

Abstract
Ether lipids (ELs), particularly plasmalogens, are essential constituents of the mammalian central nervous system. The physiological role of ELs, in vivo, however is still enigmatic. In the present study, we characterized a mouse model carrying a targeted deletion of the peroxisomal dihydroxyacetonephosphate acyltransferase gene that results in the complete lack of ELs. Investigating the cerebellum of these mice, we observed: (i) defects in foliation patterning and delay in precursor granule cell migration, (ii) defects in myelination and concomitant reduction in the level of myelin basic protein, (iii) disturbances in paranode organization by extending the Caspr distribution and disrupting axo-glial septate-like junctions, (iv) impaired innervation of Purkinje cells by both parallel fibers and climbing fibers and (v) formation of axon swellings by the accumulation of inositol-tris-phosphate receptor 1 containing smooth ER-like tubuli. Functionally, conduction velocity of myelinated axons in the corpus callosum was significantly reduced. Most of these phenotypes were already apparent at P20 but still persisted in 1-year-old animals. In summary, these data show that EL deficiency results in severe developmental and lasting structural alterations at the cellular and network level of the cerebellum, and reveal an important role of ELs for proper brain function. Common molecular mechanisms that may underlie these phenotypes are discussed.
INTRODUCTION
Zellweger syndrome, neonatal adrenoleukodystrophy, infantile Refsum disease and rhizomelic chondrodysplasia punctata (RCDP) are severe peroxisomal biogenesis disorders originating from the defect of different peroxisomal functions (1,2). These disorders have in common defects in ether lipid (EL) biosynthesis implicating severe structural brain abnormalities.
ELs are glycerophospholipids containing a long-chain alcohol at the sn-1 position. In plasmalogens (PLs), the major ELs in mammals, this alcohol is unsaturated carrying a labile vinyl ether group (3,4). In human brain, ∼70% of total phosphatidylethanolamine is plasmenylethanolamine (4,5). Other ELs, such as platelet-activating factor (PAF) and sulfogalactosyl-alkylacylglycerol (seminolipid) (6–8) are present in much lower amounts, but may reach relevant concentrations in local microdomains. Mice carrying a targeted deletion of PAF receptor (PAFR) do not reveal striking CNS dysfunctions except a weak but significant delay in embryonic granule cell precursor (GCP) migration in vitro (9,10). Furthermore, selected glycosylphosphatidylinositol (GPI)-anchored proteins also contain a long-chain alkyl ether in their GPI anchor (11,12), hence their synthesis might require the peroxisomal EL pathway.
A number of functions have been attributed to PLs, such as scavenging of reactive oxygen species (5), facilitation of membrane fusion (13), storage and release of polyunsaturated fatty acids (14) as well as effects on intracellular cholesterol distribution, endocytosis and the activity of the Na+/Ca2+ exchanger (3,15–18). Most of these functions were established in vitro and thus the role PLs play in vivo remains to be defined. In this context, mouse models defective in peroxisomal biogenesis, such as PEX2, PEX5, PEX13 or PEX7 deletions, have to be mentioned. They are distinguished by defects in multiple peroxisomal functions including EL biosynthesis and thus are particularly helpful in elucidating the importance of the peroxisomal compartment and its contributions to overall cell function (19–25).
As we became interested in previously unknown functions of ELs, we generated a mouse model specifically defective in EL biosynthesis. These mice reveal defects in reproduction, eye and lens development and optic nerve myelination. In fibroblasts of patients with isolated-EL deficiency, the structure of caveolae and clathrin-coated pits, the distribution of selected lipid raft microdomain (LRM)-associated proteins and the intracellular transport of cholesterol is altered (3,18,26).
Here, we further explore the significance of ELs for the integrity of the CNS in vivo using the clearly organized cerebellum as a model. Obvious hints for cerebellar dysfunction in these mutants were deficits observed in the vertical pole test and the rotarod suggesting cerebellar ataxia. We describe multiple aberrations caused by EL deficiency including defects in foliation, myelination, paranode organization and Purkinje cell (PC) innervation suggesting that ELs are essential constituents for development and functioning of the mammalian cerebellum. Moreover, the observed phenotypes provide valuable hints for elucidating the molecular role of ELs.
RESULTS
EL deficiency causes defects in cerebellar foliation patterning
As previously reported, dihydroxyacetonephosphate acyltransferase (DAPAT)-deficient mice were underweight and developmentally retarded (26). In CNS, these developmental anomalies were manifested by a reduction in brain weight (Supplementary Material, Fig. S1) and the size of cerebral and cerebellar hemispheres as well as foliation defects in the cerebellar vermis (Fig. 1). Normal cerebellar foliation shows 10 cortical lobules. Although the fissures determining the cardinal lobes appeared unchanged, the intraculminate, declival and uvular fissures, e.g. located between lobules IV and V and within lobules VI and IX, respectively, were poorly developed or missing in the EL-deficient cerebellum and lobe IV/V was prolonged. These alterations were observed postnatally from P10 to P45. Folia VI/VII appeared to be particularly affected. Semithin sectioning of this area at P20 also showed the reduced depth of the intracrural fissure and demonstrated the presence of GCPs in the external granule layer, which were both absent in age-matched controls (Fig. 1C–E). During cerebellar development, GCPs radially migrate from the external granule layer past the PCs finally forming the inner granule layer of the cerebellum. This process of migration and differentiation is finished in most wild-type mouse strains by P20 (27), thus the presence of GCPs in the external granule cell layer in folia VI/VII at this late developmental stage indicates a delay in GCP migration and suggests ELs to be required for the correct development of the cerebellum.
Figure 1.
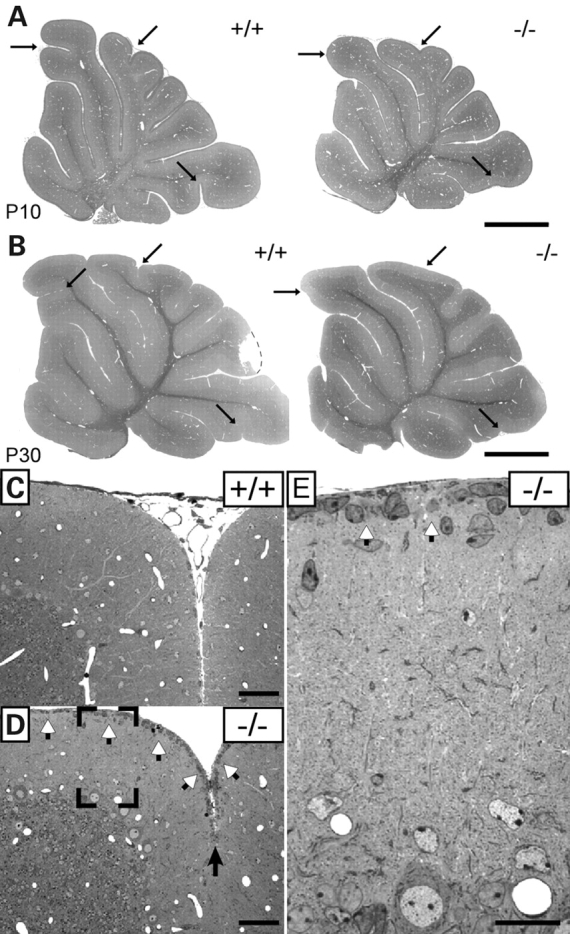
Alterations in foliation patterning (A and B) at P10 and P30 and delay in granule cell precursor migration (C–E) in control (+/+) and EL-deficient (−/−) cerebellum at P20. Arrows indicate poorly developed interculminate, declival and uvula fissures. White arrowheads in (D and E) point to granule cell precursors still residing in the external granule layer at the pial surface. Scale bars represent 1 mm (A and B), 50 µm (C and D) and 20 µm (E).
EL-deficient mice show defects in myelination and action potential propagation
By P20 myelination in the EL-deficient vermis (Bregma, lateral 0–0.5 mm) was considerably delayed particularly in folia IV/V and VI (Fig. 2), a defect that became less pronounced with advancing age. By P45, the reduced myelination in knockouts, visualized by myelin basic protein (MBP) immunofluorescence, was predominantly apparent in folium VI. For that reason, most of the following studies described here were carried out in this folium. Analysis of PC axons and their corresponding myelin sheaths at P20 using calbindin (CB) and MBP as target antigens provided evidence that, in line with our previous observations in the optic nerve, the initial non-myelinated portion of the axon was significantly extended in the knockouts (Fig. 3A–D; Supplementary Material, Movies 1and 2).
Figure 2.
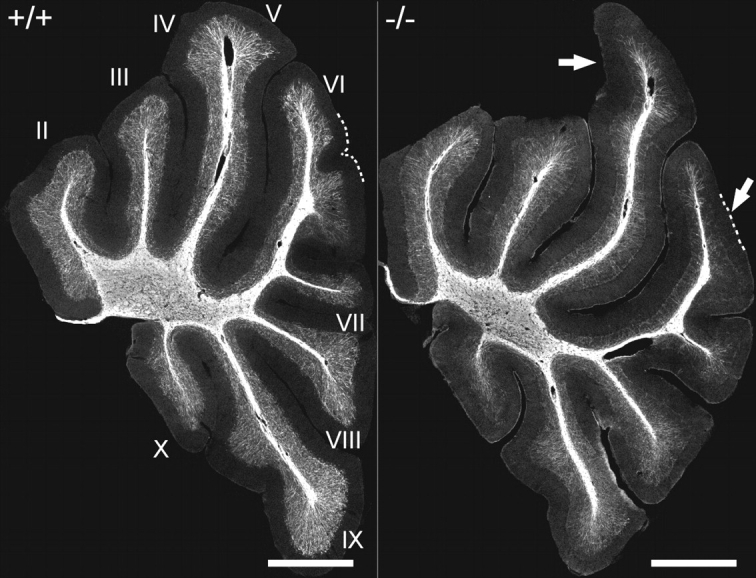
Foliation patterning and myelination in the cerebellum of P20 control (+/+) and DAPAT knockout (−/−) mice. Roman numerals indicate individual folia. Arrows mark the most conspicuous differences in foliation between controls and knockouts. Note the severe reduction in myelination especially in folium VI. The cerebellar sections correspond to Bregma 0.0–0.5 mm. Sections of six wild-type and six knockout animals were analyzed. The data of one pair of animals are representatively shown. Scale bars correspond to 1 mm.
