

Abstract
The rapid rise and application of proteomic technologies has resulted in an exponential increase in the number of proteins that have been discovered and presented as ‘potential’ biomarkers for specific diseases. Unfortunately, the number of biomarkers approved for use by the Food and Drug Administration has not risen in likewise manner. While there are a number of reasons for this discrepancy, this glut of ‘potential’ biomarkers also indicates the need for validation methods to confirm or refute their utility in clinical diagnostics. For this reason, the emphasis on developing methods to target and measure the absolute quantity of specific proteins and peptides in complex proteomic samples has grown.
Keywords: mass spectrometry, biomarker validation, targeted proteomics, multiple-reaction monitoring, AQUA, SISCAPA
INTRODUCTION
Much of the effort in proteomics over the past decade has been to design experiments and technologies that are capable of characterizing as much of the proteome as possible. As these methods were developed, the focus turned to comparative proteomics, whose goal was to identify differences in the proteome between differentially treated cells, samples obtained from healthy and diseased patients, etc. [1–4]. The results were very impressive. For example, John Yates’ group was able to apply a high-throughput proteomics approach using multidimensional chromatographic separation and tandem mass spectrometry (MS2) to profile over 2400 proteins through the four stages of the life cycle of Plasmodium falciparum [5]. P. falciparum is a protozoan parasite and the most deadly of the four Plasmodium species that cause human malaria [6]. The proteome of Plasmodium was extracted from sporozoites (the infectious form injected by the mosquito), merozoites (the stage that invades the erythrocytes), trophozoites (the form that multiplies in erythrocytes) and gametocytes (sexual stages). The sporozoite proteome appeared markedly different from the other stages as almost half of the proteins identified were unique to this stage. Less than one-third of the proteins were unique to each of the other three stages. The specificity of the analysis is reflected in the fact that only 6% of the proteins identified were found in all four stages, with commonalities representing various classes of housekeeping proteins. This level of information is pivotal for development of efficient drugs or vaccines that are able to interrupt the life cycle of this complex parasite. More recently, an experimentally similar comprehensive proteomic approach was used to study three major types of lesions associated with the pathology of multiple sclerosis [7]. Laser-capture microdissection was used to acquire cells related to acute, chronic active and chronic plaques from patients suffering from multiple sclerosis. Comprehensive proteomic profiling was able to identify well over 2000 proteins from the various lesions. Comparison of the proteomes of each lesion type resulted in the identification of tissue factor and protein C inhibitor unique to chronic active plaque samples, suggesting a dysregulation of coagulation-associated proteins in these lesions. Follow-up studies in which recombinant activated protein C was administered in vivo, showed a lessening of the severity of the disease in experimental autoimmune encephalomyelitis. Understanding the neuropathology of multiple sclerosis is essential for improved therapies. Both of these preceding studies provide excellent examples of how a global, comparative proteomic approach can discover potential targets for, in one case preventing disease, and in another, therapeutic intervention.
THE SEARCH FOR BIOMARKERS
Once tools for conducting comprehensive proteome analysis became available, much of the interest turned towards analyzing biofluids and tissues for the purpose of finding novel biomarkers of diseases, such as cancer [8]. The general premise was quite straightforward; identify as many proteins as possible in a specific type of biofluid acquired from disease-affected patients and compare them to those identified from samples acquired from matched, healthy individuals. While samples such as urine, cerebrospinal fluid and interstitial fluid have been used, the primary sample type utilized in these discovery studies was serum and plasma. Because of the unique protein content of these samples strategies for removing highly abundant proteins needed to be developed [9]. Once these methods were in place, the ability to routinely identify hundreds of proteins in these biofluids became commonplace.
One of the biggest challenges in finding biomarkers using a comprehensive mapping strategy was throughput. While the term ‘high-throughput’ is commonly used to describe proteomic biomarker discovery, it does not truly apply to cases in which liquid chromatography coupled with MS analysis is used. To obtain the identification of greater than 1000 proteins in a serum or plasma sample takes on the order of days per sample [8]. This throughput has limited the number of samples analyzed per study to rarely more than 10. A simple power calculation shows that this number of samples is woefully inadequate for any potential biomarker reaching statistical significance. This lack of statistical power is one of the reasons why the progress in validation has been so poor; there is a general lack of confidence in most of the potential biomarkers reported in the literature and down-right unbelief in others. Unless there is a major breakthrough in technology, biomarker discovery studies using comprehensive proteomic identification will remain low-throughput compared to experiments such as mRNA microarray analysis or genome sequencing. There is a realization in the biomarker discovery community that unless there is a major technological breakthrough, the problem of sample throughput will remain. This issue, however, has not prevented research into developing MS methods for validating potential biomarkers discovered in global proteomic comparisons.
