

Abstract
Two-component signaling systems involving receptor-histidine kinases are ubiquitous in bacteria and have been found in yeast and plants. These systems provide the major means by which bacteria communicate with each other and the outside world. Remarkably, very little is known concerning the extracellular ligands that presumably bind to receptor-histidine kinases to initiate signaling. The two-component agr signaling circuit in Staphylococcus aureus is one system where the ligands are known in chemical detail, thus opening the door for detailed structure–activity relationship studies. These ligands are short (8- to 9-aa) peptides containing a thiolactone structure, in which the α-carboxyl group of the C-terminal amino acid is linked to the sulfhydryl group of a cysteine, which is always the fifth amino acid from the C terminus of the peptide. One unique aspect of the agr system is that peptides that activate virulence expression in one group of S. aureus strains also inhibit virulence expression in other groups of S. aureus strains. Herein, it is demonstrated by switching the receptor-histidine kinase, AgrC, between strains of different agr specificity types, that intragroup activation and intergroup inhibition are both mediated by the same group-specific receptors. These results have facilitated the development of a global inhibitor of virulence in S. aureus, which consists of a truncated version of one of the naturally occurring thiolactone peptides.
Staphylococcus aureus infections are a major cause of morbidity and mortality in community and hospital settings. Consequently, the emergence of methicillin-resistant and, more recently, vancomycin-resistant strains of S. aureus represents an enormous threat to public health. S. aureus pathogenesis primarily involves the secretion of toxins that damage or lyse host cells or interfere with the immune system, enzymes that degrade tissue components, and cell wall-associated proteins that may be involved in adhesion and protection against host defenses. Synthesis of many of these virulence factors is controlled by a global regulatory locus, agr (1, 2). This locus contains a two-component module that is activated by a secreted autoinducing peptide (AIP) in a cell density-dependent manner. This type of regulation falls under the rubric of the ever-broadening field of quorum sensing (3), whereby a population of bacteria responds in concert when a critical cell density is reached. In the case of S. aureus, as cells enter postexponential phase, the AIP reaches a threshold concentration that turns on the agr (virulence) response. This process is mediated by the five genes in the agr locus, agrB, D, C, A, and rnaiii (Fig. 1). The P2 promoter drives the transcription of the agrB, D, C, and A genes, which provide the cytosolic, transmembrane, and extracellular components of a quorum-sensing/autoinduction circuit. The agrD gene product is a propeptide that is probably processed and secreted by AgrB, an integral membrane protein (4). The resultant mature AIP binds to the transmembrane receptor-histidine kinase coded by agrC. Binding of the AIP triggers phosphorylation of AgrC on a histidine residue. The response regulator, AgrA, accepts the phosphate group from AgrC and, in conjunction with a second transcription factor, SarA (5, 6), activates transcription from the agr P2/P3 promoters. The P3 transcript, RNAIII, mediates up-regulation of secreted virulence factors as well as down-regulation of surface proteins (7–9).
Figure 1.

The agr locus is depicted, showing the various components and their putative actions in the signaling network.
The AIP–AgrC receptor pair shows considerable interstrain sequence variation, which must have resulted from evolutionary covariation of this region of the chromosome to retain the specificity of the receptor–ligand interaction. S. aureus strains can be divided into at least four agr specificity groups (10, 11). These strains appear to compete with each other at the level of agr expression, as each AIP inhibits the agr response in strains belonging to other groups. This type of bacterial interference is unusual, because it affects the expression of a subset of genes rather than inhibiting growth (10). Because of the intergroup inhibitory effects of the AIPs, we are now redefining “AIP,” an acronym for “autoinducing peptide,” to mean the mature AgrD peptides and their synthetic or modified variants. This usage is irrespective of whether the AIP acts as an autoinducer or as an inhibitor of agr expression.
Structure–activity analysis of the AIPs from S. aureus has begun to elucidate their mechanism of action (10, 12). These peptides contain a thiolactone ring structure (10) in which the α-carboxyl group of the C-terminal amino acid is linked to the sulfhydryl group of a cysteine, which is always the fifth amino acid from the C terminus of the peptide (see example in Fig. 1) (12). There are usually three or four amino acids N-terminal to the cysteine, which are collectively referred to as the “tail” of the AIP. The high-energy thiolactone linkage appears to be necessary for activation; the corresponding lactone and lactam analogues of the Group II AIP are inactive, as is the linear version of this and other AIPs (4, 12). However, the lactam and lactone analogues (but not the linear version) are potent intergroup inhibitors, arguing that the high-energy thiolactone linkage is not required for intergroup inhibition, but that the ring structure is important for both types of activity. It has also been shown in a murine s.c. abscess model that the Group II AIP attenuates the virulence of a Group I strain, which suggests possible therapeutic value for the AIPs (12). Agr is conserved throughout the staphylococci, and studies of the Staphylococcus epidermidis AIPs have generally confirmed what is known for S. aureus (13, 14).