Figure 3.
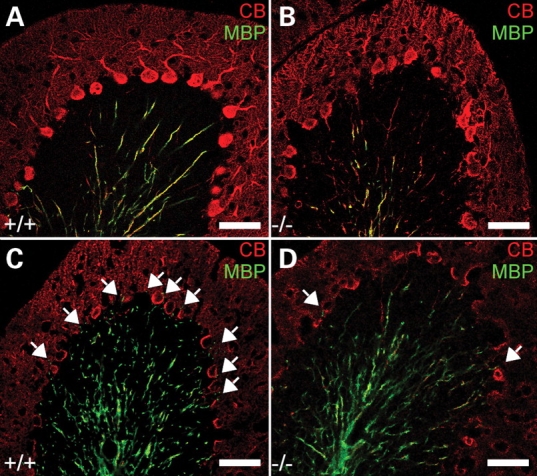
Dysmyelination of PC axons in DAPAT knockout mice. PCs and PC axons in sagittal cerebellar slices (20 µm) corresponding to Bregma 0.0–0.5 mm of controls (+/+) and EL-deficient (−/−) mice at P20 (A and B) and P45 (C and D) were stained for CB (red) and myelin (MBP, green). Note that the non-myelinated portion of the PC axon is significantly increased in EL-deficient mice. The arrows mark myelinated fibers within the PC layer. Scale bars correspond to 50 µm.
For comparison, we also investigated myelination in neocortex of P20, P45 and 8-month-old wild-type and DAPAT-deficient mice. Dysmyelination was clearly seen in the outer cortical region (Bregma 1 to −0.5 mm) at P20 and 8 months (Supplementary Material, Fig. S2), but in contrast to most folia of the cerebellar vermis, was still significant 8 months after birth. DAPAT-deficient animals also exhibited a less complex and less dense network of cortical and cerebellar myelinated axons (Fig. 2 and Supplementary Material, Fig. S2).
Dysmyelination of cerebellar and neocortical axons was accompanied by an ∼40–60% reduction in the total concentration of MBP affecting each of the MBP isoforms similarly (Fig. 4). Although the defect in cerebellar vermis myelination at P45, as observed by immunofluorescence, was less than that seen at P20, the differences between controls and P20 or P45 knockouts, as determined by western blotting, were similar (Fig. 4).
Figure 4.

Decreased MBP concentration in cerebellum (P20 and P45) and neocortex (P45) of wild-type (+/+) and DAPAT knockout (−/−) mice by gel electrophoresis and western blotting (upper panel). β-Tubulin was used as a marker for loading equal amounts of protein. The densitometric evaluation of blot signals (lower panel) was done using the Odyssey system. The tissue of four wild-type and four knockout animals was analyzed. For statistical analysis the unpaired t-test was used (**P ≤ 0.01; *P ≤ 0.05). Data are expressed as mean ± SD.
The observed dysmyelination in DAPAT-deficient animals was correlated with an ∼40% decrease in nerve conduction velocity of callosal myelinated fibers. Hippocampal Schaffer collaterals, which in rat and mouse are largely unmyelinated (28), did not show this decrease (Fig. 5). Thus, our studies suggest that ELs are essential for proper myelination and their lack causes dysmyelination and functional impairment of action potential conduction.
Figure 5.
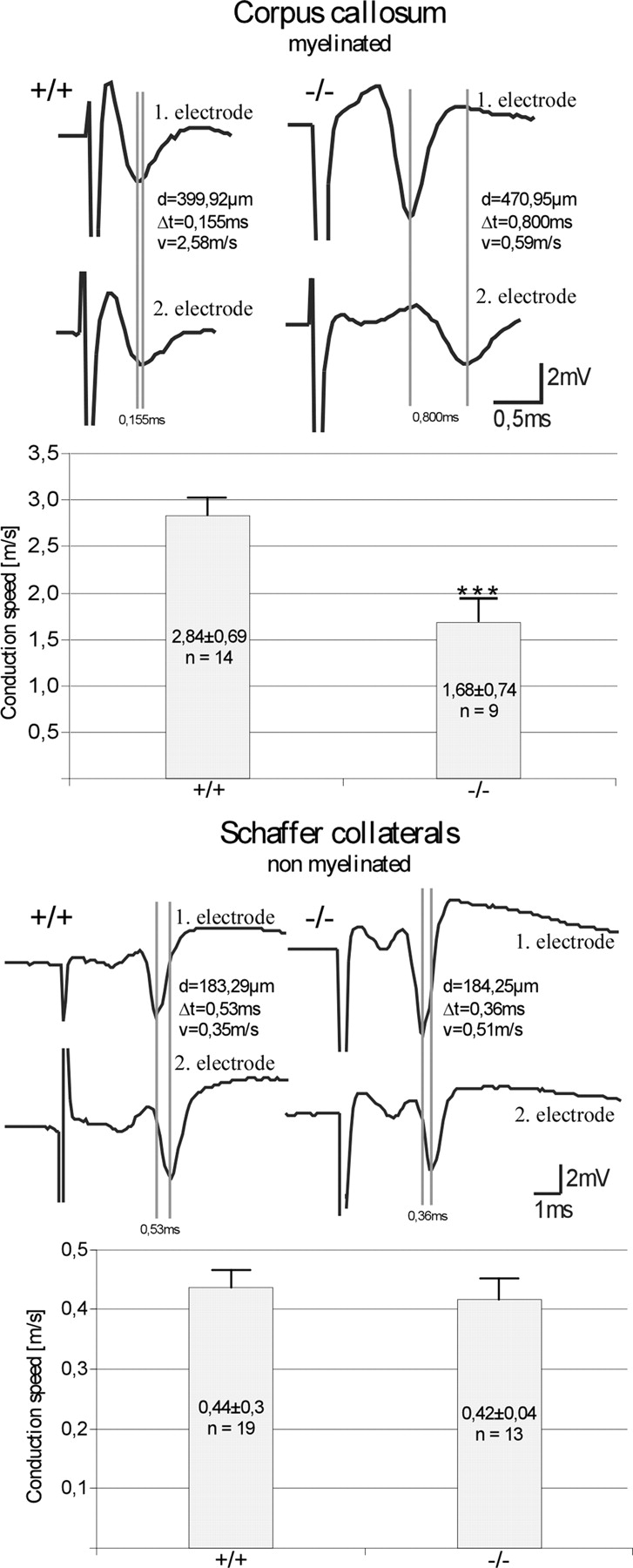
Reduced action potential conduction velocity in myelinated fibers of control (+/+) and DAPAT-deficient (−/−) mice. Evoked population spike from two different recording sites along the corpus callosum (upper panels) and the CA1 pyramidal cell layer (lower panels). Electrical stimulation was performed upstream from the first electrode within the corpus callosum (artifact truncated) and within the stratum radiatum (Schaffer collateral, artifact truncated). The vertical lines indicate the run time difference between the two recording sites (d, distance of the two recording sites; Δt, run time; v, velocity). The corresponding bar diagram shows the statistical evaluation of 9–19 independent experiments. Note the significant reduction in conduction velocity in myelinated fibers of the EL-deficient corpus callosum. The trace shown represents one with lower measured velocity. For statistical analysis, the Mann–Whitney U-test was used (***P < 0.01). Data are expressed as mean ± SEM.
EL deficiency causes increase in paranodal length and impairment of axo-glial interactions
Next, we investigated the nodal architecture in the EL-deficient mouse brain. At both sides of the node of Ranvier, lamellae of compact myelin sheaths terminate in glial loops that establish the paranodal axo-glial septate-like junctions (29). In these junctions, F3/contactin provides a platform of multiple binding sites for additional recognition molecules including contactin-associated protein (Caspr) and neurofascin (NF) 155 (30,31). First, we studied in optic nerve, cerebellum and corpus callosum the distribution of nodal Nav+ and juxtaparanodal Kv+ channels by immunofluorescence. Different to what has been described in CST knockout mice (8), the two channels appeared well separated from each other (Fig. 6A, B and F). However, visualizing paranodes by Caspr staining revealed that the length of the paranodes was significantly extended in the mutants (Fig. 6C and D). Dispersion of the Caspr signal was most pronounced in the optic nerve, but also significant in the corpus callosum and the cerebellum (Fig. 6E). Immunofluorescence studies on Caspr and F3/contactin demonstrated paranodal colocalization suggesting a dispersed distribution of both Caspr and F3/contactin within the paranodal area (Supplementary Material, Fig. S3). Quantitative evaluation of more than 700 paranodes demonstrated that paranodes in the mutants were more significantly increased in length than in width, a fact that also became apparent by calculating the ratio between paranodal length and width (Table 1).
Figure 6.
