

Abstract
We have conducted a three-stage, comprehensive single nucleotide polymorphism (SNP)-tagging association study of ESR1 gene variants (SNPs) in more than 55 000 breast cancer cases and controls from studies within the Breast Cancer Association Consortium (BCAC). No large risks or highly significant associations were revealed. SNP rs3020314, tagging a region of ESR1 intron 4, is associated with an increase in breast cancer susceptibility with a dominant mode of action in European populations. Carriers of the c-allele have an odds ratio (OR) of 1.05 [95% Confidence Intervals (CI) 1.02–1.09] relative to t-allele homozygotes, P = 0.004. There is significant heterogeneity between studies, P = 0.002. The increased risk appears largely confined to oestrogen receptor-positive tumour risk. The region tagged by SNP rs3020314 contains sequence that is more highly conserved across mammalian species than the rest of intron 4, and it may subtly alter the ratio of two mRNA splice forms.
INTRODUCTION
Genetic changes that alter the alpha oestrogen receptor (ERα) and its downstream signalling are likely to affect breast cancer susceptibility, tumour growth and response to treatment. In 1991, the locus encoding ERα was first postulated to be linked to breast cancer (1). The ESR1 gene has eight exons that span more than 300 kb of chromosome 6. It encodes a transcription factor, capable of binding oestrogen as well as oestrogen response element (ERE) DNA sequences. Oestrogen-activated ERα dimers initiate the transcription of down-stream genes through their EREs. Most of the effects of the anti-breast cancer drugs, tamoxifen and raloxifene, are through their binding to the oestrogen-binding domain of ERα and blocking these down-stream activities. Breast tumours that express ERα (receptor-positive tumours—ER+) are considered to be hormone-responsive and have better prognosis than others.
Three hundred and ninety-six single nucleotide polymorphisms (SNPs) across the ESR1 gene footprint have been collated, and haplotyped in a subset of the multiethnic cohort (MEC), (http://www.uscnorris.com/Core/DocManager/DocumentList.aspx?CID=13) and there are 286 common SNPs in Caucasians listed on HAPMAP (http://www.hapmap.org/). None of the common [minor allele frequency (MAF) > 0.05] SNPs are predicted to generate amino acid substitutions. Most published association studies on ESR1 have been confined to two SNPs (first detected with the restriction enzymes PvuII and XbaI (2)) that lie within 45 bp of each other in intron 1 of the gene. They have been variously reported to be associated with differential age-of-onset of menopause (3); body mass index, waist–hip ratio and fat mass (4); myocardial infarction and ischaemic heart disease (5) and risk of fracture (6). They have not consistently been found to be associated with risk of breast cancer (7,8). Other studies, examining more extensive ESR1 SNPs with breast cancer risk have also been inconclusive (9–12).
The MEC SNP database was generated by a combination of re-sequencing across coding exons and mining dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP/) and the Celera SNP databases within introns and upstream regulatory regions. At the commencement of this study, it was more comprehensive than HAPMAP at the ESR1 locus, and formed the basis for this large-scale breast cancer association study. Our aim was to examine the association of all known, common variants in ESR1 with risk of breast cancer using a set of tag SNPs.
RESULTS
The MEC database gives details of 227 common SNPs (MAF > 0.05) in Caucasians. A map of these SNPs, generated in Haploview (http://www.broad.mit.edu/mpg/haploview/) (Fig. 1) shows five clear haplotype blocks, and the largest of these can be further sub-divided into four regions (labelled 4a–d in Fig. 1). Within each block, many of the SNPs are highly correlated with each other. We identified a set of 68 SNPs that tagged the full set of 227 SNPs with pairwise correlation (Rp2) > 0.8, using Tagger (http://www.broad.mit.edu/mpg/tagger). To enable comparisons with previous papers that concentrated on two restriction fragment length polymorphisms (RFLPs), the SNPs creating the PvuII (rs2234693) and XbaI RFLPs (rs9340799) were forced into our set of tag SNPs. Details of the SNP tagging are shown in Supplementary Material, Table S1. Sixty of the 68 chosen tag SNPs were successfully made into Taqman assays and these efficiently tagged 96% of the common SNPs in Caucasians. These tag SNPs were all genotyped in the first stage of the study (SEARCH Set1, consisting of 2271 cases and 2280 controls from the East Anglian region of Britain). All Stage 1 results are shown in Supplementary Material, Table S2. Any SNP giving an indication of association with breast cancer at this stage (P-trend < 0.1) was eligible for further investigation (Stage 2).
Figure 1.

Haploview output. The matrix indicates the correlation coefficient between each pair of SNPs (RP2)—darker colours are equivalent to higher values. The relative positions of the ESR1 exons and haplotype blocks are shown together with the region of intron 4 tagged by SNP rs3020314.