VALIDATION OF BIOMARKERS
Once potential candidates are recognized, the key step becomes validation. The literature is bursting with manuscripts reporting the discovery of ‘potential’ biomarkers for various diseases. A critical analysis shows that the likelihood of more than a handful of these being useful in a diagnostic setting is slim. Unlike discovery, however, validation requires a much greater stringency and very few MS laboratories have the resources to conduct biomarker validation in a high-throughput manner. For example, an excellent report by Dr Steve Carr compared the number of samples and analytes that are analyzed between discovery and validation studies [10]. At the discovery level, proteomic technologies permit only a few (10–20) samples to be analyzed, however, thousands of analytes are measured in each experiment. At the validation stage, hundreds (if not thousands) of samples must be analyzed, however, only a small number (e.g. 1–10) analytes are measured. If validation is conducted using MS, a typical analysis time is on the order of 1 h per sample. Since samples cannot be run concurrently on a single instrument, validation of 1000 samples will require over 40 days of continuous operation. There is also another very important distinction between analysis of proteins at the discovery and validation stages. This distinction involves the stringency in quantitation of the analytes. In the discovery phase, measurements quantitating differences in analytes between samples are not particularly accurate or precise. The confidence level in very subtle changes in abundance (e.g. <1.5-fold difference) between analytes in two different samples is generally low for MS-based measurements obtained from complex peptide mixtures for discovery of biomarkers. This low confidence level is tolerated for the sake of throughput and knowledge that any potential marker will necessarily go through a rigorous validation before it is ever used in a clinical setting. In validation, where the goal is to focus on a handful of potential markers, the confidence level in the ability to quantify these targets needs to be very high.
Another important issue in validation is that a majority of the ‘potential’ disease-specific biomarkers that have been discovered are not specific to the disease being studied. Many of these proteins fall into the categories of acute-phase response proteins whose concentrations change in response to infection or tissue injury. Biomarkers from this class of proteins would have very low specificity for a specific disease. Other proteins that have been reported to be potential biomarkers are induced by other stresses such as diet and medication and may have absolutely no relationship to the disease of interest. Unfortunately these proteins generally rank among the highest abundant proteins in serum or plasma and therefore exasperate the dynamic range problem in the discovery of lower abundance proteins that may be bona fide biomarkers.
Correct patient recruitment is another challenge that is sometimes underappreciated in biomarker discovery and validation. The challenge is considerably greater in validation simply because a much larger number of samples need to be analyzed. Not only must patients with the correct demographics for the disease of interest be located, their medical history, lifestyle, etc. must be carefully examined and if a prospective study is being design their continued participation must be assured. Beyond patient samples, proper controls must also be acquired from individuals with similar demographics as the patients but are disease-free. Calculations need to be performed to determine the number of cases and controls that provide adequate statistical power once the results are analyzed. All of these ‘practical’ issues can be a key determinant as to the eventual success of a biomarker discovery and validation effort.
Validation of potential biomarkers requires methods capable of quantitating these proteins across thousands of clinical samples. This quantitation should be absolute, rather than relative, so that results can be compared across sample cohorts, analytical platforms and laboratories. The analytical characteristic of MS makes it ideal for quantitating the absolute amounts of molecules in complex mixtures. Mass spectrometers have been workhorses in pharmaceutical companies for quantitating specific drugs and their metabolites in complex mixtures. The mass spectrometer provides a direct (rather than averaged) signal for each metabolite in the mixture ensuring the targeted compound is being measured. In addition, the sensitivity of current mass spectrometers, which continues to increase, enables compounds in the femtogram range to be accurately and precisely quantitated [11]. Theoretically, it should be possible to quantitate specific proteins and peptides with the same degree of accuracy and precision shown for metabolites. Another advantage of using MS for biomarker validation are the potential difficulties associated with antibodies. Antibody-based assays are not readily available for discovered biomarkers. Hence, the validation process depends on the ability to develop antibodies or other assays (e.g. tissue microarrays), which limits number of potential biomarkers for validation. For some potential biomarkers, antibodies are available to test their sensitivity and specificity within a clinical sample cohort. Due to their simplicity and throughput, high quality antibodies should be considered first for validation across a large number of samples. In the case of proteins for which no useful antibody exists, a targeted MS approach could be designed and applied to test the validity of a putative biomarker. In general, while development of an MS quantitative method is cheaper than producing an affinity reagent, an antibody-based test is less expensive over the long term. As MS technology continues to improve, however, it may supplant immunoassays as the best method for measuring specific analytes in biologic samples. Validation of biomarkers that rely on MS-based absolute quantitation of peptides, however, is time consuming and low-throughput. Methods for absolute quantitation of peptides [e.g. multiple reaction monitoring (MRM)] require significant development time and only a limited number of peptide quantitation assays can be developed and utilized concurrently.
