

Abstract
Cystatin E/M (CST6) is a natural inhibitor of lysosomal cysteine proteases. Recent studies have shown that experimental manipulation of CST6 expression alters the metastatic behavior of human breast cancer cells. However, the association of CST6 with prostate cancer invasion and progression is remains unclear. Here, we show that CST6 is robustly expressed in normal human prostate epithelium while its expression is downregulated in metastatic prostate cell lines and prostate tumor tissues. Treatment of metastatic prostate cell lines with the histone deacetylase inhibitor trichostatin A resulted in significant induction of CST6 mRNA levels and increased CST6 protein expression, indicating that epigenetic silencing may play a role in the loss of CST6 expression observed in prostate cancer. CST6 overexpression in human prostate cancer cells significantly reduced in vitro cell proliferation and matrigel invasion. Furthermore, the results from a bioluminescence tumor/metastasis model showed that the overexpression of CST6 significantly inhibits tumor growth and the incidence of lung metastasis. These results suggest that the downregulation of the CST6 gene is associated with promoter histone modifications and that this association plays an important role in prostate cancer progression during the invasive and metastatic stages of the disease.
Keywords: cystatins, cathepsins, invasion, histone modification, chromatin, prostate cancer
INTRODUCTION
The dissemination of localized prostate cancer to distant tissues, including the bone, lung, and liver, represents a prominent health care burden in the aging adult male population (Shah et al., 2004). The underlying mechanisms that promote and support the metastatic spread of prostate cancer remain vague. It is clear that fundamental processes, such as cellular detachment, proliferation, migration, invasion, and angiogenesis, are essential to the spread of cancer cells and their growth at distant sites (Bogenrieder and Herlyn, 2003). One family of proteins that enables malignant progression is the family of lysosomal cysteine proteases that degrade components of the extracellular matrix (Turk et al., 2000). The impaired regulation of expression and activity of lysosomal cysteine proteases has been implicated in cancer progression (Keppler and Sloane, 1996; Roshy et al., 2003). Several groups have hypothesized that the increased proteolytic activities observed in malignant tumors are due to upregulation of proteases and/or downregulation of their endogenous inhibitors (Brunner et al., 1994; DeClerck and Imren, 1994; Konduri et al., 2002; Pulukuri et al., 2007b). Cystatins comprise a family of naturally occurring lysosomal cysteine protease inhibitors and protect cells against uncontrolled proteolysis (Abrahamson, 1994; Turk and Bode, 1991). They control the catalytic function of target proteases by forming reversible, high-affinity complexes (Alvarez-Fernandez et al., 1999).
CST6 is a newly identified member of the human cystatin gene family and demonstrates more diverse tissue distribution and target specificity than other cystatins (Sotiropoulou et al., 1997). CST6 was first identified as a RNA transcript that is downregulated in metastatic breast cancer cells but not in primary breast cancer cells (Ni et al., 1997; Sotiropoulou et al., 1997). CST6 has been shown to inhibit cathepsins B and L, which are the most important cysteine proteases implicated tumor cell invasion and metastasis (Krueger et al., 2001; Yanamandra et al., 2004). It has also been reported that constitutive expression of CST6 in human breast cancer cells (MDA MB-435S) significantly reduced in vitro cell proliferation, migration and matrigel invasion (Shridhar et al., 2004). Furthermore, the ectopic expression of CST6 in scid mice strongly delayed breast tumor growth and lowered the rate of metastases to the lung and liver (Zhang et al., 2004). Taken together, these characteristics strongly suggest an anti-tumor function of the CST6 gene in cancer progression.
Epigenetic inactivation of tumor suppressor genes (TSGs) or tumor-related genes via histone modification and DNA methylation is a common event in human cancers and tumor cell lines. Several TSGs are inactivated through these mechanisms (Esteller et al., 2001). Previous studies on CST6 suggest that DNA methylation is important for the regulation of CST6 expression in breast cancer cells and primary breast tumor tissues (Ai et al., 2006). Robertson et al. (Kim et al., 2006) demonstrated the downregulation of CST6 expression in malignant glioma by epigenetic mechanisms. A recent study uncovered that CST6 expression increased in cultured lung cancer cells after treatment with the DNA methylation inhibitor 5-aza and the histone deacetylase inhibitor TSA (Zhong et al., 2007). However, the functional relevance of histone modification and DNA methylation in the regulation of the CST6 gene expression in prostate cancer is unknown.
