

Abstract
The generation of excess reactive oxygen species (ROS) in mitochondria is responsible for much of the oxidative stress associated with ageing (aging), and mitochondrial dysfunction is part of the pathology of neurodegeneration and type 2 diabetes. Lipophilic pyridinium ions are known to accumulate in mitochondria and this paper describes a general route for the preparation of nitrone-containing N-arylpyridinium salts having a range of lipophilicities, as potential therapeutic antioxidants. The compatibility of nitrones with the Zincke reaction is the key to their synthesis. Their trapping of carbon-centred radicals and the EPR spectra of the resulting nitroxides are reported.
Graphical abstract

1. Introduction
The reactive oxygen species (ROS) generated within the mitochondria are ultimately responsible for much of the oxidative damage that leads to the neurodegeneration associated with ageing.1 ROS produced by dysfunctional mitochondria also contribute to increased risk of cardiovascular disease in people showing insulin resistance through type 2 diabetes, which is also associated with ageing.2 The life-expectancy of people is rising and the birth rate is low in most developed countries and this is leading to an ageing population so there is great interest in ensuring that people have a healthy old age. Nitrones have shown potential for the prevention and treatment of age-related diseases.3 One possible mechanism for their action is the scavenging of the free radicals that lead to oxidative damage. Nitrones 1 react with these highly reactive oxygen-centred and carbon-centred radicals (Y•) to give nitroxides 2 that are more stable and longer lived (Scheme 1).4 The nitroxides 2 can be detected by EPR spectroscopy and the hyperfine splittings observed can often be used to identify the radical that led to their formation. Thus, nitrones can be used as so-called spin traps for the study of radical processes in biological samples. Cyclic spin traps give observable nitroxides from both oxygen-centred and carbon-centred radicals. Acyclic spin traps such as N-tert-butyl-α-phenylnitrone, PBN (1 R1=Ph, R2=tBu), and N-tert-butyl-α-(N-oxypyrid-4-yl)nitrone, POBN (1 R1=N-oxypyrid-4-yl, R2=tBu), give long-lived adducts from carbon-centred radicals, and though the adducts of hydroxy and hydroperoxy radicals fragment rapidly, such traps are widely used with EPR spectroscopy5, 6 and continue to be developed for this purpose.7 Since acyclic nitrones have shown promise for the treatment of age-related diseases,3 they are also used as chemical interventions to study biological processes, and new acyclic nitrones continue to be developed as potential therapeutics.8 Stability to fragmentation would not be important to antioxidant activity if the overall pathway takes a reactive radical to benign products.
Scheme 1.

Spin trapping with nitrones.
A few nitrone spin traps 3–5 have been designed to accumulate in mitochondria to scavenge and/or detect radicals generated there (Fig. 1).9 All bear the lipophilic alkyltriphenylphosphonium (TPP) cation, which has been pioneered as a targeting group for antioxidants by the groups of Murphy and Smith working in collaboration.10 TPP cations easily permeate biological membranes and accumulate up to a thousand-fold in the mitochondrial matrix due to the large mitochondrial membrane potential across the inner mitochondrial membrane set up by the electron transport chain. Although the TPP-group is effective and relatively non-toxic, it is not the only lipophilic cation that could act as a targeting group. We considered the pyridinium ion as a lower-molecular-weight alternative, and here report the synthesis of PBN-analogues that incorporate this group.
Figure 1.
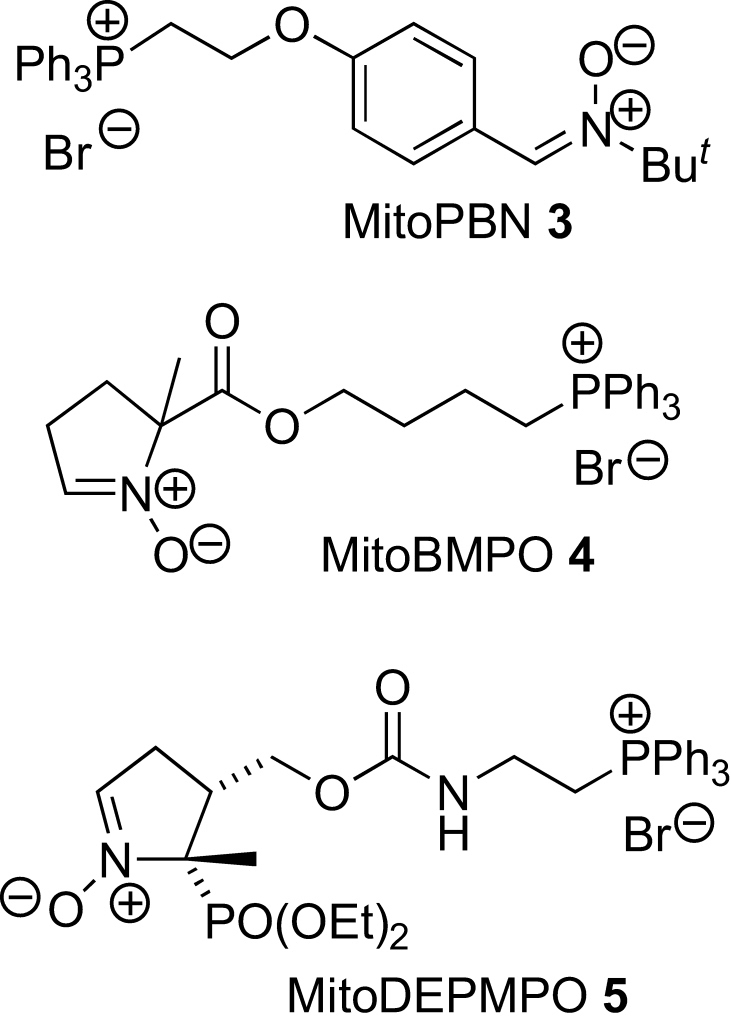
Mitochondria-targeted spin traps.
There is precedent for the use of pyridinium ions in drug candidates to improve water solubility,11 but more importantly for us, pyridinium ions bearing lipophilic groups have been shown to accumulate in mitochondria.12, 13 These include the rhodacyanine dye MKT-077 6,12 which has anticarcinoma activity, and a ceramide derivative 7 that induces mitochondrial permeabilization (through the action of the ceramide moiety) (Fig. 2).13
Figure 2.

Pyridinium salts that accumulate in mitochondria.
A few PBN-type nitrone spin traps derived from N-alkylpyridinium salts have been reported, but their use in targeting mitochondria has not been suggested. Janzen's team studied the behaviour of 2-, 3- and 4-(N-methylpyridinium) N-tert-butyl nitrones (2-MePyBN, 3-MePyBN and 4-MePyBN 8, Fig. 3).14 These spin traps were water-soluble and stable for several days in aqueous solution and the spin adducts were marginally longer lived than those of PBN and 4-PyOBN at a range of pHs.15 The water-soluble16 4-MePyBn 8 has since been used to study the chemical effects of ultrasound.17 The N-dodecyl derivative 9 was also prepared and was found to be almost insoluble in water,14 and the related lipophilic N-linoleyl derivative 10 was used by Hill and Thornalley as a membrane-bound spin trap for studying the generation of phenyl radicals when erythrocytes were treated with phenylhydrazine.18 Interestingly, the resulting spectrum was isotropic, showing that the nitroxide produced was not immobilised in the membrane.19 Nitrones 11 and 12 in which the tert-butyl group is replaced with a bulkier, more lipophilic substituent have also have also been reported.14, 20
Figure 3.