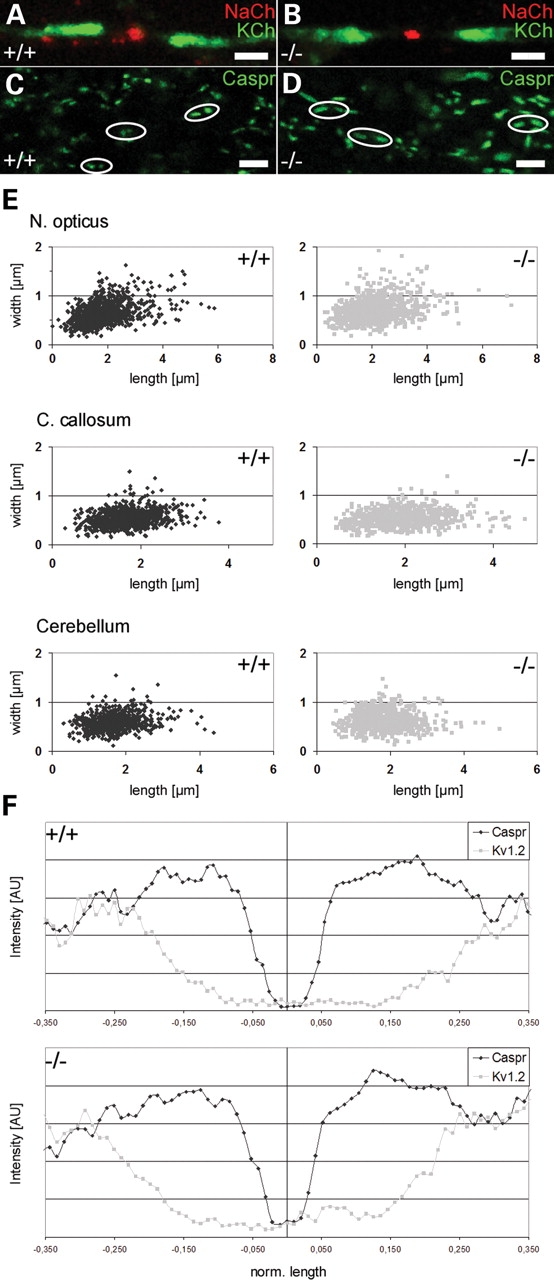
Increase in paranodal length in myelinated axons of EL-deficient mice. The distribution of Nav+ and Kv+ channels (A and B) in the optic nerve did not show differences between controls (+/+) and EL-deficient (−/−) mutants at P42. However, paranodal length, as revealed by Caspr immunofluorescence staining was significantly increased in optic nerve (C and D), corpus callosum and cerebellum of EL-deficient mice. The dispersion of the Caspr signal in optic nerve, corpus callosum and cerebellum is further demonstrated by plotting paranodal length versus paranodal width (E). Quantitative analysis of normalized distribution of Caspr and Kv1.2 signals in optic nerve, cerebellum and corpus callosum (only the data for corpus callosum are shown in F) did not show differences between controls and mutants suggesting that the relative location of Caspr and Kv1.2 in wild-types and mutants was the same. A total number of 2–4 wild-type and knockout animals was analyzed. Paranodal length and width of more than 700 paranodes of each tissue were statistically evaluated (Table 1). Scale bars correspond to 2 µm (A and B) and 5 µm (C and D).
Table 1.
Dimensions of wild-type and EL-deficient paranodes in N. opticus, C. callosum and cerebellum folium VI
| Tissue | Genotype | Length (µm) | Length >2 µm | Width (µm) | Width >1 µm | Ratio |
|---|---|---|---|---|---|---|
| N. opticus | Wt | 1.83 ± 0.00 | 33.27% | 0.64 ± 0.00 | 5.34% | 3.00 |
| Ko | 2.12 ± 0.00* | 50.73% | 0.71 ± 0.00* | 9.56% | 3.18* | |
| C. callosum | Wt | 1.68 ± 0.00 | 23.85% | 0.53 ± 0,00 | 1.37% | 3.44 |
| Ko | 1.92 ± 0.00* | 42.16% | 0.54 ± 0.00 | 0.83% | 3.83* | |
| Cerebellum | Wt | 1.71 ± 0.00 | 24.58% | 0.59 ± 0.00 | 1.54% | 3.11 |
| Ko | 1.84 ± 0.00* | 35.16% | 0.61 ± 0.00* | 2.94% | 3.33* |
More than 700 paranodes of wild-type (Wt) and EL-deficient (Ko) mice were analyzed for each tissue. Statistics were done using the unpaired t-test. Data are expressed as mean ± SEM.
*P ≤ 0.01.
As these observations indicate disorganization of the paranode, we investigated axo-glial septate-like junctions of PC axons by electron microscopy. These analyses revealed PC axons, particularly those having large diameters, to be clearly altered missing the typical transverse bands either along the entire paranode or at individual loops (Fig. 7A and B). Interglial gap junctions visualized by Cx32 immunofluorescence did not appear to be morphologically different in controls and mutants (Fig. 7C and D). From these observations, we conclude that ELs are important for the correct assembly and maintenance of PC paranodal axo-glial junctions.
Figure 7.
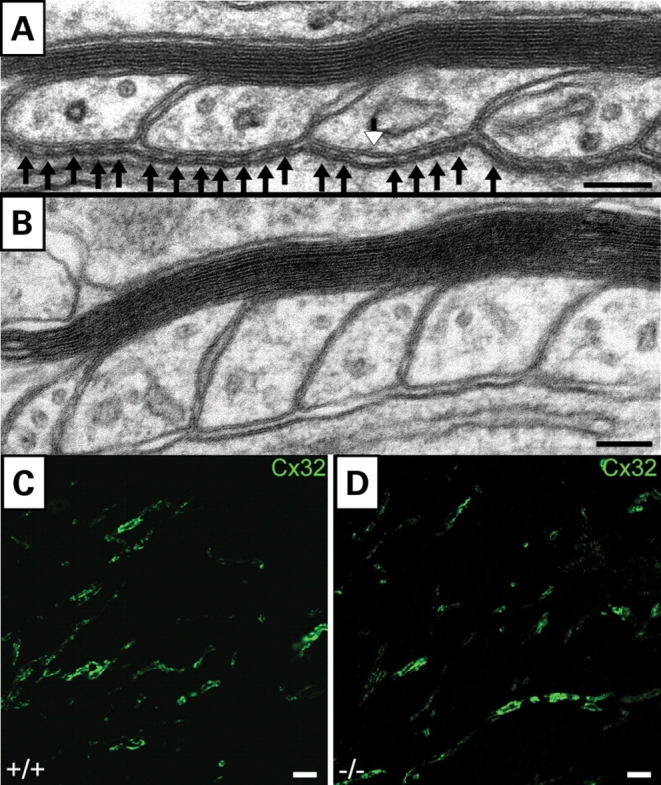
Structural alterations in PC septate-like junctions in P45 mutant cerebellum. (A) Ultrastructure of PC axo-glial loops showing individual septate-like junctions (black arrows). Occasionally, the typical transverse bands are missing (white arrowhead) or as in some paranodes (B) are completely absent. Compact myelin (A and B) seems to be intact. Gap junctions between glial loops (C and D) appear not to be affected as shown by immunofluorescence staining of Cx32 (green) in control (+/+) and EL-deficient (−/−) animals. Scale bars correspond to 100 nm (A and B) and 5 µm (C and D).
Lack of ELs impairs PC spinogenesis and synaptogenesis
To approach synaptic organization in the EL-deficient mouse, we studied PC innervation by both parallel fibers (PFs, visualized by VGluT1) and climbing fibers (CFs, visualized by VGluT2) as a model (32–35). PCs of adult, DAPAT-deficient mice revealed a ‘hyperspiny’ phenotype (36) distinguished by additional perisomatic dendrites and multiple perisomatic and proximal dendritic spines not observed in age-matched controls (Fig. 8A and B). Furthermore, striking differences were observed in synapse formation between PCs and either PFs or CFs. In controls, PFs connecting with as many PCs as possible usually formed synapses at a PF to spine ratio of 1:1 or maximally 1:2. These synapses were seen in all regions of the molecular layer with highest density in distal regions of the dendritic tree. In knockouts, PFs formed ‘synaptic clusters’ at a PF to spine ratio of 1:4 or even 1:5 that were predominantly localized near to PC somata and the innermost zone of the molecular layer (Fig. 8C–G). In more distal regions of PC dendrites, these enlarged synapses decreased in number and in the upper third of the molecular layer PF to spine ratios of 1:1 and 1:2 predominated.
Figure 8.
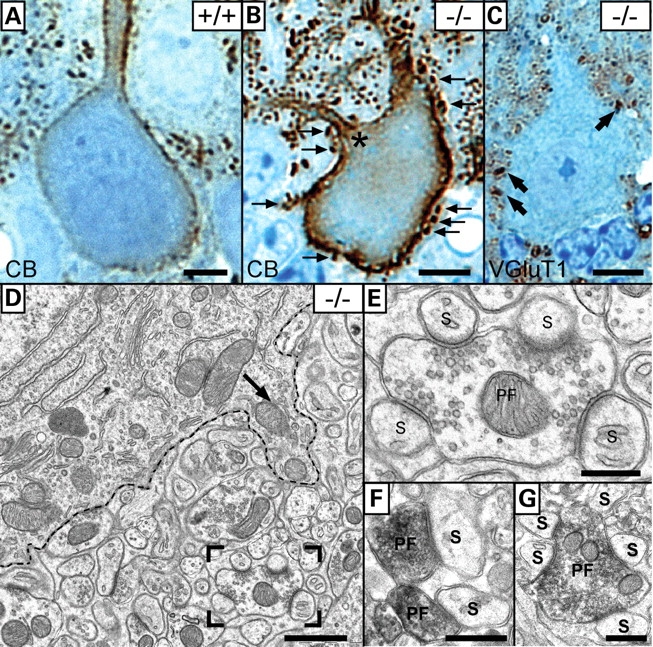
Formation of a ‘hyperspiny’ PC phenotype in El-deficient mice. (A and B) Calbindin (CB) ABC/DAB-enhanced immunohistochemical staining shows a hyperspiny PC of the mutant cerebellum (B) compared with the control (A). The hyperspiny character is distinguished by additional dendrites (asterisk in B) and spines (arrows in B) at both cell somata (arrow in D) and proximal dendrites. VGluT1 staining revealed enlarged PF synapses (‘synapse clusters’, arrow in C) that were located near the cell soma highlighted by the dashed line (D). Unusually, these enlarged PFs formed synapses with four (boxed area in D and E) and five PC spines (G) as demonstrated by VGlut1 staining at the EM level. In wild-type mice, a PF to spine ratio of 1:1 is usually maintained (F). (A–C) Semithin sections counter-stained with methylene blue–azure II. Scale bars correspond to 10 µm (A–C), 1 µm (D) and 500 nm (E–G).