DEVELOPMENT OF ABSOLUTE QUANTITATIVE METHODS
To accomplish quantitative proteomics required by biomarker validation, the development of absolute quantification (AQUA) by Dr Stephen Gygi's laboratory laid the foundation for developing methods that most laboratories use for MS-based biomarker validation [12, 13]. While AQUA was reported only a few years ago, it owes much of it genesis to stable-isotope dilution methods that have been around for decades. Dr Gygi, however, was quick to realize a similar strategy could be applied to peptides in complex mixtures. Many laboratories subsequently adopted this strategy for quantitating proteins within biofluids and other biologic samples. In an AQUA study, a peptide containing a stable-isotope labeled amino acid (e.g. lysine 13C6) is synthesized based on the sequence of a peptide of interest that is being targeted for quantitation. This ‘heavy’ peptide is spiked into the complex proteome sample and used as an internal standard for quantitation purposes. The measured absolute quantitation of the native peptide is then assumed to be reflected of the abundance of its protein of origin. This strategy has been applied to not only quantitate proteins but also their phosphorylation states by using phosphopeptide standards [13]. However, AQUA uses chemically synthesized isotopic peptides as internal standards and each of these peptides has to be synthesized, purified and optimized individually, which makes multiplex monitoring of proteins deferrable. Recently two other strategies as to how to construct internal standards were proposed and investigated. One method, referred to as QconCAT, transfected the DNA coding sequence of interested protein into Escherichia coli, which was grown in stable isotope containing media, to yield an isotope labeled protein concatamer [14]. Then the QconCAT protein was introduced into the biological samples and digested with trypsin to yield isotopic labeled peptides. The other method, protein standard absolute quantification (PSAQ), simply used in vitro-synthesized isotope labeled full-length proteins as standards for quantification [15]. By incorporating proteins as standards, more robust absolute quantitation can be achieved by introducing the standards into sample preparation as early as possible, and even more important, to easily monitoring multiple peptides derived from the same protein. To carry out global absolute quantitative proteomics, Silva et al. [16] developed a method based on the discovery that the average MS signal response for the three most intense tryptic peptides per mole of any given protein is constant within a coefficient of variation of less than ±10%. Based on this hypothesis, spiking in an internal standard protein at a known concentration to determine the universal signal response factor, absolute protein amounts for all identified proteins in a sample can thus be calculated. Although this approach is based on an empirical observation and simplifying the complexity and diversity of proteins to three representative peptides is susceptible to statistical under-representation, this easily applied and efficient approach offers an alternative to hypothesis-driven MRM-based approaches to biomarker discovery.
DETECTION AND QUANTITATION IN A COMPLEX CLINICAL SAMPLE
A key to measuring the absolute concentration of a peptide in a complex sample is knowing with absolute certainty, that the targeted molecule is being quantified. Using the known molecular mass of the peptide of interest can lead to an incorrect assignment, as the similitude of masses over all of the peptides within a proteome database is large. While adding retention time to this equation can increase the confidence of the assignment, chromatographic profiles can shift from run to run leading to an incorrect peptide being quantitated. Monitoring a specific peptide within a complex proteome sample is conducted through either selective reaction monitoring (SRM) or MRM [17]. In an SRM analysis, a single product ion derived from the MS/MS fragmentation of the parent ion is measured. While MRM is fundamentally similar, many product ions are measured, increasing the certainty of identification. In both SRM and MRM, the proteome mixture is fractionated using LC directly online with MS analysis. In SRM and MRM analysis, the elution time of the analytes of interest is generally known. At a designated retention time a specific mass-to-charge (m/z) value within the first quadrupole (Q1) region of the instrument is guided into the collision cell (Q2) and subjected to collision-induced dissociation (CID). One or more of the fragments are isolated in the Q3 region and allowed to pass onto the detector. The importance of using SRM or MRM to monitor a combination of parent and/or fragment ions of a peptide, rather than simply monitoring the parent ion of the peptide, is illustrated in Figure 1. The top inset shows the tandem MS spectrum of the peptide SGGGDLTLGLEPSEEEAPR (parent ion [M + 2H]2+ m/z 957.5), with three transition ions (m/z 914.4, 1043.5 and 1213.6) observed in the MS/MS analysis of this peptide. Single monitoring of the m/z range 957.00–958.00 (the parent ion) produces a very intense peak at ∼37 min. Two other less intense peaks are observed for peptides within this m/z range at ∼31 and 42 min. Based on the intensity of the signals, any investigator would be eager to select the peak at 37 min as the peptide of interest. However, when three known product ions (m/z 914.1, 1043.5 and 1213.6) resulting from CID of the parent ion (m/z 957.5) are monitored, it shows that only the peak at 31 min gives rise to these product ions. Therefore, the peak that elutes ∼31 min into the chromatogram is the peptide of interest. Besides the confidence that SRM and MRM provide, ensuring that the correct peptide signal is being measured, fragment ion monitoring also excludes a considerable amount of noise from the spectrum. Excluding the noise increases the sensitivity of the measurement over what could be obtained if only the parent ions were analyzed.