Pyridinium salts used as spin traps.
Nitrones derived from pyridinium salts have also been used for purposes other than spin trapping. Some N-methylpyridinium nitrones have been tested for anticancer activity,21 and a nitronyl nitroxide pyridinium salt has been prepared as a spin-labelled version of the biological co-factor nicotinamide adenine dinucleotide (NAD+).22 Related pyridinium salts have been investigated as new materials, for example, in studies of molecule-based mangnetism, and nitronyl nitroxide radical units have been combined with an N-methylpyridinium ion to allow the ionic bonding necessary for molecular packing.23
We reasoned that nitrones derived from N-arylpyridinium salts would have a greater hydrophobic surface in the region of the cation than primary alkylpyridinium salts 8–12 and would be excellent candidates for mitochondria-targeted antioxidants. Placing the electron-withdrawing nitrone on a different aromatic ring from the pyridinium unit should reduce the propensity for biological reduction of the pyridinium ion relative to nitrones 8–12.
We decided to prepare three nitrones 13–15 that have increasing lipophilicity (Fig. 4). The key reaction in the synthesis of these N-arylpyridinium salts is the Zincke reaction,24, 25 which requires a two-step procedure (Scheme 2). First a pyridine 16 is reacted with 1-chloro-2,4-dinitrobenzene 17 to give an N-(2,4-dinitrophenyl)pyridinium chloride 18, and this is then reacted with an aniline 19 to give a new N-arylpyridinium salt 20 and 2,4-dinitroaniline 21. The mechanism of this second step, which is the Zincke reaction, involves nucleophilic attack by the nitrogen atom of the aniline at C-2 of the pyridinium ion, ring opening, E–Z interconversions, ring closure, and elimination of the 2,4-dinitroaniline 21. In order for the formation of the N-(2,4-dinitrophenyl)pyridinium chloride 18 to proceed well, the pyridine must be nucleophilic, but if it is too electron-rich the Zincke reaction with the aniline 19 is impeded, particularly if the latter bears electron-withdrawing groups. Thus, the sequence will work best if R2 is electron-donating, but R1 must be neither too electron-withdrawing for the first step nor too electron-donating for the second.
Figure 4.
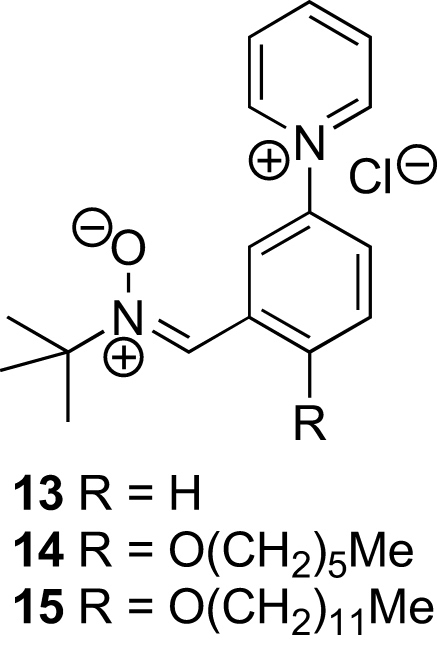
Scheme 2.

Preparation of pyridinium salts.
The first target was nitrone 13, which corresponds to the cationic head group alone. 1-Chloro-2,4-dinitrobenzene 17 reacted smoothly with pyridine to give pyridinium salt 22. Zincke reaction with the electron-rich aniline 23 in methanol was straightforward, but the resulting benzylic alcohol 24 failed to oxidise to the aldehyde 26 under a variety of conditions. The oxidations were made difficult by the poor solubility of the alcohol in organic solvents other than methanol and DMSO together with the difficulty of separating different pyridinium salts from each other and the high water solubility of the pyridinium salts (adaptations of oxidation procedures to accommodate the solubility of the pyridinium salt 24 included using manganese dioxide,26 IBX (stabilised),27 hydrogen peroxide and iron(III) bromide,28 Swern and Parikh–Doering oxidation29). Conversion into the benzylic chloride 25 with neat thionyl chloride and heating with sodium bicarbonate in DMSO30 also failed to give the desired product 26 (Scheme 3).
Scheme 3.

First approach.
Next, introduction of the aldehyde masked as an acetal was attempted (Scheme 4). 3-Nitrobenzaldehyde 27 was converted into acetal 28. Hydrogenation to give aniline 29 was carried out using platinum oxide to avoid the acidity associated with Pd–C. Aniline 29 underwent the Zincke reaction smoothly with the N-(2,4-dinitrophenyl)pyridinium salt 22 to give the pyridinium salt 30 in high yield. Unfortunately, removal of the acetal groups proved less straightforward with the 5,5-dimethyl-1,3-dioxane 30 failing to cleave in 6 M aqueous hydrochloric acid (conditions chosen for ease of isolating the water-soluble pyridinium salt product). Fortunately, at this stage an alternative route had been successful (Scheme 5). 3-Nitrobenzaldehyde 31 was converted into the N-tert-butylnitrone 32. Selective hydrogenation of the nitro group then gave aniline 33, which underwent the Zincke reaction with the N-(2,4-dinitrophenyl)pyridinium salt 22 to give the desired water-soluble nitrone 13. Some reduction of the nitrone was observed if the hydrogenation of the nitro compound was driven to completion and the over-reduced side products were difficult to remove and led to paramagnetic contaminants in the final product 13. Fortunately, the starting nitro compound 32 was easily crystallised from a mixture of nitrones 32 and 33 in ethanol–water when the hydrogenation was stopped after consuming about half the starting material, and this proved to be the best procedure.
Scheme 4.

Second approach.
Scheme 5.

Successful route.
With a route to the N-arylpyridinium head group in hand, we set about modifying it to include a lipophilic tail. Initially 4-methylpyridine 34 was converted into the 4-hexyl derivative 35 by lithiation–alkylation (Scheme 6). This readily formed an N-(2,4-dinitrophenyl)pyridinium salt 36, but the latter did not react with the aniline 33 bearing the electron-withdrawing nitrone group. Clearly, the combination of a less electron-rich pyridine and a more electron-rich aniline would be better. Therefore, salicylaldehyde 37 was converted into 2-hexyloxybenzaldehyde 38 and dodecyloxybenzaldehyde 39 in high yield. Nitration gave the corresponding nitro compounds 40 and 41 in modest yield after separation from other nitrated products. Conversion to nitrones 42 and 43 proceeded smoothly and hydrogenation, optimised through a change of catalyst, gave anilines 44 and 45, respectively. Although aniline 44 was isolated as a 5:1 mixture with nitro compound 42, this did not present a problem in the next step. Gratifyingly, the Zincke reactions between these anilines and the N-(2,4-dinitrophenyl)pyridinium salt 22 now gave the desired nitrones 14 and 15 (Scheme 7).
Scheme 6.

Scheme 7.

As expected for acyclic nitrone spin traps,4 trapping of oxygen-centred radicals did not give stable nitroxides. On the other hand, when the nitrones 13–15 were reacted with methyl radicals generated from the reaction between DMSO and hydroxyl radicals produced under Fenton conditions from hydrogen peroxide and iron(II) sulfate (Scheme 8),4 EPR spectra were obtained that were consistent with nitroxides 46–48 (Figure 5, Figure 6, Figure 7). The high field lines are broadened due to mI-dependent linewidth effects and incomplete averaging of the anisotropic components caused by restricted tumbling of the spin adducts. Interestingly, the middle doublet shows the greatest peak height for nitroxides 47 and 48, which contain the long aliphatic tails, whereas, for nitroxide 46, the lowest field doublet is tallest. A weak background signal was observed when samples of nitrone 13 were dissolved in DMSO–water (Fig. 8), presumably due to nitroxide 49 (Fig. 9), which would result from some reduction of the nitrone moiety of aniline 33 to a hydroxylamine during the hydrogenation of nitro compound 32, followed by air-oxidation to the nitroxide. Nitrone 13 produced under the optimised procedure was estimated to contain about 2% of this impurity by integration of the signal at δ 1.42 ppm in the 1H NMR spectrum, presumed to result from its tert-butyl group. The EPR signal for nitroxide 49 is visible in the EPR spectrum of nitroxide 46 (Fig. 5) because EPR spectroscopy is extremely sensitive and because the nitrone is used in excess (2.5 equiv with respect to the hydrogen peroxide) so that while the nitroxide 49 is already present, not all the nitrone 13 is converted into nitroxide 46.
Scheme 8.

Figure 5.

EPR spectrum of nitroxide 46g=2.0069, AN=14.70 G (t), AHβ=2.45 G (d).
Figure 6.

EPR spectrum of nitroxide 47g=2.0057, AN=14.83 G (t), AHβ=2.70 G (d).
Figure 7.

EPR spectrum of nitroxide 48g=2.0057, AN=14.90 G (t), AHβ=2.75 G (d).
Figure 8.

