

SUMMARY
Multipotential naïve CD4+ T cells differentiate into distinct lineages including T helper 1 (Th1), Th2, Th17, and inducible T regulatory (iTreg) cells. The remarkable diversity of CD4+ T cells begs the question whether the observed changes reflect terminal differentiation with heritable epigenetic modifications or plasticity in T cell responses. We generated genome-wide histone H3 lysine 4 (H3K4) and lysine 27 (H3K27) trimethylation maps in naïve, Th1, Th2, Th17, iTreg, and natural (n)Treg cells. We found that although modifications of signature cytokine genes (Ifng, Il4, and Il17) partially conform to the expectation of lineage commitment, critical transcription factors such as Tbx21 exhibit a broad spectrum of epigenetic states, consistent with our demonstration of T-bet and IFNγ induction in nTreg cells. Our data suggest an epigenetic mechanism underlying the specificity and plasticity of effector and regulatory T cells and also provide a framework for understanding complexity of CD4+ T helper cell differentiation.
INTRODUCTION
Depending upon signals from innate immune cells induced by various pathogenic agents, naïve CD4+ T cells differentiate into one of several effector fates including T helper 1 (Th1), Th2, and Th17 cells (Ansel et al., 2006; Murphy and Reiner, 2002; Szabo et al., 2003; Weaver et al., 2007; Zhu and Paul, 2008). In addition, other stimuli cause CD4+ T cells to become regulatory T (Treg) cells (Sakaguchi et al., 2008). These effector and regulatory CD4+ T cell lineages protect the body from infection when appropriately activated but also contribute to pathogenesis of autoimmunity and inappropriate inflammation (Reiner et al., 2007).
The signaling events that lead to polarization of CD4 subsets have become increasingly clear. Interleukin-12 (IL-12), acting via the signal transducer and activator of transcription 4 (Stat4), initiates the development of Th1 cells that produce the signature cytokine IFN-γ and express the key regulator T-box 21 (Tbx21, also called T-bet) (Afkarian et al., 2002; Kaplan et al., 1996b; Szabo et al., 2000; Thierfelder et al., 1996). The Th2 cell response is characterized by expression of IL-4 and IL-13. IL-4 activates Stat6 and induces the key transcription factor GAGA binding protein 3 (Gata3) (Kaplan et al., 1996a; Takeda et al., 1996; Zheng and Flavell, 1997). A relatively new effector CD4+ T cell lineage producing IL-17 has recently been recognized (Harrington et al., 2006; Langrish et al., 2005). In combination with transforming growth factor-beta 1 (TGFβ-1), the inflammatory cytokines IL-6 and IL-1 promote Th17 cell differentiation from naïve CD4+ T cells. IL-21 produced by Th17 cells promotes Th17 cell commitment in an autocrine manner, whereas IL-23 maintains and expands these cells in vivo. IL-6, IL-21 and IL-23 all signal through Stat3, and in combination with TGFβ-1 induce the transcription factors RORγt and RORα (Chen et al., 2006; Dong, 2008; Korn et al., 2007; Nurieva et al., 2007; Wei et al., 2007; Yang et al., 2008a; Zhou et al., 2007). In addition to Th cells, regulatory T (Treg) cells represent another important immunoregulatory subset of CD4+ T cells and both natural(n) Tregs and inducible (i) Tregs contribute to the maintenance of peripheral tolerance. The transcription factor Foxp3, essential for Treg development, maintenance, and functions, can be induced by TGFβ-1 and IL-2, as well as retinoic acid, in peripheral naïve CD4+ T cells ex vivo (Elias et al., 2008; Lohr et al., 2006; Miyara and Sakaguchi, 2007; Zheng and Rudensky, 2007).
Although transcription factors are critical in regulating cellular differentiation, epigenetic mechanisms are also key factors. The basic structural unit of eukaryotic chromatin is the nucleosome, which consists of 146 base pairs of DNA wrapped around an octamer of four core histones (H2A, H2B, H3 and H4). Posttranslational modifications of histones are implicated in regulating gene expression by controlling chromatin structure and DNA accessibility (Goldberg et al., 2007; Kouzarides, 2007). Compelling evidence suggests that histone modifications and nucleosome positioning correlate with gene transcription in T cells (Barski et al., 2007; Roh et al., 2006; Roh et al., 2005; Schones et al., 2008; Wang et al., 2008). For example, trimethylation of H3K4 and H3K79 as well as monomethylation of H3K27, H3K9 and H4K20 are associated with gene activation whereas di-and tri-methylation of H3K27 are associated with gene repression. Although H3K4me3 and H3K27me3 are usually enriched at active and inactive chromatin regions respectively, they are found to colocalize in some genomic regions (Azuara et al., 2006; Bernstein et al., 2006; Roh et al., 2006). These bivalent modifications have been suggested to poise developmental genes for either activation or repression during differentiation in embryonic stem cells (Bernstein et al., 2006). Although epigenetic information is not coded in the genomic sequence, it is maintained in a cell and its decendents and therefore contributes to the maintenance of cellular phenotype (Goldberg et al., 2007).
Previous studies have supported the notion that Th1 and Th2 cells represent true lineages by demonstrating the epigenetic modification state of a number of signature cytokine loci, including Ifng locus in Th1 cells (Hatton et al., 2006; Schoenborn et al., 2007) and Il4 locus in Th2 cells (Ansel et al., 2006). These studies confirmed that signature cytokine genes are associated with active epigenetic marks in featured lineages but with repressive epigenetic marks in the opposing lineages. These feedback and feed-forward mechanisms result in progressive, symmetric commitment to a lineage with extinction of the opposing fate. As new fates of CD4+ T cells, Treg and Th17 cells, have been recognized, some aspects of the epigenetic state of the corresponding characteristic genes have been investigated. In Th17 cells, it has been shown that H3 acetylation and H3K4me3 mark the Il17 and Il17f loci (Akimzhanov et al., 2007). In both human and mouse nTreg cells, the Foxp3 locus is found to be demethylated (Floess et al., 2007; Janson et al., 2008; Kim and Leonard, 2007). However, whether these new lineages can be viewed in the same way as the prior symmetric Th1 and Th2 cell model is far from clear. Furthermore, other than the three loci discussed, there is little information that substantiates the notion that Th1, Th2 or Th17 cells are true lineages with defined heritable cassettes of genes that contribute to their phenotypes. More importantly, recent data suggest considerable plasticity of Th17 and Treg cells with respect to their capacity to produce cytokines. For example, Th17 cells can produce IFN-γ, the signature cytokine of Th1 cells (Harrington et al., 2005); Treg cells can produce IL-17 (Xu et al., 2007); and cells expressing both IL-17F and Foxp3 are found in both Th17 and iTreg cell cultures (Yang et al., 2008b). Taken together, this information suggests a more complex picture of “lineage commitment” among T helper cell types with a considerable degree of cellular plasticity. However, it is not clear to what degree the epigenetic mechanisms contribute to potential cellular flexibility.
