

Abstract
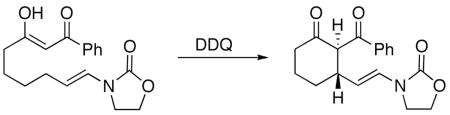
Vinyl oxazolidinones react with DDQ to form α,β-unsaturated acyliminium ions in a new method for forming electrophiles under oxidative conditions. Appended nucleophiles undergo 1,4-addition reactions with these intermediates to form cyclic vinyl oxazolidinones with good levels of diastereocontrol, highlighting a new approach to utilizing oxidative carbon–hydrogen bond functionalization to increase molecular complexity.
Converting carbon–hydrogen bonds to functional groups through oxidative transformations provides excellent opportunities for increasing molecular complexity from readily accessible substrates,1 and efforts toward expanding the scope of these processes are well warranted. We recently reported2 that benzylic and allylic ethers are oxidized by DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) to form oxocarbenium ion intermediates that react with appended nucleophiles in an efficient cyclization process. The oxidative cation formation appears to proceed (Scheme 1) through a sequence of radical cation formation followed by hydrogen atom abstraction. Based on this hypothesis we proposed an alternative entry to unsaturated carbocations that proceeds from vinyl ethers or amides. This approach benefits from improved access to radical cation intermediates because of the lower oxidation potentials of heteroatom-substituted alkenes relative to allylic ethers.3
Scheme 1.

Alternate oxidative pathways to stabilized carbocations.
Vinyl oxazolidinones have recently become an increasingly well-studied structural family.4 These compounds are easily prepared from oxazolidinones through direct condensation reactions with aldehydes,5 cross coupling reactions,6 and metal mediated additions across alkynes.7 Vinyl oxazolidinones are electron rich alkenes and react readily with electron deficient reagents such as oxidants,8 dihalides,9 metallocarbenes,4,10 and dicarbonyl compounds.11 The facile access and electron wealth of these compounds suggested that they would be excellent precursors to α,β-unsaturated acyliminium ions upon oxidative carbon–hydrogen bond activation.12 In this manuscript we demonstrate that vinyl oxazolidinones react readily with DDQ to form electrophiles that undergo 1,4-addition reactions with appended nucleophiles to form cyclohexane structures. The reactions proceed with good levels of diastereocontrol when tertiary carbons are formed. 1,3-Diketones can be used directly as nucleophiles in this process, obviating the need to preform the nucleophile.
We chose to utilize the hydroxyl group as a nucleophile in initial studies (Scheme 2). Branching was alsoincorporated at the allylic position of the vinyl oxazolidinone to prevent product oxidation. Exposing 1 to DDQ in 1,2-dichloroethane (DCE) resulted in the formation of tetrahydropyran 3 in 98% yield after 20 min. This process proceeded through the formation of α,β-unsaturated acyliminium ion 2 followed by an intramolecular 1,4-addition reaction. Secondary alcohol 4 cyclized to form tetrahydropyran 5 at −30 °C in 85% yield with modest diastereocontrol as expected based on the steric similarity between methyl and vinyl groups. Diastereocontrol was somewhat higher when this reaction was conducted in toluene rather than dichloroethane.
Scheme 2.

Allylic carbon–hydrogen bond activation from vinyl oxazolidinones.
Our initial studies in applying this approach for carbon–carbon bond formation focused on the use of enol acetate nucleophiles. These easily handled and readily accessible moities are excellent latent enolates in reactions that proceed through oxidatively-generated carbocations.13 We postulated that the ketones that arise from these reactions would suppress overoxidation by destabilizing carbocations that could form the reaction between cyclization products and DDQ. While exposing substrate 6 to DDQ resulted in substantial amounts of overoxidation, adding LiClO4 (15 mol%) to the reaction led to the formation of cyclohexanone 7 in 76% yield in 1 h (Scheme 3). Quaternary carbons can also be prepared through carbon–carbon bond formation, as illustrated by the conversion of 8 to 9. This reaction proceeded to completion within 1 h to provide the desired product in 74% yield. In contrast allylic oxazolidinone 10, which lacks heteroatom substitution on the alkene, was inert under the reaction conditions, thereby demonstrating the kinetic benefit of utilizing substrates with electron rich alkenes in these reactions.