The strongest, independently associated tag SNPs from SEARCH Set 1 were identified by multiple logistic regression and these were genotyped in Stage 2 of the association study (SEARCH Set 2, 2203 cases and 2280 controls, also drawn from the East Anglian region). To help elucidate the very strong association in haplotype block 4b, both tag SNPs from this block were studied in Stage 2 as was SNP rs2234693 (the PvuII RFLP) because of its previously reported associations. At the end of the second stage, only tag SNP rs3020314 showed an independent, statistically significant association with increased breast cancer risk. Odds ratio (OR) [ct/tt] = 1.15 (95% CI 1.05–1.26), OR [cc/tt] = 1.27 (95% CI 1.10–1.46); P(trend) = 0.00008 [Table 1].
Table 1.
Genotype Distributions of ESR1 tag SNPs after Stage 2 (SEARCH Sets 1 and 2)
| Haplotype block | rs number | Minor allele frequency | Genotype | Controls | Cases | Odds ratio (95% CI) | Trend test P-value |
|---|---|---|---|---|---|---|---|
| 2 | rs2234693 PvuII | 0.46 | TT | 1318 | 1260 | 1a | 0.48 |
| TC | 2296 | 2164 | 0.99 [0.89–1.09] | ||||
| CC | 934 | 938 | 1.05 [0.93–1.18] | ||||
| 2 | rs9340799 XbaI | 0.35 | AA | 1873 | 1682 | 1a | 0.09 |
| AG | 2048 | 1967 | 1.07 [0.98–1.17] | ||||
| GG | 526 | 521 | 1.10 [0.96–1.27] | ||||
| 3 | rs1709182 | 0.36 | TT | 1796 | 1669 | 1a | 0.08 |
| TC | 2117 | 2017 | 1.03 [0.94–1.12] | ||||
| CC | 607 | 642 | 1.14 [1.00–1.30] | ||||
| 4b | rs3020314 | 0.31 | TT | 2132 | 1869 | 1a | 0.00008 |
| TC | 1970 | 1989 | 1.15 [1.05–1.26] | ||||
| CC | 447 | 499 | 1.27 [1.10–1.46] | ||||
| 4b | rs3020317 | 0.19 | TT | 2983 | 2746 | 1a | 0.003 |
| TC | 1420 | 1427 | 1.09 [1.00–1.19] | ||||
| CC | 151 | 188 | 1.35 [1.08–1.69] | ||||
| 4c | rs926777 | 0.24 | CC | 2566 | 2401 | 1a | 0.16 |
| CA | 1722 | 1703 | 1.06 [0.97–1.15] | ||||
| AA | 266 | 271 | 1.09 [0.91–1.30] |
Values in the minor allele frequency column refer only to Controls. Values in bold denote statistically significant results.
aReferent group.
SNP rs3020314 was the tag for seven other known SNPs. All lie in a segment of DNA, approximately 18 kb in length (haplotype block 4b) that falls entirely within intron 4 of the gene (Fig. 1). All eight SNPs were therefore genotyped in SEARCH case-control Set 1 and the results were compared (Supplementary Material, Table S3). As expected, all show significant association with risk of breast cancer. Conditional logistic regression and likelihood ratio tests were used to compare genotype data from all eight SNPs. Both forward conditional regression and the likelihood ratio test indicate that rs3020314 is the best of these eight candidates, although none of the others can be definitively excluded. Reverse conditional regression leaves a combination of three other candidates (rs3020396, rs3020401 and rs1884051), indicating that no single, currently known SNP explains all the disease association seen.
SNP rs3020314 was genotyped in Stage 3, consisting of 28 more case–control studies within the international collaborative consortium, BCAC. Twenty-one European and two Asian studies, containing 53 994 and 3659 subjects, respectively, passed QC criteria for this SNP—details of the individual studies are shown in Supplementary Material, Table S4. In the Europeans, relative to the common (tt) homozygote group, ct heterozygotes have OR = 1.06 (95% CI 1.02–1.09); P = 0.004 (1 df) and cc homozygotes have OR = 1.04 (95%CI 0.98–1.10); P = 0.2 (1 df). When compared with a full model, the data fit best (P = 0.5) with a dominant mode of action for the minor c-allele OR [c-carrier/tt] = 1.05 (95% CI 1.02–1.09); P = 0.004 (1 df) (Fig. 2A). An alternative, co-dominant mode of action fits less well (P = 0.057) but cannot be formally rejected. There is significant heterogeneity, even between European studies (P = 0.002). Four studies are outliers—their 95% CIs do not include the overall risk estimate. Excluding these studies from the analysis (ABCFS, KARBAC, MARIE and SEARCH [Sets 1 and 2]—the latter being the hypothesis generating study) removes the heterogeneity (P = 0.9) and leaves the risk estimate essentially unchanged: OR [c-carrier/tt] = 1.05 (95% CI 1.00–1.09); P = 0.04 (1 df); n = 36 908 subjects. Thus, the observed heterogeneity is not generating the association.