Although epigenetic modifications undoubtedly serve as an important regulatory mechanism during CD4+ T cell fate choice, there is a paucity of information related to the epigenetic changes that occur during differentiation. Moreover, previous studies on epigenetic modifications of signature cytokine loci and the Foxp3 locus focused mainly on conserved noncoding sequences (CNS) further limiting the view of critical chromatin structures that maintain the lineage commitment. In the present study, we have used the high-throughput ChIP-Seq approach to generate genome-wide H3K4me3 and H3K27me3 maps in naïve, Th1, Th2, Th17, iTreg, and nTreg cells to gain insights into lineage commitment and plasticity in developing T cells. Our results point to surprising complexity in the potential regulation of transcription factors, cytokines, chemokines and receptors that contribute to the phenotype considered as Th cell lineages. Although some signature loci can be viewed as stable and heritable traits, other aspects of Th cells appear to be more plastic.
RESULTS
Genome-wide maps of H3K4me3 and H3K27me3 modifications in CD4+ effector and regulatory T cell lineages
To reveal epigenetic features acquired during CD4+ T cell differentiation, we generated global maps of H3K4me3 and H3K27me3 modifications using the ChIP-seq approach (Barski et al., 2007; Schones et al., 2008). We assessed these modifications in six lineages of CD4+ T cells: freshly isolated CD4+CD62L+CD44-CD25- (naïve) T cells and CD4+CD25hi nTreg cells, as well as CD4+ T cells cultured under Th1, Th2, Th17, and iTreg cell conditions (Figure 1), resulting in 12 histone modification maps. A total of 97 million short reads from all samples were uniquely aligned onto mouse genome (mms8) (Table S1). We developed a model-based algorithm for identification of genomic regions, termed as modification islands, which are significantly enriched for a histone modification (see Supplemental Information for details). Our scaling analysis indicated that the sequence reads were sufficient to identify most regions associated with the histone modifications in the genome (see Figure S1 for details). The differentiation state was verified using DNA microarray analysis of mRNA profiles in these T cell lineages and confirmed for characteristic genes, including Ifng, Il4, Il17, and Foxp3 using real-time PCR assays (Figure S2). In addition, our results of intracellular staining of those characteristic genes indicated the successful polarization of Th cell subsets (Figure S3).
Figure 1. Overview of the experimental design.

Splenic and lymph node CD4+ T cells from C57BL/6J mice were sorted for CD4+CD62L+CD44-CD25- naïve population and CD4+CD25hi natural T regulatory cells, or cultured under Th1, Th2, Th17, and iTreg cell conditions. The genome-wide distribution of H3K4me3 and H3K27me3 was determined using ChIP-seq. Gene expression profiling of the six T cell subsets was performed using DNA microarrays.
To obtain an overall picture of H3K4me3 and H3K27me3 distribution, we divided the mouse genome into four kinds of regions: proximal promoter (1 kb upstream and downstream of transcription start site), exon, intron, and intergenic regions, based on annotation of “known genes” from UCSC genome browser (http://genome.ucsc.edu) (Kent et al., 2002) (Figure 2A). Interestingly, more H3K4me3 and H3K27me3 islands were identified in naïve and nTreg cells than in Th cells. Although the majority of H3K4me3 islands were found in proximal promoter regions, ranging from 41% in nTreg cells to 60% in Th2 cells, the majority of H3K27me3 islands were found in intergenic regions. Moreover, only a small number of lineage specific islands were found for Th cells. This contrasts with the fact that 28% unique H3K4me3 and 22% unique H3K27me3 islands were found in nTreg cells (Figure 2B, Table S2), indicating the unique epigenetic features of natural regulatory T cells. Examination of H3K4me3 and H3K27me3 islands within gene bodies and their 5' and 3' 5 kb extended regions also revealed the enrichment of H3K4me3 islands near transcription start sites (Figure 2C). These results are consistent with recent observations that H3K4me3 associates extensively with proximal promoters in human T cells, as well as human and murine embryonic stem cells (Barski et al., 2007; Fouse et al., 2008; Guenther et al., 2007; Mikkelsen et al., 2007; Pan et al., 2007; Zhao et al., 2007). Interestingly, it appears that H3K27me3 modification decreased within gene bodies (Figure 2C), consistent with the observation that the majority of the H3K27me3 islands located in intergenic regions (Figure 2A).
Figure 2. Distribution of H3K4me3 and H3K27me3 islands.
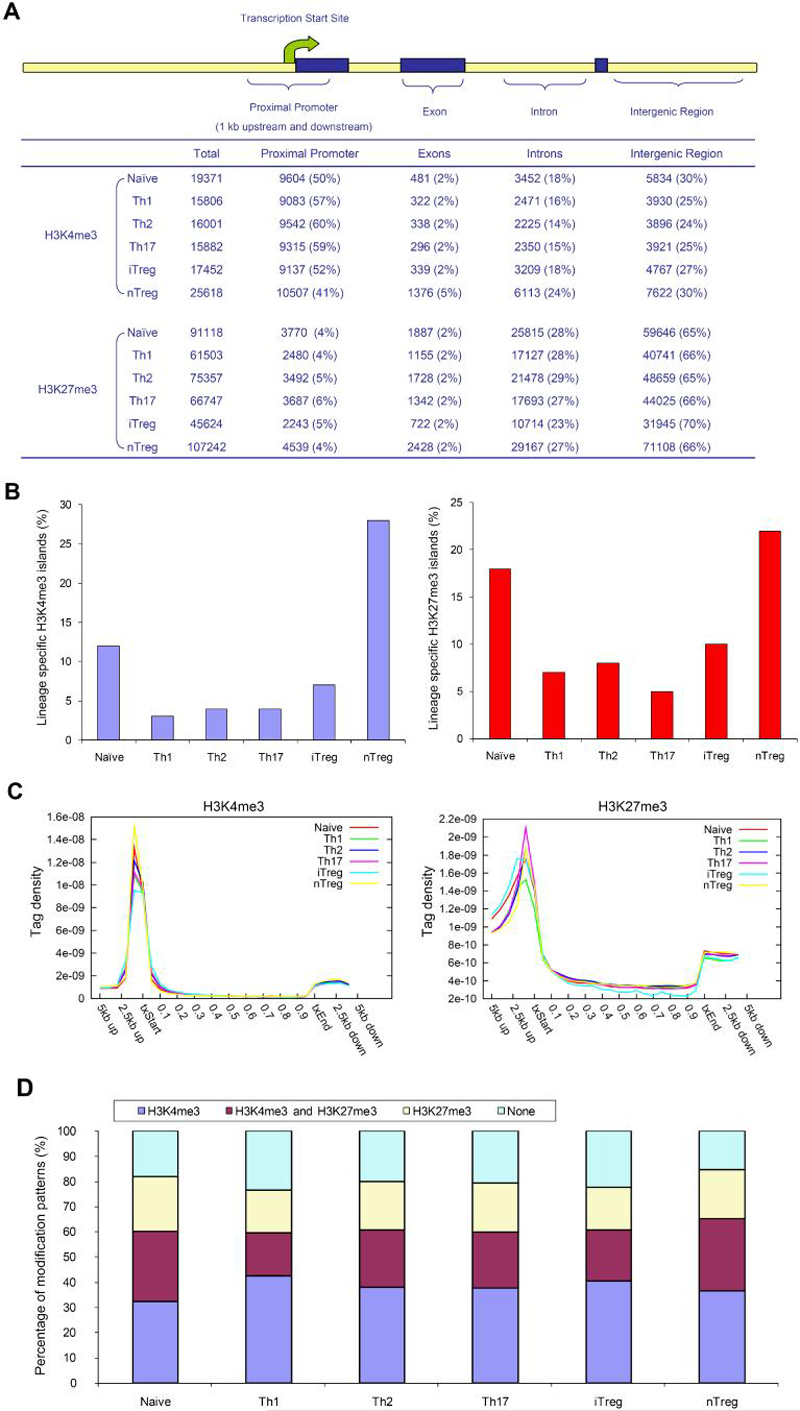
(A) The mouse genome was divided into four kinds of regions: proximal promoter (1 kb upstream and downstream of transcription start site), exon, intron, and intergenic regions. The number of islands for each sample was listed as the total identified islands (Total), followed by the islands among genomic regions with the percentage listed in the parenthesis.
