 5D) and nuclear staining with terminal deoxynucleotidyl transferase biotin-dUTP nick-end labeling (TUNEL) (Fig. 5E). Thus, these experiments demonstrate that interference of normal mitochondrial fission by forced expression of the dominant-negative Drp-1K38A mutant protects cells against expanded polyglutamine-induced mitochondrial fragmentation, the reduction of ATP levels and cell death.
Figure 5.

The dominant-negative Drp-1 mutant, Drp-1K38A, reduces mitochondrial fragmentation and cell death. HeLa cells were co-transfected with either GFP-Htt28Q or GFP-Htt74Q along with a Drp-1K38A construct or a control vector. Twenty-four hours after transfection, cells were fixed and immunostained with a Tom20 antibody and mitochondrial morphology was analyzed under a fluorescence microscope. Alternatively, the transfected cells were vitally stained with a DNA binding dye Hoechst to observe nuclear morphology and fragmentation. Mitochondrial fragmentation and cell death in these experiments were quantified by counting at least 500 GFP-Htt28Q and GFP-Htt74Q-expressing cells, respectively. (A) Overexpression of the Drp-1K38A mutant reduces GFP-HttQ74-induced mitochondrial fragmentation. Cells with or without fragmented mitochondria (see Materials and Methods) were counted using a fluorescence microscope using a 100× objective lens. The results show the percentage of cells with fragmented mitochondria in the population of GFP-expressing cells in cells transfected with or without the Drp1K38A mutant construct (*P < 0.001). (B) Similar to (A), except that the analysis was conduced in cells transfected with FLAG-tagged Htt-17Q or Htt-138Q constructs. Expression of the Drp-1K38A mutant reduces FLAG-Htt138Q induced mitochondrial fragmentation (*P < 0.05). (C) Similar to (A), but this time ATP levels were measured in cell extracts obtained after the transfections. Expression of the Drp-1K38A mutant alleviates the GFP-Htt74Q-induced reduction in ATP levels (*P < 0.01). (D) Same as (A), but this time nuclear fragmentation was measured 20 h after the transfections. Drp-1K38A reduces GFP-Htt74Q caused nuclear fragmentation (*P < 0.005). (E) Same as (A), but this time DNA damage was measured by TUNEL staining. Expression of the Drp-1K38A mutant reduces GFP-Htt74Q-induced DNA damage (*P < 0.001). The data in all the graphs shown are the mean ± SDM.
Overexpression of Mfn2 reduces polyglutamine-induced mitochondrial fragmentation, diminution of ATP levels in cells and propagation of cell death
We next examined whether promotion of mitochondrial fusion in the absence of interference of mitochondrial fission would reduce polyglutamine-induced mitochondrial fragmentation, the reduction of ATP levels and cell death. Accordingly, we co-transfected HeLa cells with either GFP-Htt28Q or GFP-Htt74Q with or without a Myc-tagged Mfn2 expression construct and then examined the extent of mitochondrial fragmentation and cell death in the transfected cells. We confirmed that the cells transfected with the GFP-tagged polyglutamine and Myc-tagged Mfn2 constructs overexpressed the appropriate size proteins by immunoblotting, as shown in Figure 6A. Immunostaining of the cells for Tom20 revealed that like the Drp-1K38A mutant, Mfn2 significantly reduced GFP-Htt74Q-induced mitochondrial fragmentation (Fig. 6B) and restored the decrease in ATP levels induced by expression of GFP-Htt74Q protein (Fig. 6C). Furthermore, overexpression of Mfn2 also reduced GFP-Htt74Q-induced cell death as monitored by an analysis of nuclear fragmentation (Fig. 6D). The protective effect of Mfn2 might be mediated through inhibition of cell death through the apoptotic pathway, since caspase-3 activation was not observed in the cells co-transfected with both GFP-Htt74Q and Myc-Mfn2 constructs (Fig. 6E). Consistent with this idea, overexpression of FLAG-tagged c-IAP (inhibitor of apoptosis-1), an anti-apoptotic molecule that inhibits caspases and targets other apoptotic related molecules for degradxation (25), also reduced caspase-3 activation following exposure of GFP-Htt74Q and GFP-Htt28Q expressing cell lines to oxidative stress (Fig. 7A–C). The inhibitory effect of c-IAP was dependent on its ubiquitin ligase activity because overexpression of a FLAG-tagged c-IAP H588A mutant, which is defective in ubiquitin ligase activity (26), was ineffective in suppressing caspase-3 activation and cell death (Fig. 7B and C).
Figure 6.

Overexpression of Mfn2 reduces expanded polyglutamine-induced mitochondrial fragmentation and cell death. HeLa cells were co-transfected with either GFP-Htt28Q or GFP-Htt74Q expression constructs together with a Myc-tagged Mfn2 expression construct or an empty vector control plasmid. Twenty-four hours after the transfection, cells were fixed and immunostained with either the anti-Tom20 antibody or Hoechst. Additionally, protein lysates were prepared from the transfected cells and analyzed for expression of various proteins by immunoblotting. Mitochondrial fragmentation and cell death was quantified by counting a minimum of 650 GFP-Htt28Q and GFP-Htt74Q-expressing cells. (A) Immunoblots showing the expression of Myc-tagged Mfn2, and GFP-tagged Htt28Q and Htt74Q proteins probed with an anti-Mfn2 antibody (upper panel, arrow head indicates Myc-tagged Mfn2 and the asterisk, endogenous Mfn2) an anti-myc antibody (second panel) and anti-GFP antibody (third panel). The actin blot (lower panel) serves to demonstrate equal protein loading. (B) Overexpression of Mfn2 reduces GFP-HttQ74-induced mitochondrial fragmentation. Cells with or without fragmented mitochondria (see Materials and Methods) were counted using a fluorescence microscope using a 100× oil objective lens. The resulting data is presented as the percentage of cells with fragmented mitochondria of the total GFP expressing cells (*P < 0.01). (C) Overexpression of Mfn2 alleviates the GFP-Htt74Q-induced reduction of ATP levels in cells (*P < 0.05). (D) Overexpression of Mfn2 reduces GFP-Htt74Q-induced nuclear fragmentation (*P < 0.001). (E) Overexpression of Mfn2 inhibits GFP-Htt74Q-induced caspase-3 activation. The data in all the graphs shown are the mean ± SDM.