CFs in wild-type mice during postnatal development are subject to a remarkable reshaping and their synaptic connections to PCs undergo notable relocations. This remodeling of CF projections involves loose contacts to several PCs in the first postnatal week. At later developmental stages, CFs first become focused to a single PC before one competing CF survives. Initially, the CF tightly surrounds the PC soma and the proximal dendritic trunk before being displaced to its final location on the intermediate apical portion of the dendritic tree (35,37). In DAPAT-deficient mice, this remodeling of CFs was severely disturbed (Fig. 9). At P20 and even at the age of 1 year, CF to PC synapses were still present in high number on PC somata and the proximal dendritic trunk. Obviously, these synapses followed an impaired relocation program resulting in an abnormal CF innervation territory and a reduced extension of the CF terminal arbor. Quantitatively, CF synapses of wild-type and mutants cover 79.00 ± 5.14 and 62.15 ± 3.55% of the entire molecular layer, respectively, demonstrating a highly significant difference in their cerebellar distribution. These data suggest that ELs are essential for the correct and dynamic address selection of both CFs and PFs. Lack of ELs leads to altered PC spinogenesis and synaptogenesis.
Figure 9.
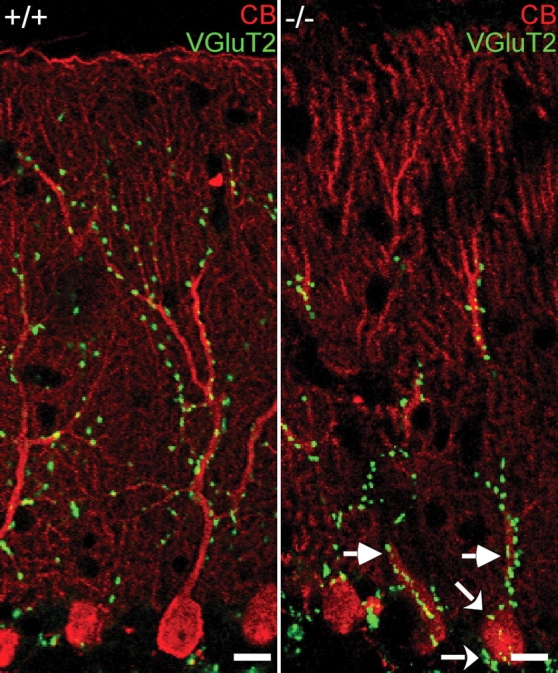
Phenotypically altered CF territory in mutant cerebellum. In mutants, CFs reside on PC somata (curved arrows) and proximal dendritic trunk (straight arrows) and occupy a severely restricted area of PC innervation. Double immunofluorescence of CB (red) and VGluT2 (green) in folium VI of a 1-year-old animal. Scale bars correspond to 10 µm.
EL-deficient PC axons generate axonal swellings accumulating IP3R1-rich smooth ER-like structures
CB staining of cerebellar PC axons frequently revealed swellings that were seen in the upper third of the granular layer already by P15 (see Supplementary Material, Fig. S4). At later developmental stages, swellings became apparent in more distal regions of the axons as well as in axon collaterals (Fig. 10A and B). Ultrastructural analysis demonstrated the accumulation of highly ordered arrays of membranous tubules closely resembling smooth ER (Fig. 10C). These axoplasmic tubules were much more extended than the smooth ER stacks regularly seen in PC somata and dendrites (Supplementary Material, Fig. S5). Immunological analysis based on the detection of protein disulfide isomerase, calnexin, BiP, rab5, early endosomal antigen, clathrin, cathepsin D, GM130, cadherin, SNAP25, synaptotagmin and α-synuclein did not allow the identification of the structures concentrated within the swellings. Characteristically, the membranes were decorated with a fuzzy coat of highly periodic alignment (Fig. 10C and D) that seemed to be composed of a protein complex of ∼10 nm in size (Fig. 11A) and resembled that described in fibroblasts transfected with inositol-1,4,5-tris-phosphate receptor type 1 (IP3R1) (38). IP3R1 is expressed in most mammalian cells and localizes to the smooth ER. It is particularly abundant in Purkinje neurons. Staining of cerebellar sections of DAPAT knockouts for IP3R1 actually revealed high immunoreactivity within the swellings suggesting the accumulation of IP3R1-rich smooth ER-like membranes (Fig. 11B).
Figure 10.
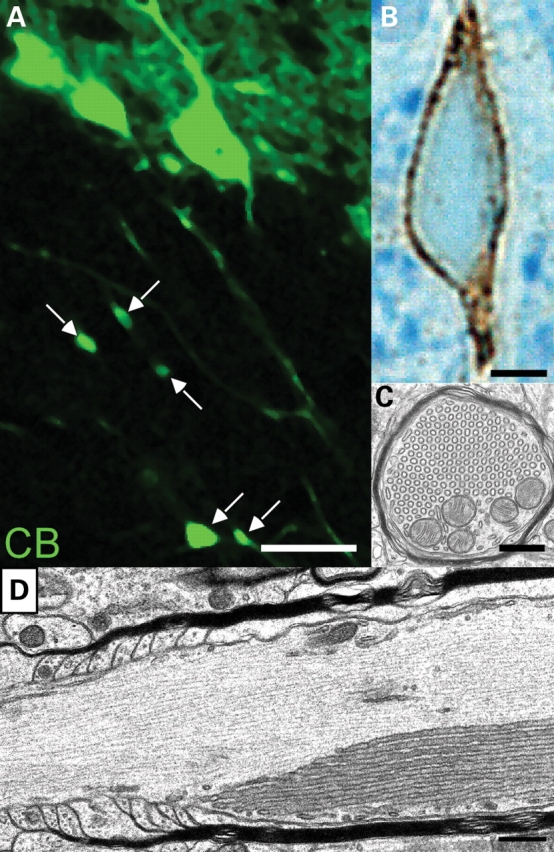
Formation of axonal swellings in PCs of folium VI of EL-deficient cerebellum at P30. PC axons were visualized by CB immunofluorescence (A) and ABC/DAB enhancement followed by osmification, Epon embedding, semithin sectioning and counter-staining with methylene blue–azure II (B). Swellings located in the granular layer were marked with arrows. The CB-positive axoplasm surrounds the smooth ER-like aggregates that are composed of tubules arranged in a hexagonal array (C). Frequently, these axon swellings were observed close to the node of Ranvier (D). A total number of 10 wild-type and 10 knockout animals was analyzed. The data of one pair of animals are representatively shown. Scale bars correspond to 20 µm (A), 2.5 µm (B) and 500 nm (C and D).
Figure 11.
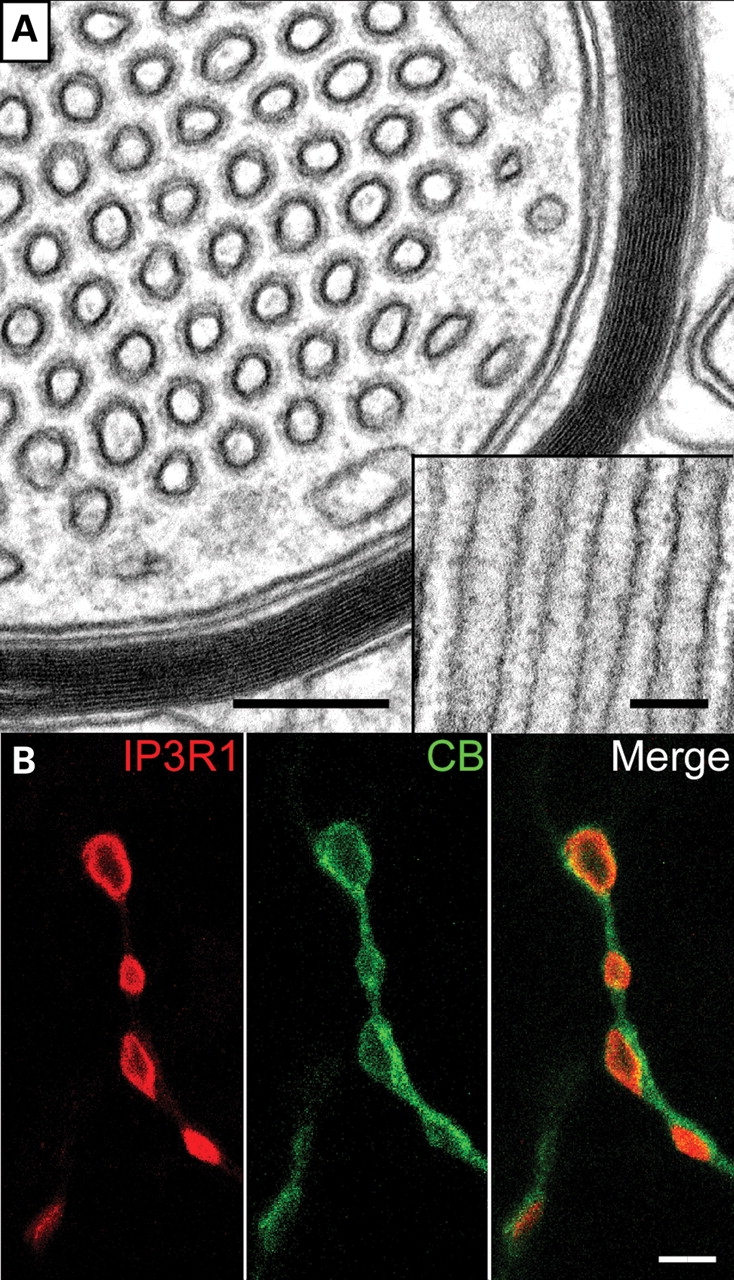
IP3R-containing smooth ER-like tubular stacks accumulating in PC axonal swellings. (A) The ultrastructural cross section reveals tubular structures of average diameter of 50–70 nm. The inset shows a longitudinal view of the tubuli. The membranes are regularly decorated by protein complexes of about 10 nm in size. (B) Localization of IP3R1 (red) in swellings of PC axons (calbindin staining in green). Scale bars correspond to 200 and 100 nm (A) and 2 µm (B).