(B) Lineage-specific islands were identified as described in Supplemental Information. The percentage of lineage-specific H3K4me3 (left) and H3K27m3 (right) islands is expressed as a proportion of all islands identified in each lineage.
(C) For each gene, uniquely mapped tags were summed in 1 kb windows for the regions from 5 kb upstream of the transcription start site (txStart) to the txStart and from the transcription end site (txEnd) to 5 kb downstream. Within the gene bodies, tags were summed in windows equal to 5% of the gene length. All tag-counts were normalized by the total number of bases in the windows and the total read number of the given library to obtain normalized tag density (Wang et al., 2008).
(D) The percentage of genes associated with H3K4me3 alone, both H3K4me3 and H3K27me3, H3K27me3 alone, or not associated with H3K4me3 or H3K27me3 for each T cell subsets.
To quantify H3K4me3 and H3K27me3 modifications for each gene, we calculated the total tag number of all islands identified within individual gene body and proximal promoter regions (gene body plus 1 kb sequence upstream of transcription start site). As shown in Figure 2D and Table S3, 32-43% and 17-22% of the 19,657 unique known genes were associated with H3K4me3 alone or H3K27me3 alone, respectively, whereas 17-28% of all genes associated with both H3K4me3 and H3K27me3 modifications. Intriguingly, a higher percentage of genes in naïve and nTreg cells were marked with H3K27me3 modification compared to other lineages, suggesting that in vitro differentiation of T cells was associated with overall reduction of H3K27me3 modification. In summary, our data indicate that the global epigenetic modification patterns of naïve and nTreg cells are distinguishable from other T helper cell subsets.
The extended phenotype of helper T cell subsets
To broaden our view of lineage commitment beyond signature cytokines, we combined H3K4me3 and H3K27me3 maps with gene expression profiling data to identify groups of genes that could characterize T cell subsets. Two hierarchical clusters were generated: the first cluster had all cytokines, chemokines, and their receptors in the murine genome (total 271 genes, see Supplemental Information for detail) (Figure S4, Table S4); the second cluster had all transcription factors in murine genome (total 782 genes, see Supplemental Information for detail) (Figure S5, Table S5). From these two clusters, we selected the genes that demonstrated high levels of lineage-specific expression and H3K4me3 modification (Figure 3). Our data revealed genes, in addition to the known Th cell signature genes, which may serve as markers of each CD4+ T cell subset. For example, in addition to IL17A, high amounts of Il1r1 and Il17re mRNA with H3K4me3 modification at their promoters were uniquely detected in Th17 cells. In both nTreg and iTreg cells cytokines and chemokine such as Il1rn, Il9, and Ccl4 and, transcription factors including TG interacting factor (Tgif) and Nuclear receptor subfamily 4, group A, member 1 (Nr4a1) were highly expressed. Interestingly, even though these genes associated with high amounts of H3K4me3 in the lineage where they were expressed, they were not necessarily associated with high amounts of H3K27me3 in the lineages where they were not expressed. Instead, the amounts of H3K27me3 in silent genes varied, depending upon the particular gene and particular lineage. For example, H3K27me3 was not detected on the Il18r1 gene in Th1 cells where it was expressed. The Il18r1 gene was associated with high amounts of H3K27me3 in Th2 cells, intermediate amounts in Th17 and nTreg cells and very low amounts in naïve and iTreg cells, even though it was not expressed in any of these cells (Figure 3). These data suggest that complex epigenetic fates exist for these interesting genes (see below for more details). In addition, many of these genes were expressed at relatively low amounts in naïve cells. In most cases, they were associated with low amounts of H3K4me3 with little or no H3K27me3. But in some cases, they were associated with both H3K4me3 and H3K27me3 marks. This is consistent with the fact that they are not permanently repressed but instead are poised for expression. Therefore, the strategy of combining gene expression and epigenetic mapping revealed epigenetic information on established signature genes and potential unreported lineage-specific genes for the T helper cells.
Figure 3. Epigenetic modifications and gene expression profiles of signature genes characterizing T cell subsets.
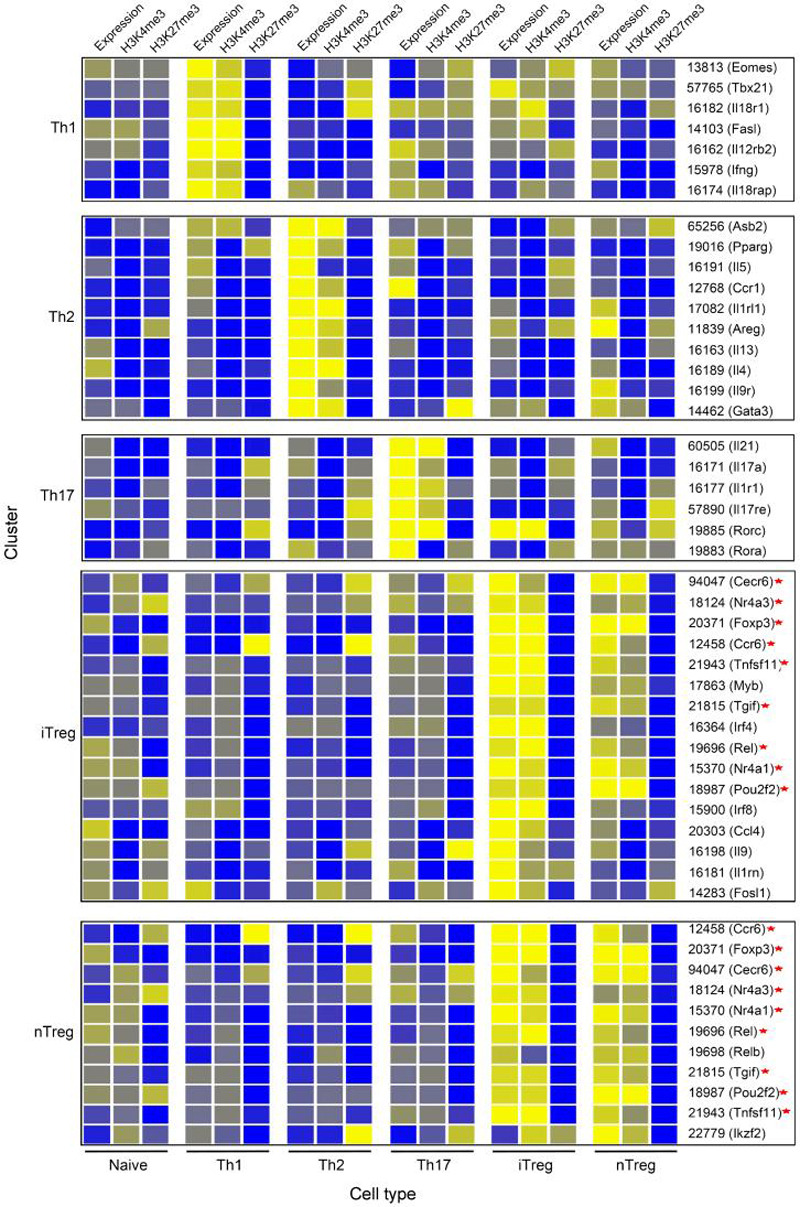
H3K4me3 and H3K27me3 modifications as well as gene expression were first normalized separately using GeneSpring software. Hierarchical clusters of signature genes for Th1, Th2, Th17, iTreg, and nTreg cells are shown. The color coding depicts the normalized value for each gene based on a scale of 0-6 with 1 as the median. The cell types for the expression and modification patterns are indicated below the panels. Each cluster is characteristic of one cell type, which is indicated on the left of the panel. The shared genes between iTreg and nTreg cells are indicated by asterisks on right.