Scheme 3.

Carbon–carbon bond formation.
We prepared several substrates to explore the scope of the process, the capacity to effect diastereoselective cyclization reactions, and the potential for expanding the scope of nucleophiles and product ring sizes in the reaction. The results of this study are shown in Table 1.
Table 1.
Oxidative cyclizations.a
| entry | substrate | product | temp (°C) | yield (%)b | dr |
|---|---|---|---|---|---|
| 1 |
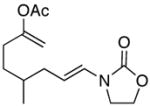 11 |
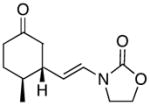 12 |
−15 | 78 | 7.3:1 |
| 2 |
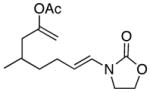 13 |
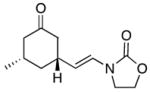 14 |
rt | 69c | 1:0 |
| 3 |
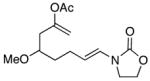 15 |
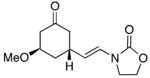 16 |
0 | 61d | 1:0 |
| 4 |
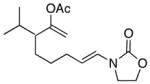 17 |
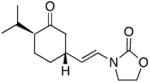 18 |
−10 | 79 | 9:1 |
| 5 |
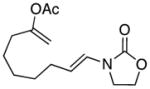 19 |
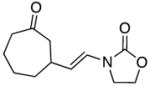 20 |
0 | 30 | - |
| 6 |
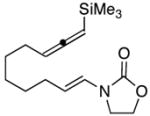 21 |
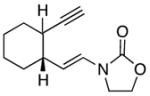 22 |
0 | 63 | 1:1 |
| 7 |
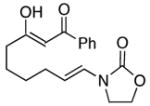 23 |
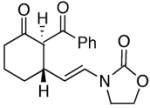 24 |
−15 | 75 | 1:0 |
| 8 |
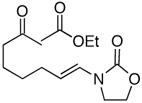 25 |
 26 |
0 | - | - |
Representative procedure: substrate, DDQ (2 equiv), LiClO4 (0.15 equiv), 2,6-dichloropyridine (2 equiv), and 4 Å MS were added to 1,2-dichloroethane at the appropriate temperature. See Supporting Information for details.
Isolated yields of purified materials. Diastereomeric ratios were determined based on isolated products or NMR-derived signal ratios of mixtures.
75% yield based on recovered starting material.
74% based on recovered starting material.
In general the reactions proceeded with good efficiency and diastereoselectivity. Notably, these are the first examples that we have observed in which carbocyclic structures have been prepared diastereoselectively through oxidative carbocation formation. The major products from substrates that contain alkyl branches (entries 1, 2, and 4) are consistent with chair-like transition states in which the alkyl groups and the α,β-unsaturated acyliminium ions occupy pseudoequatorial orientations. Interestingly the stereochemical outcome of the cyclization of methyl ether 15 (entry 3) is opposite from the cyclization of 13. This suggests that the stereochemical outcomes of these reactions are controlled by kinetics rather than thermodynamics. Cycloheptanones can be formed through this method (entry 5), though this reaction proceeds less efficiently than reactions that produce lower homologs. Substrate 19, however, contains no conformational control elements that could preorganize it for cyclization. Incorporating such constraints in the design of a cyclization substrate should improve cyclization efficiency. Silylallenes, which have proven to be excellent surrogates for propargylic anions14 in oxidative cyclization reactions,13a,b are effective nucleophiles in the synthesis of alkynylcyclohexanes (entry 6). These substrates show low levels of diastereocontrol, as expected based on the minimal gauche and transannular interactions between the linear allene and the forming ring. Entries 7 and 8 show the results of our efforts at using β-dicarbonyl compounds as nucleophiles. The use of these nucleophiles is advantageous because no additional steps are required for the preparation of a latent nucleophile, and the cyclization products contain a high degree of substitution. The resulting process is an example of a cross-dehydrogenative coupling reaction15 in which carbon–carbon bond formation occurs through a formal loss of H2. We observed that the enol content of the substrate played a critical role in the success of these reactions. β-Diketones exist largely in the enol form in non-polar solvents, as illustrated in compound 23, and are suitable nucleophiles for the cyclization reaction. β-Keto esters, however, exist largely in the dicarbonyl form and are not effective nucleophiles.