Figure 1:

Importance of monitoring transition ions when conducting targeted quantitative studies. The inset shows the tandem mass spectrometry (MS2) spectrum of the HER-2 peptide SGGGDLTLGLEPSEEEAPR. The bottom four spectra show peaks throughout the chromatograms when the parent ion and three transition ions labeled in the MS2 spectrum are monitored. Collectively presented spectra show that the peak at ∼31 min (asterisk and outlined) is the peptide of interest.
Probably the major challenge facing targeted, quantitative proteomics is that the MS signal obtained from a peptide behaves differentially depending on the matrix it is in. For example, it is well known that the ionization efficiency of a molecule is affected by its environment. Therefore, a solution only containing the peptide of interest will undoubtedly give a much more intense MS signal than the identical peptide within serum or plasma, for example. This phenomenon is illustrated in Figure 2, which shows the effect of adding increasing amounts of cell lysate on the signals obtained from a heavy isotopic version of the surrogate peptide (SGGGDLTLGLEPSEEEAPR; [M + 2H]2+ m/z 962.5) for the protein HER2 shown above. The areas of the peaks, representing three transition ions that were monitored during an MRM experiment, all show a >50% decrease when the amount of matrix added to the pure peptide is increased from 100 to 250 ng. A final addition of 1000 ng of cell lysate to the peptide solution makes each signal essentially unquantifiable. SRM and MRM methods, will play big roles in biomarker validation, however, optimization of each assay is going to be critical and most of the data will need to be determined empirically, although computational data will help in designing the starting points for these studies.
Figure 2:

Effect of matrix concentration on targeting specific peptide in complex mixture using MS. Increasing amounts of cell lysates was added to a peptide internal standard and the MS signals obtained using MRM. As the amount of matrix added increased, a concomitant decrease in the area of the peaks corresponding to the transition ions (open circle) m/z 924.5, (filled triangle) m/z 1053.5, (open triangle) m/z 1224.5, as well as their total areas (filled circle) was observed.
The above example shows a fundamental need for methods/strategies enabling targeting of specific proteins or peptides within complex proteomic mixtures: the requirement to create a mixture that is enriched for the molecule of interest. While this is a simple enough concept, in practice it is not trivial. The previous example shows the detrimental effect that only a small amount of lysate can have on a peptide signal. Therefore, the higher the enrichment, the better the assay will be. One of the most intriguing methods for enriching specific peptide was demonstrated by Leigh Anderson. This method termed SISCAPA (stable isotope standards and capture by anti-peptide antibodies) uses an isotopically labeled peptide internal standard (as with AQUA) that is spiked into an enzymatically digested complex proteome sample (Figure 3) [18]. In most demonstrations of SISCAPA, the proteome sample has been serum or plasma. The proteome sample, containing the peptide internal standard, is then passed over a column to which an antibody targeted to the peptide(s) of interest is immobilized. After washing the column, the bound components are eluted and analyzed using MS operating in an SRM or MRM mode. Since the m/z values of the peptide of interest and its stable isotope labeled internal standard is known, they can be measured directly within the chromatogram. The antibody column is then regenerated and re-used for other samples. The value in SISCAPA is that it aids in creating a mixture that is enriched for the peptide(s) of interest and enables lower abundant species to be detected since matrix effects are minimized.