Figure 7.
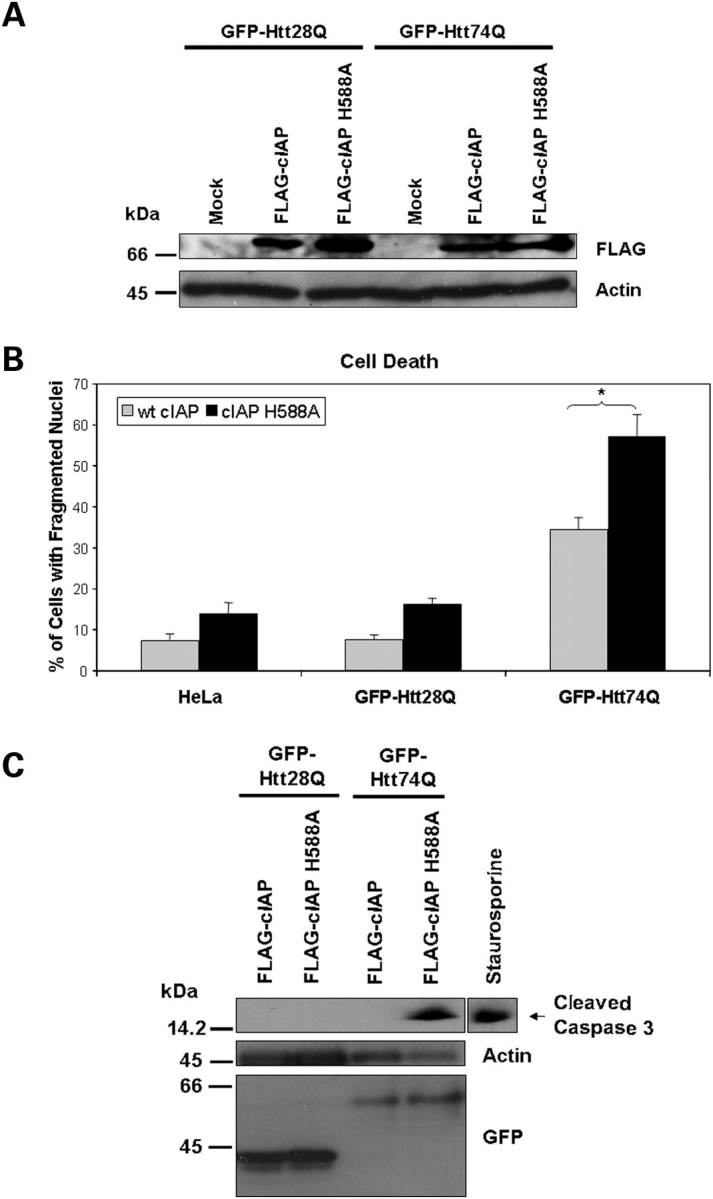
Overexpression of wild-type c-IAP but not a c-IAP H588A RING mutant suppresses expanded polyglutamine induced caspase-3 activation and cell death. GFP-28Q and GFP-74Q stable expressing HeLa cell lines were either mock transfected or transfected with either a FLAG-tagged wild-type c-IAP or a c-IAP H588A RING-finger ubiquitin ligase mutant. Following transfection, the cells were cultured in medium containing low serum (1.25% FBS) and cell death was quantified after staining with Hoechst, or alternatively protein lysates were prepared and immunoblotted for expression of the FLAG-tagged constructs or for caspase-3 activation. (A) Immunoblot of protein lystates showing expression of the FLAG-tagged c-IAP constructs (upper panel) and actin (lower panel) used as a loading control. (B) Expression of wt FLAG-tagged c-IAP reduces oxidative stress-induced cell death in GFP-Htt28Q and GFP-Htt74Q-expressing lines compared with the expression of the c-IAP-H588A mutant. The data shown are the quantification of fragmented nuclei in the parental HeLa cell line (HeLa) and the two daughter GFP-Htt28Q and GFP-Htt74Q-expressing cell lines. At least 500 cells were counted for these quantifications. Mean ± SEM (*P < 0.001). (C) Overexpression of wt FLAG-tagged c-IAP, but not the c-IAP H588A mutant inhibits oxidative stress-induced caspase-3 activation in the GFP-Htt74-expressing cell line. A HeLa cell lysate from a staurosporine treated culture (right lane) was used as a positive control to detect the caspase-3-cleaved fragment.