EPR spectrum of nitroxide 49g=2.0056, AN=16.32 G (t), AHβ=10.35 G (t).
Figure 9.
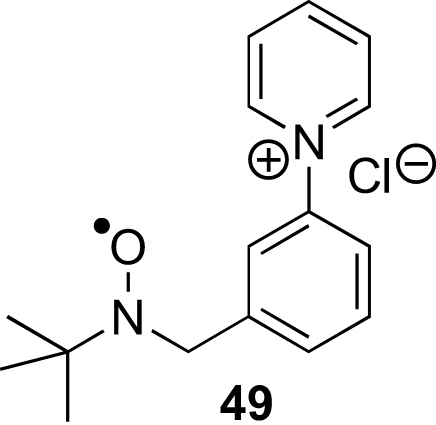
Proposed nitroxide contaminant.
In conclusion, we have reported a new type of spin trap that bears an N-arylpyridinium ion, which should cause the more lipophilic members of the family 14 and 15 to accumulate in the matrix of mitochondria to act as antioxidants there. The nitrones 14 and 15 react with methyl radicals to give nitroxides 47 and 48 that give strong EPR signals, so they could potentially be used to detect carbon-centred radicals within mitochondria. The synthesis demonstrates the selective reduction of nitro groups in the presence of nitrones, the compatibility of nitrones with the Zincke reaction conditions, and illustrates the fine balance that has to be struck with regard to electron-donating and electron-withdrawing groups in this reaction.
2. Experimental
2.1. Synthesis
All reactions under an inert atmosphere were carried out using oven dried or flame dried glassware. Solutions were added via syringe. Diethyl ether, tetrahydrofuran, dichloromethane and toluene were dried where necessary using a solvent drying system, Puresolv™, in which solvent is pushed from its storage container under low nitrogen pressure through two stainless steel columns containing activated alumina and copper. Methanol was dried by distillation from magnesium and iodine, and then stored over 3 Å molecular sieves. Reagents were obtained from commercial suppliers and used without further purification unless otherwise stated. 1H and 13C NMR spectra were obtained on a Bruker DPX/400 spectrometer operating at 400 and 100 MHz, respectively. All coupling constants are measured in hertz. DEPT was used to assign the signals in the 13C NMR spectra as C, CH, CH2 or CH3. Mass spectra (MS) were recorded on a Jeol JMS700 (MStation) spectrometer. Infra-red (IR) spectra were obtained using attenuated total reflectance (ATR) so that the IR spectrum of the compound (solid or liquid) could be directly detected (thin layer) without any sample preparation.
2.2. N-tert-Butyl-α-[3-(pyrid-1′-yl)phenyl]nitrone chloride 13
N-tert-Butyl-α-(3-aminophenyl)nitrone 33 (80 mg, 0.44 mmol) was added to a stirred solution of N-(2′,4′-dinitrophenyl)pyridinium chloride 22 (42 mg, 0.15 mmol) in anhydrous methanol (4 mL) under argon at rt. After 2 h the resulting red mixture was heated to reflux for 18 h until the red colour disappeared. The mixture was cooled, diluted with H2O and washed with EtOAc until no further colour was removed from the aqueous layer. The aqueous portion was concentrated in vacuo to give pyridinium salt 13 as a brown oil (40 mg, 96%). δH (400 MHz, MeOD): 1.68 (9H, s, 3×CH3), 7.87 (1H, t, J 8.0 Hz, H-5), 7.97 (1H, ddd, J 8.1, 2.4 and 0.9 Hz, H-6), 8.21 (1H, s, CH N), 8.37 (2H, dd, J 7.9 and 6.9 Hz, H-3′ and H-5′), 8.49 (1H, dt, J 7.9 and 1.4 Hz, H-4), 8.87 (1H, tt, J 7.9 and 1.3 Hz, H-4′), 9.16 (1H, dd, J 2.0 and 1.8 Hz, H-2), 9.35 (2H, dd, J 6.9 and 1.4 Hz, H-2′ and H-6′). δC (100 MHz, MeOD): 28.37 (CH3), 73.17 (C), 125.14 (CH), 127.13 (CH), 129.67 (CH), 131.72 (CH), 132.00 (CH), 133.42 (CH), 134.57 (C), 144.38 (C), 146.12 (CH), 148.18 (CH). LRMS (FAB+) 255 [M+ (pyridinium cation), 100%]. HRMS: 255.1501, C16H19N2O requires 255.1497. νmax (ATR) 3074 (CH), 2980 (CH), 2934 (CH), 1628 (Ar), 1583 (Ar), 1472 (Ar), 1190 (nitrone) cm−1.
2.3. N-tert-Butyl-α-[2-hexyloxy-5-(pyrid-1′-yl)phenyl]nitrone chloride 14
α-(5-Amino-2-hexyloxyphenyl)-N-tert-butylnitrone 44 [556 mg of a mixture (5:1 mole ratio) of nitrones 44 and 42, 1.56 mmol] was added to a stirred solution of N-(2′,4′-dinitrophenyl)pyridinium chloride 22 (180 mg, 0.64 mmol) in anhydrous methanol (14 mL) under argon at rt. After 2 h the resulting red mixture was heated to reflux for 18 h until the red colour disappeared. The mixture was cooled, diluted with H2O and washed with DCM until no further colour was removed from the aqueous layer. The aqueous portion was concentrated in vacuo to give 14 as an orange oil (189 mg, 76%). δH (400 MHz, MeOD): 0.93 (3H, t, J 6.8 Hz, CH3), 1.36–1.44 (4H, m, 2×CH2), 1.52–1.60 (11H, m, 3×CH3 and CH2), 1.87–1.94 (2H, m, CH2), 4.24 (2H, t, J 6.2 Hz, CH2), 7.40 (1H, d, J 9.0 Hz, H-3), 7.90 (1H, dd, J 9.0 and 3.0 Hz, H-4), 8.32 (1H, s, CH N), 8.34 (2H, dd, J 7.9 and 6.8 Hz, H-3′ and H-5′), 8.76 (1H, tt, J 7.9 and 1.3 Hz, H-4′), 9.23 (2H, dd, J 6.9 and 1.3 Hz, H-2′ and H-6′), 9.62 (1H, d, J 3.0 Hz, H-6). δC (100 MHz, MeOD): 14.54 (CH3), 23.86 (CH2), 27.11 (CH2), 28.49 (CH3), 30.13 (CH2), 32.80 (CH2), 70.90 (CH2), 73.38 (C), 114.15 (CH), 122.28 (C), 124.92 (CH), 126.49 (CH), 128.91 (CH), 129.76 (CH), 136.89 (C), 146.03 (CH), 147.71 (CH), 160.45 (C). LRMS (FAB+) 355 [M+ (pyridinium cation), 100%]. HRMS: 355.2389, C22H31O2N2 requires 355.2386. νmax (ATR) 2924 (CH), 2855 (CH), 1626 (aromatic), 1479 (aromatic), 1462 (aromatic), 1271 (nitrone), 1230 (C–O stretch) cm−1.
2.4. N-tert-Butyl-α-[2-dodecyloxy-5-(pyrid-1′-yl)phenyl]nitrone chloride 15
α-(5-Amino-2-dodecyloxyphenyl)-N-tert-butylnitrone 45 (517 mg, 1.38 mmol) was added to a stirred solution of N-(2′,4′-dinitrophenyl)pyridinium chloride 22 (156 mg, 0.55 mmol) in anhydrous methanol (12 mL) under argon at rt. After 2 h the resulting red mixture was heated to reflux for 18 h until the red colour disappeared. The mixture was cooled, diluted with H2O and washed with DCM until no further colour was removed from the aqueous layer. The aqueous portion was concentrated in vacuo to give nitrone 15 as an orange oil (129 mg, 49%). δH (400 MHz, MeOD): 0.94 (3H, t, J 6.7 Hz, CH3), 1.34–1.67 (27H, m, 3×CH3+9×CH2), 1.93–2.00 (2H, m, CH2), 4.30 (2H, t, J 6.2 Hz, CH2), 7.46 (1H, d, J 9.0 Hz, H-3), 7.95 (1H, dd, J 9.0 and 3.0 Hz, H-4), 8.33 (2H, dd, J 7.8 and 6.8 Hz, H-3′ and H-5′), 8.34 (1H, s, CH N), 8.82 (1H, tt, J 7.8 and 1.3 Hz, H-4′), 9.30 (2H, dd, J 6.9 and 1.3 Hz, H-2′ and H-6′), 9.69 (1H, d, J 3.0 Hz, H-6). δC (100 MHz, MeOD): 14.48 (CH3), 23.74 (CH2), 27.31 (CH2), 28.39 (CH3), 30.06 (CH2), 30.45 (CH2), 30.47 (CH2), 30.71 (CH2), 30.76 (CH2), 30.79 (CH2), 33.79 (CH2), 70.73 (CH2), 73.22 (C), 113.97 (CH), 122.21 (C), 124.76 (CH), 126.17 (CH), 128.70 (CH), 129.61 (CH), 136.76 (C), 145.91 (CH), 147.56 (CH), 160.28 (C). LRMS (FAB+) 439 [M+ (pyridinium cation), 100%]. HRMS: 439.3322, C28H43O2N2 requires 439.3325. νmax (ATR) 2920 (CH), 2850 (CH), 1626 (aromatic), 1481 (aromatic), 1468 (aromatic), 1271 (nitrone), 1238 (C–O stretch) cm−1.