Figure 2.
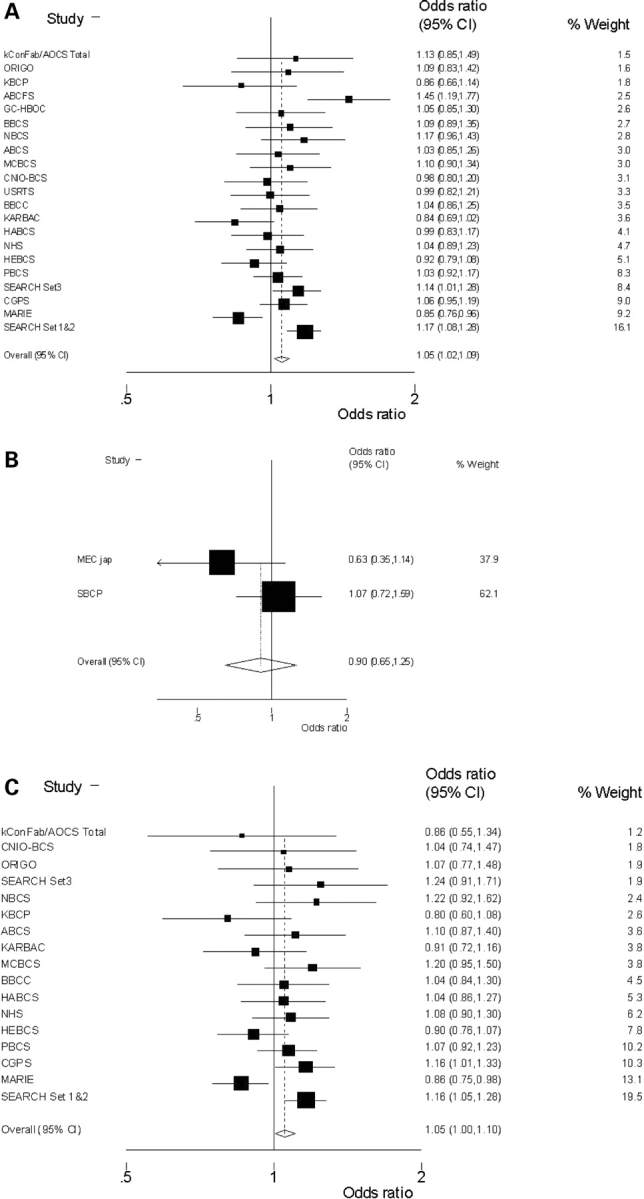
Forrest plots. Results from meta-analysis of SNP rs3020314, using a dominant model, in (A) 21 invasive breast cancer case control studies from Europe and (B) 2 studies from Asia. (C) The subset of 17 studies (all European) with recorded ER+ tumours.
From the two Asian studies there is no apparent association of SNP rs3020314 with breast cancer risk, [OR = 0.90 (95% CI 0.65–1.25); P = 0.5 (dominant model) (Fig. 2B) although the CIs include those of the European studies. The c allele has a similar frequency among all European control subjects and is the minor allele (mode = 0.33) whereas in Asian controls it is the major allele with a frequency of 0.8.
From the subset of 17 studies (all European) in which ER status has been determined the data indicate that any increased risk may be confined to ER-positive tumours (ER+, Fig. 2C) OR (ER+) = 1.05 (95% CI 1.00–1.10); P = 0.04 (1 df); P(heterogeneity = 0.04 (n = 10 777 ER+ tumours versus 24 836 controls). One study, MARIE, is again an outlier and removing this from the meta-analysis leaves: OR (ER+) = 1.08 (95% CI 1.03–1.14); P = 0.002 (1 df); P(heterogeneity) = 0.4 (n = 31 169 subjects). This tag SNP shows no evident association with risk of developing ER− tumours (n = 3 269 ER− tumours versus 24 836 controls): OR (ER−) = 1.00 (95% CI 0.93–1.08); P = 0.9 (1 df), although there is no significant difference between the risk estimates for ER+ and ER− tumours [P(interaction) = 0.29].
We have conducted preliminary association studies of SNP rs3020314 with levels of two ESR1 splice-forms in B-lymphocytes from 33 subjects and ER+ breast tumours from 30 subjects. Results from both suggest that increasing c-allele dosage may be associated with increasing levels of an RNA splice-form that lacks ESR1 exon 5 (Δ5ERα relative to full-length ESR1 mRNA. This trend reaches significance (Kruskal–Wallis test, P = 0.01) in the B-lymphoctyes (Fig. 3).
Figure 3.