Figure 3:
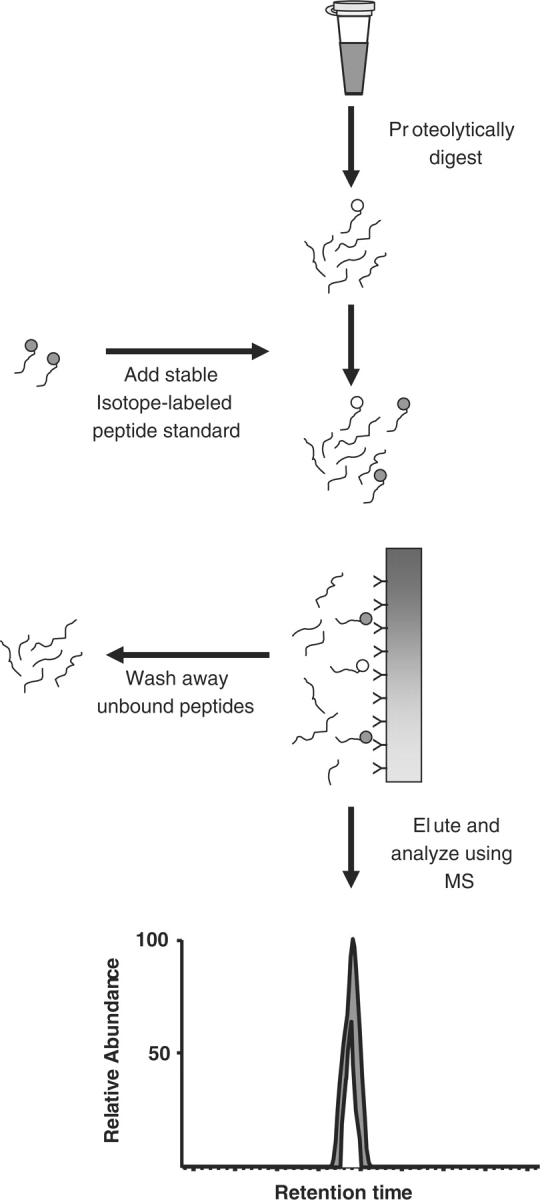
Schematic of stable isotope standards and capture by anti-peptide antibodies (SISCAPA) for targeted quantitative proteomic analysis. In SISCAPA, the proteome from a complex sample (such as plasma) is digested into peptides. A known quantity of stable-isotope labeled peptide corresponding to a peptide of interest in the digested proteome sample is added. The mixture is passed across an affinity column to which an antibody directed against the target peptide is coupled. After washing to remove unbound peptides, the specifically bound peptides are eluted and analyzed using MS. Comparison of the peak areas of the native and internal standard peptide is used to calculate the absolute amount of the native peptide in the clinical sample.
One of the earliest studies showing the ability to target specific peptides in a complex biofluid sample was conducted by Hunter and Anderson [19]. This study used immunodepleted plasma in which the six most abundant proteins were removed. The sample was then analyzed by LC–MS/MS using a multiplexed MRM for tryptic peptides representing 53 high and medium abundance proteins in human plasma. The within-run coefficients of variation (CVs) for quantitating the peptides was between 2% and 22% were for 47 out of the 53 proteins that were targeted. While immunodepletion of six high abundance proteins significantly improved CVs compared with whole plasma, the targeted analytes could still be detected in raw plasma. However, immunodepletion prior to LC–MS/MS analysis resulted in more precise measurements. Proteins present at concentrations less than 1 µg/ml could be reliably quantitated using this MRM method.
Recently, Steve Carr's laboratory published an in-depth analysis describing the use of MRM to quantitate six individual proteins that had been spiked into serum samples [20]. In this study, human female serum was spiked with five proteins derived from non-human sources and the male-specific protein, prostate-specific antigen (PSA).
Isotopically labeled internal peptide standards were used for absolute quantitation and one of the major goals was to limit the sample preparation required to quantitatively measure each of the analytes. Obviously one of the major drawbacks of using MRM is the low-throughput compared to immunoassays. While MRM methods may never match the speed of an immunoassay, nevertheless, it is important to maximize their throughput as much as possible. This study demonstrated the ability to quantitate the six proteins with sensitivities in the 1–10 ng/ml range and concentrations spanning a linear range of two orders of magnitude. The limit of quantitation (LOQ) was in the low to mid-pg/ml range. This sensitivity and LOQ was approximately three orders of magnitude better than what can be quantitated when do direct MRM/MS analysis of plasma. While one of the objectives of this study was to keep the sample preparation minimal, it was found that using a limited SCX peptide separation strategy resulted in a 10–40-fold improvement in the LOQ.
QUANTITATIVE ANALYSIS OF SPECIFIC PROTEINS IN FORMALIN-FIXED PARAFFIN-EMBEDDED (FFPE) TISSUES
The development of a quantitative method to measure a potential biomarker in easily acquired biofluids, such as serum, plasma and urine, has obvious advantages. Unfortunately, these fluids may not be proximal to the disease site resulting in any useful biomarker becoming diluted at the point of sample acquisition. This dilution factor can interfere with the ability to analytically measure any difference in levels between samples acquired from disease-affected and control patients. Sometimes it is necessary to assay the affected tissue directly as the targeted biomarker may not be secreted or shed into circulation at a level that is detectable using modern technologies [21]. Recently, there has been an appreciation that directly assaying affected tissues or proximal fluids provides the greatest chance of success in discovering and validated useful biomarkers. A recent study analyzed paired cancerous and normal clinical tissue samples taken from patients with colorectal adenocarcinoma [22]. A combination of heparin affinity fractionation, 2D–PAGE and MS2 identified 32 low abundance proteins that differed in abundance between the two sample types. The overexpression of proteasome subunit beta type 7 (PSB7), peroxiredoxin-1 (PRDX1) and signal recognition particle 9 (SRP9) that was observed in the global proteomic screen was validated by western blot analysis of patients with colon adenocarcinomas when compared to samples acquired from patients with lung adenocarcinomas. The overexpression of PSB7, PRDX1 and SRP9 was further confirmed using immunohistochemistry.