Because CST6 is downregulated via epigenetic mechanisms in a number of cancers where it regulates proliferation, invasion and metastasis, we asked whether it could play a causative role in prostate tumor development. We found that CST6 is downregulated in prostate cancer tissues and that CST6 was induced in metastatic prostate cell lines by HDAC inhibitors. Furthermore, we show that restoration of CST6 expression in metastatic prostate cancer cells significantly inhibits prostate tumor growth and the incidence of lung metastasis in vivo. Our results have implications for the molecular diagnosis and treatment of prostate cancer metastases.
RESULTS
CST6 expression in human prostate cancer
Previous studies have shown that CST6 expression is lost in both cultured human breast cancer cell lines and primary breast malignancies (Ai et al., 2006). However, to date, CST6 expression has not been investigated in prostate cancer. To clarify the clinical significance of the CST6 gene in prostate cancer, we evaluated the expression level of CST6 protein in human prostate cancer tissues using immunohistochemistry (IHC). CST6 expression was classified as negative, weak positive and strong positive in 17 (56.1%), 21 (38.2%) and 4 (5.7%) tumor tissue samples, respectively (Figure 1A). There was a significantly lower level of CST6 protein expression in the tumors than in the normal tissue samples (P <0.01, Figure 1A) and the representative pictures were presented Figure 1B. We also detected strong immunostaining in the majority of PIN lesions, which represent precursors of prostate cancer (data not shown). To further confirm these observations, we performed immunoblot analysis of four-paired human normal prostate and tumor tissue samples with known levels of CST6 (Figure 1C). It was clear that the tumor tissue samples exhibited a downregulation of CST6 protein expression as compared with the normal prostate tissue, which was consistent with the level of CST6 protein expression determined by immunohistochemical staining. We also examined mRNA expression of twenty-paired human normal prostate and tumor tissue samples using real-time PCR analysis (Figure 1D). Consistent with the results of the IHC and immunoblotting, CST6 mRNA was significantly downregulated in the majority of tumor samples when compared to their normal counterparts (Figure 1D). Since a decrease in the amount of CST6 relative to cathepsin B could function to favor tumor progression, we were also interested in analyzing cathepsin B protein expression in human normal prostate and tumor tissue samples. Although either no or weak cathepsin B immunoreactivity was detected in normal prostate tissues, strong cathepsin B protein staining was observed in the majority of prostate tumors (P <0.01, Supplemental Figure S1).
Figure 1. Expression of CST6 in human prostate tissue samples.

(A) Compared with normal prostate tissue, the overall expression level of CST6 in the prostate cancer tissue was significantly lower (P < 0.001). Tissue sections were prepared from formalin-fixed, paraffin-embedded specimens of normal and tumor human prostate tissues. Immunostaining was carried out using a specific anti-human CST6 antibody. CST6 expression in normal human prostate tissue (32 cases) and prostate cancer (42 cases) was analyzed.
(B) Representative immunostaining photographs were taken at different magnifications: a, normal human prostate showing CST6 in epithelial cells; b, high-power view of (a) showing membrane staining of CST6; c, lack of CST6 staining in prostate cancer tissues; d, high-power view of (c) showing lack of CST6 expression in prostate cancer tissues (p < 0.001); T, tumor cells; N, normal prostate epithelial cells.
(C) Total cell extracts were further prepared from four-paired normal prostate (N) and prostate tumor tissue (T) specimens. The 14-kDa unglycosylated and 17-kDa glycosylated forms of CST6 protein expression was determined by immunoblot analysis. MDA-MB-231 cells (C) that express CST6 were used as a positive control for the specificity of the antibody. The level of CST6 protein expression was significantly lower in tumor tissue than in normal tissue, which was indicated by the ratio of CST6/GAPDH (bottom).