There are two remarkable features of this system. First, the AIPs show major sequence diversity yet explicitly cross-inhibit agr activation with IC50s that are in the same range as the EC50s of the cognate AIPs. Second, analogs with oxygen or nitrogen in place of sulfur in the ring structure (i.e., the lactone or lactam vs. the thiolactone) are potent intergroup inhibitors, but neither activate nor inhibit their cognate receptors, at least when tested at concentrations a hundred-fold higher than the activating and inhibiting concentrations of the natural ligands. In earlier studies (4, 10, 12), agr activation and inhibition were studied in strains containing the chromosomal agr locus, sometimes by using a plasmid-carried agrP3∷β-lactamase (blaZ) reporter. In these studies, the endogenous peptide was produced and its concentration varied over time, which made precise studies of activation and inhibition difficult. Furthermore, the experimental design precluded the separation of the agr locus from any other possibly group-specific factors that might have a role in inhibition or activation. In the present study, we demonstrate that the group specificity of the response to the AIP (inhibition or activation) is, in fact, determined solely by the AIP–receptor interaction. In other words, AIP-specific inhibition as well as activation occurs at the level of the receptor, AgrC. These results have led to the development of a variant AIP that inhibits autologous as well as heterologous agr expression and is therefore a global inhibitor of the virulence response in S. aureus. This variant, a truncated Group II peptide lacking the tail, is proposed to act by interfering with AIP binding to the receptor.
Materials and Methods
Bacterial Strains and Growth Conditions.
Bacterial strains used are listed in Table 1. S. aureus cells were grown in CYGP broth (15) with shaking at 37°C. Overnight cultures on GL plates (15) were routinely used as inocula. Cell growth was monitored with a Klett–Summerson colorimeter with a green (540-nm) filter (Klett, Long Island City, NY) or a THERMOmax microplate reader (Molecular Devices) at OD650. Antibiotics used were ampicillin (100 μg/ml) for Escherichia coli and erythromycin (10 μg/ml) and tetracycline (5 μg/ml) for S. aureus. Biotyping of staphylococci was performed at the Tisch Clinical Microbiology Laboratory, New York University Medical School, which confirmed the species classification of the non-aureus strains.
Table 1.
Strains and plasmids
| Strains or plasmids | References | Comments |
|---|---|---|
| S. aureus | ||
| RN4220 | 15 | Mutant of 8325-4 that accepts foreign DNA |
| RN6390b | 15 | Group I prototype |
| RN6734 | 15 | Group I strain, φ-13 lysogen of 6390b |
| RN6911 | 8 | Group I RN6390b, with tetM replacing agr (agr-null) |
| RN7206 | 8 | Group I prototype, RN6734, with tetM replacing agr |
| RN6607 | 10 | Group II prototype |
| RN9120 | Unpublished | Group II with tetM replacing agr (agr-null) |
| RN8465 | 10 | Group III prototype |
| RN4850 | Unpublished | Group IV prototype from University of Minnesota strain KG* |
| RN9121 | Unpublished | Group IV with tetM replacing agr (agr-null) |
| RN9033 | This study | RN6911 with pRN7035 |
| CA1-I (RN9222) | This study | RN6911 with pRN7062 |
| CA1-I (RN9365) | This study | RN7206 with pRN7062 |
| CA1-II (RN9366) | This study | RN9120 with pRN7062 |
| CA2-II (RN9372) | This study | RN9120 with pRN7105 |
| CA2-I (RN9367) | This study | RN7206 with pRN7105 |
| CA4-IV (RN9371) | This study | RN9121 with pRN7107 |
| CA4-I (RN9380) | This study | RN7206 with pRN7107 |
| S. epidermidis | ||
| RN2375 | This study | Clinical isolate |
| S. warnerii | ||
| RN3178 | This study | Clinical isolate |
| E. coli | ||
| DH5α | Cloning strain | |
| Plasmids | ||
| pRN7035 | This study | Vector containing only agr-P3∷blaZ fusion |
| pRN7062 | This study | Vector containing group I agrCA and agr-P3∷blaZ |
| pRN7105 | This study | Vector containing group II agrCA and agr-P3∷blaZ |
| pRN7107 | This study | Vector containing group IV agrCA and agr-P3∷blaZ |
Kindly provided by Dr. Patrick Schlievert.
Construction of Strains and Plasmids.