One issue related to the analysis of tissues, however, is their scarce availability. Obtaining tissue is very invasive and it is difficult to get a sufficient number of in vivo procured patient samples for discovery and validation studies. This limited availability issue might have been overcome with recent developments in the analysis of FFPE tissues using MS. In the past couple of years, a number of investigators have shown the ability to identify hundreds of proteins within FFPE tissues. Most of the studies have compared proteomes from different types of FFPE tissues for the purpose of discovering potential disease-related biomarkers. The group of Dr David Han at the University of Connecticut, however, has taken this type of analysis one-step further [23]. In a study comparing normal and prostate tissue, this group was able to identify 428 proteins from FFPE tissues of prostate cancer using MS. In the next experiment, they devised a method to specifically measure the levels of PSA, a known marker of prostate cancer, in FFPE tissue. They selected a PSA-specific peptide (LSEPAELTDAV*K) that had been detected in the global analysis of the tissues and had a synthetic version of this peptide containing a heavy isotope amino acid residue (valine-d8) prepared. One hundred femtomoles of this ‘heavy’ peptide was added as an internal standard to a tryptically digested proteome extracted from FFPE tissue sections. The amount of PSA was quantified in five normal controls and in 15 cancerous prostate tissues. The amount of PSA quantified directly from various prostate and normal tissue sections ranged from 0.5 to 140 pg. While the levels of PSA between normal (healthy) patients and those with low-, medium- and high-grade prostate cancer were not statistically significant, probably due to the limited number of samples analyzed, the general trend was to find increasing levels of PSA in more advanced tumors. These results demonstrate the ability to target specific biomarkers, such as PSA, for the purpose of accurately quantitating their levels in FFPE tissues aid in increasing the specificity and sensitivity in diagnosis of prostate cancer. The value of this method is it provides an absolute concentration of a protein, unlike immunohistochemistry (IHC), allowing accurate comparison across an infinite number of tumors.
THE USE OF TISSUE MICROARRAYS IN BIOMARKER VALIDATION
One of the most exciting developments in clinical proteomics are tissue microarrays (TMA) [24, 25]. Tissue microarrays essentially permit IHC to be performed concurrently on tens to hundreds of tissue specimen. In designing a TMA experiment, regions of interest are cored from archived tissues (either fresh frozen or FFPE). These cores, which are typically 0.6–2.0 mm in diameter, are sectioned into thin slices, which are transferred onto glass slides for automated IHC analysis. The IHC results are assessed manually or automated and can be directly linked to other available clinical data that has been acquired on the patient samples. The obvious advantage of a TMA is the high degree of precision and throughput that it provides for the analysis of these clinically important samples.
A recent study illustrates how TMA can be used for protein level validation of results obtained at the peptide level. In this study, cells were extracted from normal squamous epithelial tissue as well as poorly- (PD), mildly- (MD) and well-differentiation (WD) head and neck squamous cell carcinoma (HNSCC) tumors [26]. The quantitative estimate of the proteins within the normal sample and each tumor type was determined by the number of peptides identified for the specific protein. While a number of differences were seen among the cells, vimentin, which was upregulated in the tumor tissues, was of particular interest. To confirm these peptide-based results at an intact protein level, the same tissue types were interrogated using a TMA with an antibody directed against vimentin. Ten normal tissue sections were used, while for the PD, MD and WD HNSCC tumors between 33 and 105 tissues were analyzed. None of the 10 normal tissues stained for vimentin showed positive immunoreactivity, while ∼60–70% of the tumors showed positive staining for this protein, validating the peptide results at an intact protein level. As they continue their meteoric development, TMA will undoubtedly play a major role in validating many of the MS results that are still dominated by discovery at the peptide level.
CONCLUSIONS AND FUTURE DIRECTIONS
Presently, there are two things that we know about biomarker discovery: (i) MS-based discovery studies have identified a huge number of ‘potential’ biomarkers for specific diseases and (ii) unless there is a major breakthrough in technology, the confidence level in many of these potential biomarkers will remain low as the present sample throughput is low and prevents repetitive investigations. Regardless of the confidence level in this pool of potential biomarkers, investigators have moved forward in developing methods that will be used in future validation studies. This scenario is reminiscent of how comparative proteomic studies evolved. The first steps were simply the development of methods to identify large numbers of proteins. The major concern was not on the organism being analyzed or on the ability to compare the results to other proteomes. The amount of starting material was not a major factor either. Over the years, however, all of these issues became of greater importance and now comparative proteomics has matured to a stage at which even cells that are laser capture microdissected from tissue specimens are compared [20, 23, 26]. In many ways the same process is being followed in developing targeted methods to validate biomarkers. Presently, the focus is on developing MS-based MRM scanning methods combined with internal standards, to measure the absolute quantity of known proteins within complex clinical samples. Most of the efforts have targeted high abundance proteins in biofluids, biofluids spiked with varying concentrations of standard proteins, or already utilized biomarkers (e.g. PSA) in tissues. The point is, all of this effort is necessary to optimize the methods and technology for actual biomarker candidates to be critically validated.