2.5. N-(2′,4′-Dinitrophenyl)pyridinium chloride 22
Pyridine (4.0 mL, 49 mmol) and 1-chloro-2,4-dinitrobenzene (10.01 g, 49.4 mmol) were heated together at 95 °C for 1 h. The resulting yellow solid was triturated with acetone until no further colour was removed, to give pyridinium salt 22 as an off-white solid (12.6 g, 91%). Mp 193–195 °C. δH (400 MHz, MeOD): 8.39 (1H, d, J 8.7 Hz, H-6′), 8.47 (2H, dd, J 7.9 and 6.8 Hz, H-3 and H-5), 8.97 (1H, dd, J 8.7 and 2.5 Hz, H-5′), 9.02 (1H, tt, J 7.9 and 1.3 Hz, H-4), 9.31 (1H, d, J 2.5 Hz, H-3′), 9.40 (2H, dd, J 6.9 and 1.3 Hz, H-2 and H-6). δC (100 MHz, MeOD): 121.35 (CH), 128.01 (CH), 130.20 (CH), 132.01 (CH), 138.72 (C), 143.05 (C), 146.08 (CH), 148.79 (CH), 149.00 (C). LRMS (FAB+) 246 [M+ (pyridinium cation), 100%]. HRMS: 246.0514, C11H8N3O4 requires 246.0515. νmax (KBr) 3117, 3057, 1610 (aromatic), 1542 (NO2), 1473 (aromatic), 1342 (NO2) cm−1. 1H NMR and 13C NMR data consistent with literature data obtained in (CD3)2SO.31
2.6. N-(3′-Hydroxymethylphenyl)pyridinium chloride 24
3-Aminobenzyl alcohol 23 (2.74 g, 22.3 mmol) was added to a stirred solution of N-(2′,4′-dinitrophenyl)pyridinium chloride 22 (2.50 g, 8.90 mmol) in anhydrous methanol (60 mL) under argon at rt. After 24 h, the resulting red mixture was heated to reflux for 48 h until the red colour disappeared. The mixture was cooled, diluted with H2O and washed with EtOAc until no further colour was removed from the aqueous layer. The aqueous layer was concentrated in vacuo to give a brown solid that was recrystallised from iPrOH–acetone to give pyridinium salt 24 as brown cubes (1.08 g, 55%). Mp 107–109 °C. δH (400 MHz, MeOD): 4.83 (2H, s, CH2OH), 7.75–7.79 (3H, m, H-4′, H-5′ and H-6′), 7.85 (1H, br s, H-2′), 8.33 (2H, dd, J 7.8 and 6.8 Hz, H-3 and H-5), 8.84 (1H, tt, J 7.8 and 1.3 Hz, H-4), 9.30 (2H, dd, J 6.9 and 1.4 Hz, H-2 and H-6). δC (100 MHz, MeOD): 62.41 (CH2), 121.96 (CH), 122.54 (CH), 128.07 (CH), 129.14 (CH), 130.14 (CH), 143.08 (C), 144.59 (CH), 145.13 (C), 146.39 (CH). LRMS (FAB+) 186 [M+ (pyridinium cation), 100%]. HRMS: 186.0916, C12H12NO requires 186.0919. νmax (ATR) 3295 (OH), 2916 (CH), 2851 (CH), 1614 (aromatic), 1471 (aromatic) cm−1.
2.7. N-(3′-Chloromethylphenyl)pyridinium chloride 25
A mixture of N-(3′-hydroxymethylphenyl)pyridinium chloride 24 (300 mg, 1.61 mmol) and SOCl2 (3.0 mL, 40.6 mmol) was heated at 95 °C under argon for 18 h. The reaction was cooled and the excess SOCl2 was quenched by slow addition of H2O. The reaction mixture was washed with CHCl3 and the aqueous portion was concentrated in vacuo to give pyridinium salt 25 as a brown oil (310 mg, 96%). δH (400 MHz, MeOD): 4.88 (2H, s, CH2Cl), 7.79–7.89 (3H, m, H-4′, H-5′ and H-6′), 8.01 (1H, s, H-2′), 8.36 (2H, m, H-3 and H-5), 8.86 (1H, t, J 7.7 Hz, H-4), 9.32 (2H, d, J 6.1 Hz, H-2 and H-6). δC (100 MHz, MeOD): 45.49 (CH2), 125.42 (CH), 125.77 (CH), 129.66 (CH), 132.12 (CH), 132.85 (CH), 142.41 (CH), 144.53 (C), 146.12 (C), 148.10 (CH). LRMS (FAB+) 204 [M+ (35Cl, pyridinium cation), 100%], 206 [M+ (37Cl, pyridinium cation), 33%]. HRMS: 204.0579 and 206.0553. C12H1135ClN requires 204.0580 and C12H1137ClN requires 206.0553. νmax (ATR) 3032 (CH), 2958 (CH), 1627 (aromatic), 1473 (aromatic) cm−1.
2.8. 5,5-Dimethyl-2-(3′-nitrophenyl)-1,3-dioxane 28
3-Nitrobenzaldehyde 27 (1.00 g, 6.62 mmol), 2,2-dimethyl-1,3-propanediol (1.41 g, 13.6 mmol) and p-toluenesulfonic acid (24 mg, 0.12 mmol) were dissolved in anhydrous toluene (23 mL). The reaction was heated under argon in a Dean–Stark apparatus at 140 °C for 24 h. Upon completion, the mixture was cooled, washed with NaHCO3 (×3), H2O and brine. The organic extracts were combined, dried (MgSO4), and concentrated in vacuo to give acetal 28 as a yellow oil (1.50 g, 95%) that solidified on standing. Mp 46–48 °C. δH (400 MHz, CDCl3): 0.81 (3H, s, CH3), 1.27 (3H, s, CH3), 3.67 (2H, d, J 10.6 Hz, CH2O–), 3.78 (2H, d, J 10.1 Hz, CH2O–), 5.45 (1H, s, CHO2), 7.53 (1H, t, J 8.0 Hz, H-5′), 7.81–7.83 (1H, m, H-4′), 8.18 (1H, ddd, J 1.0, 2.3 and 8.2 Hz, H-6′), 8.37 (1H, t, J 1.9 Hz, H-2′). δC (100 MHz, CDCl3): 21.86 (CH3), 23.07 (CH3), 30.31 (C), 77.70 (CH2), 99.92 (CH), 121.62 (CH), 123.69 (CH), 129.30 (CH), 132.49 (CH), 140.61 (C), 148.23 (C). LRMS (CI+) 238 [(M+H)+, 78%], 79 (100). HRMS: 238.1078, C12H16NO4 requires (M+H)+, 238.1079. νmax (ATR) 2955 (CH), 2870 (CH), 1529 (NO2), 1460 (aromatic), 1348 (NO2), 1082 (C–O stretch) cm−1. 1H NMR and mp not in agreement with literature.32
2.9. 2-(3′-Aminophenyl)-5,5-dimethyl-1,3-dioxane 29
5,5-Dimethyl-2-(3′-nitro-phenyl)-[1,3]dioxane 28 (825 mg, 3.48 mmol) and platinum(IV) oxide (16 mg, 5 mol %) were dissolved in ethyl acetate (16.5 mL). The solution was flushed with hydrogen then placed under a hydrogen atmosphere and stirred at rt for 20 h. The catalyst was removed by filtration through cotton wool and the solution was concentrated in vacuo to give amine 29 as an orange-brown solid (720 mg, 100%). δH (400 MHz, CDCl3) 0.78 (3H, s, CH3), 1.30 (3H, s, CH3), 3.58–3.64 (4H, m, NH2+CH2), 3.74–3.77 (2H, m, CH2), 5.30 (1H, s, CHO2), 6.61 (1H, ddd, J 0.9, 2.4 and 7.9 Hz, H-6′), 6.83 (1H, t, J 2.0 Hz, H-2′), 6.86–6.89 (1H, m, H-4′), 7.13 (1H, t, J 7.8 Hz, H-5′). δC (100 MHz, MeOD): 21.84 (CH3), 23.06 (CH3), 30.19 (C), 77.58 (CH2), 101.78 (CH), 112.67 (CH), 115.61 (CH), 116.31 (CH), 129.18 (CH), 139.55 (C), 146.53 (C). LRMS (EI+) 207 (M+•, 90%), 121 (100). HRMS: 207.1263, C12H17NO2 requires 207.1259. νmax (ATR) 3381 (NH2), 3360 (NH2), 2957 (CH), 1620 (NH2 bend), 1462 (aromatic), 1385, 1094 (C–O stretch) cm−1; mp 60–61 °C.
2.10. N-[3′-(5″,5″-Dimethyl-1″,3″-dioxan-2″-yl)phenyl]pyridinium chloride 30