Reduction of Drp-1 levels in Caenorhabditis elegans by RNA interference reduces the motility defect seen in a worm model of HD
We next examined whether reduction in mitochondrial fission might be beneficial in preventing toxicity of expanded Htt proteins expressed in animals. For this investigation, we used two C. elegans lines that we had engineered to express the GFP-Htt28Q or GFP-Htt74Q fusion proteins in muscle cells of the worm (20). In these worms, the expression of the GFP-tagged Htt-fusion proteins results in a polyglutamine length-dependent reduction in worm motility, which can be measured by counting the number of body bends flexed by the animals over a fixed period of time (20). Using these lines, we measured motility in animals that were fed Escherichia coli transformed with plasmids to either knock down Drp-1 levels by RNA interference (RNAi) or not. An immunoblot of extracts prepared from these worms confirmed that the worms that had been targeted for genetic interference of Drp-1 had greater than 70% reduction in Drp-1 protein levels compared with worms in which Drp-1 had not been targeted for disruption (Fig. 8A). Interestingly, genetic interference of Drp-1 led to an increase in expression of the GFP-Htt28Q and GFP-Htt74Q fusion proteins in the worms, by an unknown mechanism. Despite this elevation, GFP-Htt28Q- and GFP-Htt74Q-expressing worms in which Drp-1 levels were reduced displayed increased motility compared with their counterparts that were fed bacteria transformed with the L4440 vector alone (Fig. 8B). Motility in the GFP-Htt28Q-expressing line was restored to that seen in non-transgenic C. elegans lines [approximately 32 body bends per minute (20)], whereas in the GFP-Htt74Q-expressing worms, motility improved 200% over that seen in the absence of Drp-1 knock-down. These results suggest that inhibition of mitochondrial fusion is beneficial for reducing the toxicity of expanded Htt-containing polyglutamine proteins in animals.
Figure 8.

Knock-down of Drp-1 levels in C. elegans by RNAi reduces the motility defect seen in lines expressing GFP-Htt polyglutamine fusion proteins. (A) Immunoblot analysis of C. elegans GFP-Htt28Q and GFP-Htt74Q lines showing reduction of Drp-1 proteins levels using the bacterial RNAi feeding protocol. Equal amounts of protein extract prepared from GFP-Htt28Q and GFP-Htt74Q worms that were fed either L4440 vector (control) or L4440-containing Drp-1 to induce genetic interference of Drp-1 (Drp-1 RNAi) or not, respectively, were immunoblotted with either a Drp-1 antibody (upper panel), a GFP antibody (middle panel) or an antibody that recognizes an unknown Protein-X, used as a control for protein loading (lower panel). (B) Motility assays of GFP-Htt28Q and GFP-Htt74Q worm lines that were fed bacteria transformed with plasmids to either induce genetic interference of Drp-1 or not. Note that genetic interference of Drp-1 by RNAi significantly reduces the motility defect seen in both GFP-Htt28Q and GFP-Htt74Q expressing worms (*P < 0.05).
GST-Htt-fusion proteins containing an expanded polyglutamine tract interact preferentially with mitofusin proteins
The mechanisms by which expanded polyglutamine proteins induce fragmentation of mitochondria are not known. We hypothesized that it might stem from increased binding and interference of the function of mitofusin proteins. Accordingly, we conducted GST pulldown assays examining whether mitofusin proteins contained in HeLa cell lysates bind differentially to GST Htt-fusion proteins containing different lengths of polyglutamine repeats. By these assays, we found that incubation of HeLa lysates prepared from cells transfected with a Myc-Mfn2 construct with either recombinant GST or GST-Htt28Q or GST-Htt74Q proteins resulted in stronger binding of Myc-Mfn2 with GST-Htt74Q fusion protein than with GST-Htt28Q or GST proteins (Fig. 9, top and middle panels). Interestingly, we also detected binding between endogenous Mfn2 with the GST-Htt74Q fusion protein, but not the other GST proteins (Fig. 9, middle panel). Unfortunately, we could not study binding of Mfn1 protein in these assays because we did not have access to a good Mfn1 antibody. Nevertheless, our results suggest that expanded polyglutamine proteins might induce an increase in mitochondrial fragmentation and cell death by binding and interfering with Mfn2 protein function.
Figure 9.

Expanded polyglutamine proteins interact with mitofusin proteins. Bacterial expressed GST, GST-HttQ28 and GST-HttQ74 fusion proteins were purified by GS-agarose affinity chromatography. Next extracts were prepared from HeLa cells transfected with the myc-tagged Mfn2 expression construct, which were then passed over the GS-agarose columns, repeatedly. After extensive washing, the proteins that bound to the column were analyzed by immunoblotting. Upper panel: proteins immunoblotted with an anti-myc antibody reveal that the myc-tagged mfn2 protein binds preferentially to GST-HttQ74 fusion protein, but not GST alone or GST-HttQ28 fusion protein. Middle panel: proteins immunoblotted with an anti-Mfn2 antibody reveal preferential binding of both endogenous (arrow head) and exogenous myc-tagged (asterisk) Mfn2 proteins with GST-HttQ74 protein. Lower panel: immunoblot of the lysates with a anti-GST antibody detects the appropriate size GST proteins used for the bindings assays.