The stereochemical outcome of the cyclization of ether substrate 15 illustrates the role that electrostatic effects can play in conformational equilibria (Scheme 4). The lone pairs on the oxygen of the equatorial methoxy group are separated from the electrophilic site by a greater distance in conformation 27 than the lone pairs of the axial methoxy group and the electrophilic site in conformation 28. Since an axially-oriented methoxy group is only minimally disfavored relative to an equatorially-oriented methoxy group through steric effects, this electrostatic stabilization is sufficient to promote cyclization through conformation 28. As a comparison we calculated (AM1) the distance between the oxygens of the methoxy groups and the corresponding carbons of axial- and equatorial-3-methoxy cyclohexanone (O → C5). The axial oxygen is 2.92 Å from C5 while the distance from the equatorial oxygen is 3.75 Å. Similar explanations have been proposed to describe the conformational preferences of cyclic alkoxy-substituted oxocarbenium ions.16
Scheme 4.

Conformational control through electrostatic effects.
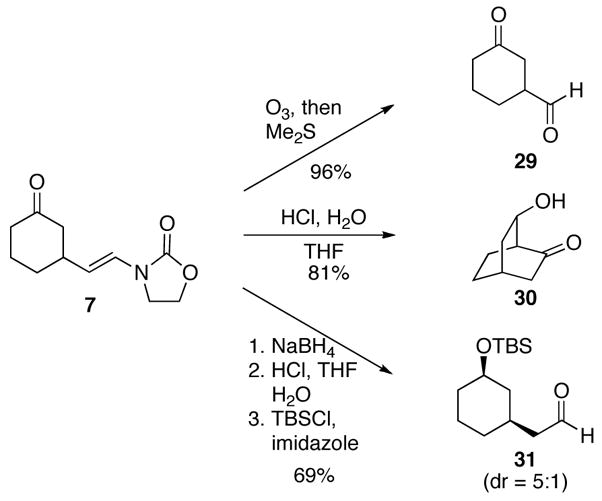
Vinyl oxazolidinones can serve as substrates for a number of transformations, providing significant flexibility to the types of structures that can ultimately be prepared through this sequence. Representative examples of these transformations are shown in Scheme 5. Ozonolytic cleavage of oxazolidinone 7 followed by reduction with Me2S provides a 96% yield of aldehyde 29. Exposing 7 to aqueous HCl results in vinyl oxazolidinone hydrolysis and subsequent aldol reaction to form the known17 bridged bicycle 30 in an 81% yield. The ketone can be reduced by NaBH4 to provide the alcohol as a 5:1 mixture of diastereomers in 93% yield. No reduction of the oxazolidinone was observed under these conditions. Acidic hydrolysis of the vinyl oxazolidinone followed by alcohol silylation yielded known18 aldehyde 31.
Scheme 5.
Vinyl oxazolidinone functionalization.