A shuttle vector, pRN7035, was used that contains plasmid replicons for E. coli and S. aureus along with antibiotic resistance cassettes: ampicillin for E. coli and erythromycin for S. aureus. This plasmid was constructed by cloning a PCR product containing the 180-nt agr P2P3 region into the pUC polylinker site of plasmid, pRN7034, which contains a β-lactamase reporter gene. In the new construct, the P3 promoter is fused to the β-lactamase reporter gene. Plasmids pRN7062, pRN7105, and pRN7107 were constructed by cloning PCR fragments containing agrC, agrA, and downstream termination signals from the various groups, into the PstI or PstI/SphI sites of the pUC polylinker of pRN7035. Genomic DNA from a prototypical strain of each group was used as template in these reactions. The following PAGE-purified primers (Integrated DNA Technologies, Coralville, IA) were used in the PCR reactions: for pRN7062, forward primer, 5′-CCAACTGCAGGAAGTACCAAAAGAATTAACACAA (PstI site underlined), reverse primer, 5′-TTATACTGCAGACGTTTGCCAACATTACAAGAGG (PstI site underlined), for pRN7105, forward primer, 5′-TTTGAACTGCAGAAAGTACCCGCTGAATTAACG (PstI site underlined), reverse primer, 5′-GGTGAAGCATGCAGTTTGCCAACATTACAAGAGGTTGAACAAGCATTTTAA, (SphI site underlined), for pRN7101, forward primer, 5′ TCTTAACTGCAGAAGTTGAAATACCTAAAGAATTAA CTCAATTACACG (PstI site underlined), reverse primer, 5′-GGTGAAGCATGCAGTTTGCCAACATTACAAGAGGTTGAACAAGCATTTTAA, (SphI site underlined). Plasmid DNA from the corresponding E. coli DH5α derivative was introduced into the restriction-defective S. aureus strain, RN4220, by the protoplast method (15). Plasmids were then moved onto various genetic backgrounds by phage transduction (15). Note that RN9365 (CA1-I) (Table 1) is an additional strain with Group I agrC and agrA in a different Group I strain background. This strain was tested, and the results for RN9222 (CA1–1) were reproduced in this strain.
Preparation of AIP-Containing Supernatants.
S. aureus strains were grown in CYGP broth with shaking at 37°C for 9 h starting with an inoculum of ≈3 × 107 cells/ml in 10 ml of broth. Cells were removed by centrifugation at 4°C, and the supernatant was filtered (0.22-μm filter, Gelman). The filtrate was stored at −80°C and used as a source of AIP. The group-specific activity of each supernatant correlated with that of the corresponding synthetic peptide.
Synthetic Peptides.
All peptides used in this study have been previously described (12),
except for the Group II truncated thiolactone peptide,
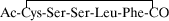 , which was synthesized by using
the transthioesterification method (12). After reverse-phase HPLC
purification of the peptide, the product was characterized as the
expected N-acetylated thiolactone-peptide by electrospray MS
and two-dimensional 1H NMR spectroscopy: observed
mass = 580.0 ± 1.0 Da, predicted (average) mass = 579.7
Da; 1H NMR (400 mHz,
DMSO-d6) ∂ 0.71 (Leu-4, γ−CH), 1.07 (Leu-4,
δ−CH), 1.30 (Leu-4, β-CH), 2.79 and 3.17 (Cys-1, β-CH), 2.95 and
3.30 (Phe-5, β-CH), 3.61 (Ser-3, β-CH), 3.65 (Ser-2, β-CH), 4.08
(Leu-4, α−CH), 4.24 (Ser-2, α−CH), 4.27 (Cys-1, α−CH), 4.29
(Ser-3, α−CH), 4.67 (Phe-5, α−CH), 7.14–7.31 (Phe-5, Ar), 8.04
(Ser-3, NH), 8.18 (Leu-4, NH), 8.31 (Ser-2, NH), 8.34 (Cys-1, NH), 8.98
(Phe-5, NH) ppm.
, which was synthesized by using
the transthioesterification method (12). After reverse-phase HPLC
purification of the peptide, the product was characterized as the
expected N-acetylated thiolactone-peptide by electrospray MS
and two-dimensional 1H NMR spectroscopy: observed
mass = 580.0 ± 1.0 Da, predicted (average) mass = 579.7
Da; 1H NMR (400 mHz,
DMSO-d6) ∂ 0.71 (Leu-4, γ−CH), 1.07 (Leu-4,
δ−CH), 1.30 (Leu-4, β-CH), 2.79 and 3.17 (Cys-1, β-CH), 2.95 and
3.30 (Phe-5, β-CH), 3.61 (Ser-3, β-CH), 3.65 (Ser-2, β-CH), 4.08
(Leu-4, α−CH), 4.24 (Ser-2, α−CH), 4.27 (Cys-1, α−CH), 4.29
(Ser-3, α−CH), 4.67 (Phe-5, α−CH), 7.14–7.31 (Phe-5, Ar), 8.04
(Ser-3, NH), 8.18 (Leu-4, NH), 8.31 (Ser-2, NH), 8.34 (Cys-1, NH), 8.98
(Phe-5, NH) ppm.