N-2,4-Dinitrophenyl pyridinium chloride 22 (333 mg, 1.18 mmol) was dissolved in anhydrous methanol (12 mL) and amine 29 (613 mg, 2.96 mmol) was added. The reaction was stirred under argon at rt for 20 h and then heated at reflux for 3 h until the red colour disappeared. The mixture was cooled, diluted with H2O and washed with EtOAc until no further colour was removed from the aqueous layer. The aqueous portion was concentrated in vacuo to give pyridinium salt 30 as a yellow oil (326 mg, 90%). δH (400 MHz, MeOD): 0.87 (3H, s, CH3), 1.30 (3H, s, CH3), 3.81 (4H, s, 2×CH2), 5.65 (1H, s, CHO2), 7.80 (1H, t, J 7.6 Hz, H-5′), 7.86 (1H, ddd, J 8.0, 2.4 and 1.3 Hz, H-4′ or H-6′), 7.91 (1H, dt, J 7.6 and 1.5 Hz, H-4′ or H-6′), 7.98–8.00 (1H, m, H-2′), 8.32 (2H, dd, J 7.9 and 6.9 Hz, H-3 and H-5), 8.83 (1H, tt, J 1.4 and 7.9 Hz, H-4), 9.29 (2H, dd, J 6.9 and 1.4 Hz, H-2 and H-6). δC (100 MHz, MeOD): 21.91 (CH3), 23.32 (CH3), 78.57 (CH2), 101.09 (CH), 123.45 (CH), 125.73 (CH), 129.53 (CH), 130.66 (CH), 131.58 (CH), 143.29 (C), 144.38 (C), 146.20 (CH), 147.97 (CH). LRMS (FAB+) 270 [M+ (pyridinium cation), 100%]. HRMS: 270.1488, C17H20NO2 requires 270.1494. νmax (ATR) 3007 (CH), 2949 (CH), 2868 (CH), 1628 (aromatic), 1473 (aromatic) cm−1.
2.11. N-tert-Butyl-α-(3-nitrophenyl)nitrone 32
3-Nitrobenzaldehyde 31 (500 mg, 3.31 mmol), N-(tert-butyl)hydroxylammonium acetate (740 mg, 4.96 mmol) and sodium hydrogen carbonate (417 mg, 4.96 mmol) were dissolved in ethanol (40 mL). The reaction was heated, with stirring, at 70 °C for 48 h. The reaction mixture was poured into H2O (120 mL) and left to stand for 1 h. The bright yellow crystals that formed were filtered off to give nitrone 32 as yellow, feathery crystals (641 mg, 92%). Mp 102–103 °C (lit.33 108–110 °C). δH (400 MHz, CDCl3): 1.62 (9H, s, 3×CH3), 7.55 (1H, t, J 8.1 Hz, H-5), 7.69 (1H, s, CH N), 8.18 (1H, dd, J 8.1 and 1.7 Hz, H-4), 8.57 (1H, dt, J 8.0 and 1.48 Hz, H-6), 9.17 (1H, t, J 1.7 Hz, H-2). δC (100 MHz, CDCl3): 28.31 (CH3), 71.99 (C), 123.18 (CH), 124.29 (CH), 127.94 (CH), 129.46 (CH), 132.53 (C), 133.99 (CH), 148.18 (C). LRMS (EI+) 222 (M+•, 10%), 84 (30%), 57 (C4H9+, 100%). HRMS: 222.1003, C11H14N2O3 requires 222.1004. νmax (KBr) 2985 (CH), 2940 (CH), 1556 (aromatic), 1522 (NO2), 1415 (aromatic), 1366 (NO2), 1339 (nitrone) cm−1. 1H NMR data consistent with literature data obtained in (CD3)2SO.33
2.12. N-tert-Butyl-α-(3-aminophenyl)nitrone 33
N-tert-Butyl-α-(3-nitrophenyl)nitrone 32 (500 mg, 2.34 mmol) and platinum(IV) oxide (11 mg, 5 mol %) were dissolved in ethyl acetate (10 mL). The solution was flushed with hydrogen then placed under a hydrogen atmosphere and stirred at rt for 45 min. The catalyst was removed by filtration through Celite and the solution was concentrated in vacuo to give a 1:1.4 mixture of the product 33 and starting material 32. The starting material 32 was removed by precipitation from EtOH–H2O (×2) as a crystalline solid. The supernatant was concentrated to give a 10:1 mixture of nitrones 33 and 32 as a yellow oil (108 mg, 40%; 81% based on recovered SM). δH (400 MHz, CDCl3): 1.49 (9H, s, 3×CH3), 3.73 (2H, br s, NH2), 6.63 (1H, ddd, J 7.9, 2.4 and 0.9 Hz, H-4), 7.07 (1H, t, J 7.9 Hz, H-5), 7.19 (1H, d, J 7.8 Hz, H-6), 7.37 (1H, s, CH N), 8.00 (1H, t, J 1.9 Hz, H-2). Material was carried on to next stage with no further purification or analysis due to potential instability.
2.13. 4-Hexylpyridine 35
LDA (3.6 mL of 2 M in THF–heptane–ethylbenzene, 7.2 mmol) was added dropwise over 10 min to a stirred solution of 4-picoline 34 (0.5 mL, 5.34 mmol) in anhydrous THF (5 mL) under argon at −78 °C. After stirring for a further 30 min at −78 °C, a solution of 1-bromopentane (0.44 mL, 3.6 mmol) in anhydrous THF (5 mL) was added dropwise over 5 min and the mixture allowed to warm to rt and stirred for 20 h. Saturated aqueous NH4Cl solution (10 mL) and H2O (10 mL) were added and the mixture was extracted with EtOAc (×2). The combined organic extracts were with H2O, dried (MgSO4) and concentrated in vacuo to give a yellow oil. The crude residue was chromatographed on SiO2 using EtOAc–hexane (1:9) as the eluent to give 4-hexylpyridine 35 as a yellow oil (579 mg, 100%). Rf=[EtOAc–hexane (3:7)]: 0.23. δH (400 MHz, CDCl3): 0.87 (3H, t, J 6.8 Hz, CH3), 1.23–1.34 (6H, m, 3×CH3), 1.57–1.64 (2H, m, CH2), 2.58 (2H, t, J 7.6 Hz, CH2), 7.10 (2H, d, J 5.9 Hz, H-3 and H-5), 8.47 (2H, d, J 5.8 Hz, H-2 and H-6). δC (100 MHz, CDCl3): 14.17 (CH3), 22.65 (CH2), 28.95 (CH2), 30.37 (CH2), 31.70 (CH2), 35.37 (CH2), 124.08 (CH), 149.52 (CH), 152.12 (C). LRMS (EI+) 163 (M+•, 30%), 93 [M+•−CH3(CH2)2CH CH2, 100]. HRMS: 163.1358, C11H17N requires 163.1361. νmax (ATR) 2955 (CH), 2928 (CH), 2857 (CH), 1603 (aromatic), 1415 (aromatic) cm−1. 1H NMR data agree with literature.34
2.14. 4-Hexyl-N-(2′,4′-dinitrophenyl)pyridinium chloride 36
4-Hexylpyridine 35 (2.27 g, 13.9 mmol) and 1-chloro-2,4-dinitrobenzene (5.62 g, 27.8 mmol) were heated together at 95 °C for 48 h. The reaction was cooled, dissolved in H2O and washed with EtOAc until no further colour was removed from the aqueous layer. The aqueous portion was concentrated in vacuo to give 36 as a dark brown oil (4.29 g, 84%). δH (400 MHz, MeOD): 1.02 (3H, t, J 7.0 Hz, CH3), 1.44–1.60 (6H, m, 3×CH2), 1.94–2.01 (2H, m, CH2), 3.24 (2H, t, J 7.6 Hz, CH2), 8.36 (2H, d, J 6.4 Hz, H-3 and H-5), 8.44 (1H, d, J 8.7 Hz, H-6′), 8.98 (1H, dd, J 8.6 and 2.3 Hz, H-5′), 9.29 (1H, d, J 2.4 Hz, H-3′), 9.31 (2H, d, J 6.5 Hz, H-2 and H-6). δC (100 MHz, MeOD): 14.46 (CH3), 23.53 (CH2), 29.92 (CH2), 30.79 (CH2), 32.59 (CH2), 37.24 (CH2), 124.08 (CH), 129.12 (CH), 131.20 (CH), 132.92 (CH), 139.96 (C), 144.56 (C), 146.11 (CH), 150.82 (C), 169.12 (C). LRMS (FAB+) 330 [M+ (pyridinium cation), 100%]. HRMS: 330.1452, C17H20O4N3 requires 330.1454. νmax (ATR) 2928 (CH), 2859 (CH), 1610 (aromatic), 1537 (NO2), 1462 (aromatic), 1342 (NO2) cm−1.
2.15. 2-Hexyloxybenzaldehyde 38
1-Bromohexane (4.7 mL, 0.034 mol) was added to a solution of salicylaldehyde (3.0 mL, 0.028 mol) and K2CO3 (4.647 g, 0.034 mol) in DMF (40 mL). The reaction was heated, with stirring, at 130 °C for 20 h. The reaction was cooled, filtered and diluted with H2O. The mixture was extracted with EtOAc (×3) and the combined organic extracts were washed with 1 M KOH. The organic extracts were dried (MgSO4) and concentrated in vacuo to give aldehyde 38 as a yellow oil (5.768 g, 99%). δH (400 MHz, CDCl3): 0.86–0.89 (3H, m, CH3), 1.27–1.33 (4H, m, 2×CH2), 1.41–1.47 (2H, m, CH2), 1.76–1.83 (2H, m, CH2), 4.01 (2H, t, J 6.4 Hz, CH2), 6.93 (1H, d, J 8.4 Hz, H-3), 6.94 (1H, t, J 7.5 Hz, H-5), 7.47 (1H, ddd, J 8.5, 7.4 and 1.8 Hz, H-4), 7.78 (1H, dd, J 7.7 and 1.8 Hz, H-6), 10.48 (1H, s, CHO). δC (100 MHz, CDCl3): 14.00 (CH3), 22.57 (CH2), 25.72 (CH2), 29.02 (CH2), 31.50 (CH2), 68.46 (CH2), 112.47 (CH), 120.38 (CH), 124.81 (C), 128.05 (CH), 135.92 (CH), 161.56 (C), 189.78 (CH). LRMS (EI+) 206 (M+•, 15%), 122 (30%), 85 (65%), 83 (100%). HRMS: 206.1309, C13H18O2 requires 206.1307. νmax (ATR) 2955 (CH), 2859 (CH), 1688 (C O), 1599 (aromatic), 1456 (aromatic), 1240 (C–O stretch) cm−1. Literature reports microanalysis only.35