(D) Quantitative mRNA expression of CST6 in human prostate tumor (green) compared to normal adjacent tissue of the same individual (red, connected by a line). RNA was isolated and reverse transcribed and SYBR Green real-time PCR carried out with CST6-specific primers. Significant downregulation of CST6 expression is observed in tumor tissues as compared to respective normal adjacent tissues.
CST6 expression in prostate cancer cell lines
As observed in human prostate cancer specimens, a similar lack of CST6 expression was observed when screening a panel of human prostate cancer cell lines. Three of four prostate cancer cell lines (LNCaP, PC3, and PC3-M) had markedly decreased or absent CST6 expression at the mRNA level, whereas decreased CST6 expression was observed in the DU145 prostate cancer cell line (Figure 2A, top). In contrast, high CST6 expression was observed in the RWPE1 cell line, which derived from normal prostate epithelium. This pattern of expression was also seen at the protein level (Figure 2A, bottom). Moreover, the prostate cancer cell lines had more consistent expression of cystatin C at both the mRNA (Figure 2A, top) and protein (Figure 2A, bottom) levels.
Figure 2. Expression of CST6 in human prostate epithelial and cancer cells, and effect of trichostatin A (TSA).

(A) RT-PCR (top) and immunoblot (bottom) analysis of CST6 in prostate cancer cell lines. Cystatin C was used as loading control for RNA and protein analysis.
(B) CST6 mRNA levels in control and TSA-treated LNCaP, PC3 and PC3-M cells were analyzed by semiquantitative RT-PCR (top). CST6 mRNA levels (223-bp amplicon) in control and 5-aza-treated LNCaP, PC3 and PC3-M cells were analyzed by semiquantitative RT-PCR (bottom). Cystatin C mRNA was amplified as a loading control and expression standard.
(C) Real-time RT-PCR analysis of CST6 mRNA expression in LNCaP, PC3 and PC3-M cells treated with TSA or 5-aza. RNA was isolated and reverse transcribed and SYBR Green real-time PCR carried out with CST6-specific primers. Columns, mean of three independent experiments; bars, SD.
(D)Immunoblot analysis of CST6 protein expression in LNCaP, PC3 and PC3-M cells with or without TSA treatment. The CST6 protein expression was determined by immunoblot analysis. Cystatin C was used as a loading control to check for equal loading of the gel.
Histone deacetylase inhibitors induce CST6 expression in human prostate cancer cell lines
Loss of gene expression during cancer progression can occur through a number of mechanisms, including mutation, loss of heterozygosity, and epigenetic silencing. Epigenetic transcriptional downregulation can occur through histone deacetylation and hypermethylation of the gene promoter, resulting in tightly packed chromatin and decreased transcription factor access. CST6 protein expression in prostate cancer cell lines correlated with CST6 mRNA levels (Figure 2A), suggesting regulation of CST6 expression at the transcriptional level. As it is known that CST6 can be silenced by epigenetic alterations (Ai et al., 2006; Kim et al., 2006; Zhong et al., 2007; Qiu et al., 2008; Schagdarsurengin et al., 2007; Veena et al., 2008), we examined the effects of the histone deacetylase inhibitor trichostatin A (TSA) and DNA methylation inhibitor 5-aza-2′-deoxycytidine (5-aza) on the re-expression of CST6 in prostate cancer cell lines by both semiquantitative reverse transcription (RT)-PCR and quantitative real-time RT-PCR (qRT-PCR). In all three prostate cancer cell lines with no CST6 expression (LNCaP, PC3 and PC3-M; Figure 2A), treatment with TSA for 20 h resulted in a robust induction of CST6 mRNA levels (Figure 2B, top). The induction of CST6 mRNA by TSA was dose-dependent in all the examined cell lines (Figure 2C). We then investigated whether the resulting increases in mRNA level were sufficient to increase CST6 protein levels. As expected, a corresponding increase in CST6 protein levels was also observed after treatment of LNCaP, PC3 and PC3-M cells with TSA (Figure 2D). SB, a HDAC inhibitor differing from TSA in structure, showed similar results in all the examined cell lines (Supplemental Figure S2). These results indicate that the effect of TSA on CST6 expression can be extended to other HDAC inhibitor SB. They also suggest that the induction of CST6 expression by HDAC inhibitors was not confined to a single type of human prostate cancer cell line model.