Agr Autoinduction and Inhibition Assay.
Agr-null derivatives were used with a plasmid containing a constant agr P2P3∷β-lactamase (blaZ) fusion plus a group-specific agrAC module. Assays were performed with CYGP cultures in early exponential phase (≈2 × 108 cells/ml). Culture supernatants (1/10 volume) or synthetic peptides in 10 mM phosphate buffer at pH 5.8 (concentration of stock solution determined by UV absorbance) were added, and cultures were incubated with shaking at 37°C for an additional 60 or 90 min, followed by determination of agr activation by β-lactamase assay (4). For inhibition tests, the activator and inhibitor were added simultaneously. EC50 and IC50 values were calculated from the sigmoidal dose-response curves by using nonlinear regression analysis with the program prism (GraphPad, San Diego). All assays were performed at least in duplicate.
Reverse-Phase HPLC Analysis of δ−Toxin Levels.
A modification of the assay originally described by Otto and coworkers (13) was used to quantify δ−toxin in supernatants from various strains. Early exponential-phase cultures were treated with increasing concentrations of synthetic peptides and then allowed to grow for 9 h. Supernatants, obtained by centrifugation, were analyzed by reverse-phase HPLC on a Hewlett–Packard 1100 series instrument with diode array detection. Typically, 400-μl aliquots of supernatant were injected onto a 1-ml Pharmacia Resource PHE column, which was eluted at 1 ml/min as follows: isocratic at 35% buffer B in buffer A for 3 min, which allowed the majority of the UV-absorbing material to wash through the column, followed by a linear gradient of 35–50% B in A over 20 min, where A = 0.1% trifluoroacetic acid (TFA) in water and B = 0.1% TFA in 90% acetonitrile/10% water. This optimized gradient achieved excellent separation of δ−toxin from other components (>95% pure by HPLC and MS), which permitted direct quantitation by peak integration. δ−Toxin is usually N-formylated, and the additional 29 Da of the N-formyl group was included in the calculated mass. We observed the following masses on purified fractions of δ−toxin: S. aureus Group III, observed mass = 3,007.1 ± 2.2 Da, predicted (average) mass = 3,006.5 Da; S. epidermidis strains, observed mass = 2,848.7 ± 1.5 Da, predicted (average) mass = 2,848.4 Da, and Staphylococcus warnerii strains, observed mass = 2,900.1 ± 0.8 Da, predicted (average) mass = 2,899.4 Da.
Results
Reconstitution of agr Signaling in agr-Null Host Backgrounds.
Shuttle vectors containing agrC and agrA under control of the agr-P2 promoter, along with a β-lactamase reporter gene driven by the agr-P3 promoter, were introduced into agr-null strains (see strains, Table 1). We reasoned, on the basis of prior studies (4, 10), that these constructs would be sufficient for signaling to occur in the agr-null backgrounds in response to exogenously added AIP. This was confirmed as shown in Table 2 and by example in Fig. 2. Two strains were used in these experiments, RN9222 (CA1-I) and RN9372 (CA2-II), containing the cloned agrCA/β-lactamase in the corresponding agr-null S. aureus strain (all reconstituted strains are provided with a descriptor in the format CAx-y, which indicates Group x agrC and agrA on the Group y agr-null background). In both cases, dose-dependent activation of the agr response was observed on addition of the cognate synthetic AIP (for example, see Fig. 2A). Moreover, the calculated EC50 values for agr activation in the two reconstituted strains were similar (Table 2). No dose-dependent agr activation was observed in the control strains, RN9033 and RN9416, which contain the vector alone (for example, see Fig. 2A).
Table 2.