DISCUSSION
In our efforts to delineate future approaches aimed to understand the molecular role of ELs in the CNS, we studied cerebellar phenotypes in the EL-deficient mouse. Macroscopically, we observed an ∼20% reduction in brain size and alterations in cerebellar foliation. The complexity in foliation patterning varies in different wild-type mouse strains. In the C57Bl/6 strain, for example, patterning is less complex than in the FVB/N or in the 129 strain (21,39). As the DAPAT-deficient mice were created from 129-derived R1 embryonic stem cells, their background genotype is C57Bl/6×R1 (26,40). However, at P20, when foliation in controls has reached mature state (39) in more than 15 DAPAT-deficient brains, we found a single dominating pattern clearly less differentiated than that of wild-type C57Bl/6 (Fig. 1A and B), suggesting that the differences in foliation were due to EL deficiency rather than variations in genetic background.
The presence of GCPs in the external granule layer at P20 indicates a delay in radial migration of these cells. GCP migration has been studied in great detail and a large number of molecules and conditions regulating radial GCP movement have been identified (41,42). Among these brain-derived neurotrophic factor, neurotrophin-3, neuregulin, stromal cell-derived factor 1α, ephrin-B2, tenascins and PAF are implicated in early postnatal migration. PAF exerts its effect by activating PAFR and PAFR-deficient GCPs and PAFR antagonist-treated GCPs exhibit reduced migration. Interestingly, PAFR activity and the signaling of most factors controlling GCP migration seem to be linked to their sorting to LRMs (43–47). Thus, correct assembly of and/or targeting of signaling receptors to LRMs may be an important clue in understanding the role of ELs in GCP migration.
Assembly of and sorting to LRMs may also be important for our understanding of the paranodal phenotypes of EL-deficient mice. At paranodes axo-glial septate-like junctions are assembled by the formation of protein complexes involving F3/contactin, Caspr and NF155. F3/contactin, a GPI-anchored axonal membrane protein, interacts with the integral membrane protein Caspr and also associates with glial NF155 (48–51). Through protein 4.1B, the axoplasmic portion of Caspr connects this multimeric paranodal protein complex to the axonal actin cytoskeleton (52). These and other paranodal components including myelin and lymphocyte protein (MAL), NrCam, CGT and CST were deleted in mice and their phenotypes analyzed (50,53–55). The targeted deletion of NrCam and ankyrin B results in disorganized lens fibers and formation of cataract (56), a phenotype present in RCDP and all EL-deficient mice. Moreover, F3/contactin ablation and EL deficiency strikingly share several common phenotypes, e.g. cerebellar ataxia, disrupted junctional attachments, reduced nerve conduction velocity, defects in controlling PF fiber orientation and significant reduction of granule cell post-synaptic area, as well as persisting abnormal axon varicosities (48,57). Besides being involved in the organization of septate-like junctions, F3/contactin is also proposed to mediate PF axon guidance and neuronal interactions (50,57). These common features between EL and F3/contactin deficiency are tempting to address a role of ELs in sorting and functioning of GPI-anchored proteins (26,58). Candidate proteins are those bearing an ether-bonded long-chain alcohol at the sn-1 position of the GPI moiety and depending on the peroxisomal steps of EL biosynthesis (59). Thy-1 is such a protein, and its mistargeting in EL-deficient thymocytes and fibroblasts was recently observed (Jayachandran et al., personal communication). The fact that promotor activation of the F3/contactin gene in post-mitotic granule neurons and PCs peaks at P6 and exactly coincides with the postnatal induction of EL biosynthesis further supports the idea of an EL–F3/contactin interrelationship (60,61). In addition to F3/contactin, both NF155 and MAL also recruit to LRMs (55) and are essential for maintenance of proper axo-glial interactions in the CNS. Both galactosylceramide and sulfatide are major constituents of LRMs. However, lack of these glycosphingolipids affects nodal organization particularly the distribution of Kv+ channels different to EL deficiency suggesting that these lipids occupy distinct functional membrane subdomains. Although the mode of action of ELs is still not clear, ELs are essential for CNS paranodal organization (53,54,62,63). In a working hypothesis, we propose ELs to be required for the proper assembly of neural LRMs and/or targeting and function of alky/acyl type GPI-anchored proteins.
Axon guidance of CFs and PFs is a highly dynamic process including various steps of synaptic remodeling. During postnatal development, each CF initially contacts several PCs at their proximal compartment. By P10, terminal arbors of CFs start to retract and a single CF remains targeted to perisomatic spines on a single PC. Thereafter, the terminal arbor is displaced from its perisomatic location upward to intermediate and apical dendrites (35,37). ELs seem to be required for this territory selection, as CF terminals in EL-deficient mice failed to ascend during development as far along proximal dendrites as they do in wild-type mice, and both the PF innervation territory was proximally expanded and single PFs frequently synapsed with three to five spines.
Recently, it has been suggested that the synaptic innervation pattern along the proximal-to-distal dendritic axis primarily depends on the balancing activities of CF and PF synapses to PCs. GluRδ2-dependent synapses can support heterosynaptic competition, i.e. competition between PF and CF synapses, in favor of PFs, whereas voltage-dependent Ca2+ channel (VDCC) α1A-dependent synapses support this competition in favor of CFs. In case one activity is lost, the innervation territory of the corresponding axon terminals becomes reduced while that of the competitor axons expands (64,65). PC innervation in EL-deficient mice strikingly resembles that of the VDCCα1A knockout. Interestingly, this phenotype distinguished by various structural changes, including axonal swellings, was also described as hyperspiny in mice with experimental CF lesions and appeared in spontaneous α1A mutants as well as in mice carrying a myosin Va missense mutation (34,66–68). Deficiency in myosin Va has been connected to impaired transport into PC spines of IP3R-containing smooth ER stacks suggesting that the observed defect in innervation is based on impaired Ca2+ signaling possibly generating ER stress (69,70).
The formation of axonal swellings in EL-deficient PC axons by accumulating IP3R1-rich smooth ER-like membranes thus may be the result of paranodal disorganization causing impaired axonal transport and/or disturbance of cellular and organellar Ca2+ homeostasis. In addition to the administration of mGluR agonists (71), various other conditions are known to cause axonal swellings, including deletion of glial cyclic nucleotide phosphodiesterase (72), F3/contactin (48), Caspr, and CGT (50). Axonal swellings were also reported to occur in the course of several neurodegenerative diseases such as cerebrospinal ataxia, Alzheimer’s disease and amyotrophic lateral sclerosis (50). Although the reasons for the development of axonal swellings are not clearly understood, some indications point to a defect in anterograde axonal transport. The molecular link for this may be the association of axo-glial junctions with the axonal actin cytoskeleton. Predominantly, the cytoplasmic portion of Caspr is involved in this interaction that also implicates protein 4.1B, αII and βII spectrin, as well as ankyrin B (50,52). Thus, disruption of axo-glial junctions initiated at heminodes of PC axons (Supplementary Material, Fig. S3) may disturb cytoskeletal functions resulting in abnormal paranodal accumulations of cell organelles.
On the other hand, increased concentrations in extracellular Glu may activate NMDA and mGlu receptors leading to enhanced intracellular Ca2+. As Na+/Ca2+ exchange activity seems to be strongly affected by PLs and plasmalogenic phosphatidic acid (15), lack of ELs may result in impaired Ca2+ extrusion, intracellular Ca2+ overload and ER stress. Na+/Ca2+ exchangers are widely distributed in brain including myelinated axons (73), and exchange activity that is regulated by the F-actin cytoskeleton has been proposed to be linked to Ca2+ accumulations within internal stores such as the ER and mitochondria (74). In summary, our results suggest that in addition to raft stability and GPI anchor synthesis future studies on EL functions should focus on the contributions of ELs to intracellular Ca2+ homeostasis and functions of the actin cytoskeleton.
MATERIAL AND METHODS
Animals
The DAPAT-null mice were generated as previously described (26). The animals were maintained in the breeding facilities of the IBF of University of Heidelberg and kept on a normal diet that did not contain detectable amounts of ELs. All the procedures were approved by Animal Care Committee.
Antibodies
The following antibodies were used (dilutions for immunohistochemistry): rat anti-MBP (Abcam, Cambridge, UK; 1:500), mouse anti-Caspr (Neuromab, Davis, USA; 1:2000), mouse anti-voltage-gated potassium channel 1.2 (Kv 1.2; Neuromab; 1:200), mouse anti-F3/contactin (Neuromab; 1:200), mouse anti-F3/contactin (Neuromab, 1:200), rabbit anti-Na+ channel III–IV loop (NaCh; Millipore, Schwalbach; 1:100), rabbit anti-inositol-tris-phosphate-receptor isotype I (IP3R1; Abcam; 1:100), mouse anti-CB (Swant; 1:2000), rabbit anti-CB (Swant, Bellinzona, Switzerland; 1:2000), rabbit anti-vesicular glutamate transporter 2 (VGluT2; Synaptic Systems, Goettingen, Germany; 1:2000), rabbit anti-VGluT1 (Synaptic Systems; 1:7000), mouse anti-Rab5 (BD Transduction Laboratories, Franklin Lakes, USA; 1:100), mouse anti-early endosome antigen 1 (EEA1, BD Transduction Laboratories; 1:100), goat anti-clathrin heavy chain (Santa Cruz, Heidelberg; 1:100), anti goat‐cathepsin D (Santa Cruz; 1:100), rabbit anti-pan-cadherin (Sigma, Munich; 1:100), rabbit anti-SNAP25 (Sigma; 1:100), mouse anti-alpha-synuclein (BD Transduction Laboratories; 1:1000), rabbit anti-synaptotagmin (Sigma; 1:100), mouse anti-calnexin (Stressgen/Biomol, Hamburg, 1:200), mouse anti-BiP (Stressgen/Biomol, Hamburg, 1:200), rabbit anti-connexin 32 (Cx32, Zymed/Invitrogen, Karlsruhe, 1:250). Both mouse anti-GM130 (1:100) and rabbit anti-protein disulfide isomerase (1:500) were a gift from Dr F. Wieland.