We have shown that vinyl oxazolidinones are excellent substrates for DDQ-mediated oxidative α,β-unsaturated acyliminium ion formation. The importance of alkene electron density is apparent through the significantly higher reactivity of vinyl oxazolidinones relative to allyl oxazolidinones. Cyclization through carbon–oxygen or carbon–carbon bond formation was demonstrated. Successful carbon–carbon bond formation was observed with enol acetate, silylallene, and β-diketone nucleophiles, with β-diketones being partularly attractive because they can be used without modification in cross-dehydrogenative coupling reactions. Diastereocontrol in the cyclizations is good to excellent when a chiral center is present in the substrate. The vinyl oxazolidinone groups in the reaction products can engage in a wide range of reactions to broaden the scope of products that are available through this method.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (GM062924) and the National Science Foundation (CHE—139851) for generous support of this work.
Footnotes
Supporting Information Available Schemes for substrate preparations, cyclization protocols, and complete characterization data for all substrates and products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For representative examples, see: Mukaiyama T, Hayashi Y, Hashimoto Y. Chem Lett. 1986:1627.Hayashi Y, Mukaiyama T. Chem Lett. 1987:1811.Murahashi SI, Komiya N, Terai H, Nakae T. J Am Chem Soc. 2003;125:15312. doi: 10.1021/ja0390303.Ying BP, Trogden BG, Kohlman DT, Liang SX, Xu YC. Org Lett. 2004;6:1523. doi: 10.1021/ol036314j.Zhang Y, Li CJ. J Am Chem Soc. 2006;128:4242. doi: 10.1021/ja060050p.Catino AJ, Nichols JM, Nettles BJ, Doyle MP. J Am Chem Soc. 2006;128:5648. doi: 10.1021/ja061146m.Chen MS, White MC. Science. 2007;318:783. doi: 10.1126/science.1148597.Niu M, Yin Z, Fu H, Jiang Y, Zhao Y. J Org Chem. 2008;73:3961. doi: 10.1021/jo800279j.Cheng D, Bao W. Adv Synth Catal. 2008;350:1263.Zhao L, Li CJ. Angew Chem, Int Ed. 2008;47:7075. doi: 10.1002/anie.200801367.Lin S, Song CX, Cai GX, Wang WH, Shi ZJ. J Am Chem Soc. 2008;130:12901. doi: 10.1021/ja803452p.
- 2.(a) Tu W, Liu L, Floreancig PE. Angew Chem, Int Ed. 2008;47:4184. doi: 10.1002/anie.200706002. [DOI] [PubMed] [Google Scholar]; (b) Tu W, Floreancig PE. Angew Chem, Int Ed. 2009;48:4567. doi: 10.1002/anie.200901489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rathore R, Kochi JK. Tetrahedron Lett. 1994;35:8577. [Google Scholar]
- 4.Carbery DR. Org Biomol Chem. 2008;6:3455. doi: 10.1039/b809319a. [DOI] [PubMed] [Google Scholar]
- 5.Song Z, Lu T, Hsung RP, Al-Rashid ZF, Ko C, Tang Y. Angew Chem, Int Ed. 2007;46:4069. doi: 10.1002/anie.200700681. [DOI] [PubMed] [Google Scholar]