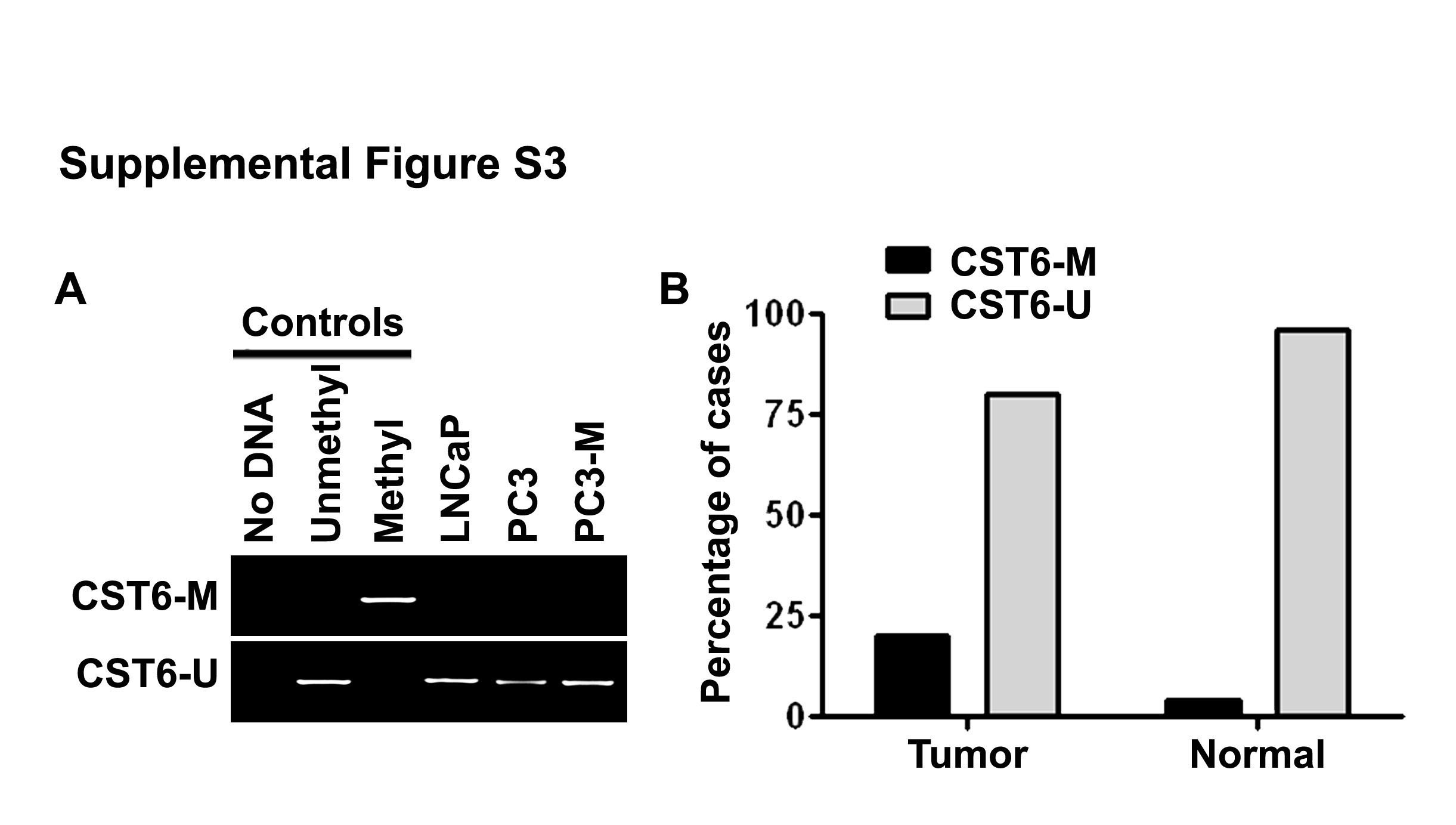
Because it is know that CST6 can be silenced by promoter DNA methylation (Ai et al., 2006; Kim et al., 2006; Schagdarsurengin et al., 2007), we examined the effects of the DNA methylation inhibitor 5-aza on the re-activation of CST6 in prostate cancer cell lines by both RT-PCR and qRT-PCR. However, treatment with higher doses (1–10 μM) of 5-aza for 5 days did not restore the expression of CST6 in all cell lines analyzed (Figure 2B, bottom and Figure 2C). The absence of re-expression was associated with the appearance of unmethylated CST6 promoter in LNCaP, PC3 and PC3-M cells by MSP (Supplemental Figure S3A). To determine whether CST6 inactivation by promoter methylation is characteristic of human prostate tumors, we performed MSP analysis with primer sets targeting the sequence around the most frequently methylated sites in a panel of 30 human prostate tumors and 30 normal prostate tissues. Only unmethylated and no methylated DNA was detected in 24 of 30 human prostate tumors (Supplementary Figure S3B). These results show that methylation levels are significantly lower in prostate tumor samples. The low frequency of methylation in primary tumors and induction of CST6 expression in all cell lines only with TSA but not demethylating agent 5-aza strongly suggests that promoter histone modifications play an important role in prostate cancer progression.