Activation and inhibition by AIPs and an AIP analog
| RN9222 (CA1-I) | RN9366 (CA1-II) | RN9372 (CA2-II) | RN9367 (CA2-I) | |
|---|---|---|---|---|
| Activation EC50, nM | ||||
| Group I AIP | 40 ± 9 | 23 ± 7 | — | — |
| Group II AIP | — | — | 34 ± 6 | 28 ± 14 |
| Inhibition IC50, nM* | ||||
| Group I AIP | — | — | 26 ± 7 | 135 ± 52 |
| Group II AIP | 90 ± 30 | 78 ± 12 | — | — |
| RN9222 (CA1-I)* | RN9372 (CA2-II)* | RN8465 (Group III)† | RN9371 (CA4-IV)‡ | |
| Inhibition IC50, nM for Group II AgrD truncated thiolactone | ||||
| Group II AIP, truncated | 272 ± 67 | 209 ± 39 | 10 ± 1 | 188 ± 50 |
All values include an error of ±SEM determined from at least two assays.
These values were obtained with a constant concentration, 100 nM, of the autologous (activating) AIP.
† All results determined for the Group III strain RN8465 were from HPLC analysis of δ toxin. All other results were from β-lactamase assays.
‡ Group IV supernatant was used as a source of activating AIP in these analyses.
Figure 2.

Synthetic thiolactone peptides are biologically active in reconstituted strains. Shown are representative data for activation (A) and inhibition (B) of the agr response by synthetic AIPs. Degree of activity based on β-lactamase activity is shown as a plot of Vinit (initial velocity) vs. peptide concentration. (A) Activation of the agr response in RN9222 (CA1-I) by Group I AIP. The RN9033 (vector alone) control is shown. (B) Inhibition of the agr response in RN9222 (CA1-I) by Group II AIP in the presence of activating Group I AIP at 100 nM.
Reconstitution of Cross-Strain Inhibition in the Absence of AgrB.
As previously demonstrated (10), S. aureus culture supernatants inhibit agr activation in heterologous strains. Our prior study by using a reporter gene assay in strains containing the endogenous agr locus (12) demonstrated that synthetic AIPs could reproduce these effects. However, the site of action for the inhibition seen with these peptides was not determined. Although the most likely site seemed to be AgrC, the diversity of sequences among inhibitory peptides and their analogs suggested that some mechanism other than competitive blocking of activator binding could be responsible. One possibility would be binding to the putative processing–secretion factor, AgrB, causing interference with production or secretion of the activator. Alternatively, the peptides could bind to any range of targets on or within the cells and could interfere with the agr signaling pathway up- or downstream of AgrC activation.
As shown in Fig. 2B, RN9222 (CA1-I) was grown with Group I AIP at 100 nM along with various concentrations of the Group II AIP. A dose-dependent inhibition response was seen with an IC50 value of 90 ± 30 nM (Table 2), thus demonstrating that inhibition occurs in the absence of AgrB. Similar experiments were performed with RN9372 (CA2-II), yielding an IC50 for inhibition by the group I AIP of 26 ± 7 nM (Table 2). These results, in two different agr specificity groups, confirm that AgrB cannot be the target of inhibition, as the new strains lack AgrB.
The agrAC Two-Component Module Is Necessary and Sufficient for Group-Specific Activation and Inhibition.
We next asked whether AgrC determines the group specificity of the AIP response. This question was addressed by testing the two-component module in agr-null host backgrounds derived from different agr groups. Transfer of the group-specific signaling phenotype with agrCA would argue against any other group-specific determinant. Accordingly, two strains, RN9366 (CA1-II) and RN9367 (CA2-I), were tested for their ability to respond to supernatants from agr± strains, as well as to Group I and II AIPs.
The two strains were compared with the appropriate controls, RN9222 (CA1-I) and RN9372 (CA2-II), for activation by post-exponential phase supernatants from a series of agr± strains and by AIPs. As illustrated in Fig. 3, only supernatant from the Group II agr wild-type strain, RN6607, was able to activate β-lactamase expression in RN9372 (CA2-II) and RN9367 (CA2-I). In addition, similar EC50 values for activation were obtained for both strains by using Group II AIP (Table 2). Analogous results were obtained by using RN9222 (CA1-I) and RN9366 (CA1-II): these were activated by wild-type Group I supernatants (RN6734) and not by Group II supernatant or by agr-null supernatants from either group (data not shown). Moreover, similar EC50 values for activation were obtained for both strains by using the Group I AIP (Table 2). Therefore, the group specificity of agr signaling does not depend on the host background. Furthermore, the conversion of an agr-null Group I strain, RN7206 into a Group II responsive strain, RN9367 (CA2-I), by the insertion of plasmid-encoded Group II agrC and agrA (and vice versa) suggests strongly that the group specificity of the agr response depends on the expression of the two-component module containing agrC and agrA.
Figure 3.

A representative example of activation of RN9372 (CA2-II) and RN9367 (CA2-I) by postexponential supernatants collected from Group 1, 2, and 4 agr± cells. Data were collected as β-lactamase activity (Vinit in mOD/min) and then normalized to percentage activation.