- 6.(a) Jiang L, Job GE, Klapars A, Buchwald SL. Org Lett. 2003;5:3667. doi: 10.1021/ol035355c. [DOI] [PubMed] [Google Scholar]; (b) Pan X, Cai Q, Ma D. Org Lett. 2004;6:1809. doi: 10.1021/ol049464i. [DOI] [PubMed] [Google Scholar]; (c) Brice JL, Meerdink JE, Stahl SS. Org Lett. 2004;6:1845. doi: 10.1021/ol0494360. [DOI] [PubMed] [Google Scholar]; (d) Bolshan Y, Batey RA. Angew Chem, Int Ed. 2008;47:2109. doi: 10.1002/anie.200704711. [DOI] [PubMed] [Google Scholar]
- 7.Goossen LJ, Rauhaus JE, Deng G. Angew Chem, Int Ed. 2005;44:4042. doi: 10.1002/anie.200462844. [DOI] [PubMed] [Google Scholar]
- 8.(a) Xiong H, Hsung RP, Shen L, Hahn JM. Tetrahedron Lett. 2002;43:4449. [Google Scholar]; (b) Adam W, Bosio SG, Wolff BT. Org Lett. 2003;5:819. doi: 10.1021/ol0273194. [DOI] [PubMed] [Google Scholar]; (c) Sivaguru J, Saito H, Poon T, Omonuwa T, Franz R, Jockusch S, Hooper C, Inoue Y, Adam W, Turro NJ. Org Lett. 2005;7:2089. doi: 10.1021/ol0502230. [DOI] [PubMed] [Google Scholar]; (d) Aciro C, Davies SG, Garner AC, Ishii Y, Key M-S, Ling KB, Prasad RS, Roberts PM, Rodriguez-Solla H, O’Leary-Steele C, Russel AJ, Sanganee HJ, Savory ED, Smith AD, Thomson JE. Tetrahedron. 2008;64:9320. [Google Scholar]
- 9.Ko C, Hsung RP, Al-Rashid ZF, Feltenberger JB, Lu T, Yang JH, Wei Y, Zificsak CA. Org Lett. 2007;9:4459. doi: 10.1021/ol701768n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu T, Song Z, Hsung RP. Org Lett. 2008;10:541. doi: 10.1021/ol702824s. [DOI] [PubMed] [Google Scholar]
- 11.Gohier F, Bouhadjera K, Faye D, Gaulon C, Maisonneuve V, Dujardin G, Dhal R. Org Lett. 2007;9:211. doi: 10.1021/ol062626l. [DOI] [PubMed] [Google Scholar]
- 12.For related examples, see: Magnus P, Lacour J. J Am Chem Soc. 1992;114:767.Evans PA, Longmire JM, Modi DP. Tetrahedron Lett. 1995;36:3985.Magnus P, Lacour J, Evans PA, Rigollier P, Tobler H. J Am Chem Soc. 1998;120:12486.
- 13.Seiders JR, II, Wang L, Floreancig PE. J Am Chem Soc. 2003;125:2406. doi: 10.1021/ja029139v.Wang L, Seiders JR, II, Floreancig PE. J Am Chem Soc. 2004;126:12596. doi: 10.1021/ja046125b.Jung HH, Seiders JR, II, Floreancig PE. Angew Chem, Int Ed. 2007;46:8464. doi: 10.1002/anie.200702999.. For the initial report of using enol acetates as latent ketone enolates, see: Mukaiyama T, Izawa T, Saigo K. Chem Lett. 1974:323.
- 14.Masse CE, Panek JS. Chem Rev. 1995;95:1293. [Google Scholar]
- 15.Li CJ. Acc Chem Res. 2009;42:335. doi: 10.1021/ar800164n. [DOI] [PubMed] [Google Scholar]
- 16.(a) Woods RJ, Andrews CW, Bowen JP. J Am Chem Soc. 1992;114:859. [Google Scholar]; (b) Miljkovic M, Yeagley D, Deslongchamps P, Dory YL. J Org Chem. 1997;62:7597. [Google Scholar]; (c) Dudley TJ, Smoliakova IP, Hoffmann MR. J Org Chem. 1999;64:1247. [Google Scholar]; (d) Ayala L, Lucero CG, Romero JAC, Tabacco SA, Woerpel KA. J Am Chem Soc. 2003;125:15521. doi: 10.1021/ja037935a. [DOI] [PubMed] [Google Scholar]
- 17.Tzvetkov NT, Schmoldt P, Neumann B, Stammler HG, Mattay J. Tetrahedron: Asymmetry. 2006;17:993. [Google Scholar]
- 18.Ren L, Crudden CM. J Org Chem. 2002;67:1746. doi: 10.1021/jo001751m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.