TSA induces accumulation of acetylated histones in chromatin-associated with the CST6 gene
A number of studies have previously shown that TSA induces the accumulation of acetylated histones in human cancer cells (Marks et al., 2001a; Marks et al., 2001b; Richon et al., 2000; Sambucetti et al., 1999). We first verified that inhibition of histone deacetylation by the HDAC inhibitor TSA altered global histone acetylation in the human prostate cancer cell lines LNCaP, PC3 and PC3-M. Immunoblot analysis using antibodies to acetylated histones H3 and H4 demonstrated that acetylation at histones H3 and H4 was substantially increased by TSA (Figure 3A and B). Next, we performed the ChIP assay to examine the effect of HDAC inhibitors on the pattern of acetylation of histones associated with the CST6 gene. Chromatin fragments from LNCaP, PC3 and PC3-M prostate cancer cells cultured with or without TSA for 20 h were immunoprecipitated with antibodies to acetylated histones H3 or H4. DNA from the immunoprecipitates was isolated, and PCR was performed using CST6 promoter primers (Figure 3C). Acetylation of histones H3 and H4 associated with the CST6 promoter region in all three human prostate cancer cell lines was undetectable before TSA treatment. However, we observed remarkable increases in acetylation of histones H3 and H4 in the promoter region of all three prostate cancer cell lines after treatment with TSA (Figure 3D). The accumulation of acetylated histones H3 and H4 confirmed that histone deacetylation was involved in the transcriptional repression of CST6. We also carried out PCR on the same set of immunoprecipitated DNA fractions for β-actin promoter as a control. The relative levels of acetylated histones H3 and H4 at the β-actin promoter was similar in all TSA-treated and untreated cells (Figure 3D).
Figure 3. TSA induces accumulation of acetylated histones H3 and H4 in chromatin associated with the CST6 gene.

(A) Nuclear extracts were isolated from control and TSA-treated LNCaP, PC3 and PC3-M cells, and immunoblot analysis was performed using anti-acetyl histone H3, anti-acetyl histone H4, and histone H3 antibodies. Histone H3 was utilized as a loading control.
(B) Densitometric analysis of immunoblots in (A) from control and TSA-treated cell lines. Data are normalized to H3, averaged, and expressed as percentage of control (CTL=1).
(C) Schematic representation of the CST6 promoter region and the location of primers used for PCR amplification in the ChIP assay. Bent arrow, transcriptional start site (+).
(D) Chromatin fragments from LNCaP, PC3 and PC3-M cells cultured with (+) or without (−) TSA for 20 h were immunoprecipitated with antibody to acetylated (Ac) histones H3 and H4 or control normal rabbit serum (NRS). PCR primers for the CST6 and β-actin promoters were used to amplify the DNA isolated from the immunoprecipitated chromatin as described in Materials and Methods. Note the CST6 promoter specific primers amplified PCR fragment of 157-bp.