The next question was whether cross-strain inhibition also proceeds through the AgrC/AgrA two-component cascade. If so, a Group II agr null strain (RN6607) reconstituted with Group I agrC and agrA, i.e., RN9366 (CA1-II), should be inhibited by Group II agr+ supernatants or by Group II AIP. Alternatively, if inhibition were to involve a group-specific factor outside of the agr locus, then inhibition would depend on the host background in which AgrC and AgrA are expressed. The former alternative was shown to be correct, as very similar IC50s for inhibition by the Group II AIP in RN9366 (CA1-II) and RN9222 (CA1-I) were obtained (Table 2). The same was true for inhibition of RN9372 (CA2-II) and RN9367 (CA2-I) by Group I AIP, albeit with a slight difference in the IC50s (Table 2). It is apparent that inhibition by the AIP depends on which agrC and agrA genes are expressed and not on the genetic background in which they are expressed, thereby indicating that the site of inhibition must be within the Agr A-C module. Because AgrC is a variable transmembrane receptor (16) and AgrA is highly conserved, it is suggested that the inhibitors bind to and act at AgrC.
Synthesis and Testing of a Global Inhibitor of Virulence in S. aureus.
Previous structure–activity studies have identified key regions within the Group II AIP required for differential agr activation and inhibition (12). Changes in the ring structure of the AIP could affect either activation or inhibition of the agr response or both, whereas changes in the tail region of the molecule affected only activation. We reasoned that a truncated AIP analog containing only the thiolactone ring structure and lacking the activating tail might be able to bind its own AgrC receptor, because of conservation of the critical thiolactone moiety, but would not have the tail necessary for activation. This analog would therefore be a good inhibitor not only in a crossgroup manner but also within the same group. Converting a receptor agonist into an antagonist of its autologous group as well as an inhibitor of heterologous groups would represent a global inhibitor of virulence in S. aureus. This would comprise a key step in the development of an agr-based therapeutic.
A truncated Group II thiolactone peptide was synthesized by using the transthioesterification approach (12) and tested for its ability to activate and inhibit the agr response in Group I–IV strains. The Group I, II, and IV experiments used the β-lactamase reporter assay, whereas the Group III strain was tested by an HPLC assay described below. The truncated Group II AIP had no detectable activation activity for the four groups. Furthermore, the peptide was an inhibitor in the nanomolar range of the agr response in all four groups (Table 2), indicating that deletion of the tail region of the peptide does indeed convert the AIP from an agonist into an antagonist of self activation as well as retaining intergroup inhibition.
The β-lactamase reporter assay could not be used for Group III, because of the production of β-lactamase by all of the available strains. However, we were able to demonstrate inhibition of Group III S. aureus by the full-length and truncated Group II AIP by HPLC analysis of δ−toxin production (13). δ−Toxin is a ≈25-aa hemolytic peptide encoded by RNAIII, and the level of this translation product is proportional to the level of RNAIII in the cell (13). Early exponential-phase cultures were treated with increasing concentrations of truncated Group II AIP and then allowed to grow for 9 h. Supernatants were collected and the amounts of δ−toxin quantitated. Inhibition by the full-length Group II AIP was demonstrated with an IC50 value of 17 ± 13 nM. A representative curve for inhibition by the truncated Group II AIP is shown in Fig. 4, with an IC50 value of 10 ± 1 nM (Table 2).
Figure 4.

The Group III strain, RN8465, was grown in the presence of differing concentrations of the truncated Group II analog. A representative inhibition curve derived from reversed-phase HPLC analysis of δ−toxin levels is shown. Quantitation of δ−toxin levels was obtained by integration of peak area from the HPLC trace.
The truncated thiolactone peptide is therefore a potent inhibitor for all four agr specificity groups of S. aureus. We have also begun to test the activity of AIPs in alternative staphylococcal species by using HPLC analysis of δ−toxin production. Our preliminary results thus far show only weak inhibition (in the micromolar range) of S. epidermidis strain RN2375 by the full-length and truncated group II AIPs. However, strong inhibition (in the nanomolar range) by the truncated group II AIP was seen with the S. warnerii strain RN3178, with an IC50 value of 20 ± 12 nM.