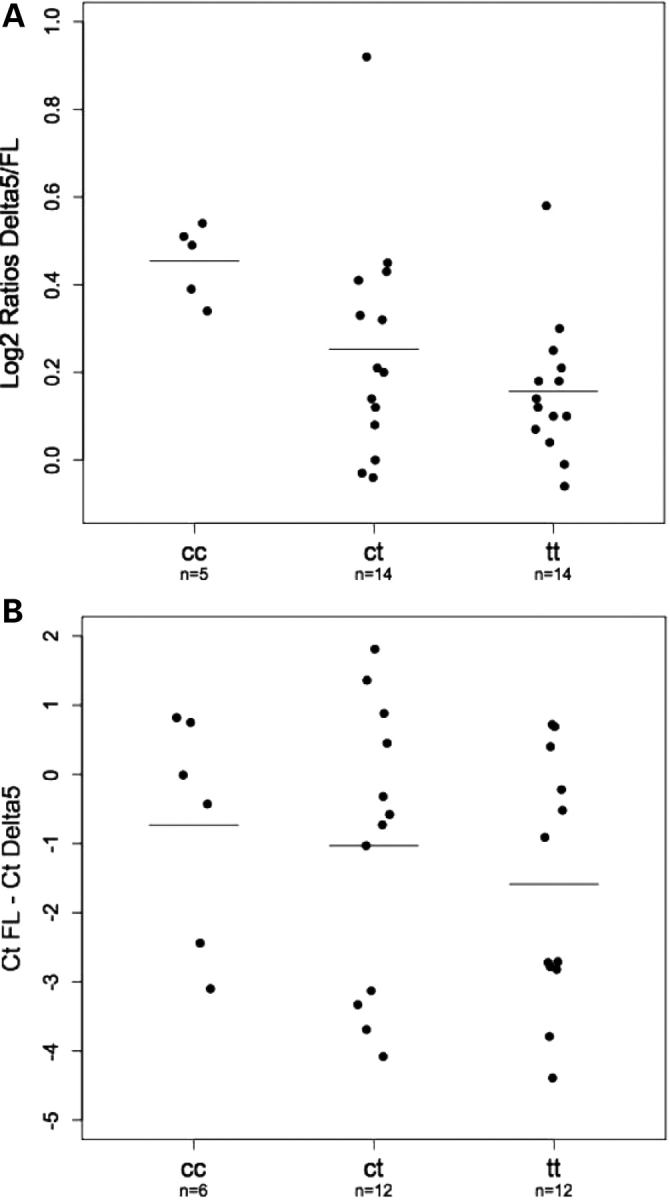
Ratios of RNA isoforms by genotype. (A) Plots showing ratios of Δ5ERα(Delta5) to full-length (FL) RNA, determined by gel fluorescence intensity in B-lymphocytes from 33 subjects who had also been genotyped for SNP rs3021314. Genotypes and number of subjects (n) in each class are shown beneath each plot, horizontal bars indicate mean values. (B) Plots showing relative amounts of Delta5 to FL RNA, determined by real-time PCR, in 30 ER+ breast tumours from subjects whose germline DNA had been genotyped for SNP rs3021314. Genotypes and number of subjects (n) in each class are shown beneath each box, and horizontal bars indicate mean values. Relative amounts are shown as the difference in PCR cycle number needed to reach a given intensity threshold (CtFL− CtDelta5). Negative values indicate that the FL form was more abundant than the Δ5ERα and vice versa.
DISCUSSION
This comprehensive SNP-tagging study of the ESR1 gene reveals no large associations of common gene variants in ESR1 with breast cancer risk. We report a marginally significant association of an SNP tagging a conserved region of intron 4 with an ∼5% increase in risk of developing ER+ tumours. There is significant heterogeneity between studies in this meta-analysis of BCAC results and the reason for this is not yet clear.
This intronic region was not re-sequenced during the construction of the MEC database and it is quite possible an, as yet undiscovered, variant is the cause of the apparent association. It is conceivable that the heterogeneity would be reduced if a causative variant could be identified and genotyped. Tag SNP rs3020314 was chosen to tag known variants in haplotype block 4b and it may not be a good tag for the putative causative variant in all the populations within BCAC. However, regardless of tagging efficiency, this study, albeit with >55 000 subjects, had only 12% power to detect the effect of an allele, such as this (MAF = 0.3; OR = 1.05) at a significance level (P < 10−7) suitable for genome-wide association studies (GWAS). Since the majority of existing breast cancer case–control sets are already part of BCAC, it is likely to be some time before a substantially larger study can definitively confirm or refute this marginally significant association. Unsurprisingly, given this study power calculation, a recent GWAS (13) that included many of the same BCAC participants failed to detect this association. SNP rs3020314 was well tagged (r2 = 0.73) but the GWAS tag SNP did not reach the significance level (P = 0.05) required to progress beyond the initial phase of that study. The level of significance (P = 0.004) at the end of Stage 3 does not exceed the Bonferroni-corrected significance threshold, which would be P = 0.0008, for 60 independent tests. However, this is an overly conservative threshold, as the 60 tag SNPs were not independent, but were correlated with each other to varying degrees. The known biology of ERα increases the prior probability of an ESR1 variant being associated with breast cancer risk but, even so, it remains possible that this is a false-positive finding. Assuming it is real, our detection of this association had an element of luck or ‘winners curse’—the OR estimate of ∼1.2 from Phase 1 of this study [Supplementary Material, Table S2A] was demonstrated to be an overestimate in Phase 3. We would have had <30% power, in Phase 1, to detect the true OR (1.05) at the significance level (P = 0.05) needed for progression to Phase 2.