CST6 decreases proliferation and invasive potential of the prostate cancer cells
To investigate the functional consequences of CST6 in prostate cancer, we analyzed the effects of restoring CST6 expression in PC3 cells. A full-length CST6-expressing vector was constructed and transfected to the highly invasive, CST6-negative cell line PC3. We observed a high expression level of CST6 in each of the four CST6-transfected cells (CST6-4, CST6-12, CST6-13, CST6-18) but not in parental (mock) or empty vector-transfected cells as detected by RT-PCR (Figure 4A, top) and immunoblotting (Figure 4A, bottom). The clone CST6-13 with the highest CST6 expression at the mRNA and protein levels was selected for further experiments. We used the MTT assay to assess the effect of restoring CST6 expression on PC3 prostate cancer cell proliferation. Restoring CST6 expression in PC3 cells resulted in a 58.5% inhibition of proliferation as compared to the empty vector-transfected PC3 cells (Figure 4B). Differences in invasion between CST6-expressing cells and control PC3 cells were evaluated using the matrigel invasion assay. We found that PC3 mock cells or empty vector-transfected PC3 cells significantly invaded through the matrigel; however, CST6-overexpressing cells showed a substantial reduction in invasive capacity (Figure 4C). Representative pictures are shown in Figure 4D. Furthermore, to extend these findings to an additional prostate cancer cell line, LNCaP cells were stably transfected with the CST6 expression vector and tested for proliferation and invasion. Similar to the results described above, LNCaP cells stably expressing CST6 exhibited decreased proliferation and invasion when compared with LNCaP cells that did not express CST6 (data not shown).
Figure 4. Overexpression of CST6 reduces proliferation and invasion of PC3 prostate cancer cells.

(A) RT-PCR (top) and immunoblot (bottom) analyses of PC3 cells stably transfected with mock, CST6 expression vector or empty vector. CST6 expression at both the mRNA and protein levels was detected only in PC3 cells stably expressing CST6 clones (CST6-4, CST6-12, CST6-13, CST6-18), but not from empty vector clone or mock. Cystatin C was used as loading control for RNA and protein analysis.
(B) Proliferation of PC3 cells stably transfected with either CST6 expression vector or controls (mock or empty vector-transfected cells) was revealed by MTT assay. Each bar represents triplicate analyses of mean ± S.D. where the significant difference from controls is represented by an asterisk (*, p < 0.05).
(C) Comparison of the in vitro invasive potentials of PC3 cells stably transfected with mock, CST6 expression vector, or empty vector. A representative number of invading cells through the matrigel were counted under the microscope in five random fields at a 200× magnification. Each bar represents the mean ±S.D. of five fields counted. Significant difference from controls (mock or siControl-transfected cells) is indicated by an asterisk (*, p < 0.05).
(D) Representative invasion photographs from PC3 cells stably transfected with mock, CST6 expression vector, or empty vector as described in (C).
(E) mRNA (left) and protein (right) levels of cathepsins in PC3 cells stably transfected with mock, CST6 expression vector or empty vector. The number under each band is expressed as a percentage of mock control, normalized by the corresponding cystatin C level. Cystatin C was used as loading control for RNA and protein analysis.
Previous studies by our group (Konduri et al., 2002; Yanamandra et al., 2004) and others (Krueger et al., 2001; Shridhar et al., 2004; Sotiropoulou et al., 1997) have established that cystatin is downregulated with increasing malignancy and that this downregulation is associated with an imbalance between the production of cathepsins and cystatins and a shift towards a pro-proteolytic state with an invasive phenotype. To determine whether CST6 was mediating their effects through the regulation of cathepsin levels, we assessed the levels of cathepsins at the mRNA level using RT-PCR and at the protein level using immunoblot analysis. Transfection with CST6 suppressed the expression of cathepsin B at both the mRNA and protein levels in PC3 cells (Figure 4E). However, the overexpression of CST6 did not change the mRNA or protein expression levels of other lysosomal proteases, such as cathepsins D, L or H (Figure 4E). We also confirmed these results by real-time qRT-PCR (Supplemental Figure S4A).