It is still uncertain, however, what role MS will play in biomarker validation. While the technology will definitely be developed for absolute quantitation of potential biomarkers, MS will compete against the use of antibodies. MS does have certain advantages; direct measurement of the specific analyte so that cross-reactivity is a non-issue, ability to measure any analyte as long as it ionizes effectively, the ability to easily differentiate modified peptides from their unmodified forms, etc. MS, however, also has definitive disadvantages compared to an antibody-based method: throughput, ease of use, initial capital equipment cost, etc. Of these the two major advantages that antibody-based methods have are throughput and ease of use. While a single MS experiment targeting a specific analyte can be run in a matter of minutes, such experiments cannot be run concurrently as with an ELISA. In a clinical setting, tens to hundreds of assays need to be run everyday. The primary method of increasing the number of samples that can be analyzed using MS is to increase the number of available mass spectrometers. This quickly becomes cost prohibitive. One of the major arguments in the MS community for using MS-based validation methods is the lack of highly specific antibodies for many proteins. This fact is true, however, what cannot be ignored is that a number of useful antibodies do exist and there are a number of world-wide initiatives to manufacture high quality antibodies for every protein in the human proteome [27, 28]. As their number increases, the use of antibody-based validation methods will also increase.
Thus, is developing targeted quantitative proteomic methods a waste of time? Of course not. With the complexity of the human proteome, there is no guarantee that an affinity reagent can be made to react specifically with every predicted protein isoform (e.g. post-translational modifications, splice variants, single-nucleotide polymorphisms). The cost associated with validating a potential biomarker can be enormous. Targeted MS methods can help to minimize the cost by acting as a first screen to determine if the biomarker has any chance of being validated in a larger study. There will always be a need for non-biomarker related quantitative studies and MS is an excellent tool for filling this need. While there is still a lot of work to do to optimize methods in the discovery of potential biomarkers, investigators have looked to the future and have realized the need for targeted quantitative methods for proteomic profiling of clinical specimens [29]. While it is still early, investigators have made tremendous progress in this important analytical area.
Acknowledgements
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under Contract NO1-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the United States Government.
Biographies
Xiaoying Ye is the Postdoctoral Fellow of the Laboratory of Proteomics and Analytical Technologies at SAIC-Frederick Inc., NCI-Frederick. Her current research interests involve the development of isotope-based and label-free methods for quantitative proteomics and the application of these techniques to cancer biomarker research and protein–protein interactions.
Josip Blonder is the Head of the Section for Quantitative Proteomics, Laboratory of Proteomics and Analytical Technology at SAIC-Frederick Inc., NCI-Frederick. His current research interests include clinical proteomics, cancer biomarker discovery and membrane proteomics.
Timothy D. Veenstra is the Director of the Laboratory of Proteomics and Analytical Technologies at SAIC-Frederick, Inc., NCI-Frederick. His research interests include biomarker discovery and developing methods for quantitative measurement of small molecules in tissue sections.
References
- Wu CC, MacCoss MJ, Howell KE, et al. Metabolic labeling of mammalian organisms with stable isotopes for quantitative proteomic analysis. Anal Chem. 2004;76:4951–9. doi: 10.1021/ac049208j. [DOI] [PubMed] [Google Scholar]
- Ishihama Y, Oda Y, Tabata T, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteomics. 2005;4:1265–72. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- Fang R, Elias DA, Monroe ME, et al. Differential label-free quantitative proteomic analysis of Shewanella oneidensis cultured under aerobic and suboxic conditions by accurate mass and time tag approach. Mol Cell Proteomics. 2006;5:714–25. doi: 10.1074/mcp.M500301-MCP200. [DOI] [PubMed] [Google Scholar]
- Gygi SP, Rist B, Aebersold R. Measuring gene expression by quantitative proteome analysis. Curr Opin Biotechnol. 2000;11:396–401. doi: 10.1016/s0958-1669(00)00116-6. [DOI] [PubMed] [Google Scholar]
- Florens L, Washburn MP, Raine JD, et al. A proteomic view of the Plasmodium falciparum life cycle. Nature. 2002;419:520–6. doi: 10.1038/nature01107. [DOI] [PubMed] [Google Scholar]