Epigenetic modification patterns indicate alternative transcript usage and functional regulatory elements
Many genes encode multiple transcripts via alternative transcription start sites. We used the H3K4me3 and H3K27me3 maps to identify genes that use alternative transcripts in a lineage specific manner and found that Runx3 was within the list. The Runx3 gene can be transcribed from either the distal or proximal promoter, resulting in protein products that have unique 19- or 6-amino-acid N-terminal sequences, respectively. It was reported that CD4+ and CD8+ T cells use the proximal and distal promoter respectively (Egawa et al., 2007). Our H3K4me3 and H3K27me3 data are consistent with the previous evidence that CD4+ T cells, especially naïve, Th2, Th17, iTreg, and nTreg cell lineages, use the proximal promoter of Runx3 (Figure 4A), whereas the distal promoter appears to be blocked by a cluster of H3K27me3 signals located between the distal and proximal promoters in these five lineages. Surprisingly, Th1 cells showed strong H3K4me3 signals at both the distal and proximal promoters and diminished H3K27me3 signals between the two promoter regions, suggesting that both of these two promoters may be used. Indeed, we detected a ~40 fold increase in expression of transcript from the distal promoter in Th1 cells as compared to other lineages, even though the sum of the short and long transcripts was comparable in all five lineages (Figure 4B). Given that Runx3 is involved in a feed-forward regulatory circuit that activates Ifng and silences Il4 (Djuretic et al., 2007), our data suggests that the isoform encoded by the distal promoter may be selectively relevant to the function of Th1 cells and CD8+ cells, but not other non-IFN-γ-producing cells.
Figure 4. Lineage specific alternative transcript usage and functional regulatory elements.
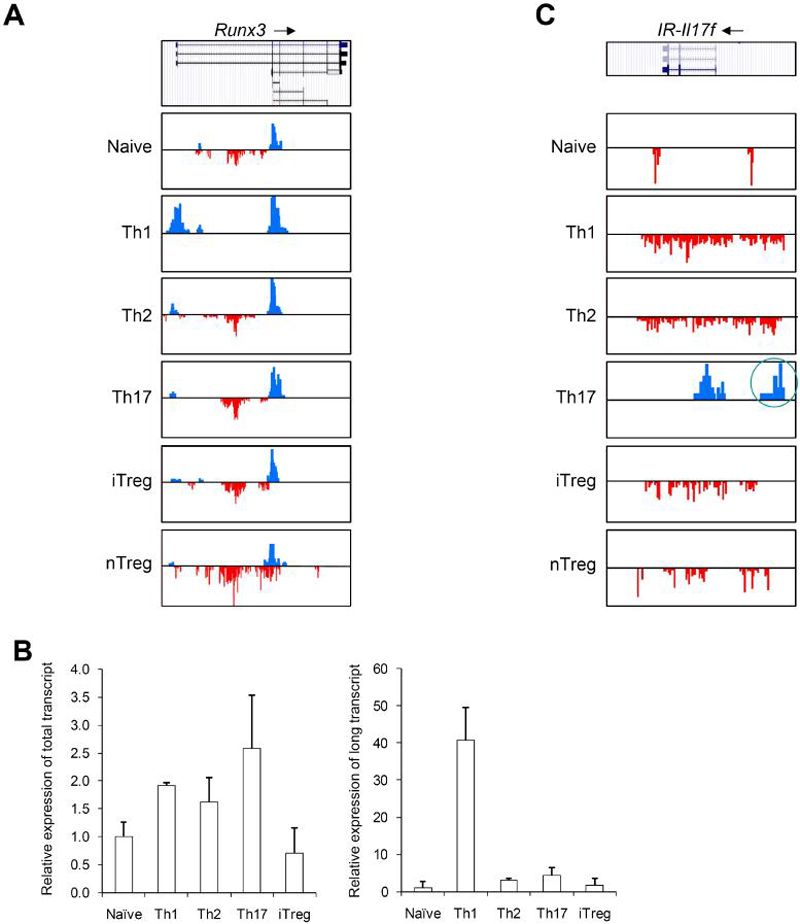
(A) Distribution of H3K4me3 and H3K27me3 islands along the Runx3 genomic region (Chr4, 134389000-134451000) in six T cell subsets is shown. All figures with views of H3K4me3 and H3K27me3 distribution are labeled such that the arrow represents the direction of gene transcription. Gene structure is downloaded from UCSC Genome Browser and only tags on islands are shown. The islands labeled in blue represent H3K4me3. The islands labeled in red represent H3K27me3. Scales are kept constant among cell types.
(B) Real-time PCR was performed on RNAs from naïve, Th1, Th2, Th17, and iTreg cells using primer sets that recognize both the long and short forms (left panel) or only the long form (right panel) of Runx3 transcripts.
(C) Distribution of H3K4me3 and H3K27me islands along the Intergenic Region upstream of Il17f (IR-Il17f) (Chr1, 20,755,000-20,780,000) in six T cell subsets is shown. The intergenic H3K4me3 island is highlighted by a red circle.
Non-promoter H3K4me3 and H3K27me3 islands can indicate the presence of enhancers and/or silencers (Barski et al., 2007; Wang et al., 2008). To discover potential lineage-specific regulatory elements, we identified all lineage specific intergenic islands (Table S5). One interesting region is the intergenic H3K4me3 island located about 8 kb upstream of the Il17f promoter in Th17 cells (Figure 4C). This island correlated with the expression of the Il17f gene and contained multiple transcription factor target sites including STATs, ETS1 etc., suggesting that it may serve as an enhancer to regulate the transcription of the Il17f gene in Th17 cells.
In summary, our genome-wide data on H3k4me3 and H3K27me3 revealed cassettes of genes, alternative transcripts, and regulatory elements that program the CD4+ lineage phenotype.