Downregulation of CST6 upregulates cathepsin B and induces proliferation/invasion
To further confirm that cathepsin B is upregulated upon CST6 loss and induces proliferation and invasion, an endogenously high CST6 expressing epithelial prostate cell line, RWPE1, was transfected with a siRNA directed against CST6 (siCST6). Although cystatin C and several other cathepsins are expressed in RWPE1 cells, only cathepsin B mRNA expression was significantly upregulated upon inhibition of CST6 (Figure 5A, left). Immunoblot analysis confirmed that the CST6-specific siRNA downregulated CST6 expression and corroborated the mRNA data that CST6 inhibition induces cathepsin B expression (Figure 5A, right). We also further confirmed these results by real-time qRT-PCR (Supplemental Figure S4B). The scrambled siRNA (siCTL) did not have an effect on CST6 or cathepsin B expression. CST6 knockdown by siCST6 was specific as it did not affect cystatin C expression. Importantly, we have found that CST6 knockdown resulted in a significant increase in the proliferation when compared with the siCTL-transfected RWPE1 cells (Figure 5B). Finally, the effect of CST6 knockdown on invasive ability of RWPE1 cells was determined using the Matrigel invasion assay. As shown in Figure 5(C and D), knockdown of CST6 significantly induced the invasive potential of RWPE1 cells. Taken together, these results suggest that cathepsin B plays a key role in mediating both proliferation and invasion when CST6 is downregulated.
Figure 5. Downregulation of CST6 induces proliferation and invasion via upregulation of cathepsin B.

(A) mRNA (left) and protein (right) levels of cathepsins in RWPE1 cells transfected with mock, scramble control siRNA (siCTL) or CST6 siRNA (siCST6). Presence of the 223-bp CST6 mRNA amplicon and two forms of CST6 protein is evident. The number under each band is expressed as a percentage of mock control, normalized by the corresponding cystatin C level. Cystatin C was used as loading control for RNA and protein analysis.
(B) Proliferation of RWPE1 cells transfected with mock, siCTL or siCST6 was revealed by MTT assay. Each bar represents triplicate analyses of mean ± S.D. where the significant difference from controls is represented by an asterisk (*, p < 0.05).
(C) Comparison of the in vitro invasive potentials of RWPE1 cells transfected with mock, siCTL or siCST6. A representative number of invading cells through the matrigel were counted under the microscope in five random fields at a 200× magnification. Each bar represents the mean ±S.D. of five fields counted. Significant difference from controls (mock or siControl−transfected cells) is indicated by an asterisk (*, p < 0.05).
(D) Representative invasion photographs from RWPE1 cells transfected with mock, siCTL or siCST6 as described in (C).
CST6 inhibits tumor growth and metastasis in nude mice
We tested whether the reduction in proliferation and invasiveness conferred by CST6 in PC3 cells under in vitro conditions could be extended to an in vivo tumor model system. To produce orthotopic tumors in nude mice, PC3 cells stably expressing either empty vector (control) or CST6-expressing vector with luciferase reporter were injected into the prostate of immunodeficient mice. Tumor progression was monitored in mice using the Xenogen in vivo imaging system. We obtained photon counts from the tumor region on days 10, 20 and 40 (Figure 6A, left). The mice injected with PC3 cells stably transfected with CST6 developed smaller prostate tumors by day 40 when compared with the mice inoculated with cells transfected with the empty vector (Supplemental Figure S5A). The presence of significant growth differences in prostate tumors between control and mice injected with CST6-overexpressing cells were confirmed by autopsy after imaging (Figure 6A, right and 6B). To determine the effect of CST6 on tumor metastasis, we dissected the lungs from each mouse and photon counts were recorded. The lungs derived from control mice injected with empty vector-transfected cells had high photon counts. In contrast, we observed a marked reduction (~ 3.9-fold) in the incidence of lung metastasis when primary tumor cells stably transfected with CST6 were seeded in host mice (Figure 6C). Representative pictures were shown in Supplemental Figure S5 (B). RT-PCR analysis confirmed that CST6-expressing prostate tumors had significantly decreased cathepsin B mRNA levels than control groups (Figure 6D). These results clearly show that CST6 expression downregulates cathepsin B and significantly inhibit both primary tumor growth and the in vivo incidence of lung metastasis.