DISCUSSION
Here, we demonstrate that N-terminal Htt proteins containing polyglutamine tracts in the pathological range (74 or 138 glutamine repeats) induce greater mitochondrial fragmentation than proteins containing polyglutamine tracts in the non-pathological range (17 or 28 glutamine repeats). In addition, we found that cells expressing the expanded polyglutamine proteins contain lower levels of ATP, suggesting that expanded polyglutamine proteins interfere with ATP generation by mitochondria. Our results of the changes in fluorescence of a photoactivatable GFP protein localized in mitochondria suggest that expanded polyglutamine proteins reduce mitochondria movement and fusion. Remarkably, our results have demonstrated that methods that drive mitochondrial fusion, namely overexpression of the dominant-negative mitochondria fission protein Drp-1K38A, or overexpression of mitochondria fusion-promoting protein Mfn2, not only prevented mitochondrial fragmentation, but that they also restored ATP levels and reduced cell death induced by expanded polyglutamine proteins. As further proof of this principle, we found that reduction in Drp-1 by RNAi, which is expected to reduce mitochondrial fusion, reduced the motility defect associated with expression of expanded Htt-polyglutamine fusion proteins in a C. elegans model of HD. These results might have important implications for both understanding the mechanisms and preventing the toxicity of expanded polyglutamine proteins involved in HD.
There are several other studies that suggest that mitochondria dysfunction might be involved in HD pathogenesis. For example, studies utilizing cell, animal and human subjects have shown that HD is associated with a reduction in ATP levels in cells, increased lactate production, impairment of calcium homeostasis and enhanced oxidative stress, all of which can be linked to mitochondria dysfunction (6,7,27–29). However, how mitochondria become dysfunctional in HD remains largely unknown, although a recent study suggests that the N-terminal portion of the Htt polypeptide, which is the portion of the protein we used in our study, might play an instrumental role in binding and inhibiting mitochondria transport (30). Our results support this idea and furthermore suggest that the N-terminal Htt protein containing an expanded polyglutamine tract, but not those containing a short polyglutamine tract, binds preferentially to Mfn proteins, which are localized to the outer mitochondrial membrane. We speculate that the increased binding of expanded Htt proteins with Mfn proteins probably compromises the normal function of Mfn proteins resulting in increased fragmentation of mitochondria. Recently, it was reported that increased mitochondria fragmentation is observed in cells in which the death promoting protein bax is activated (24), and in oxidative stress-induced cells (16). It is conceivable that excessive fragmentation of mitochondria results in the release of proapoptotic proteins that are normally sequestered in mitochondria, such as cytochrome c, triggering caspase-dependent cell death cascades.
The notion that expanded polyglutamine proteins increase cytotoxicity by enhancing mitochondrial fragmentation is supported by our results showing that overexpression of either Drp-1K38A, a dominant-negative mutant of Drp1, or Mfn2 not only reduces mitochondrial fragmentation, but also suppresses ATP depletion, caspase activation and cell death. There is growing evidence suggesting that proteins involved in regulating fission and fusion of mitochondria are essential for cell survival, division and differentiation (12). Downregulation of Drp1 or overexpression of Drp-1K38A, a dominant-negative Drp1, inhibits apoptotic cell death (14,31). It is interesting to note that Drp1 might also mediate caspase-independent cell death (32). Similarly, there is strong evidence that indicates that Mfn2, a mitochondrial fusion protein, plays an important role in cell survival. Mutations in Mfn2 have been linked to autosomal dominant Charcot–Marie–Tooth type 2A disease, a peripheral neuropathy (13,33). Moreover, Chen et al. (19) demonstrated that Mfn2, but not Mfn1, is required for differentiation and survival of Purkinje cells in vivo. Finally, Mfn2 was also showed to protect neuronal cells against oxidative stress-induced cell death (18,34).
The two methods we used to enhance mitochondria fusion are quite different in the molecular mechanisms by which Mfn2 and Drp-1K38A are thought to stimulate mitochondria fusion (19,35). This observation leads us to speculate that methods that simply tip the balance towards mitochondria fusion, rather than fission, might have a beneficial role in HD therapy. A practical illustration of the utility of such a therapy is our demonstration that inhibition of mitochondrial fission in a C. elegans model of HD can reduce the toxicity of expanded Htt proteins. Obviously, any method that interferes with the normal dynamics of mitochondrial fusion and fission would have to be carefully regulated so as not to drive the dynamics too far in the opposite direction, which might lead to other abnormalities.
We are especially intrigued by the known properties of Mfn2 in conferring protection against cell death, which might explain its protective role against expanded polyglutamine proteins described here. One possibility is that Mfn2 might interfere with the apoptotic function of Bax. During apoptosis, Bax appears to co-localize with both Mfn2 and Drp1 (36). Studies have shown that overexpression of Mfn2 can interfere and block the apoptotic action of Bax (34). A particularly interesting finding leading us to believe that Mfn proteins might indeed play an important role in HD pathogenesis was our demonstration that expanded polyglutamine repeats bind more strongly with Mfn2 proteins than GST proteins containing 28 polyglutamine repeats. One simple possibility we propose is that this binding interferes with the normal function of Mfn proteins in mitochondria fusion. In support of this idea, overexpression of Mfn2 rescued polyglutamine-induced mitochondria fragmentation. Another possibility that we cannot discount is that expanded polyglutamine proteins might somehow cause cytotoxicity by impeding mitochondria movement. Consistent with this idea, we found that mitochondria in cell expressing expanded polyglutamine proteins were indeed compromised in movement. This is in accord with other studies that have shown mutant huntingtin proteins impair mitochondrial trafficking (30,37,38). Regardless of the mechanism by which expanded polyglutamine increase mitochondria fission, our results clearly demonstrate that methods to modulate mitochondria fusion and fission, particularly those that promote fusion, might have a beneficial role in treatment of HD.