Signature genes do not conform to a simple view of lineage commitment
If the helper T cell subsets represented distinct, stable lineages, one might predict that the signature genes corresponding to their phenotype would have marks consistent with expression in respective lineage and conversely, marks of repression in all other fates. We therefore first examined epigenetic modification patterns for Ifng, Il4, and Il17, the defining lineage markers for Th1, Th2, and Th17 cells. As shown in Figure 5A and Figure S6, H3K4me3 was associated with Ifng, Il4, Il17, and Il21 in the respective lineages, consistent with previous data and our expectations. However, the pattern of H3K27me3 modification was more complicated. As previously reported (Schoenborn et al., 2007), the Ifng promoter region and gene body were marked by H3K27me3 in Th2 cells (Figure 5A). In addition, in naïve, Th2, Th17, and iTreg cells the 3' untranslated region (UTR) of the Ifng gene was preferentially marked by H3K27me3. Of note however, was the absence of H3K27me3 modifications in nTreg cells.
Figure 5. H3K4me3 and H3K27me3 modifications of signature cytokine and transcription factor genes.
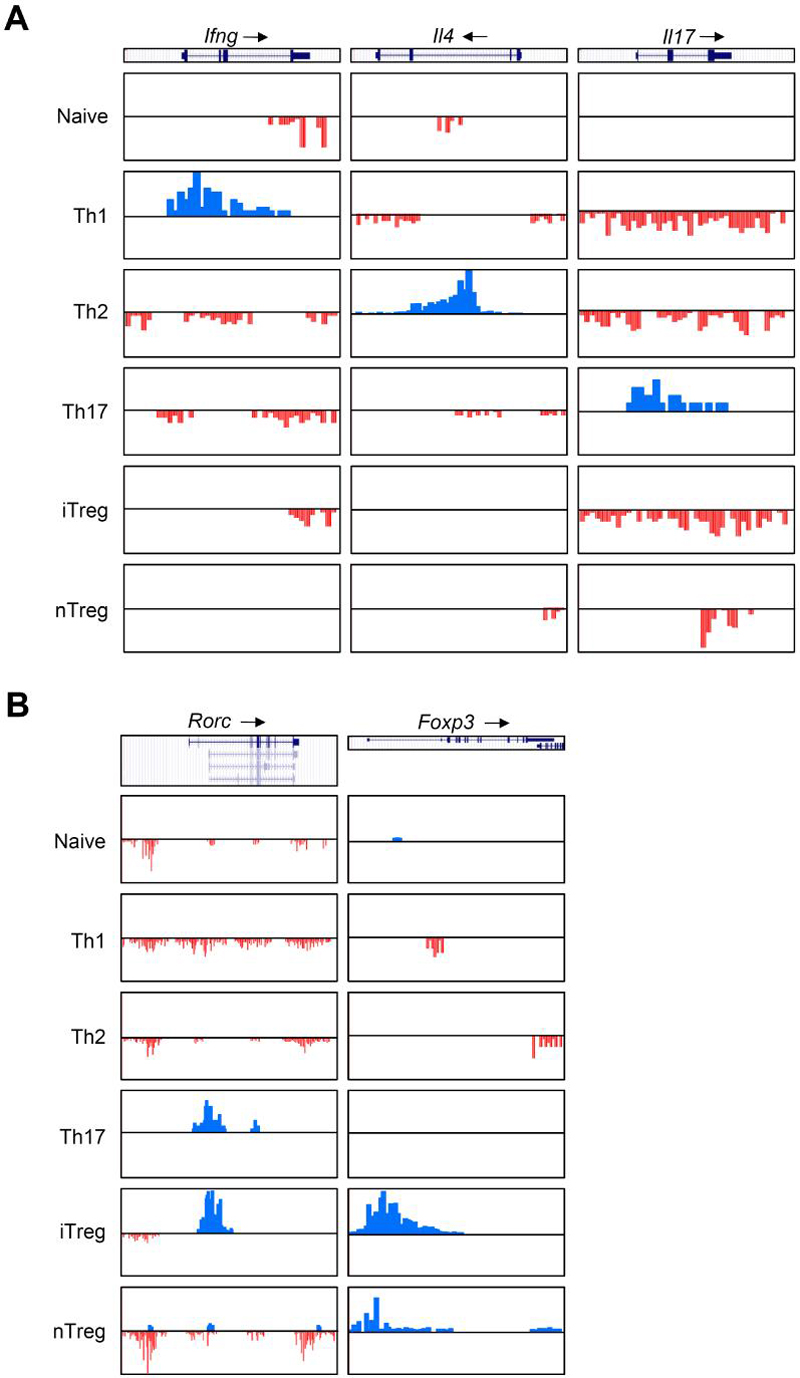
(A) H3K4me3 (blue) and H3K27me3 (red) modifications on Ifng (Chr10, 117842000-117850000), Il4 (Chr11, 53455000-53465000), and Il17 (Chr1, 20714000-20722000) loci.
(B) H3K4me3 and H3K27me3 modifications on Rorc (Chr3, 94443000-94493000) and Foxp3 (ChrX, 6735380-6753000) loci.
As expected, H3K27me3 marks were evident in the Il4 promoter and 3' UTR in Th1 cells. However, only relatively low levels of H3K27me3 were detected in the Il4 locus in Th17 and nTreg cells. Moreover, no detectable levels of H3K27me3 were found in the locus in iTreg cells (Figure 5A). Interestingly, the Il17 and Il21 loci were extensively marked by H3K27me3 in Th1, Th2, and iTreg and nTreg cells, suggesting that these genes are strongly repressed in these cells. It is also interesting to note that although the Ifng and Il4 promoters were associated with H3K27me3 in naïve cells, the Il17 promoter region associated with no detectable levels of H3K27me3 in naïve cells.
In summary, these data validate the existing information on epigenetic modifications of the Ifng, Il4, and Il17 loci and the Il21 locus is marked in the expected manner. However, we also find that the signature gene is not always repressed by H3K27me3 modification in the lineages where it is not expressed; instead, the H3K27me3 pattern is both gene- and lineage-specific, suggesting more complexity than initially envisioned in standard models of Th differentiation.
Epigenetic modifications of “master regulators” suggest plasticity of effector T cells
Stat family transcription factors are critical for helper cell differentiation; however, their expression is not sufficient to drive lineage commitment. Consistent with their ubiquitous expression patterns, we found that most Stats were marked in their promoter regions by H3K4me3 without substantial H3K27me3 signals in all T cell subtypes. Small increases in H3K4me3 marks were associated with Stat4 and Stat5 loci in Th1 and iTreg cells, respectively (Figure S7). In contrast, weak H3K27me3 marks were present in the Stat4 locus in naïve and Th2 cells and the Stat5 loci in Th2 and iTreg cells (Figure S7). This is notable in that Stat4 is differentially expressed in Th1 and Th2 cells (Usui et al., 2006). Interestingly, H3K4m3 marks indicated that two isoforms of Stat5b may be differentially expressed in different lineages, suggesting a potential role of Stat5b isoforms in lineage fate decision.