The putative causative variant would be expected to be within haplotype block 4b of ESR1, a region delimited by SNPs rs3003917 and rs3003925 within intron 4 (Fig. 1). Examination of the sequence conservation between mouse and man shows that haplotype block 4b is significantly more highly conserved (7947 conserved bp/17 991 bp=44%) than the sequence of the rest of intron 4 (15 470 conserved bp/49 156 bp=31%, χ2 = 936, P = 1.2 × 10−205, data not shown). This suggests that, within ESR1 intron 4, haplotype block 4b is more likely to contain functionally important DNA than the other blocks. This intronic location raises the possibility that the functional variant may affect ESR1 splicing. One documented splice variant, Δ5ERα, has a deletion of exon 5 that generates a premature stop codon in exon 6 and thus a receptor lacking the hormone-binding and gene-activation domains (14). Although in need of confirmation, our preliminary experiments indicate a trend for the Δ5ERα form to be more abundant, relative to full-length RNA, in B-lymphocytes with increasing doses of the risk-associated c allele (Fig. 3A). There is a similar, although non-significant, trend in ER+ tumours (Fig. 3B). The Δ5ERα form, which cannot bind oestrogen, has been shown to be capable of constitutively activating a reporter construct in the absence of oestrogen (14). This hints at a potential mechanism for the association with breast cancer risk.
MATERIALS AND METHODS
Patients and controls
For Stages 1 and 2 of this study, cases were drawn from the SEARCH (breast) collection (Sets 1 and 2), an ongoing population based study, Controls were randomly selected from the Norfolk component of EPIC (European Prospective Investigation of Cancer). Details of all 28 BCAC case–control studies, used in Stage 3, are given in Supplementary Material, Table S4.
Selection of tagging snps
SNP identification within the ESR1 gene has been carried out in Caucasians as part of studies in the MEC. (http://www.uscnorris.com/Core/DocManager/DocumentList.aspx?CID=13). We aimed to define a set of tagging SNPs such that all known, common SNPs had an estimated Rp2 of >0.8 with at least one tag SNP. We used the Tagger programme (http://www.broad.mit.edu/mpg/tagger/) to select SNPs using the aggressive pairwise tagging. This program identifies tagging SNPs regardless of the haplotype block structure.
Taqman genotyping (Stages 1 and 2)
Genotyping was carried out using Taqman® according to the manufacturer’s instructions. Primers and probes were supplied directly by Applied Biosystems as Assays-by-Design™. All assays were carried out in 384-well plates. Each plate included negative controls (with no DNA) and positive controls duplicated on a separate quality control plate. Plates were read on the ABI Prism 7900 using the Sequence Detection Software (Applied Biosystems). Failed genotypes were not repeated. Assays in which the genotypes of duplicate samples did not show >95% concordance were discarded and replaced with alternative assays with the same tagging properties.
QC across BCAC
Twenty-six studies used Taqman (with Assays supplied centrally) and three (ABCFS, kConFab/AOCS & MARIE) used Sequenom iPlex genotyping platforms. The following QC criteria were applied across all BCAC studies: DNA samples that failed to PCR in more than one previous BCAC studies were excluded from analysis. After exclusion of these samples, the call-rate for SNP rs3020314 had to be >95% of all attempted samples (one study was excluded due to call rate <94%). All studies contained duplicate samples that showed >98% concordance between called genotypes. The genotype distributions in the Control samples had to conform to those expected under Hardy–Weinberg equilibrium (HWE). Representative cluster plots were examined and scored on scale of 1–4 for tightness of the clusters—studies with scores <2 were excluded (three studies were excluded due to diffuse clusters and low fit to HWE, P < 0.05). All genotyping centres were provided with an identical test plate of 94 samples (Coriell plate). For this plate, call-rates had to be >95% and concordance rates >98% (one study was excluded due to call-rate −65%). A further study was excluded because it could not supply sufficient data for QC to be adequately assessed.