Immunohistochemistry
The brain was fixed by transcardial perfusion with 4% paraformaldehyde (PFA) in 0.1 m phosphate buffer (PB), pH 7.4 containing 2% polyvinylpyrrolidone 25 (PVP; Merck, Darmstadt) for 30 min. Cryoprotection was achieved by immersing the brain in increasing concentrations of sucrose (10, 20 and 30%) at 4°C. After embedding in OCT-mounting medium (Tissue-Tek®, Sakura, Zoeterwoude) using isopentane precooled by liquid nitrogen, 20 µm thick coronal sections of cerebral hemispheres and mid- and parasagittal sections through the cerebellar vermis were prepared using the Leica Cryostat CM 3050S. Corresponding sections of wild-type and EL-deficient brains were selected using the Paxinos atlas (75). For visualization of sodium channels, fresh brain and optic nerve were fixed by immersion in PB containing 1% PFA for 1.5 h. Samples were kept in 30% sucrose for cryoprotection over night, frozen and processed as described above.
Immunostaining of 20 µm thick cryostat sections was performed by a free-floating procedure. Sagittal cryostat sections of optical nerve and vermis were mounted onto SuperFrost® Plus object slides. Before antibody labeling, sections were blocked and permeabilized with 10% bovine serum albumin (BSA; Applichem, Darmstadt) or 10% goat serum (Sigma) containing 0.3% Triton X-100 (Roche Applied Science, Mannheim) in PBS for 1 h. Subsequently, sections were incubated with primary antibodies diluted in the corresponding blocking medium for 24 h at 4°C. FITC-, TRITC-, Cy2- or Cy3-labeled secondary antibodies (Sigma) were applied for 1 h. In case of using two monoclonal mouse primary antibodies, immunodecoration was done sequentially by blocking free binding sites with mouse anti-human IgG (50 mg/ml) and goat anti-mouse F(ab) (Dianova, Hamburg) in PBS containing 1% BSA. Sections were mounted in Mowiol containing 1% p-phenylenediamine (Sigma) or DakoCytomation fluorescent mounting medium (Dako, Glostrup, Denmark). Sections were analyzed using fluorescence microscopy (Zeiss Axiovert 200M) or confocal laser scanning microscopy (Zeiss LSM510 Meta). Overview pictures and distance measurements were made using the Cell^P software of the Olympus BX81 microscope.
Electron microscopy
Following anesthesia, the brain was fixed by transcardial perfusion with 2.5% glutaraldehyde (GA) in 0.1 m Na-cacodylate buffer, pH 7.6 containing 2% PVP and 0.05% CaCl2. 60–150 µm thick mid- and parasagittal sections of the vermis were prepared using a Dosaka DTK-1000 microslicer. Samples were postfixed in 1.5% reduced osmium tetroxide for 30 min followed by cacodylate-buffered 1.5% osmium tetroxide for 1 h and stained en bloc in 1% uranyl acetate for 30 min. After dehydration in graded ethanol, the samples were embedded in Epon using Aclar strips. Semithin sections were stained with a modified Richardson methylene blue–azure II solution. Ultrathin sections were stained with lead citrate and analyzed using a Zeiss EM 906E.
For immunolight and electron microscopy, the cerebellum was fixed by transcardial perfusion with 4% PFA and 0.05% GA in 0.1 m PB, pH 7.6 containing 1% PVP for 20 min followed by an additionally 10 min with 4% buffered PFA containing 2% PVP. For pre-embedding immunolabeling 50 µm thick sagittal microslicer sections of the vermis were prepared and subjected to cryoprotection in increasing concentrations of sucrose (10, 20 and 30%, 2.3 m containing 10% PVP). Subsequently, slices were frozen in isopentane precooled by liquid nitrogen for permeabilization and rinsed in reverse ordered sucrose concentrations. Permeabilized sections were incubated with anti-VGluT1, anti-VGluT2 or CB overnight followed by biotinylated secondary antibodies enhanced with the ABC-Kit (Vector Labs, Burlingame, USA) and DAB according to the manufacturer's instruction. Osmification and Epon embedding of the immunolabeled samples were performed as described above.
Blotting
Tissue homogenates (1:10) were prepared in 0.32 m sucrose, 1 mm EDTA containing an EDTA-free complete protease inhibitor mix (Roche Applied Science, Mannheim).
Protein content was determined as described (76). TCA-precipitated proteins were solubilized in 3 m Tris followed by an equal amount of SDS-sample buffer. Proteins were resolved on a 10–20% Tris–Tricine gel (Anamed, Groß-Bieberau) and transferred onto PVDF membranes. MBP and tubulin were visualized using rat anti-MBP (Abcam; 1:1000) and mouse anti-tubulin (Sigma; 1:2000) antibodies, respectively. After staining with far red fluorescent secondary antibodies (Invitrogen, Karlsruhe; 1:5000) proteins were quantified by the Odyssey infrared imaging system (LI-COR, Bad Homburg).
Electrophysiology
Conduction velocity was analyzed in 450 µm thick acutely prepared brain slices from control and EL-deficient mice. Mice were anesthetized with ether, decapitated and the brains were rapidly removed and immersed in ice-cold artificial cerebrospinal fluid (ACSF; containing NaCl 129 mm, KCl 3 mm, MgSO4 1.8 mm, CaCl2 1.6 mm, glucose 10 mm, NaH2PO4 1.25 mm, NaHCO3 21 mm; pH 7.4). We prepared horizontal and coronal slices for recordings from the frontal cortex including hippocampus. After sectioning, slices were transferred to a Haas-type interface recording chamber where they rested for at least 1 h at 35 ± 1°C (77).
Field potentials were recorded with ACSF-filled glass microelectrodes at two different sites along the CA1 pyramidal cell layer or at two different locations along the corpus callosum. Population spikes were elicited by electrical stimulation of the Schaffer collateral pathway or the corpus callosum with bipolar platinum/iridium wire electrodes. Data were low-pass filtered at 3 kHz, digitized at 10 kHz, and analyzed off-line with the Signal program (CED, Cambridge, UK).
Electrode distances were determined by filling both electrodes with NHS-fluorescein (Pierce Biotechnology, Erembodegem-Alsast, Belgium) and reinserting them at the recording positions. After 5 min of incubation, the distance between the recording sites was measured under the fluorescence microscope. Conduction velocity was calculated as the ratio between distance and time interval between the two successive population spikes (averaged from five recordings).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work was supported by grants of the German Research Foundation (DFG, Ju 166/3-1 and Go 432/2-1) and the FP6 European Union Project ‘Peroxisome’ (LSGH-CT-2004-512018).
ACKNOWLEDGEMENTS
The authors thank Ingrid Kuhn-Krause for excellent technical assistance. The monoclonal antibody Kv1.2 K+ channel, clone K14/16 and anti-Caspr, clone K65/35 were obtained from the UC Davis/NINDS/NIMH NeuroMab Facility, supported by NIH grant U24NS050606 and maintained by Department of Pharmacology, School of Medicine, University of California, Davis, CA 95616, USA. We also thank Drs Felix Wieland and Klaus Willecke for antibodies.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Moser H.W. Molecular genetics of peroxisomal disorders. Front. Biosci. 2000;5:D298–D306. doi: 10.2741/moser. [DOI] [PubMed] [Google Scholar]
- 2.Wanders R.J., Waterham H.R. Peroxisomal disorders I: biochemistry and genetics of peroxisome biogenesis disorders. Clin. Genet. 2005;67:107–133. doi: 10.1111/j.1399-0004.2004.00329.x. [DOI] [PubMed] [Google Scholar]
- 3.Gorgas K., Teigler A., Komljenovic D., Just W.W. The ether lipid-deficient mouse: tracking down plasmalogen functions. Biochim. Biophys. Acta. 2006;1763:1511–1526. doi: 10.1016/j.bbamcr.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 4.Nagan N., Zoeller R.A. Plasmalogens: biosynthesis and functions. Prog. Lipid Res. 2001;40:199–229. doi: 10.1016/s0163-7827(01)00003-0. [DOI] [PubMed] [Google Scholar]
- 5.Zoeller R.A., Morand O.H., Raetz C.R. A possible role for plasmalogens in protecting animal cells against photosensitized killing. J. Biol. Chem. 1988;263:11590–11596. [PubMed] [Google Scholar]
- 6.Diagne A., Fauvel J., Record M., Chap H., Douste-Blazy L. Studies on ether phospholipids. II. Comparative composition of various tissues from human, rat and guinea pig. Biochim. Biophys. Acta. 1984;793:221–231. doi: 10.1016/0005-2760(84)90324-2. [DOI] [PubMed] [Google Scholar]
- 7.Han X., Holtzman D.M., McKeel D.W., Jr Plasmalogen deficiency in early Alzheimer’s disease subjects and in animal models: molecular characterization using electrospray ionization mass spectrometry. J. Neurochem. 2001;77:1168–1180. doi: 10.1046/j.1471-4159.2001.00332.x. [DOI] [PubMed] [Google Scholar]
- 8.Honke K., Zhang Y., Cheng X., Kotani N., Taniguchi N. Biological roles of sulfoglycolipids and pathophysiology of their deficiency. Glycoconj. J. 2004;21:59–62. doi: 10.1023/B:GLYC.0000043749.06556.3d. [DOI] [PubMed] [Google Scholar]