In contrast to Stats, transcription factors like T-bet, GATA-3, RORγt, and Foxp3 are able to promote and maintain at least some aspects of the lineage phenotypes. Foxp3, which is required for iTreg and nTreg cell functions, was indeed associated with strong H3K4me3 marks in these cells. Interestingly, although H3K27me3 was detected in the Foxp3 gene in Th1 and Th2 cells as expected, it was not detected in Th17 and naïve cells (Figure 5B), indicating that distinct epigenetic states exist for the gene in different non-expressing lineages. This is consistent with the recent report that Foxp3 can be expressed in Th17 cells (Zhou et al., 2008). The Rorc gene, which is required for Th17 development, associated with prominent H3K4me3 marks in Th17 cells and H3K27me3 marks in other Th cells (Figure 5B). However, our data also revealed strong H3K4me3 in iTreg cells (Figure 5B), which was consistent with high amounts of Rorc mRNA in the cells (Figure 3). Intriguingly, nTreg cells exhibited a different pattern of modification (Figure 5B) in that both H3K4me3 and H3K27me3 marks are present; again this would allow for co-expression of Rorγt and Foxp3.
Examination of other genes encoding key transcription factors revealed that Tbx21 was associated with strong H3K4me3 but not H3K27me3 in Th1 cells (Figure 6). Similarly, we found strong H3K4me3 but not H3K27me3 marks the Gata3 promoter in Th2 cells (Figure 6). It appears that complete polarization requires two rounds of culture because H3K27me3 was detected at the Gata3 promoter in Th2 cells after only one round of culture (Figure S8). The H3K4me3 mark was consistent with the prominent expression of these transcription factors in the expected lineage (data not shown). Interestingly, Tbx21 and Gata3 were also associated with H3K4me3 in the promoter regions and wide-spread H3K27me3 signals in non-expressing lineages. Colocalization of H3K4me3 and H3K27me3 is an indication of bivalent domains, which serve to poise key developmental genes for lineage-specific activation or repression in pluripotent cells such as embryonic stem cells (Azuara et al., 2006; Bernstein et al., 2006). These data suggest that the Tbx21 and Gata3 genes may similarly be “poised” for expression.
Figure 6. H3K4me3 and H3K27me3 modifications of key transcription factor genes.

(A) H3K4me3 (blue) and H3K27me3 (red) modifications on Tbx21 (Chr11, 96894000-96939000) and Gata3 (Chr2, 9770000-9830000) loci.
(B,C) Freshly isolated nTreg cells were repolarized under neutral condition (ThN) and Th1 conditions for 72 hours. Proportions of Foxp3- and IFN-γ-producing cells (B) and T-bet (C) were determined by intracellular staining and flow cytometry. The data are representative of two independent experiments.
Transcription factors known to regulate CD4 differentiation include zinc finger and BTB domain containing 7B (Zbtb7b, also called Th-POK or c-Krox), thymocyte selection-associated high mobility group box (Tox), runt related transcription factor 1 (Runx1), Runx3 and Gata3 (Bosselut, 2004; He and Kappes, 2006). As shown in Figure S9, high levels of H3K4me3, but not H3K27me3, associated with Zbtb7b promoters in all six lineages (Figure S9A), which correlated with high-level expression of Zbtb7b in all six lineages (Figure S9B). In contrast, epigenetic modification patterns of Tox and Runx1 are more complex (Figure S9A).
Thus, none of the “master regulators” had precisely the lineage-specific epigenetic modifications observed in the signature cytokine genes Ifng and Il17. This raised the possibility that bivalent genes such as Tbx21 and Gata3 could serve as flexible switches that retain a certain degree of plasticity of CD4+ effector T cells. Based on the lack of H3K27me3 marks in the Ifng locus and the bivalent marks associated with the Tbx21 locus in nTreg cells, we sought to determine if IFN-γ could be induced in these cells. To this end, we isolated nTreg from Foxp3-GFP reporter mice. (> 99% pure) and cultured the cells under optimal Th1 conditions. As indicated in Figure 6B, 99.7% freshly isolated nTreg cells expressed FOXP3, but no IFN-γ. In addition, low T-bet expression were detected in freshly isolated nTreg cells (Figure 6C). Surprisingly, after culturing these cells with IL-12 for only 72 hours, we detected expression of IFN-γ and T-bet, even while Foxp3 expression remained (Figures 6B and 6C). These results argue that in non-IFN-γ-producing cells, the expression of the Tbx21 gene can be poised by bivalent modifications, allowing plasticity of the cells with respect to IFN-γ expression.
DISCUSSION
In response to infectious challenge, the CD4+ helper T cells clonally expand and adapt multiple fates, which are preserved in subsequent rounds of divisions (Reiner and Seder, 1999). In this respect, CD4+ T cell subsets are thought to be distinct lineages, as defined by the cytokines they produce (Zhu and Paul, 2008). Early models provided a dualistic view of fate determination with two mutually exclusive outcomes - Th1 and Th2 cells. In this symmetric view, subsets stably differentiate to produce their signature cytokine and extinguish expression of the opposing cytokine. Extensive studies have carefully defined epigenetic modifications of Ifng and Il4-Il13-Il5 loci. These data solely based on cytokine loci support the previous concept of T cell subsets as stable lineages, with little capacity for de-differentiation. However, recent studies have revealed new subsets (Treg and Th17 cells) and increased heterogeneity of CD4+ T cells with more flexibility in their responses. It is tempting to try to fit these new fates in symmetrical models akin to dualistic, Th1-Th2 model. However, it is unclear what aspects of T cell biology conform to a more rigid “fate determination” model versus a malleable view that allows for more adaptive responses.
Previously, it has not been possible to obtain a global view that allows one to begin to define aspects of the phenotype of helper T cells that are firmly fixed and those that provide plasticity. Insofar as epigenetic modifications help to define heritable features, new technology permits large-scale dissection of this problem. Our genome-wide maps of H3K4me3 and H3K27me3 confirm many aspects of previous lineage commitment model, but also provide much unexpected information on alternative models. Specifically, the data provide an explanation for the unanticipated plasticity of Treg cells.
Consistent with previous findings, we generally found good correlation between gene expression and H3K4me3 modification of signature genes. However, we found that the association with H3K27me3 signals is both gene- and cell-specific when the gene is not expressed. The Ifng locus is a good example where repressive marks can help to maintain extinction of expression in lineages that do not secrete this cytokine. H3K27me3 was detected at the Il4 gene in naïve, Th1, and Th17. In contrast, nTreg and cells had little or no repressive marks in the Il4 gene. These data suggest that the Il4 gene may not be subject to permanent repression by epigenetic mechanisms in Treg cells and therefore, may have the potential to be expressed. This is consistent with a recent report that decreased Foxp3 expression in Tregs resulted in preferential Th2 differentiation (Wan and Flavell, 2007).
Although helper T cell subsets are defined by their signature cytokines, the subsets also have characteristic patterns of expression of receptors, chemokines and other products. The expectation might be that these genes would have epigenetic marks congruent with those of the signature cytokine genes. Some genes like the Il18r1 had a strong H3K27me3 mark in Th2 cells as one might expect. Similarly, Il21, which is expressed by Th17 cells, had prominent H3K27me3 marks in Th1 and Th2 cells. However, in many other cases, the situation was more complex. In some cases, repressive marks were lacking. This argues that the extended phenotype of a subset may be subject to more flexibility than one might have assumed.