Discussion
Two-component systems are the major means by which bacteria sense a large variety of environmental stimuli. In most cases, the activating ligands for the receptors are unknown (17). The agr system is one of a few cases where the ligands have been chemically characterized, as thiolactone peptides (AIPs), and the system is unique in that AIPs from heterologous strains are naturally occurring inhibitors of agr signaling. In this study, we have begun to analyze the mechanism by which naturally occurring AIP sequence variants inhibit agr activation. Although the agr locus is conserved throughout the staphylococci, the agr autoinducing peptides and their cognate receptors have radically diverged, generating at least four autoinduction specificity groups in S. aureus. In general, the AIP activates agr expression in strains belonging to the same group as the producing organism and inhibits agr expression in organisms belonging to any other group. It is very possible that this divergence causes intergroup interference, leading to environmental isolation, and is at least partially responsible for speciation within the staphylococci. Similar divergence within the structurally similar comAP locus, required for competence in Bacilli, is also thought to be responsible for speciation in these organisms (18).
Structure–activity analysis has revealed two key features of the AIP–receptor interaction in S. aureus that are critical for the understanding of the present results: (i) Derivative peptides of the Group II AIP with a lactone or lactam replacing the essential thiolactone bond showed substantial loss of intragroup agr activation activity (at least 100-fold less potent) but retention of cross-inhibition activity. These derivatives, however, did not inhibit activation of the cognate receptor by the native thiolactone peptide (did not self inhibit). (ii) Substitution of any amino acid residue within the C-terminal thiolactone ring affected either activation and/or inhibition activity, whereas substitution of any of the four residues in the N-terminal linear section of the peptide decreased or eliminated activation but did not decrease inhibition. We have previously hypothesized that activation and inhibition by native AIPs must therefore involve different mechanisms—either different modes of interaction with the AIP receptor, AgrC, or interaction with cellular components other than AgrC, e.g., AgrB.
This latter possibility was clearly ruled out by means of plasmid constructs containing a β-lactamase reporter driven by the (conserved) agr P3 promoter and including the P2 driven agrA + C modules from different specificity groups. Placement of these plasmids in agr-null derivatives of the agr specificity group corresponding either to that of the cloned module or to that of other groups enabled the direct in vivo analysis of AIP-specific activation/inhibition in the absence of AgrB or of any endogenous AIP. Because these tests demonstrated the predicted cross-group inhibition, qualitatively in parallel with previous results obtained with the endogenous agr loci intact, it is clear that AgrB is not the site of inhibition. Since these tests also demonstrated that crossgroup inhibition occurs independently of host background, and with similar dose responses, it is also clear that the only determinant of agr group specificity is the AgrA + C module. Although we have not tested AgrA and C independently, AgrA is highly conserved among agr specificity groups within and between species, whereas the receptor portion of AgrC is highly variable; thus there is little doubt that AgrC is the key determinant. Although there are certainly other cellular factors involved in agr signaling, our results indicate that none of these determines group specificity.
The assay method used in this study is different from that previously used (12), in enabling an assessment of AIP activity in the absence of skewing effects of the endogenous peptide. The values obtained here are a more accurate reflection of the correct physiological values, because we can precisely deliver and quantify the amount of AIP in the system. The inhibition studies for the agr-null Group I and II strains reconstituted with the AgrA + C modules were performed with activator at 100 nM, which was determined to be a saturating but not oversaturating dose of activator. This generated maximal activation against which to test various concentrations of inhibiting heterologous AIPs. This differs from the natural situation, where the concentration of the endogenously produced peptide appears to be less than 100 nM, on the basis of comparison of the measured IC50 values with those of a previous study by using endogenously produced peptide as the source of activator (12).
The apparent difference in the roles of the ring and tail moieties of the Group II AIP could be interpreted to mean that the ring structure is responsible for recognition of and binding to the putative ligand pocket of the receptor, and that the tail region is responsible for initiating the signaling process. It was reasoned that removal of the tail region might render a ligand that should be unable to activate AgrC signaling but should be a strong inhibitor of its cognate AgrC as well as of the heterologous AgrCs. This was confirmed by using a synthetic Group II AIP lacking the tail but retaining the thiolactone structure. This peptide inhibited not only the Group II receptor but also those of Groups I, III, and IV (which, based on our current understanding of agr grouping, represents global agr inhibition in S. aureus). Furthermore, the truncated analog weakly inhibited δ−toxin production in the S. epidermidis strain, RN2375, but strongly inhibited δ−toxin production in the S. warnerii strain, RN3178. These results suggest that the truncated thiolactone Group II AIP may serve as the model for a global inhibitor of staphylococcal agr activation, which may have significant clinical utility.