- 9.Ishii S., Shimizu T. Platelet-activating factor (PAF) receptor and genetically engineered PAF receptor mutant mice. Prog. Lipid Res. 2000;39:41–82. doi: 10.1016/s0163-7827(99)00016-8. [DOI] [PubMed] [Google Scholar]
- 10.Tokuoka S.M., Ishii S., Kawamura N., Satoh M., Shimada A., Sasaki S., Hirotsune S., Wynshaw-Boris A., Shimizu T. Involvement of platelet-activating factor and LIS1 in neuronal migration. Eur. J. Neurosci. 2003;18:563–570. doi: 10.1046/j.1460-9568.2003.02778.x. [DOI] [PubMed] [Google Scholar]
- 11.Ikezawa H. Glycosylphosphatidylinositol (GPI)-anchored proteins. Biol. Pharm. Bull. 2002;25:409–417. doi: 10.1248/bpb.25.409. [DOI] [PubMed] [Google Scholar]
- 12.Kinoshita T., Inoue N. Dissecting and manipulating the pathway for glycosylphos-phatidylinositol-anchor biosynthesis. Curr. Opin. Chem. Biol. 2000;4:632–638. doi: 10.1016/s1367-5931(00)00151-4. [DOI] [PubMed] [Google Scholar]
- 13.Glaser P.E., Gross R.W. Plasmenylethanolamine facilitates rapid membrane fusion: a stopped-flow kinetic investigation correlating the propensity of a major plasma membrane constituent to adopt an HII phase with its ability to promote membrane fusion. Biochemistry. 1994;33:5805–5812. doi: 10.1021/bi00185a019. [DOI] [PubMed] [Google Scholar]
- 14.Chilton F.H., Murphy R.C. Remodeling of arachidonate-containing phosphoglycerides within the human neutrophil. J. Biol. Chem. 1986;261:7771–7777. [PubMed] [Google Scholar]
- 15.Hale C.C., Ebeling E.G., Hsu F.F., Ford D.A. The selective activation of the cardiac sarcolemmal sodium–calcium exchanger by plasmalogenic phosphatidic acid produced by phospholipase D. FEBS Lett. 1998;422:247–251. doi: 10.1016/s0014-5793(98)00017-9. [DOI] [PubMed] [Google Scholar]
- 16.Kubota M., Nakane M., Nakagomi T., Tamura A., Hisaki H., Shimasaki H., Ueta N. Regional distribution of ethanolamine plasmalogen in the hippocampal CA1 and CA3 regions and cerebral cortex of the gerbil. Neurosci. Lett. 2001;301:175–178. doi: 10.1016/s0304-3940(01)01631-7. [DOI] [PubMed] [Google Scholar]
- 17.Munn N.J., Arnio E., Liu D., Zoeller R.A., Liscum L. Deficiency in ethanolamine plasmalogen leads to altered cholesterol transport. J. Lipid Res. 2003;44:182–192. doi: 10.1194/jlr.m200363-jlr200. [DOI] [PubMed] [Google Scholar]
- 18.Thai T.P., Rodemer C., Jauch A., Hunziker A., Moser A., Gorgas K., Just W.W. Impaired membrane traffic in defective ether lipid biosynthesis. Hum. Mol. Genet. 2001;10:127–136. doi: 10.1093/hmg/10.2.127. [DOI] [PubMed] [Google Scholar]
- 19.Brites P., Motley A.M., Gressens P., Mooyer P.A., Ploegaert I., Everts V., Evrard P., Carmeliet P., Dewerchin M., Schoonjans L., et al. Impaired neuronal migration and endochondral ossification in Pex7 knockout mice: a model for rhizomelic chondrodysplasia punctata. Hum. Mol. Genet. 2003;12:2255–2267. doi: 10.1093/hmg/ddg236. [DOI] [PubMed] [Google Scholar]
- 20.Faust P.L., Hatten M.E. Targeted deletion of the PEX2 peroxisome assembly gene in mice provides a model for Zellweger syndrome, a human neuronal migration disorder. J. Cell Biol. 1997;139:1293–1305. doi: 10.1083/jcb.139.5.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Faust P.L., Su H.M., Moser A., Moser H.W. The peroxisome deficient PEX2 Zellweger mouse: pathologic and biochemical correlates of lipid dysfunction. J. Mol. Neurosci. 2001;16:289–297. doi: 10.1385/JMN:16:2-3:289. [DOI] [PubMed] [Google Scholar]
- 22.Hulshagen L., Krysko O., Bottelbergs A., Huyghe S., Klein R., Van Veldhoven P.P., De Deyn P.P., D’Hooge R., Hartmann D., Baes M. Absence of functional peroxisomes from mouse CNS causes dysmyelination and axon degeneration. J. Neurosci. 2008;28:4015–4027. doi: 10.1523/JNEUROSCI.4968-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janssen A., Gressens P., Grabenbauer M., Baumgart E., Schad A., Vanhorebeek I., Brouwers A., Declercq P.E., Fahimi D., Evrard P., et al. Neuronal migration depends on intact peroxisomal function in brain and in extraneuronal tissues. J. Neurosci. 2003;23:9732–9741. doi: 10.1523/JNEUROSCI.23-30-09732.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krysko O., Hulshagen L., Janssen A., Schutz G., Klein R., De Bruycker M., Espeel M., Gressens P., Baes M. Neocortical and cerebellar developmental abnormalities in conditions of selective elimination of peroxisomes from brain or from liver. J. Neurosci. Res. 2007;85:58–72. doi: 10.1002/jnr.21097. [DOI] [PubMed] [Google Scholar]
- 25.Maxwell M., Bjorkman J., Nguyen T., Sharp P., Finnie J., Paterson C., Tonks I., Paton B.C., Kay G.F., Crane D.I. Pex13 inactivation in the mouse disrupts peroxisome biogenesis and leads to a Zellweger syndrome phenotype. Mol. Cell. Biol. 2003;23:5947–5957. doi: 10.1128/MCB.23.16.5947-5957.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodemer C., Thai T.P., Brugger B., Kaercher T., Werner H., Nave K.A., Wieland F., Gorgas K., Just W.W. Inactivation of ether lipid biosynthesis causes male infertility, defects in eye development and optic nerve hypoplasia in mice. Hum. Mol. Genet. 2003;12:1881–1895. doi: 10.1093/hmg/ddg191. [DOI] [PubMed] [Google Scholar]
- 27.Sudarov A., Joyner A.L. Cerebellum morphogenesis: the foliation pattern is orchestrated by multi-cellular anchoring centers. Neural Dev. 2007;2:26. doi: 10.1186/1749-8104-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersen P., Silfvenius H., Sundberg S.H., Sveen O., Wigstrom H. Functional characteristics of unmyelinated fibres in the hippocampal cortex. Brain Res. 1978;144:11–18. doi: 10.1016/0006-8993(78)90431-6. [DOI] [PubMed] [Google Scholar]
- 29.Spiegel I., Peles E. Cellular junctions of myelinated nerves (Review) Mol. Membr. Biol. 2002;19:95–101. doi: 10.1080/09687680210130009. [DOI] [PubMed] [Google Scholar]
- 30.Falk J., Bonnon C., Girault J.A., Faivre-Sarrailh C. F3/contactin, a neuronal cell adhesion molecule implicated in axogenesis and myelination. Biol. Cell. 2002;94:327–334. doi: 10.1016/s0248-4900(02)00006-0. [DOI] [PubMed] [Google Scholar]
- 31.Hedstrom K.L., Rasband M.N. Intrinsic and extrinsic determinants of ion channel localization in neurons. J. Neurochem. 2006;98:1345–1352. doi: 10.1111/j.1471-4159.2006.04001.x. [DOI] [PubMed] [Google Scholar]
- 32.Hashimoto K., Kano M. Postnatal development and synapse elimination of climbing fiber to Purkinje cell projection in the cerebellum. Neurosci. Res. 2005;53:221–218. doi: 10.1016/j.neures.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 33.Hioki H., Fujiyama F., Taki K., Tomioka R., Furuta T., Tamamaki N., Kaneko T. Differential distribution of vesicular glutamate transporters in the rat cerebellar cortex. Neuroscience. 2003;117:1–6. doi: 10.1016/s0306-4522(02)00943-0. [DOI] [PubMed] [Google Scholar]
- 34.Sotelo C. Cerebellar synaptogenesis: what we can learn from mutant mice. J. Exp. Biol. 1990;153:225–249. doi: 10.1242/jeb.153.1.225. [DOI] [PubMed] [Google Scholar]
- 35.Yuste R., Bonhoeffer T. Genesis of dendritic spines: insights from ultrastructural and imaging studies. Nat. Rev. 2004;5:24–34. doi: 10.1038/nrn1300. [DOI] [PubMed] [Google Scholar]
- 36.Sotelo C., Arsenio-Nunes M.L. Development of Purkinje cells in absence of climbing fibers. Brain Res. 1976;111:289–295. doi: 10.1016/0006-8993(76)90782-4. [DOI] [PubMed] [Google Scholar]
- 37.Sugihara I. Organization and remodeling of the olivocerebellar climbing fiber projection. Cerebellum. 2006;5:15–22. doi: 10.1080/14734220500527385. [DOI] [PubMed] [Google Scholar]
- 38.Takei K., Mignery G.A., Mugnaini E., Sudhof T.C., De Camilli P. Inositol 1,4,5-trisphosphate receptor causes formation of ER cisternal stacks in transfected fibroblasts and in cerebellar Purkinje cells. Neuron. 1994;12:327–342. doi: 10.1016/0896-6273(94)90275-5. [DOI] [PubMed] [Google Scholar]
- 39.Sillitoe R.V., Joyner A.L. Morphology, molecular codes, and circuitry produce the three-dimensional complexity of the cerebellum. Ann. Rev. Cell Dev. Biol. 2007;23:549–577. doi: 10.1146/annurev.cellbio.23.090506.123237. [DOI] [PubMed] [Google Scholar]
- 40.Nagy A., Rossant J., Nagy R., Abramow-Newerly W., Roder J.C. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc. Natl Acad. Sci. USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gressens P. Pathogenesis of migration disorders. Curr. Opin. Neurol. 2006;19:135–140. doi: 10.1097/01.wco.0000218228.73678.e1. [DOI] [PubMed] [Google Scholar]
- 42.Komuro H., Yacubova E. Recent advances in cerebellar granule cell migration. Cell. Mol. Life Sci. 2003;60:1084–1098. doi: 10.1007/s00018-003-2248-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bollinger C.R., Teichgraber V., Gulbins E. Ceramide-enriched membrane domains. Biochim. Biophys. Acta. 2005;1746:284–294. doi: 10.1016/j.bbamcr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 44.Meyer S., Orso E., Schmitz G., Landthaler M., Vogt T. Lubrol-RAFTs in melanoma cells: a molecular platform for tumor-promoting ephrin-B2-integrin-beta1 interaction. J. Invest. Dermatol. 2007;127:1615–1621. doi: 10.1038/sj.jid.5700778. [DOI] [PubMed] [Google Scholar]