In contrast to signature cytokine genes, the epigenetic modifications of transcription factors expressed in CD4+ T cells are far from simple and do not conform to the pattern observed for the signature cytokine genes. Although strong H3K27me3 signals were detected in all the Th populations at well-known bona fide master regulators for embryonic stem cells (such as Nanog and Oct4) and other cell fates (such as MyoD and Pax5), the master regulators for the T cell subsets did not simply acquire repressive marks in opposing lineages. Rather our data show that T-bet and Gata3 were marked with both H3K4me3 and H3K27me3 modifications. This pattern contrasts sharply with that of Zbtb7b and Stat family transcription factors, which were typically associated with unopposed H3K4me3 marks in their promoter region. However, not all transcription factors involved in CD4+ T cell specification had unopposed H3K4me3 marks. Despite their general importance for CD4+ T cell development, Tox and Runx1 also acquired H3K27me3 marks with retained H3K4me3 association. The bivalent modification of these transcription factors suggests that they are “poised” for expression and may contribute to CD4+ T cell plasticity when alternative responses are necessary. In this respect, polarized T cells might not be hardwired for certain effector or regulatory functions. T cell subsets may appear to be “terminally differentiated” based on epigenetic marks on cytokine loci. However, the bivalent modification of key transcription factors suggests more dynamic regulation than previously envisioned, which may reverse the suppression of signature cytokines and promote plasticity of helper T cells.
Although Foxp3 has been argued to serve as a master regulator, its epigenetic modification is quite different from that of T-bet and Gata3 and also contrasts with that of Rorc. Previous work has demonstrated DNA demethylation, H3K4me3 modification and H3 acetylation in the Foxp3 promoter in CD25+ nTregs (Floess et al., 2007; Kim and Leonard, 2007). However, we found no evidence of significant H3K27me3 association in the Foxp3 promoter in any of the populations we studied. This is consistent with the notion that Foxp3 may be expressed more widely and transiently than initially conceived (Xu et al., 2007; Yang et al., 2008b; Zhou et al., 2008).
Although the prominent repressive marks in the Ifng and Il4 loci in Th2 and Th1 cells argue for little flexibility, the finding of bivalent marks of the Tbx21 locus associated with the absence of H3K27me3 marks in the Ifng locus in Treg cells suggested that it should be possible to induce IFN-γ production in these cells. In fact, this was the case. This implies that Treg cells may not be as “stable” as previously assumed; as such, this finding has important implications for the therapeutic use of Treg cells. Of note, there was minimal H3K27me3 association with the Il4 locus, associated with bivalent domains in the Gata3 locus in Treg cells. This suggests that it may also be possible to induce IL-4 expression in Treg cells; indeed, as discussed above, there are circumstances where this occurs (Wan and Flavell, 2007). In contrast, the Ifng gene is associated with H3K27me3 in Th2 and Th17 cells. Similarly, Il17 locus is strongly repressed in polarized Th1, Th2, iTreg and nTreg cells. We know that some cells produce both IL-17 and IFN-γ, but we do not know where these cells come from. The fact that the Tbx21 locus is bivalent might suggest that it will be possible to convert an IL-17 producing cell into an IFN-γ producer. Because both the Rorc and Il17 loci have repressive marks in Th1 cells, it may be more difficult to induce IL-17 production in a polarized Th1 cell. In fact, recent data suggests that this is the case. Recent work also suggests that under some circumstances, Foxp3 and IL-17 can be co-expressed (Zhou et al., 2008). The finding that the Foxp3 locus has no repressive marks in Th17 cells is also consistent with this observation.
In summary, the present study not only provides an initial blueprint of the epigenetic modifications that occur during differentiation of naïve T cells into various T helper cell fates but also provides a new view of the complexity of differentiating T cells. What emerges is not a simple picture of terminally differentiated cells but rather a limited cassette of genes that conform to this rigid model. In contrast to models in which CD4+ T cells might have one of two diametrically opposed fates (Th1 vs. Th2, Treg vs Th17), the present data provide a more flexible view, at least partially due to the bivalent master regulators and broad spectrum of epigenetic states of other critical regulators in opposing lineages. Our data suggest an epigenetic mechanism underlying the specificity and plasticity of effector and regulatory T cells and also provide a framework for understanding complexity of CD4+ T helper cell differentiation.
EXPERIMENTAL PROCEDURES
Mice, Isolation of Cells and Cell Culture
C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME). Animals were handled and housed in accordance with the guidelines of the NIH Animal Care and User Committee. Splenic and lymph node CD4+ T cells from C57BL/6J mice were prepared by MACS purification using CD4 microbeads (Miltenyi Biotec) and the purity was usually 90-95%. Naïve T cells were obtained by surface staining with anti-CD4, anti-CD62L, anti-CD44, and anti-CD25. The CD4+CD62L+CD44-CD25- population was isolated by flow cytometry cell sorting with a Mo-Flo cell sorter (Dako, Carpinteria, CA). Natural T regulatory cells were obtained by surface staining with anti-CD4 and anti-CD25, followed by Mo-Flo sorting for CD4+CD25high population. For T helper cell cultures, T-depleted APC were prepared by incubating splenocytes with anti-Thy1.2 and rabbit complement (Cedarlane Laboratories Limited) at 37°C for 45 min, and then irradiated at 30 Gy (3000 rad). CD4+ T cells were first co-cultured with irradiated APC at a 1:5 ratio, in the presence of anti-CD3 (1 μg/ml) and anti-CD28 (3 μg/ml) together with different combinations of antibodies and cytokines for 72 hours: for Th1 conditions, anti-IL-4 (10 μg/ml), IL-12 (10 ng/ml) and IL-2 (100 U/ml); for Th2 conditions, anti-IFN-γ (10 μg/ml) and anti-IL-12 (10 μg/ml), IL-4 (5000 U/ml) and IL-2 (100 U/ml); for Th17 conditions, anti-IL-4 (10μg/ml), anti-IFN-γ (10 μg/ml), anti-IL-12 (10 μg/ml), TGF-β (5 ng/ml), IL-6 (10 ng/ml) and IL-1β (10 ng/ml); for iTreg conditions, anti-IL-4 (10 μg/ml), anti-IFN-γ (10 μg/ml), TGF-β (5 ng/ml) and IL-2 (100 U/ml). After first round culture, all Th cells other than iTreg condition, were further expended in IL-2 (100 U/ml) for 4 days, followed by the second round of 72 hour culture using the same Th culture conditions as the first round.
ChIP and ChIP-Seq
ChIP-Seq experiments were performed as described previously (Barski et al., 2007). Briefly, T cells were treated with MNase to generate approximately 80% mononucleosomes and 20% dinucleosomes. Chromatin from 2 × 107 cells was used for each ChIP experiment, which yielded approximately 100 ng of DNA. Antibodies against histone H3K4me3 (ab8580, Abcam) and H3K27me3 (07-449, Upstate) were used. The ChIP DNA fragments were blunt-ended, ligated to the Solexa adaptors, and sequenced using the Illumina 1G Genome Analyzer.