MATERIALS AND METHODS
Plasmid DNA constructs
Mammalian cytomegalovirus (CMV) expression constructs encoding either GFP, GFP-Htt28Q or GFP-Htt74Q were described previously (20,39). Mammalian CMV expression constructs encoding FLAG-tagged Htt17Q and FLAG-Htt138Q fusion proteins were generously provided by Dr Shengyun Fang (UMBI) and were described previously (23). The latter constructs were used generate untagged versions of the proteins by subcloning an N-terminal NcoI fragment of Htt into our own CMV expression plasmid, resulting in constructs encoding untagged-Htt17Q and untagged-Htt138 encompassing codons 1–549 of Htt. To express GST-Htt exon 1 fusion proteins containing different lengths of polyglutamine repeats in bacteria, pCMV-EGFP-Htt28Q or pCMV-EGFP-Htt74Q expression plasmids were each digested with BglII and EcoRI restriction enzymes and the resulting fragments encoding human Htt exon 1 containing either 28Q or 74Q, but devoid of GFP sequences, were fused in-frame and downstream of GST in the pGST-T1 expression vector (Pharmacia) between its BamHI and EcoRI sites. The same Htt28Q and Htt74Q exon 1 fragments was also inserted between the BglII and EcoRI sites in a mRFP-C1 expression plasmid to make CMV mammalian expression constructs encoding mRFP fused at the N-termini of Htt28Q or mRFP-Htt74 fusion proteins, respectively. The mitochondrial-matrix-targeted photoactivatable GFP (mito-PAGFP), the dominant-negative Drp1 mutant, Drp-1K38A, and myc-tagged Mnf2 plasmid constructs have been described previously (36). The cIAP constructs were described previously (26); the only difference was that the constructs were tagged with a FLAG epitope rather than the original Myc-tag.
Cell culture and transfection
HeLa cells were grown at 37°C on glass cover slips in 6- or 24-well culture plates containing DMEM medium supplemented with 10% FBS. They were transfected with plasmid DNAs using either the calcium phosphate co-precipitation method (20) or using Lipofectamine 2000 reagent (Invitrogen). Both transfections lasted for 4 h and were conducted in medium lacking serum, after which it was replaced with a medium containing either 10% or 1.25% FBS, as indicated. For H2O2 treatment, cells were exposed to 25 μm H2O2 in complete medium for 30 min. For serum deprivation treatment, cells were incubated in a medium containing DMEM supplemented with 1.25% FBS for at least 18 h.
Stable cell lines expressing Htt-fusion proteins
Cells stably expressing either GFP-Htt28Q or GFP-Htt74Q have been described previously (20). Cells were cultured on glass cover slips in six-well culture plates in complete medium (DMEM supplemented with 10% FBS) and exposed to H2O2 or serum deprivation as described above.
Quantification of cell death
Cell death was quantified by two methods: by counting the number of GFP or RFP expressing cells that were also positive for TUNEL staining or by counting the number of GFP or RFP expressing cells whose membrane integrity had been destroyed as indicative by their failure to exclude staining with the DNA dye Hoechst 33342 (20). TUNEL staining of fixed cells was performed with the DeadEnd colorimetric TUNEL staining kit (Promega). Cells whose membrane integrity had been destroyed were identified by adding Hoechst 33342 (1 µg/ml final conc.) to the culture medium for 20 min prior to examination by live fluorescence microscopy with a Zeiss 100 inverted microscope the number of GFP or RFP expressing cells that exhibited nuclear DNA staining.
Immunocytochemical staining of mitochondria
We followed the published procedure (40) to fix cells and stain mitochondria. Briefly, cells grown on glass cover slips were fixed with 4% paraformaldehyde for 30 min, permeabilized with 0.15% Triton X-100 for 15 min and then blocked with 10% bovine serum albumin for 45 min at room temperature. The cover slips were then incubated with a mouse monoclonal anti-Tom20 (1:250; BD Biosciences, NJ, USA), followed by incubation with goat anti-mouse Alexa Fluor 546 antibody (1:200; Molecular Probes, Eugene, OR, USA). Immunostained cells were observed with a fluorescence microscope (Zeiss Axiovert 200) using a 100× oil objective and images were captured with a Hamamatsu camera using the C-imaging software. We utilized the method described by Barsoum et al. (16) to categorize cells into those that contained fragmented or non-fragmented mitochondria. In this procedure, cells that contain elongated tubular mitochondria were scored as possessing non-fragmented mitochondria, whereas cells that contained ≥90% small, round mitochondria were scored as possessing fragmented mitochondria. Expression of untagged Htt17Q and Htt138Q proteins were detected with a rabbit polyclonal antibody (sc8767) specific for the N-terminal portion of Htt protein (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA).
Electron microscopy
Cells grown on 35 mm plastic dishes were fixed in 2% glutaraldehyde in 0.1 m cacodylate with 3% sucrose and 3 mm CaCl2 and postfixed for 1 h with reduced osmium tetroxide. The cells were dehydrated through graded ethanol changes and embedded in Eponate 12 Resin (Ted Pella, Redding, CA, USA). Thin sections cut on a Reichert-Jung E microtome were placed on 200 mesh copper grids and stained with uranyl acetate followed by lead citrate. The grids were examined on a Philips CM 120 transmission electron microscope with a Gatan Orius SC 1000 4 digital camera.