RNA isoform studies
RNA was extracted from breast tumours and B-lymphocytes were collected from healthy donors, using Trizol (Invitrogen) according to the manufacturer’s instructions, followed by DNase I treatment. cDNA was prepared using Reverse Transcription Kit (Applied Biosystems) with random primers according to the manufacturer’s instructions.
For the B-lymphocyte assay PCR primers were designed in ESR1 exon 4 (Forward: 5′-GAGACATGAGAGCTGCCAACC-3′) and exon 6 (Reverse: 5′-CCACCATGCCCTCTACACATT-3′), to simultaneously amplify the full length (FL −417 bp) and Δ5ERα (Delta 5–278 bp) spliced forms. One per cent of the cDNA obtained was amplified in a buffer containing 0.5 U of Platinum Taq (Invitrogen), 1× PCR buffer, 2 mm of MgCl2, 200 mm of dNTPs and 0.3 mm of each primer. Cycling conditions were: 94°C for 5 min followed by 35 cycles of 94°C for 30 s, 59°C 30 s and 72°C for 1 min, with a final extension step of 72°C for 5 min. PCR products were separated by electrophoresis in 2% Nusieve 3:1 agarose (Cambrex) gels and stained with ethidium bromide. Intensities of the FL and Delta 5 bands were determined using Molecular Imaging Kodak 1D software (v 3.5.4), and ratios were calculated and plotted on a log2 scale. The ER+ breast tumours were analysed by real-time PCR using the PCR primers as described above, and MGB-probes (Applied Biosystems: 6-FAM-CCTCCATGATCAGGTCC-MGB for the FL form and VIC-TGCCAGGAACCAGGG-MGB, for Delta5). Reactions were carried out in triplicate on each sample according to the manufacturer’s instructions. Ct values were exported using the SWDS 2.1.2 software, and the ratio of the two forms was calculated as (CtFL− CtDelta5).
Statistical methods
For each SNP, deviation of genotype frequencies in controls from HWE was assessed by a χ2 test with one degree of freedom (1df). Genotype risks were estimated as ORs with 95% CIs, and significance levels were calculated using logistic regression. Additive, dominant and recessive models for the mode of action of associated SNPs were compared by likelihood ratio tests. The relative effects of correlated SNPs were compared by forward and reverse conditional logistic regression as well as likelihood ratio tests. Meta-analysis was carried out and Forrest plots generated using the Metan command within Stata™ (http://www.stata.com).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
FUNDING
This work has been predominantly funded by Cancer Research-UKgrants: C490/A11021 and C20/A3084 for SEARCH, C1287/A7497 for BCAC.
ACKNOWLEDGEMENTS
ABCS acknowledges Laura Van’t Veer, Rob Tollenaar, Flora van Leeuwen and other members of the BOSOM project, the support of Dr H.B. Bueno-de-Mesquita for organizing the release of control DNA, and financial support by the Dutch Cancer Society (NKI 2001-2423 and 2007-3839), the Cancer Genomics Center (Dutch Genomics Initiative) and the Ministry of Health, Welfare and Sports of The Netherlands. ABCFS is part of the Breast Cancer Family Registry supported by the National Cancer Institute, National Institutes of Health under RFA-CA-06-503 and R01-CA121245-01A2, and through cooperative agreements with members of the Breast Cancer Family Registry and P.I.s, including Cancer Care Ontario (U01 CA69467), Northern California Cancer Center (U01 CA69417) and University of Melbourne (U01 CA69638). J.H. is an Australia Fellow of the National Health and Medical Research Council (NHMRC) and Melissa Southey is an NHMRC Senior Research Fellow. BBCS—the British Breast Cancer Study and the Mammography Oestrogens and Growth Factors Study are funded by Cancer Research-UK and Breakthrough Breast Cancer. We acknowledge NHS funding to the NIHR Biomedical Research Centre. CGPS was funded by the Chief Physician Johan Boserup and Lise Boserups Fund the Copenhagen County Research Fund and the Danish Medical Research Council. CNIO-BCS was partially funded by the Genome Spain Foundation and the Marato Foundation. GENICA was supported by the German Human Genome Project and funded by the Federal Ministry of Education and Research (BMBF) grants 01KW9975/5, 01KW9976/8, 01KW9977/0 and 01KW0114. We thank Hiltrud Brauch for conceptual design and coordination of the study, Beate Pesch, Volker Harth and Thomas Brüning for patient recruitment and collection of epidemiologic data, as well as Susanne Haas and Hans-Peter Fischer for histopathological data. GC-HBOC was supported by the Deutsche Krebshilfe. We thank Claus R. Bartram