The observation that an AIP agonist of the agr response (i.e., the intragroup AIP) can be converted into an antagonist of that response by removing the tail (i.e., the truncated AIP) provides further compelling evidence that AgrC is the molecular locus of activation and inhibition. These results also suggest that other less dramatic modifications in the AIP tail might lead to the generation of global inhibitors of the agr response. Several questions remain to be answered in this system. For example, we have yet to fully account for why the lactam and lactone Group II AIP analogs are potent intergroup inhibitors but are not potent self activators or inhibitors. Clearly, this points to the thiolactone linkage being an important determinant of self activation or inhibition. We have previously speculated that the acylating nature of the thioester group might play a role in these processes and, although the current study does not address this issue specifically, it does identify AgrC as the likely target of such a modification, should it take place. Future studies will now address this issue, as well as whether activation and inhibition occur within the same binding pocket of AgrC.
Acknowledgments
We thank Hope Ross, Ehab Khalil, Julio Camarero, Graham Cotton, and others in the Novick and Muir labs for their advice and support. We thank Mike Goger for assistance with the NMR experiments. This work was supported by National Institutes of Health (NIH) MSTP grant GM07739 (G.J.L.), the Burroughs–Welcome Fund (T.W.M.), and the NIH (AI 42783, R.P.N.).
Abbreviations
- agr
accessory gene regulator
- AIP
agr-autoinducing or -inhibiting peptide
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Recsei P, Kreiswirth B, O'Reilly M, Schlievert P, Gruss A, Novick R P. Mol Gen Genet. 1986;202:58–61. doi: 10.1007/BF00330517. [DOI] [PubMed] [Google Scholar]
- 2.Peng H L, Novick R P, Kreiswirth B, Kornblum J, Schlievert P. J Bacteriol. 1988;170:4365–4372. doi: 10.1128/jb.170.9.4365-4372.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bassler B L. Curr Opin Microbiol. 1999;2:582–587. doi: 10.1016/s1369-5274(99)00025-9. [DOI] [PubMed] [Google Scholar]
- 4.Ji G, Beavis R C, Novick R P. Proc Natl Acad Sci USA. 1995;92:12055–12059. doi: 10.1073/pnas.92.26.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheung A L, Projan S J. J Bacteriol. 1994;176:4168–4172. doi: 10.1128/jb.176.13.4168-4172.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chien Y, Cheung A L. J Biol Chem. 1998;273:2645–2652. doi: 10.1074/jbc.273.5.2645. [DOI] [PubMed] [Google Scholar]
- 7.Morfeldt E, Taylor D, von Gabain A, Arvidson S. EMBO J. 1995;14:4569–4577. doi: 10.1002/j.1460-2075.1995.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Novick R P, Ross H F, Projan S J, Kornblum J, Kreiswirth B, Moghazeh S. EMBO J. 1993;12:3967–3975. doi: 10.1002/j.1460-2075.1993.tb06074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Benito Y, Kolb F A, Romby P, Lina G, Etienne J, Vandenesch F. RNA. 2000;6:668–679. doi: 10.1017/s1355838200992550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ji G, Beavis R, Novick R P. Science. 1997;276:2027–2030. doi: 10.1126/science.276.5321.2027. [DOI] [PubMed] [Google Scholar]
- 11.Jarraud S, Lyon G J, Figueiredo A M S, Lina G, Vandenesch F, Jerome E, Muir T, Novick R P. J Bacteriol. 2000;182:6517–6522. doi: 10.1128/jb.182.22.6517-6522.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayville P, Ji G, Beavis R, Yang H, Goger M, Novick R P, Muir T W. Proc Natl Acad Sci USA. 1999;96:1218–1223. doi: 10.1073/pnas.96.4.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otto M, Sussmuth R, Vuong C, Jung G, Gotz F. FEBS Lett. 1999;450:257–262. doi: 10.1016/s0014-5793(99)00514-1. [DOI] [PubMed] [Google Scholar]
- 14.Otto M, Sussmuth R, Jung G, Gotz F. FEBS Lett. 1998;424:89–94. doi: 10.1016/s0014-5793(98)00145-8. [DOI] [PubMed] [Google Scholar]
- 15.Novick R P. Methods Enzymol. 1991;204:587–636. doi: 10.1016/0076-6879(91)04029-n. [DOI] [PubMed] [Google Scholar]
- 16.Lina G, Jarraud S, Ji G, Greenland T, Pedraza A, Etienne J, Novick R P, Vandenesch F. Mol Microbiol. 1998;28:655–662. doi: 10.1046/j.1365-2958.1998.00830.x. [DOI] [PubMed] [Google Scholar]
- 17.Hoch J. Curr Opin Microbiol. 2000;3:165–170. doi: 10.1016/s1369-5274(00)00070-9. [DOI] [PubMed] [Google Scholar]
- 18.Tortosa P, Dubnau D. Curr Opin Microbiol. 1999;2:588–592. doi: 10.1016/s1369-5274(99)00026-0. [DOI] [PubMed] [Google Scholar]