Photoactivation studies of mitochondria
We utilized an expression construct encoding a photoactivatable GFP protein that is targeted to the mitochondria matrix (mito-PAGFP) (24) to study fusion, fission and movement of mitochondria in living cells over time. The mito-PAGFP protein, when expressed in cells, can be photoactivated by focal illumination with 405 nm light, enabling mitochondria containing the photoactivated GFP protein to be visualized under the microscope. Furthermore, by time-lapse fluorescence microscopy, it is possible to track movement, fusion and fission of mitochondria (24). Accordingly, we grew HeLa cells on cover glasses and co-transfected them with the mito-PAGFP construct together with either mRFP-tagged Htt28Q or Htt74Q constructs using Lipofectamine 2000 (Invitrogen). Approximately 20 h after transfection, a cover glass containing the transfected cells was placed in a 37°C FCS2 temperature-regulated chamber (Bioptechs, Inc., Butler, PA, USA) where all subsequent imaging of the cells was done using a 100× oil objective and a Hamamatsu Orca camera attached to an inverted Zeiss microscope. Transfected cells were identified by expression of the different mRFP-tagged proteins, and an ROI of these cells was photoactivated with 405 nm light for 5–10 s. Immediately after photoactivation, GFP fluorescence in the whole cell was monitored over a period of several minutes. The images were analyzed using the C-imaging software to detect fusion, fission and movement of mitochondria. Changes in fluorescence intensity in mitochondria and other regions of the cell were quantified using the C-imaging software.
ATP measurement
ATP measurement was performed with an ATP determination kit (Invitrogen) by following the manufacturer’s instruction and normalized against measured total protein of the cell lysates.
Immunoprecipitation, immunoblotting and GST pulldown assay
Immunoprecipitation and immunoblotting were performed as described before (41). The antibodies used for western blotting were a rabbit polyclonal anti-GFP (1:3000, generated to recombinant GST–GFP fusion protein by us), a rabbit polyclonal anti-RFP (1:3000, generated to recombinant GST–RFP fusion protein by us), a rabbit polyclonal anti-Mfn2 (1:1000), a rabbit polyclonal (sc-32898, Santa Cruz Biotechnology) and a mouse monoclonal anti-Drp1 (DLP-1; 1:1000, BD Biosciences), a mouse monoclonal anti-Mfn1 (clone 3C9, Novus Biologicals, CO, USA) or a goat polyclonal anti-actin (1:2000) antibody (Santa Cruz Biotechnology). GST pulldown assays were performed as described previously (23). Briefly, GST, GST-Htt28Q and GST-Htt74Q fusion proteins were expressed in BL21 E. coli cells by IPTG induction and immobilized onto glutathione-agarose beads. Next, HeLa cells were transfected with or without myc-tagged Mnf2 and the cells were lyzed in buffer containing 0.5% Triton X-100 and protease inhibitor cocktails in PBS. The protein–glutathione–agarose bead complexes were incubated with HeLa cell lysate supernatants transfected with myc-tagged Mfn2. The beads were washed with pulldown buffer (0.5% Triton X-100 in PBS) and the bound proteins eluted with SDS–protein gel loading buffer. The eluted proteins were separated by SDS–PAGE and transferred to nitrocellulose membrane and immunoblotted with a rabbit polyclonal anti-Mfn2 antibody (34) or a mouse monoclonal anti-myc antibody (Santa Cruz Biotechnology, Inc.) or an anti-GST antibody.
C. elegans methods
Nematodes were maintained and grown under standard cultivation conditions (42). The generation and characterization of transgenic lines expressing GFP-tagged Htt28Q or Htt74Q fusion proteins in muscle cells of the worm were described previously (20).
Expression of Drp-1 was knocked down using the bacteria-induced RNAi feeding protocol used in worms (43,44). The RNAi clone, T12E12.4, containing Drp-1 cloned in L4440 vector was obtained from Open Biosystems (Huntsville, AL, USA) and both this and the L4440 vector alone were transformed into E. coli strain HT115 (DE3), using standard methods. Individual colonies from the resulting transformants were inoculated into Luria broth containing ampicillin (50 µg/ml) and tetracycline (12.5 µg/ml) and grown overnight at 37°C. Cultures were diluted 1:100 and allowed to grow to an OD600 of approximately 0.4 after which time isopropyl β-d-thiogalactoside (IPTG) was added to 0.4 mm and incubation was continued for another 4 h to induce synthesis of double-stranded RNA. Bacteria were then seeded onto NGM plates containing ampicillin and IPTG and allowed to dry overnight.
For motility assays, individual animals were picked to fresh plates and allowed to feed on the IPTG-induced bacteria for 48 h after which time the number of body bends propagated in a 1 min interval of continuous movement was counted for 50 independent animals using a Leica dissection stereomicroscope. Statistical significance of the results was determined by a t-test.
Preparation of protein lysates and immunoblotting were performed as described previously (20). Antibodies against GFP and Drp-1 were obtained from Roche Applied Science (Indianapolis, IN, USA) and Santa Cruz Biotechnology, respectively. An unknown protein, which we called Protein-X, whose expression was not altered by the RNAi procedure was used as a control for protein loading.
Statistical analysis
To perform statistical analysis, Student’s t-test will be used for comparison of parametric data from cell populations and one-way analysis of variance will be applied. Data shown is the mean ± standard deviation of the mean (SDM) and P < 0.05 are considered as statistically significant.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work was supported in part by grants from the National Institutes on Health (NIA AG016839 and GM 066287) to M.J.M.