2.16. 2-Dodecyloxybenzaldehyde 39
1-Iodododecane (8.38 mL, 0.034 mol) was added to a solution of salicylaldehyde (3.0 mL, 0.028 mol) and K2CO3 (4.647 g, 0.034 mol) in DMF (40 mL). The reaction was heated, with stirring, at 130 °C for 20 h. The reaction was cooled, filtered and diluted with H2O. The mixture was extracted with EtOAc (×3) and the combined organic extracts were washed with 1 M KOH. The organic extracts were dried (MgSO4) and concentrated in vacuo to give aldehyde 39 as a yellow solid, which melts upon handling (8.0295 g, 99%). δH (400 MHz, CDCl3): 0.87 (3H, t, J 6.2 Hz, CH3), 1.22–1.35 (16H, m, 8×CH2), 1.43–1.47 (2H, m, CH2), 1.79–1.86 (2H, m, CH2), 4.05 (2H, t, J 6.4 Hz, CH2), 6.94–6.99 (2H, m, H-4 and H-5), 7.48–7.52 (1H, m, H-3), 7.81 (1H, d, J 7.6 Hz, H-6), 10.50 (1H, s, CHO). δC (100 MHz, CDCl3): 14.19 (CH3), 22.77 (CH2), 26.13 (CH2), 29.16 (CH2), 29.43 (CH2), 29.54 (CH2), 29.64 (CH2), 29.67 (CH2), 29.71 (CH2), 29.73 (CH2), 31.99 (CH2), 68.57 (CH2), 112.54 (CH), 120.48 (CH), 124.94 (C), 128.20 (CH), 135.96 (CH), 161.64 (C), 189.90 (CH). LRMS (EI+) 290 (M+•, 60%), 122 (100). HRMS: 290.2244, C19H30O2 requires 290.2246. νmax (ATR) 2922 (CH), 2853 (CH), 1688 (C O), 1599 (aromatic), 1458 (aromatic), 1240 (C–O stretch) cm−1.
2.17. 2-Hexyloxy-5-nitrobenzaldehyde 40
A mixture of fuming nitric acid (100%, d=1.52, 7 mL) and concentrated sulfuric acid (18.1 M, 7 mL) was cooled, with stirring, to −10 °C. 2-Hexyloxybenzaldehyde 38 (5.55 g, 269 mmol) was added dropwise to the mixture. The reaction was allowed to warm to rt. After 1 h the reaction was poured onto ice. The precipitate formed was filtered, washed with H2O and dissolved in Et2O. The ether solution was washed with H2O, saturated aqueous NaHCO3 (×3) and again with H2O. The organic extracts were dried (MgSO4) and concentrated in vacuo to give a yellow solid. This solid was recrystallised three times from Et2O–hexane to give aldehyde 40 as off-white needles (1.49 g, 22%). Mp 61–62 °C (lit.36 66–70 °C). δH (400 MHz, CDCl3): 0.91 (3H, t, J 6.8 Hz, CH3), 1.35–1.38 (4H, m, 2×CH2), 1.47–1.52 (2H, m, CH2), 1.87–1.94 (2H, m, CH2), 4.21 (2H, t, J 6.4 Hz, CH2), 7.10 (1H, d, J 9.2 Hz, H-3), 8.41 (1H, dd, J 9.2 and 2.8 Hz, H-4), 8.69 (1H, d, J 2.8 Hz, H-6), 10.47 (1H, s, CHO). δC (100 MHz, CDCl3): 14.11 (CH3), 22.65 (CH2), 25.71 (CH2), 28.90 (CH2), 31.52 (CH2), 70.00 (CH2), 113.00 (CH), 124.64 (CH), 124.66 (C), 130.78 (CH), 141.49 (C), 165.39 (C), 187.77 (CH). LRMS (EI+) 251 (M+•, 85%), 84 (65), 43 (100). HRMS: 251.1157, C13H17NO4 requires 251.1158. νmax (ATR) 2951 (CH), 2911 (CH), 2843 (CH), 1688 (C O), 1609 (aromatic), 1512 (NO2), 1427 (aromatic), 1339 (NO2), 1273 (C–O stretch) cm−1.
2.18. 2-Dodecyloxy-5-nitrobenzaldehyde 41
A solution of 2-dodecyloxybenzaldehyde 39 (3.79 g, 13 mmol) in concentrated sulfuric acid (3 mL) was cooled, with stirring, to −10 °C. A mixture of 70% nitric acid (1.3 mL) and concentrated sulfuric acid (18.1 M, 1.3 mL) was also cooled, with stirring, to −10 °C. The acid mixture was added dropwise to the aldehyde solution and the mixture stirred at rt for 15 min. The reaction mixture was poured onto ice. The aqueous layers were extracted with EtOAc (×3). The combined organic extracts were washed with H2O (×3), dried (MgSO4) and concentrated in vacuo to give an orange oil. Chromatography on SiO2 using 5% EtOAc–hexane (1:19) as the eluent gave 41 as a yellow oil (1.141 g, 34%). Rf=0.40 [EtOAc–hexane (1:4)]. δH (400 MHz, CDCl3): 0.83 (3H, t, J 6.5 Hz, CH3), 1.22–1.35 (16H, m, 8×CH2), 1.44–1.51 (2H, m, CH2), 1.85–1.92 (2H, m, CH2), 4.20 (2H, t, J 6.5 Hz, CH2), 7.10 (1H, d, J 9.2 Hz, H-3), 8.36 (1H, dd, J 9.2 and 2.8 Hz, H-4), 8.60 (1H, d, J 2.8 Hz, H-6), 10.42 (1H, s, CHO). δC (100 MHz, CDCl3): 14.14 (CH3), 22.72 (CH2), 25.94 (CH2), 28.86 (CH2), 29.30 (CH2), 29.38 (CH2), 29.54 (CH2), 29.59 (CH2), 29.66 (CH2), 31.62 (CH2), 31.94 (CH2), 69.99 (CH2), 113.04 (CH), 124.31 (CH), 124.54 (C), 130.64 (CH), 141.32 (C), 165.35 (C), 187.58 (CH). LRMS (EI+) 335 (M+•, 22%), 318 (45), 97 (49), 85 (70), 71 (88), 57 (C4H9+, 100). HRMS: 335.2096, C19H29NO4 requires 335.2097. νmax (ATR) 2924 (CH), 2853 (CH), 1692 (C O), 1609 (aromatic), 1589 (aromatic), 1522 (NO2), 1466 (aromatic), 1341 (NO2), 1271 (C–O stretch) cm−1.
2.19. N-tert-Butyl-α-(2-hexyloxy-5-nitrophenyl)nitrone 42
2-Hexyloxy-5-nitrobenzaldehyde 40 (490 mg, 1.95 mmol), N-(tert-butyl)hydroxylammonium acetate (437 mg, 2.93 mmol) and sodium hydrogen carbonate (246 mg, 2.93 mmol) were dissolved in ethanol (20 mL). The reaction was heated, with stirring, at 70 °C for 72 h. The reaction mixture was poured into H2O (100 mL) and left to stand for 1 h. The resulting precipitate was filtered and dissolved in EtOAc. The solution was washed with H2O (×2) and brine, dried (MgSO4) and concentrated in vacuo to give a yellow solid. The solid was recrystallised from Et2O–hexane to give nitrone 42 as yellow cubes (610 mg, 97%). Mp 102–103 °C. δH (400 MHz, CDCl3): 0.92 (3H, t, J 6.7 Hz, CH3), 1.34–1.39 (4H, m, 2×CH2), 1.46–1.54 (2H, m, CH2), 1.63 (9H, s, 3×CH3), 1.85–1.92 (2H, m, CH2), 4.14 (2H, t, J 6.4 Hz, CH2), 6.95 (1H, d, J 9.2 Hz, H-3), 8.16 (1H, s, CH N), 8.27 (1H, dd, J 9.1 and 2.6 Hz, H-4), 10.24 (1H, d, J 2.6 Hz, H-6). δC (100 MHz, CDCl3): 14.12 (CH3), 22.73 (CH2), 25.85 (CH2), 28.36 (CH3), 28.98 (CH2), 31.55 (CH2), 69.48 (CH2), 72.04 (C), 110.32 (CH), 120.54 (C), 123.33 (CH), 124.05 (CH), 126.87 (CH), 141.25 (C), 160.95 (C). LRMS (EI+) 322 (M+•, 20%), 266 (M+•−C4H8, 70), 57 (C4H9+, 100). HRMS: 322.1896, C17H26N2O4 requires 322.1893. νmax (ATR) 2951 (CH), 2982 (CH), 1609 (aromatic), 1580 (aromatic), 1516 (NO2), 1466 (aromatic), 1343 (NO2), 1273 (nitrone), 1248 (C–O stretch) cm−1.
2.20. N-tert-Butyl-α-(2-dodecyloxy-5-nitrophenyl)nitrone 43