for patient recruitment and Sandrine Tchatchou for genotyping. HABCS acknowledges the support of Profs. Michael Bremer and Johann Hinrich Karstens at the Clinics of Radiation Oncology and of Prof. Peter Hillemanns at the Clinics of Obstetrics and Gynaecology, Hannover Medical School, for providing patient samples and infrastructure. HEBCS was supported by the Academy of Finland (110663), Helsinki University Central Hospital Research Fund, the Sigrid Juselius Fund and the Finnish Cancer Society. We thank Kati Kämpjärvi for her contribution to the molecular analyses and Drs Kirsimari Aaltonen, Carl Blomqvist and Kristiina Aittomäki for their help in patient sample and data collection. KARBAC was supported by the Swedish Cancer Society, the Gustav V Jubilee Foundation, and the Bert von Kantzow foundation. kConFab is supported by the National Breast Cancer Foundation, the National Health and Medical Research Council of Australia and the Cancer Councils of Queensland, New South Wales, Western Australia, South Australia and Victoria. We thank kConFab nurses, staff of the Familial Cancer Clinics, Heather Thorne, Eveline Niedermayr, Helene Holland and Gillian Dite for their contribution and Study of kConFab [funded by the National Health and Medical Research Council of Australia (NHMRC grants 145684 and 288704)] for supplying some data. AOCS, which provided control samples only for the kConFab study, was supported by the U.S. Army Medical Research and Materiel Command under DAMD17-01-1-0729, the National Health and Medical Research Council of Australia (199600), the Cancer Council Tasmania and Cancer Foundation of Western Australia. The AOCS Management Group members include David Bowtell, Adele Green, Penny Webb, Anna DeFazio and Dorota Gertig. The genotyping and analysis were supported by grants from the National Health and Medical Research Council (NHMRC). We thank Jonathan Beesley for laboratory assistance. Georgia Chenevix-Trench is an NHMRC Senior Principal Research Fellow. MARIE was supported by the Deutsche Krebshilfe e.V. (Grant number 70-2892-Br I) and the German Cancer Research Center (DKFZ). We thank Dr Tracy Slanger and Elke Mutschelknauss for organization and conduct of the field work and Dr Ramona Salazar for the genotyping and Ursula Eilber, Sabine Behrens and Belinda Kaspereit for competent technical support. MCBCS was supported by NIH/NCI Breast Cancer SPORE P50 CA116201, NIH CA122340 and U.S. Department of Defense grant W81XWH-04-1-0588. MCCS is supported by the Australian National Health and Medical Research Council (grants 209057, 251533, 396414, 504711). Cohort recruitment and follow-up is funded by The Cancer Council Victoria. We want to thank Letitia Smith for her contribution to the genotyping. NHS was funded by the National Institutes of Health through CA098233, CA065725 and UO1-CA98233. We thank the NHS investigators, staff and participants. ORIGO was funded by the Dutch Cancer Society and coordinated by Peter Devilee, Jan Klijn and Rob Tollenaar; We thank Jannet Blom,Elly Krol-Warmerdam and Petra Huijts for patient recruitment and collection of clinical data. PBCS thank Stephen Chanock from the Advanced Technology Center and the Division of Cancer Epidemiology and Genetics, Mark Sherman from the Division of Cancer Epidemiology and Genetics, NCI, USA; N. Szeszenia-Dabrowska and Beata Peplonska of the Nofer Institute of Occupational Medicine; and W. Zatonski of the Department of Cancer Epidemiology and Prevention, Cancer Center and M. Sklodowska-Curie Institute of Oncology, 02-781 Warsaw, Poland for their contribution to the Polish Breast Cancer Study. The content of this manuscript does not necessarily reflect the views or policies of the NCI or any of the collaborating centers in the CFR, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government. SASBAC thank all the subjects who participated. We also give special thanks to Li Yuqing and Carine Bonnard for performing genotyping analysis. This work was funded by the Agency for Science & Technology and Research of Singapore (A*STAR), the National Institute of Health (grant number R01 CA 104021), and the Susan J. Kommen Foundation. SEARCH owe particular thanks to the EPIC Management team for access to control DNA; Ms Inma Spiteri for polysomal RNA, Prof. Carlos Caldas for tumour samples and the Eastern Cancer Registry and Intelligence Unit for data. We would like to acknowledge the support of The University of Cambridge, Hutchison Whampoa Ltd and Cancer Research-UK. A.M.D. is supported by CR-UK and P.D.P.P. is a Senior Clinical Research Fellow of CR-UK.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Zuppan P., Hall J.M., Lee M.K., Ponglikitmongkol M., King M.C. Possible linkage of the estrogen receptor gene to breast cancer in a family with late-onset disease. Am. J. Hum. Genet. 1991;48:1065–1068. [PMC free article] [PubMed] [Google Scholar]