ACKNOWLEDGEMENTS
We thank the members of the Monteiro laboratory for their critical comments and suggestions. We are extremely grateful to Carol A. Cooke (John Hopkins University) for conducting the EM. We would also like to thank Drs. David Rubinsztein (University of Cambridge), Marian DiFiglia (Massachusetts General Hospital) and Shengyun Fang (UMBI) for kindly providing Htt expression constructs and Dr. Shengyun Fang for the wt c-IAP and mutant c-IAP H588A expression plasmids.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Imarisio S., Carmichael J., Korolchuk V., Chen C.W., Saiki S., Rose C., Krishna G., Davies J.E., Ttofi E., Underwood B.R., et al. Huntington’s disease: from pathology and genetics to potential therapies. Biochem. J. 2008;412:191–209. doi: 10.1042/BJ20071619. [DOI] [PubMed] [Google Scholar]
- 2.The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 3.Bates G. Huntingtin aggregation and toxicity in Huntington’s disease. Lancet. 2003;361:1642–1644. doi: 10.1016/S0140-6736(03)13304-1. [DOI] [PubMed] [Google Scholar]
- 4.Gatchel J.R., Zoghbi H.Y. Diseases of unstable repeat expansion: mechanisms and common principles. Nat. Rev. Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- 5.Beal M.F. Mitochondria take center stage in aging and neurodegeneration. Ann. Neurol. 2005;58:495–505. doi: 10.1002/ana.20624. [DOI] [PubMed] [Google Scholar]
- 6.Puranam K.L., Wu G., Strittmatter W.J., Burke J.R. Polyglutamine expansion inhibits respiration by increasing reactive oxygen species in isolated mitochondria. Biochem. Biophys. Res. Commun. 2006;341:607–613. doi: 10.1016/j.bbrc.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 7.Jenkins B.G., Koroshetz W.J., Beal M.F., Rosen B.R. Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized 1H NMR spectroscopy. Neurology. 1993;43:2689–2695. doi: 10.1212/wnl.43.12.2689. [DOI] [PubMed] [Google Scholar]
- 8.Mann V.M., Cooper J.M., Javoy-Agid F., Agid Y., Jenner P., Schapira A.H. Mitochondrial function and parental sex effect in Huntington’s disease. Lancet. 1990;336:749. doi: 10.1016/0140-6736(90)92242-a. [DOI] [PubMed] [Google Scholar]
- 9.Brouillet E., Hantraye P., Ferrante R.J., Dolan R., Leroy-Willig A., Kowall N.W., Beal M.F. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc. Natl Acad. Sci. USA. 1995;92:7105–7109. doi: 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seong I.S., Ivanova E., Lee J.M., Choo Y.S., Fossale E., Anderson M., Gusella J.F., Laramie J.M., Myers R.H., Lesort M., et al. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum. Mol. Genet. 2005;14:2871–2880. doi: 10.1093/hmg/ddi319. [DOI] [PubMed] [Google Scholar]
- 11.Panov A.V., Gutekunst C.A., Leavitt B.R., Hayden M.R., Burke J.R., Strittmatter W.J., Greenamyre J.T. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat. Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- 12.Chan D.C. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 2006;125:1241–1252. doi: 10.1016/j.cell.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 13.Westermann B. Molecular machinery of mitochondrial fusion and fission. J. Biol. Chem. 2008;283:13501–13505. doi: 10.1074/jbc.R800011200. [DOI] [PubMed] [Google Scholar]
- 14.Youle R.J., Karbowski M. Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell. Biol. 2005;6:657–663. doi: 10.1038/nrm1697. [DOI] [PubMed] [Google Scholar]
- 15.Smirnova E., Griparic L., Shurland D.L., van der Bliek A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barsoum M.J., Yuan H., Gerencser A.A., Liot G., Kushnareva Y., Graber S., Kovacs I., Lee W.D., Waggoner J., Cui J., et al. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J. 2006;25:3900–3911. doi: 10.1038/sj.emboj.7601253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan H., Gerencser A.A., Liot G., Lipton S.A., Ellisman M., Perkins G.A., Bossy-Wetzel E. Mitochondrial fission is an upstream and required event for bax foci formation in response to nitric oxide in cortical neurons. Cell Death Differ. 2007;14:462–471. doi: 10.1038/sj.cdd.4402046. [DOI] [PubMed] [Google Scholar]
- 18.Jahani-Asl A., Cheung E.C., Neuspiel M., MacLaurin J.G., Fortin A., Park D.S., McBride H.M., Slack R.S. Mitofusin 2 protects cerebellar granule neurons against injury-induced cell death. J. Biol. Chem. 2007;282:23788–23798. doi: 10.1074/jbc.M703812200. [DOI] [PubMed] [Google Scholar]
- 19.Chen H., McCaffery J.M., Chan D.C. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell. 2007;130:548–562. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 20.Wang H., Lim P.J., Yin C., Rieckher M., Vogel B.E., Monteiro M.J. Suppression of polyglutamine-induced toxicity in cell and animal models of Huntington’s disease by ubiquilin. Hum. Mol. Genet. 2006;15:1025–1041. doi: 10.1093/hmg/ddl017. [DOI] [PubMed] [Google Scholar]
- 21.Terada K., Kanazawa M., Yano M., Hanson B., Hoogenraad N., Mori M. Participation of the import receptor Tom20 in protein import into mammalian mitochondria: analyses in vitro and in cultured cells. FEBS Lett. 1997;403:309–312. doi: 10.1016/s0014-5793(97)00070-7. [DOI] [PubMed] [Google Scholar]