- 45.Nguyen D.H., Taub D. CXCR4 function requires membrane cholesterol: implications for HIV infection. J. Immunol. 2002;168:4121–4126. doi: 10.4049/jimmunol.168.8.4121. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki S., Numakawa T., Shimazu K., Koshimizu H., Hara T., Hatanaka H., Mei L., Lu B., Kojima M. BDNF-induced recruitment of TrkB receptor into neuronal lipid rafts: roles in synaptic modulation. J. Cell Biol. 2004;167:1205–1215. doi: 10.1083/jcb.200404106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang X.L., Xiong W.C., Mei L. Lipid rafts in neuregulin signaling at synapses. Life Sci. 2004;75:2495–2504. doi: 10.1016/j.lfs.2004.04.036. [DOI] [PubMed] [Google Scholar]
- 48.Boyle M.E., Berglund E.O., Murai K.K., Weber L., Peles E., Ranscht B. Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron. 2001;30:385–397. doi: 10.1016/s0896-6273(01)00296-3. [DOI] [PubMed] [Google Scholar]
- 49.Collard J.F., Cote F., Julien J.P. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature. 1995;375:61–64. doi: 10.1038/375061a0. [DOI] [PubMed] [Google Scholar]
- 50.Garcia-Fresco G.P., Sousa A.D., Pillai A.M., Moy S.S., Crawley J.N., Tessarollo L., Dupree J.L., Bhat M.A. Disruption of axo-glial junctions causes cytoskeletal disorganization and degeneration of Purkinje neuron axons. Proc. Natl Acad. Sci. USA. 2006;103:5137–5142. doi: 10.1073/pnas.0601082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poliak S., Peles E. The local differentiation of myelinated axons at nodes of Ranvier. Nat. Rev. 2003;4:968–980. doi: 10.1038/nrn1253. [DOI] [PubMed] [Google Scholar]
- 52.Ogawa Y., Schafer D.P., Horresh I., Bar V., Hales K., Yang Y., Susuki K., Peles E., Stankewich M.C., Rasband M.N. Spectrins and ankyrinB constitute a specialized paranodal cytoskeleton. J. Neurosci. 2006;26:5230–5239. doi: 10.1523/JNEUROSCI.0425-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bosio A., Bussow H., Adam J., Stoffel W. Galactosphingolipids and axono-glial interaction in myelin of the central nervous system. Cell Tiss. Res. 1998;292:199–210. doi: 10.1007/s004410051051. [DOI] [PubMed] [Google Scholar]
- 54.Coetzee T., Fujita N., Dupree J., Shi R., Blight A., Suzuki K., Popko B. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 1996;86:209–219. doi: 10.1016/s0092-8674(00)80093-8. [DOI] [PubMed] [Google Scholar]
- 55.Schaeren-Wiemers N., Bonnet A., Erb M., Erne B., Bartsch U., Kern F., Mantei N., Sherman D., Suter U. The raft-associated protein MAL is required for maintenance of proper axon–glia interactions in the central nervous system. J. Cell Biol. 2004;166:731–742. doi: 10.1083/jcb.200406092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.More M.I., Kirsch F.P., Rathjen F.G. Targeted ablation of NrCAM or ankyrin-B results in disorganized lens fibers leading to cataract formation. J. Cell Biol. 2001;154:187–196. doi: 10.1083/jcb.200104038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berglund E.O., Murai K.K., Fredette B., Sekerkova G., Marturano B., Weber L., Mugnaini E., Ranscht B. Ataxia and abnormal cerebellar microorganization in mice with ablated contactin gene expression. Neuron. 1999;24:739–750. doi: 10.1016/s0896-6273(00)81126-5. [DOI] [PubMed] [Google Scholar]
- 58.Honsho M., Yagita Y., Kinoshita N., Fujiki Y. Isolation and characterization of mutant animal cell line defective in alkyl-dihydroxyacetonephosphate synthase: localization and transport of plasmalogens to post-Golgi compartments. Biochim. Biophys. Acta. 2008;1783:1857–1865. doi: 10.1016/j.bbamcr.2008.05.018. [DOI] [PubMed] [Google Scholar]
- 59.Houjou T., Hayakawa J., Watanabe R., Tashima Y., Maeda Y., Kinoshita T., Taguchi R. Changes in molecular species profiles of glycosylphosphatidylinositol anchor precursors in early stages of biosynthesis. J. Lipid Res. 2007;48:1599–1606. doi: 10.1194/jlr.M700095-JLR200. [DOI] [PubMed] [Google Scholar]
- 60.De Benedictis L., Bizzoca A., Corsi P., Albieri I., Consalez G.G., Gennarini G. Activation profile of the F3/contactin gene in the developing mouse cerebellum. Mol. Cell Neurosci. 2006;32:403–418. doi: 10.1016/j.mcn.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 61.Snyder F., Hibbs M., Malone B. Enzymic synthesis of O-alkyl glycerolipids in brain and liver of rats during fetal and postnatal development. Biochim. Biophys. Acta. 1971;231:409–411. doi: 10.1016/0005-2760(71)90156-1. [DOI] [PubMed] [Google Scholar]
- 62.Honke K., Hirahara Y., Dupree J., Suzuki K., Popko B., Fukushima K., Fukushima J., Nagasawa T., Yoshida N., Wada Y., et al. Paranodal junction formation and spermatogenesis require sulfoglycolipids. Proc. Natl Acad. Sci. USA. 2002;99:4227–4232. doi: 10.1073/pnas.032068299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ishibashi T., Dupree J.L., Ikenaka K., Hirahara Y., Honke K., Peles E., Popko B., Suzuki K., Nishino H., Baba H. A myelin galactolipid, sulfatide, is essential for maintenance of ion channels on myelinated axon but not essential for initial cluster formation. J. Neurosci. 2002;22:6507–6514. doi: 10.1523/JNEUROSCI.22-15-06507.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ichikawa R., Miyazaki T., Kano M., Hashikawa T., Tatsumi H., Sakimura K., Mishina M., Inoue Y., Watanabe M. Distal extension of climbing fiber territory and multiple innervation caused by aberrant wiring to adjacent spiny branchlets in cerebellar Purkinje cells lacking glutamate receptor delta 2. J. Neurosci. 2002;22:8487–8503. doi: 10.1523/JNEUROSCI.22-19-08487.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miyazaki T., Hashimoto K., Shin H.S., Kano M., Watanabe M. P/Q-type Ca2+ channel alpha1A regulates synaptic competition on developing cerebellar Purkinje cells. J. Neurosci. 2004;24:1734–1743. doi: 10.1523/JNEUROSCI.4208-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rhyu I.J., Abbott L.C., Walker D.B., Sotelo C. An ultrastructural study of granule cell/Purkinje cell synapses in tottering (tg/tg), leaner (tg(la)/tg(la)) and compound heterozygous tottering/leaner (tg/tg(la)) mice. Neuroscience. 1999;90:717–728. doi: 10.1016/s0306-4522(98)00518-1. [DOI] [PubMed] [Google Scholar]
- 67.Rhyu I.J., Oda S., Uhm C.S., Kim H., Suh Y.S., Abbott L.C. Morphologic investigation of rolling mouse Nagoya (tg(rol)/tg(rol)) cerebellar Purkinje cells: an ataxic mutant, revisited. Neurosci. Lett. 1999;266:49–52. doi: 10.1016/s0304-3940(99)00254-2. [DOI] [PubMed] [Google Scholar]
- 68.Takagishi Y., Hashimoto K., Kayahara T., Watanabe M., Otsuka H., Mizoguchi A., Kano M., Murata Y. Diminished climbing fiber innervation of Purkinje cells in the cerebellum of myosin Va mutant mice and rats. Dev. Neurobiol. 2007;67:909–923. doi: 10.1002/dneu.20375. [DOI] [PubMed] [Google Scholar]
- 69.Langford G.M. Myosin-V, a versatile motor for short-range vesicle transport. Traffic. 2002;3:859–865. doi: 10.1034/j.1600-0854.2002.31202.x. [DOI] [PubMed] [Google Scholar]
- 70.Paschen W., Mengesdorf T. Endoplasmic reticulum stress response and neurodegeneration. Cell Calcium. 2005;38:409–415. doi: 10.1016/j.ceca.2005.06.019. [DOI] [PubMed] [Google Scholar]
- 71.Banno T., Kohno K. Conformational changes of the smooth endoplasmic reticulum are facilitated by l-glutamate and its receptors in rat Purkinje cells. J. Comp. Neurol. 1998;402:252–263. [PubMed] [Google Scholar]
- 72.Lappe-Siefke C., Goebbels S., Gravel M., Nicksch E., Lee J., Braun P.E., Griffiths I.R., Nave K.A. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- 73.Steffensen I., Waxman S.G., Mills L., Stys P.K. Immunolocalization of the Na(+)–Ca2+ exchanger in mammalian myelinated axons. Brain Res. 1997;776:1–9. doi: 10.1016/s0006-8993(97)00868-8. [DOI] [PubMed] [Google Scholar]
- 74.Condrescu M., Opuni K., Hantash B.M., Reeves J.P. Cellular regulation of sodium–calcium exchange. Ann. N. Y. Acad. Sci. 2002;976:214–223. doi: 10.1111/j.1749-6632.2002.tb04744.x. [DOI] [PubMed] [Google Scholar]
- 75.Franklin K., Paxinos G. The Mouse Brain in Stereotaxic Coordinates. San Diego: Academic Press; 1997. CF, 1997. [Google Scholar]
- 76.Daughaday W.H., Lowry O.H., Rosebrough N.J., Fields W.S. Determination of cerebrospinal fluid protein with the Folin phenol reagent. J. Lab. Clin. Med. 1952;39:663–665. [PubMed] [Google Scholar]
- 77.Nimmrich V., Maier N., Schmitz D., Draguhn A. Induced sharp wave-ripple complexes in the absence of synaptic inhibition in mouse hippocampal slices. J. Physiol. 2005;563:663–670. doi: 10.1113/jphysiol.2004.079558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.