- 2.Hill S.M., Fuqua S.A., Chamness G.C., Greene G.L., McGuire W.L. Estrogen receptor expression in human breast cancer associated with an estrogen receptor gene restriction fragment length polymorphism. Cancer Res. 1989;49:145–148. [PubMed] [Google Scholar]
- 3.Weel A.E., Uitterlinden A.G., Westendorp I.C., Burger H., Schuit S.C., Hofman A., Helmerhorst T.J., van Leeuwen J.P., Pols H.A. Estrogen receptor polymorphism predicts the onset of natural and surgical menopause. J. Clin. Endocrinol. Metab. 1999;84:3146–3150. doi: 10.1210/jcem.84.9.5981. [DOI] [PubMed] [Google Scholar]
- 4.Okura T., Koda M., Ando F., Niino N., Ohta S., Shimokata H. Association of polymorphisms in the estrogen receptor alpha gene with body fat distribution. Int. J. Obes. Relat. Metab. Disord. 2003;27:1020–1027. doi: 10.1038/sj.ijo.0802378. [DOI] [PubMed] [Google Scholar]
- 5.Schuit S.C., Oei H.H., Witteman J.C., Geurts van Kessel C.H., van Meurs J.B., Nijhuis R.L., van Leeuwen J.P., de Jong F.H., Zillikens M.C., Hofman A., et al. Estrogen receptor alpha gene polymorphisms and risk of myocardial infarction. J. Am. Med. Assoc. 2004;291:2969–2977. doi: 10.1001/jama.291.24.2969. [DOI] [PubMed] [Google Scholar]
- 6.Ioannidis J.P., Ralston S.H., Bennett S.T., Brandi M.L., Grinberg D., Karassa F.B., Langdahl B., van Meurs J.B., Mosekilde L., Scollen S., et al. Differential genetic effects of ESR1 gene polymorphisms on osteoporosis outcomes. J. Am. Med. Assoc. 2004;292:2105–2114. doi: 10.1001/jama.292.17.2105. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez-Zuloeta Ladd A.M., Vasquez A.A., Rivadeneira F., Siemes C., Hofman A., Stricker B.H., Pols H.A., Uitterlinden A.G., van Duijn C.M. Estrogen receptor alpha polymorphisms and postmenopausal breast cancer risk. Breast Cancer Res. Treat. 2007;107:415–419. doi: 10.1007/s10549-007-9562-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kjaergaard A.D., Ellervik C., Tybjaerg-Hansen A., Axelsson C.K., Gronholdt M-L.M., Grande P., Jensen G.B., Nordestgaard B.G. Estrogen receptor alpha polymorphism and risk of cardiovascular disease, cancer and hip fracture. Circulation. 2007;115:861–871. doi: 10.1161/CIRCULATIONAHA.106.615567. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez L.P., Milne R.L., Barroso E., Cuadros M., Arias J.I., Ruibal A., Benitez J., Ribas G. Estrogen and progesterone receptor gene polymorphisms and sporadic breast cancer risk: a Spanish case-control study. Int. J. Cancer. 2006;119:467–471. doi: 10.1002/ijc.21847. [DOI] [PubMed] [Google Scholar]
- 10.Gold B., Kalush F., Bergeron J., Scott K., Mitra N., Wilson K., Ellis N., Huang H., Chen M., Lippert R., et al. Estrogen receptor genotypes and haplotypes associated with breast cancer risk. Cancer Res. 2004;64:8891–8900. doi: 10.1158/0008-5472.CAN-04-1256. [DOI] [PubMed] [Google Scholar]
- 11.Wang J., Higuchi R., Modugno F., Li J., Umblas N., Lee J., Lui L.Y., Ziv E., Tice J.A., Cummings S.R., Rhees B. Estrogen receptor alpha haplotypes and breast cancer risk in older Caucasian women. Breast Cancer Res. Treat. 2007;106:273–280. doi: 10.1007/s10549-007-9497-8. [DOI] [PubMed] [Google Scholar]
- 12.Einarsdottir K., Darabi H., Li Y., Low Y-L., Li Y-Q., Bonnard C., Sjolander A., Czene K., Wedren S., Liu E.T., et al. ESR1 and EGF genetic variation in relation to breast cancer risk and survival. Breast Cancer Res. 2008;10:R15. doi: 10.1186/bcr1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Easton D.W., Pooley K.A., Dunning A.M., Pharoah P.D.P., Thompson D., Ballinger D.G., Struewing J.P., Morrison J., Field H., Luben R., et al. Genome-wide association study identifies novel breast cancer loci. Nature. 2007;447:1087–1093. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuqua S.A.W., Fitzgerald S.D., Chamness G.C., Tandon A.K., McDonnell D.P., Nawaz Z., O’Malley B.W., McGuire W.L. Variant human.breast tumour receptor with constitutive activity. Cancer Res. 1991;51:105–109. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.