- 22.Goping I.S., Millar D.G., Shore G.C. Identification of the human mitochondrial protein import receptor, huMas20p. Complementation of delta mas20 in yeast. FEBS Lett. 1995;373:45–50. doi: 10.1016/0014-5793(95)01010-c. [DOI] [PubMed] [Google Scholar]
- 23.Yang H., Zhong X., Ballar P., Luo S., Shen Y., Rubinsztein D.C., Monteiro M.J., Fang S. Ubiquitin ligase Hrd1 enhances the degradation and suppresses the toxicity of polyglutamine-expanded huntingtin. Exp. Cell Res. 2007;313:538–550. doi: 10.1016/j.yexcr.2006.10.031. [DOI] [PubMed] [Google Scholar]
- 24.Karbowski M., Arnoult D., Chen H., Chan D.C., Smith C.L., Youle R.J. Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J. Cell Biol. 2004;164:493–499. doi: 10.1083/jcb.200309082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srinivasula S.M., Ashwell J.D. IAPs: what’s in a name? Mol. Cell. 2008;30:123–135. doi: 10.1016/j.molcel.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y., Fang S., Jensen J.P., Weissman A.M., Ashwell J.D. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science. 2000;288:874–877. doi: 10.1126/science.288.5467.874. [DOI] [PubMed] [Google Scholar]
- 27.Gu M., Gash M.T., Mann V.M., Javoy-Agid F., Cooper J.M., Schapira A.H. Mitochondrial defect in Huntington’s disease caudate nucleus. Ann. Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 28.Koroshetz W.J., Jenkins B.G., Rosen B.R., Beal M.F. Energy metabolism defects in Huntington’s disease and effects of coenzyme Q10. Ann. Neurol. 1997;41:160–165. doi: 10.1002/ana.410410206. [DOI] [PubMed] [Google Scholar]
- 29.Ferrante R.J., Andreassen O.A., Jenkins B.G., Dedeoglu A., Kuemmerle S., Kubilus J.K., Kaddurah-Daouk R., Hersch S.M., Beal M.F. Neuroprotective effects of creatine in a transgenic mouse model of Huntington’s disease. J. Neurosci. 2000;20:4389–4397. doi: 10.1523/JNEUROSCI.20-12-04389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orr A.L., Li S., Wang C.E., Li H., Wang J., Rong J., Xu X., Mastroberardino P.G., Greenamyre J.T., Li X.J. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 2008;28:2783–2792. doi: 10.1523/JNEUROSCI.0106-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frank S., Gaume B., Bergmann-Leitner E.S., Leitner W.W., Robert E.G., Catez F., Smith C.L., Youle R.J. The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev. Cell. 2001;1:515–525. doi: 10.1016/s1534-5807(01)00055-7. [DOI] [PubMed] [Google Scholar]
- 32.Bras M., Yuste V.J., Roue G., Barbier S., Sancho P., Virely C., Rubio M., Baudet S., Esquerda J.E., Merle-Beral H., et al. Drp1 mediates caspase-independent type III cell death in normal and leukemic cells. Mol. Cell. Biol. 2007;27:7073–7088. doi: 10.1128/MCB.02116-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zuchner S., Mersiyanova I.V., Muglia M., Bissar-Tadmouri N., Rochelle J., Dadali E.L., Zappia M., Nelis E., Patitucci A., Senderek J., et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot–Marie–Tooth neuropathy type 2A. Nat. Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 34.Neuspiel M., Zunino R., Gangaraju S., Rippstein P., McBride H. Activated mitofusin 2 signals mitochondrial fusion, interferes with Bax activation, and reduces susceptibility to radical induced depolarization. J. Biol. Chem. 2005;280:25060–25070. doi: 10.1074/jbc.M501599200. [DOI] [PubMed] [Google Scholar]
- 35.Arnoult D., Rismanchi N., Grodet A., Roberts R.G., Seeburg D.P., Estaquier J., Sheng M., Blackstone C. Bax/Bak-dependent release of DDP/TIMM8a promotes Drp1-mediated mitochondrial fission and mitoptosis during programmed cell death. Curr. Biol. 2005;15:2112–2118. doi: 10.1016/j.cub.2005.10.041. [DOI] [PubMed] [Google Scholar]
- 36.Karbowski M., Lee Y.J., Gaume B., Jeong S.Y., Frank S., Nechushtan A., Santel A., Fuller M., Smith C.L., Youle R.J. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1 and Mfn2 during apoptosis. J. Cell Biol. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trushina E., Dyer R.B., Badger J.D., II, Ure D., Eide L., Tran D.D., Vrieze B.T., Legendre-Guillemin V., McPherson P.S., Mandavilli B.S., et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang D.T., Rintoul G.L., Pandipati S., Reynolds I.J. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol. Dis. 2006;22:388–400. doi: 10.1016/j.nbd.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 39.Narain Y., Wyttenbach A., Rankin J., Furlong R.A., Rubinsztein D.C. A molecular investigation of true dominance in Huntington’s disease. J. Med. Genet. 1999;36:739–746. doi: 10.1136/jmg.36.10.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Karbowski M., Neutzner A., Youle R.J. The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J. Cell Biol. 2007;178:71–84. doi: 10.1083/jcb.200611064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H., Monteiro M.J. Ubiquilin interacts and enhances the degradation of expanded-polyglutamine proteins. Biochem. Biophys. Res. Commun. 2007;360:423–427. doi: 10.1016/j.bbrc.2007.06.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Timmons L., Fire A. Specific interference by ingested dsRNA. Nature. 1998;395:854. doi: 10.1038/27579. [DOI] [PubMed] [Google Scholar]
- 44.Timmons L., Court D.L., Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263:103–112. doi: 10.1016/s0378-1119(00)00579-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.