2-Dodecyloxy-5-nitrobenzaldehyde 41 (1.41 g, 4.40 mmol), N-(tert-butyl)hydroxylammonium acetate (984 mg, 6.60 mmol) and sodium hydrogen carbonate (554 mg, 6.60 mmol) were dissolved in ethanol (40 mL). The reaction was heated, with stirring, at 70 °C for 48 h. The reaction mixture cooled, diluted with H2O and extracted with EtOAc (×3). The combined organic extracts were washed with H2O (×2) and brine (×1), dried (MgSO4) and concentrated in vacuo to give nitrone 43 as a yellow solid (1.60 g, 90%). Mp 39–40 °C. δH (400 MHz, CDCl3): 0.88 (3H, t, J 6.4 Hz, CH3), 1.27–1.40 (16H, m, 8×CH2), 1.47–1.52 (2H, m, CH2), 1.63 (9H, s, 3×CH3), 1.85–1.92 (2H, m, CH2), 4.13 (2H, t, J 6.4 Hz, CH2), 6.93 (1H, d, J 9.2 Hz, H-3), 8.09 (1H, s, CH N), 8.24 (1H, dd, J 9.1 and 2.7 Hz, H-4), 10.28 (1H, d, J 2.7 Hz, H-6). δC (100 MHz, CDCl3): 14.25 (CH3), 22.81 (CH2), 26.18 (CH2), 28.39 (CH3), 29.03 (CH2), 29.40 (CH2), 29.46 (CH2), 29.68 (CH2), 29.73 (CH2), 29.75 (CH2), 29.77 (CH2), 32.03 (CH2), 69.45 (CH2), 72.01 (C), 110.27 (CH), 120.72 (C), 122.70 (CH), 123.92 (CH), 126.69 (CH), 141.28 (C), 160.87 (C). LRMS (EI+) 406 (M+•, 12%), 350 (70), 318 (65), 182 (73), 57 (C4H9+, 100). HRMS: 406.2829, C23H38N2O4 requires 406.2832. νmax (ATR) 2955 (CH), 2920 (CH), 2851 (CH), 1607 (aromatic), 1518 (NO2), 1464 (aromatic), 1339 (NO2), 1271 (nitrone), 1244 (C–O stretch) cm−1.
2.21. α-(5-Amino-2-hexyloxyphenyl)-N-tert-butylnitrone 44
N-tert-Butyl-α-(2-hexyloxy-5-nitrophenyl)nitrone 42 (613 mg, 1.90 mmol) and palladium hydroxide (20% on carbon, 66 mg, 5 mol %) were dissolved in ethyl acetate (9.5 mL). The solution was flushed with hydrogen then placed under a hydrogen atmosphere and stirred at rt for 30 min. The catalyst was removed by filtration through Celite and the solution was concentrated in vacuo to give a 5:1 mixture of α-(5-amino-2-hexyloxyphenyl)-N-tert-butylnitrone 44 and nitrone 42 as a yellow oil (556 mg, approx. 82% yield of nitrone 44). Data derived for nitrone 44: δH (400 MHz, CDCl3): 0.87 (3H, t, J 7.1 Hz, CH3), 1.29–1.34 (4H, m, 2×CH2), 1.40–1.47 (2H, m, CH2), 1.56 (9H, s, 3×CH3), 1.70–1.77 (2H, m, CH2), 3.45 (2 h, br s, NH2), 3.88 (2H, t, J 6.4 Hz, CH2), 6.64–6.69 (2H, m, H-5 and H-6), 8.01 (1H, s, CH N), 8.78 (1H, d, J 2.6 Hz, H-2)]. Material was carried on to next stage with no further purification or analysis.
2.22. α-(5-Amino-2-dodecyloxyphenyl)-N-tert-butylnitrone 45
N-tert-Butyl-α-(2-dodecyloxy-5-nitrophenyl)nitrone 43 (563 mg, 1.38 mmol) and palladium hydroxide (20% on carbon, 48 mg, 5 mol %) were dissolved in ethyl acetate (7.5 mL). The solution was flushed with hydrogen then placed under a hydrogen atmosphere and stirred at room temperature for 50 min. The catalyst was removed by filtration and the solution was concentrated in vacuo to give 45 as a brown solid (517 mg, 99%). δH (400 MHz, CDCl3): 0.87 (3H, t, J 6.3 Hz, CH3), 1.21–1.34 (16H, m, 8×CH2), 1.42–1.47 (2H, m, CH2), 1.59 (9H, s, 3×CH3), 1.73–1.78 (2H, m, CH2), 3.40 (2H, br s, NH2), 3.91 (2H, t, J 6.4 Hz, CH2), 6.69–6.73 (2H, m, H-5 and H-6), 8.05 (1H, s, CH N), 8.82 (1H, d, J 2.3 Hz, H-2). The material was carried onto next stage with no further purification or analysis.
2.23. EPR spectroscopy
Iron(II) sulfate (100 μL of a 1 mM aqueous solution) and hydrogen peroxide (100 μL of a 1 mM aqueous solution) were added to a solution of the nitrone 13, 14 or 15 in DMSO (100 μL of a 2.5 mM solution in the case of nitrone 13 and a 10 mM solution for nitrones 14 and 15). The solution [0.83 mM nitrone 13 or 3.33 mM nitrone 14 or 15, 0.33 mM hydrogen peroxide, 0.33 mM iron(II) sulfate in water–DMSO (2:1)] was then immediately transferred to a quartz flat cell and placed in the EPR spectrometer for analysis. Spectra were acquired on a Bruker e-scan™ bench-top EPR machine with a permanent magnet and a magnetic sweep circuit (centre of field=0.345 T, sweep width 25 mT) operating at a frequency of 9.8 GHz (X-band). Acquisition parameters: RG 3.99×103, 2.76 mW, MA 0.5 G. Hyperfine couplings were derived from simulations using WINEPR SimFonia™.
Acknowledgements
The Wellcome Trust for funding. SPARC and the BBSRC for the purchase of the bench-top EPR spectrometer used. Ruth Edge (EPR National Service, University of Manchester) for advice on EPR spectra.
References and notes
- 1.Lin M.T., Beal M.F. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 2.Kim J., Wei Y., Sowers J.R. Circ. Res. 2008;102:401–414. doi: 10.1161/CIRCRESAHA.107.165472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Floyd R.A. Aging Cell. 2006;5:51–57. doi: 10.1111/j.1474-9726.2006.00189.x. [DOI] [PubMed] [Google Scholar]
- 4.Rosen G.M., Britigan B.E., Halpern H.J., Pou S. OUP; Oxford: 1999. Free Radicals: Biology and Detection by Spin Trapping. [Google Scholar]
- 5.Recent reviews of spin-trapping in biological systems:; (a) Swartz H.M., Khan N., Khramtsov V.V. Antioxidants and Redox Signaling. 2007;9:1757–1771. doi: 10.1089/ars.2007.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Polovka M. J. Food Nutr. Res. 2006;45:1–11. [Google Scholar]
- 6.Recent uses of spin-trapping with acyclic nitrones in chemical systems include:; (a) Rosenau T., Kloser E., Gille L., Mazzini F., Netscher T. J. Org. Chem. 2007;72:3268–3281. doi: 10.1021/jo062553j. [DOI] [PubMed] [Google Scholar]; (b) Usuki T., Nakanishi K., Ellestad G.A. Org. Lett. 2006;8:5461–5463. doi: 10.1021/ol062061t. [DOI] [PubMed] [Google Scholar]; (c) Gigmes D., Gaudel-Siri A., Marque S.R.A., Bertin D., Tordo P., Astolfi P., Greci L., Rizzoli C. Helv. Chim. Acta. 2006;89:2312–2326. [Google Scholar]; (d) Sueishi Y., Yoshioka D., Yoshioka C., Yamamoto S., Kotake Y. Org. Biomol. Chem. 2006;4:896–901. doi: 10.1039/b515682c. [DOI] [PubMed] [Google Scholar]
- 7.(a) Gamliel A., Afri M., Frimer A.A. Free Radical Biol. Med. 2008;44:1394–1405. doi: 10.1016/j.freeradbiomed.2007.12.028. [DOI] [PubMed] [Google Scholar]; (b) Bardelang D., Charles L., Finet J.P., Jicsinszky L., Karoui H., Marque S.R.A., Monnier V., Rockenbauer A., Rosas R., Tordo P. Chem.—Eur. J. 2007;13:9344–9354. doi: 10.1002/chem.200700369. [DOI] [PubMed] [Google Scholar]; (c) Caldwell S.T., Quin C., Edge R., Hartley R.C. Org. Lett. 2007;9:3499–3502. doi: 10.1021/ol071285o. [DOI] [PubMed] [Google Scholar]