- Griffith KS, Lewis LS, Mali S, Parise E. Treatment of malaria in the United States: a systematic review. JAMA. 2007;297:2264–77. doi: 10.1001/jama.297.20.2264. [DOI] [PubMed] [Google Scholar]
- Han MH, Hwang SI, Roy DN, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451:1076–81. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- Issaq HJ, Xiao Z, Veenstra TD. Serum and plasma proteomics. Chem Rev. 2007;107:3601–20. doi: 10.1021/cr068287r. [DOI] [PubMed] [Google Scholar]
- Zolotarjova N, Martosella J, Nicol G, et al. Differences among techniques for high-abundant protein depletion. Proteomics. 2005;5:3304–13. doi: 10.1002/pmic.200402021. [DOI] [PubMed] [Google Scholar]
- Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotech. 2006;24:971–83. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- Zhou M, Veenstra TD. Mass spectrometry: m/z 1983–2008. Biotechniques. 2008;44:667–70. doi: 10.2144/000112791. [DOI] [PubMed] [Google Scholar]
- Gerber SA, Rush J, Stemman O, et al. Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc Natl Acad Sci USA. 2003;100:6940–5. doi: 10.1073/pnas.0832254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkpatrick DS, Gerber SA, Gygi SP. The absolute quantification strategy: a general procedure for the quantification of proteins and post-translational modifications. Methods. 2005;35:265–73. doi: 10.1016/j.ymeth.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Pratt JM, Simpson DM, Doherty MK, et al. Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nat Protoc. 2006;2:1029–43. doi: 10.1038/nprot.2006.129. [DOI] [PubMed] [Google Scholar]
- Brun V, Dupuis A, Adrait A, et al. Isotope-labeled protein standards: toward absolute quantitative proteomics. Mol Cell Proteomics. 2007;6:2139–49. doi: 10.1074/mcp.M700163-MCP200. [DOI] [PubMed] [Google Scholar]
- Silva JC, Gorenstein MV, Li GZ, et al. Absolute quantification of cardiac troponin T by means of liquid chromatography/triple quadrupole tandem mass spectrometry. Mol. Cell. Proteomics. 2006;5:144–56. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- Ahmed N, Thornalley PJ. Quantitative screening of protein biomarkers of early glycation, advanced glycation, oxidation and nitrosation in cellular and extracellular proteins by tandem mass spectrometry multiple reaction monitoring. Biochem Soc Trans. 2003;31:1417–22. doi: 10.1042/bst0311417. [DOI] [PubMed] [Google Scholar]
- Anderson NL, Anderson NG, Haines LR, et al. Mass spectrometric quantitation of peptides and proteins using stable isotope standards and capture by anti-peptide antibodies (SISCAPA) J Proteome Res. 2004;3:235–44. doi: 10.1021/pr034086h. [DOI] [PubMed] [Google Scholar]
- Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5:573–88. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- Keshishian H, Addona T, Burgess M, et al. Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol Cell Proteomics. 2007;6:2212–29. doi: 10.1074/mcp.M700354-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prieto DA, Ye X, Veenstra TD. Proteomic analysis of traumatic brain injury: the search for biomarkers. Expert Rev. Proteomics. 2008;5:283–91. doi: 10.1586/14789450.5.2.283. [DOI] [PubMed] [Google Scholar]
- Rho JH, Qin S, Wang JY, Roehrl MH. Proteomic expression analysis of surgical human colorectal cancer tissues: up-regulation of PSB7, PRDX1, and SRP9 and hypoxic adaptation in cancer. J Proteome Res. 2008;7:2959–72. doi: 10.1021/pr8000892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SI, Thumar J, Lundgren DH, et al. Direct cancer tissue proteomics: a method to identify candidate cancer biomarkers from formalin-fixed paraffin-embedded archival tissues. Oncogene. 2007;26:65–76. doi: 10.1038/sj.onc.1209755. [DOI] [PubMed] [Google Scholar]
- Kononen J, Bubendorf L, Kallioniemi A, et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med. 1998;4:844–47. doi: 10.1038/nm0798-844. [DOI] [PubMed] [Google Scholar]
- Takikita M, Chung JY, Hewitt SM. Tissue microarrays enabling high-throughput molecular pathology. Curr Opin Biotechnol. 2007;18:318–25. doi: 10.1016/j.copbio.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Patel V, Hood BL, Molinolo AA, et al. Proteomic analysis of laser-captured paraffin-embedded tissues: a molecular portrait of head and neck cancer progression. Clin Cancer Res. 2008;14:1002–14. doi: 10.1158/1078-0432.CCR-07-1497. [DOI] [PubMed] [Google Scholar]
- Kaiser J. PROTEOMICS: Public-Private Group Maps Out Initiatives. Science. 2002;296:827. doi: 10.1126/science.296.5569.827. [DOI] [PubMed] [Google Scholar]
- Blow N. Antibodies: the generation game. Nature. 2007;447:741–4. doi: 10.1038/447741a. [DOI] [PubMed] [Google Scholar]
- Johann DJ, Jr, Blonder J. Biomarker discovery: tissues versus fluids versus both. Expert Rev Mol Diagn. 2007;7:473–5. doi: 10.1586/14737159.7.5.473. [DOI] [PubMed] [Google Scholar]