The sequence reads and expression data have been deposited in the NCBI Short Reads Archive (GSE14254 and GSE14308). Detailed strategies for data analysis can be found in Supplemental Information.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Warren Leonard for critical reading of the manuscript and J. Simone for technical help with cell sorting. The gene expression analysis using the Affymetrix microarrays was performed by the Gene Expression Core Facilities of NHLBI and NIAMS. This research was supported by the Intramural Research Programs of NIAID, NHLBI and NIAMS.
REFERENCES
- Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, Murphy KM. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. 2002;3:549–557. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- Akimzhanov AM, Yang XO, Dong C. Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J Biol Chem. 2007;282:5969–5972. doi: 10.1074/jbc.C600322200. [DOI] [PubMed] [Google Scholar]
- Ansel KM, Djuretic I, Tanasa B, Rao A. Regulation of Th2 differentiation and Il4 locus accessibility. Annu Rev Immunol. 2006;24:607–656. doi: 10.1146/annurev.immunol.23.021704.115821. [DOI] [PubMed] [Google Scholar]
- Azuara V, Perry P, Sauer S, Spivakov M, Jorgensen HF, John RM, Gouti M, Casanova M, Warnes G, Merkenschlager M, Fisher AG. Chromatin signatures of pluripotent cell lines. Nat Cell Biol. 2006;8:532–538. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- Bosselut R. CD4/CD8-lineage differentiation in the thymus: from nuclear effectors to membrane signals. Nat Rev Immunol. 2004;4:529–540. doi: 10.1038/nri1392. [DOI] [PubMed] [Google Scholar]
- Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L, O'Shea JJ. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci U S A. 2006;103:8137–8142. doi: 10.1073/pnas.0600666103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol. 2007;8:145–153. doi: 10.1038/ni1424. [DOI] [PubMed] [Google Scholar]
- Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- Egawa T, Tillman RE, Naoe Y, Taniuchi I, Littman DR. The role of the Runx transcription factors in thymocyte differentiation and in homeostasis of naive T cells. J Exp Med. 2007;204:1945–1957. doi: 10.1084/jem.20070133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM, O'Shea JJ. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood. 2008;111:1013–1020. doi: 10.1182/blood-2007-06-096438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouse SD, Shen Y, Pellegrini M, Cole S, Meissner A, Van Neste L, Jaenisch R, Fan G. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG complex, and histone H3 K4/K27 trimethylation. Cell Stem Cell. 2008;2:160–169. doi: 10.1016/j.stem.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- Harrington LE, Mangan PR, Weaver CT. Expanding the effector CD4 T-cell repertoire: the Th17 lineage. Curr Opin Immunol. 2006;18:349–356. doi: 10.1016/j.coi.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Hatton RD, Harrington LE, Luther RJ, Wakefield T, Janowski KM, Oliver JR, Lallone RL, Murphy KM, Weaver CT. A distal conserved sequence element controls Ifng gene expression by T cells and NK cells. Immunity. 2006;25:717–729. doi: 10.1016/j.immuni.2006.09.007. [DOI] [PubMed] [Google Scholar]
- He X, Kappes DJ. CD4/CD8 lineage commitment: light at the end of the tunnel? Curr Opin Immunol. 2006;18:135–142. doi: 10.1016/j.coi.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Janson PC, Winerdal ME, Marits P, Thorn M, Ohlsson R, Winqvist O. FOXP3 promoter demethylation reveals the committed Treg population in humans. PLoS ONE. 2008;3:e1612. doi: 10.1371/journal.pone.0001612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996a;4:313–319. doi: 10.1016/s1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996b;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HP, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med. 2007;204:1543–1551. doi: 10.1084/jem.20070109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr J, Knoechel B, Abbas AK. Regulatory T cells in the periphery. Immunol Rev. 2006;212:149–162. doi: 10.1111/j.0105-2896.2006.00414.x. [DOI] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyara M, Sakaguchi S. Natural regulatory T cells: mechanisms of suppression. Trends Mol Med. 2007;13:108–116. doi: 10.1016/j.molmed.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- Pan G, Tian S, Nie J, Yang C, Ruotti V, Wei H, Jonsdottir GA, Stewart R, Thomson JA. Whole-genome analysis of histone h3 lysine 4 and lysine 27 methylation in human embryonic stem cells. Cell Stem Cell. 2007;1:299–312. doi: 10.1016/j.stem.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Reiner SL, Sallusto F, Lanzavecchia A. Division of labor with a workforce of one: challenges in specifying effector and memory T cell fate. Science. 2007;317:622–625. doi: 10.1126/science.1143775. [DOI] [PubMed] [Google Scholar]
- Reiner SL, Seder RA. Dealing from the evolutionary pawnshop: how lymphocytes make decisions. Immunity. 1999;11:1–10. doi: 10.1016/s1074-7613(00)80076-x. [DOI] [PubMed] [Google Scholar]
- Roh TY, Cuddapah S, Cui K, Zhao K. The genomic landscape of histone modifications in human T cells. Proc Natl Acad Sci U S A. 2006;103:15782–15787. doi: 10.1073/pnas.0607617103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh TY, Cuddapah S, Zhao K. Active chromatin domains are defined by acetylation islands revealed by genome-wide mapping. Genes Dev. 2005;19:542–552. doi: 10.1101/gad.1272505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Schoenborn JR, Dorschner MO, Sekimata M, Santer DM, Shnyreva M, Fitzpatrick DR, Stamatoyannopoulos JA, Wilson CB. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat Immunol. 2007;8:732–742. doi: 10.1038/ni1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G, Zhao K. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132:887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. 2003;21:713–758. doi: 10.1146/annurev.immunol.21.120601.140942. [DOI] [PubMed] [Google Scholar]
- Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, Nakanishi K, Yoshida N, Kishimoto T, Akira S. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, Sangster MY, Vignali DA, Doherty PC, Grosveld GC, Ihle JN. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. 1996;382:171–174. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- Usui T, Preiss JC, Kanno Y, Yao ZJ, Bream JH, O'Shea JJ, Strober W. T-bet regulates Th1 responses through essential effects on GATA-3 function rather than on IFNG gene acetylation and transcription. J Exp Med. 2006;203:755–766. doi: 10.1084/jem.20052165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan YY, Flavell RA. Regulatory T-cell functions are subverted and converted owing to attenuated Foxp3 expression. Nature. 2007;445:766–770. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- Wei L, Laurence A, Elias KM, O'Shea JJ. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem. 2007;282:34605–34610. doi: 10.1074/jbc.M705100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725–6729. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, Kuchroo VK, Hafler DA. IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature. 2008a doi: 10.1038/nature07021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008b;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XD, Han X, Chew JL, Liu J, Chiu KP, Choo A, Orlov YL, Sung WK, Shahab A, Kuznetsov VA, et al. Whole-genome mapping of histone h3 lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell Stem Cell. 2007;1:286–298. doi: 10.1016/j.stem.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–596. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol. 2007;8:457–462. doi: 10.1038/ni1455. [DOI] [PubMed] [Google Scholar]
- Zhou L, Ivanov, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- Zhou L, Lopes JE, Chong MM, Ivanov, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, et al. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–1569. doi: 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.