- 8.Recent examples include:; (a) Durand G., Poeggeler B., Boeker J., Raynal S., Polidori A., Pappolla M.A., Hardeland R., Pucci B. J. Med. Chem. 2007;50:3976–3979. doi: 10.1021/jm0706968. [DOI] [PubMed] [Google Scholar]; (b) Kim S., Vilela G.V.M.de.A., Bouajila J., Dias A.G., Cyrino F.Z.G.A., Bouskela E., Costa P.R.R., Nepveu F. Bioorg. Med. Chem. 2007;15:3572–3578. doi: 10.1016/j.bmc.2007.02.033. [DOI] [PubMed] [Google Scholar]; (c) Asanuma T., Yasui H., Inanami O., Waki K., Takahashi M., Iizuka D., Uemura T., Durand G., Polidori A., Kon Y., Pucci B., Kuwabara M. Chem. Biodivers. 2007;4:2253–2267. doi: 10.1002/cbdv.200790184. [DOI] [PubMed] [Google Scholar]; (d) Sklavounou E., Hay A., Ashraf N., Lamb K., Brown E., MacIntyre A., George W.D., Hartley R.C., Shiels P.G. Biochem. Biophys. Res. Commun. 2006;347:420–427. doi: 10.1016/j.bbrc.2006.06.087. [DOI] [PubMed] [Google Scholar]; (e) Ortial S., Durand G., Poeggeler B., Polidori A., Pappolla M.A., Böker J., Hardeland R., Pucci B. J. Med. Chem. 2006;49:2812–2820. doi: 10.1021/jm060027e. [DOI] [PubMed] [Google Scholar]
- 9.(a) Hardy M., Chalier F., Ouari O., Finet J.P., Rockenbauer A., Kalyanaraman B., Tordo P. Chem. Commun. 2007:1083–1085. doi: 10.1039/b616076j. [DOI] [PubMed] [Google Scholar]; (b) Hardy M., Rockenbauer A., Vasquez-Vivar J., Felix C., Lopez M., Srinivasan S., Avadhani N., Tordo P., Kalyanaraman B. Chem. Res. Toxicol. 2007;20:1053–1060. doi: 10.1021/tx700101d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Xu Y., Kalyanaraman B. Free Radical Res. 2007;41:1–7. doi: 10.1080/10715760600911147. [DOI] [PubMed] [Google Scholar]; (d) Murphy M.P., Echtay K.S., Blaikie F.H., Asin-Cayuela J., Cocheme H.M., Green K., Buckingham J.A., Taylor E.R., Hurrell F., Hughes G., Miwa S., Cooper C.E., Svistunenko D.A., Smith R.A., Brand M.D. J. Biol. Chem. 2003;278:48534–48545. doi: 10.1074/jbc.M308529200. [DOI] [PubMed] [Google Scholar]
- 10.Murphy M.P., Smith R.A. Annu. Rev. Pharmacol. Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 11.For example:; (a) Casey J.R., Morgan P.E., Vullo D., Scozzafava A., Mastrolorenzo A., Supuran C.T. J. Med. Chem. 2004;47:2337–2347. doi: 10.1021/jm031079w. [DOI] [PubMed] [Google Scholar]; (b) Pastorekova S., Casini A., Scozzafava A., Vullo D., Pastorek J., Supuran C.T. Bioorg. Med. Chem. 2004;14:869–874. doi: 10.1016/j.bmcl.2003.12.029. [DOI] [PubMed] [Google Scholar]; (c) Springer D.M., Luh B.-Y., Goodrich J.T., Bronson J.J. Bioorg. Med. Chem. 2003;11:281–292. doi: 10.1016/s0968-0896(02)00335-8. [DOI] [PubMed] [Google Scholar]; Springer D.M., Luh B.-Y., Goodrich J.T., Bronson J.J. Bioorg. Med. Chem. 2003;11:265–280. doi: 10.1016/s0968-0896(02)00336-x. [DOI] [PubMed] [Google Scholar]; (d) Springer D.M., Luh B.-Y., Bronson J.J. Bioorg. Med. Chem. Lett. 2001;11:797–802. doi: 10.1016/s0960-894x(01)00060-9. [DOI] [PubMed] [Google Scholar]
- 12.Koya K., Li Y., Wang H., Ukai T., Tatsuta N., Kawakami M., Shishido T., Chen L.B. Cancer Res. 1996;56:538–543. [PubMed] [Google Scholar]
- 13.Novgorodov S.A., Szule Z.M., Luberto C., Jones J.A., Bielawski J., Bielawski A., Hannun Y.A., Obeid L.M. J. Biol. Chem. 2005;280:16096–16105. doi: 10.1074/jbc.M411707200. [DOI] [PubMed] [Google Scholar]
- 14.Janzen E.G., Dudley R.L., Shetty R.V. J. Am. Chem. Soc. 1979;101:243–245. [Google Scholar]
- 15.Janzen E.D., Kotake Y., Hinton R.D. Free Radical Biol. Med. 1992;12:169–173. doi: 10.1016/0891-5849(92)90011-5. [DOI] [PubMed] [Google Scholar]
- 16.Janzen E.G., West M.S., Kotake Y., DuBose C.M. J. Biochem. Biophys. Methods. 1996;32:183–190. doi: 10.1016/0165-022x(96)00008-5. [DOI] [PubMed] [Google Scholar]
- 17.Misik V., Miyoshi N., Riesz P. J. Phys. Chem. 1995;99:3605–3611. [Google Scholar]
- 18.Hill H.A.O., Thornalley P.J. Can. J. Chem. 1982;60:1528–1531. [Google Scholar]
- 19.Hay A., Burkitt M.J., Jones C.M., Hartley R.C. Arch. Biochem. Biophys. 2005;435:336–346. doi: 10.1016/j.abb.2004.12.030. For an example of trapping radicals in membranes see: [DOI] [PubMed] [Google Scholar]
- 20.Sár C.P., Hideg E., Vass I., Hideg K. Bioorg. Med. Chem. Lett. 1998;8:379–384. doi: 10.1016/s0960-894x(98)00033-x. [DOI] [PubMed] [Google Scholar]
- 21.(a) Schulze W., Gutsche W., Vater W., Oertel B., Böhm K.J., Unger E., Werner W. Die Pharmazie. 1990;45:686–687. [PubMed] [Google Scholar]; (b) Schulze W., Gutsche W., Jungstan W. Arzneim. Forsch. 1967;17:605–607. [PubMed] [Google Scholar]; (c) Schulze W. J. Prakt. Chem. 1962;17:24–34. [Google Scholar]
- 22.Abdallah M.A., Andre J.J., Biellmann J.-F. Bioorg. Chem. 1977;6:157–163. [Google Scholar]
- 23.Hayakawa K., Shiomi D., Ise T., Sato K., Takui T. J. Mater. Chem. 2006;16:4146–4154. [Google Scholar]
- 24.Cheng W.-C., Kurth M.J. Org. Prep. Proced. Int. 2002;34:587–608. [Google Scholar]
- 25.Recent examples include:; (a) Yamaguchi I., Higashi H., Shigesue S., Shingai S., Sato M. Tetrahedron Lett. 2007;48:7778–7781. [Google Scholar]; (b) Nguyen T.M., Sanchez-Salvatori M.del.R., Wypych J.-C., Marazano C. J. Org. Chem. 2007;72:5916–5919. doi: 10.1021/jo0707582. [DOI] [PubMed] [Google Scholar]; (c) Kearney A.M., Vanderwal C.D. Angew. Chem., Int. Ed. 2006;45:7803–7806. doi: 10.1002/anie.200602996. [DOI] [PubMed] [Google Scholar]; (d) Viana G.H.R., Santos I.C., Alves R.B., Gil L., Marazano C., Gil R.P.F. Tetrahedron Lett. 2005;46:7773–7776. [Google Scholar]
- 26.Fatiadi A.J. Synthesis. 1976:65–104. [Google Scholar]
- 27.Ozanne A., Pouységu L., Depernet D., François B., Quideau S. Org. Lett. 2003;5:2903–2906. doi: 10.1021/ol0349965. [DOI] [PubMed] [Google Scholar]
- 28.Martín S.E., Garrone A. Tetrahedron Lett. 2003;44:549–552. [Google Scholar]
- 29.Tidwell T.T. Org. React. 1990;39:297–572. [Google Scholar]
- 30.Helms A., Heiler D., McLendon G. J. Am. Chem. Soc. 1992;114:6227–6238. [Google Scholar]
- 31.Claramunt R.M., Elguero J. Collect. Czech. Chem. Commun. 1981;46:584–596. [Google Scholar]
- 32.Bandgar B.P., Gaikwad N.B. Monatsh. Chem. 1998;129:719–722. [Google Scholar]
- 33.Hinton R.D., Janzen E.G. J. Org. Chem. 1992;57:2646–2651. [Google Scholar]
- 34.Oszczapowicz J., Pines H. J. Org. Chem. 1972;37:2799–2806. [Google Scholar]
- 35.U.S. Patent 2,930,731, Upjohn Co. March 29, 1960.
- 36.Collins R.F., Davis M. J. Chem. Soc. C. 1966:2196–2201. doi: 10.1039/j39660002196. [DOI] [PubMed] [Google Scholar]