Figure 6. CST6 overexpression inhibits prostate tumor growth and metastasis in nude mice.

(A) PC3 cells stably expressing luciferase reporter with either empty vector (control) or CST6 expression vector were injected into mouse prostate, and luciferase activity was recorded for each mouse. Photon counts of orthotopic prostate tumors on days 10, 20, and 40 (left). Comparison of dissected prostate tumors in (A) from mice 40 days after cell implantation. Each bar represents the mean tumor weight ± S.D. of six animals per group. Significant difference from control group is indicated by an asterisk (*, p < 0.05) (right).
(B) Prostate tumors in (A), from mice 40 days after cell implantation, were excised and photographed.
(C) Quantification of luciferase activities from either each control mouse (left) or each CST6-expressing mouse (right).
(D) RNA samples extracted from prostate tumors (6 animals/group) were analyzed using RT-PCR for CST6 and cathepsin B expression levels. GAPDH mRNA was amplified as a loading control and expression standard.
DISCUSSION
As the role of CST6 in prostate cancer progression is not clearly elucidated and the mechanisms regulating CST6 expression are not understood, we examined the functional relevance of CST6 expression and tumor invasiveness and investigated histone modification and DNA methylation in the regulation of CST6 gene expression. Our study using clinical prostate samples shows that the majority of the prostate cancer tissues (n=42) have weak or no expression of CST6 when compared with BPH or normal prostate tissues (Figure 1). Additionally, we have shown that expression of CST6 is downregulated in the prostate cancer cell lines (Figure 2A). These findings provide further insight into earlier observations that CST6 was frequently downregulated during the progression of human breast, lung and brain cancers (Ai et al., 2006; Kim et al., 2006; Zhong et al., 2007).
In addition to DNA methylation, another epigenetic mechanism that frequently controls the transcriptional regulation of genes is the acetylation/deacetylation of chromosomal histones associated with target genes (Cameron et al., 1999). In the present study, we have provided evidence for the first time that HDAC inhibitors can reactivate CST6 expression in the human prostate cancer cell lines LNCaP, PC3 and PC3-M (Figure 2). CST6 gene silencing was found to correlate with hypoacetylation of histones H3 and H4 at their promoters in all cell lines (Figure 3). A previous report describes that DNA methylation of the promoter is responsible for the transcriptional silencing of CST6 in breast cancer cell lines (Ai et al., 2006). In that report, the silenced CST6 gene was reactivated by treatment with 5 μm of 5-aza for 2 days. Another report showed that both 5-aza and TSA induced CST6 gene expression in all tested lung cancer cell lines whereas treatment the of lung cancer cell line NCI-H358 with the HDAC inhibitor TSA failed to reactivate CST6 gene expression (Zhong et al., 2007). In comparison, we found that the epigenetic mechanisms for silencing CST6 are somewhat different in prostate cancer cells. In the prostate cancer cell lines, inhibition of HDAC by TSA was sufficient to restore the acetylation of histone and re-expression of CST6. Treatment with 5-aza did not restore the expression of CST6, which is consistent with the finding that the CST6 promoter is unmethylated in these cells. These data indicate that both histone modifications and DNA methylation can act independently or cooperatively to silence the CST6 in different types of cancers. The reason for different mechanisms being involved in the silencing of CST6 in cancer development is currently unknown and may rely on the tissue and/or organ specificity of certain chromatin modifying enzymes or protein effectors that read the epigenetic modifications (Hake et al., 2004).
One of the key steps in the process of cancer invasion and metastasis is the degradation of the extracellular matrix. Several lysosomal cysteine proteases are involved in the degradation of extracellular matrix components; as such, these proteases have long been considered of potential importance during tumor cell invasion and metastasis (Keppler and Sloane, 1996; Roshy et al., 2003; Turk et al., 2000). CST6 belongs to a family of naturally occurring inhibitors of lysosomal cysteine proteases (Abrahamson, 1994; Alvarez-Fernandez et al., 1999; Turk and Bode, 1991). In this study, we show that CST6 specifically regulates the proliferation and invasive behavior of prostate cancer cells. The level of expression of CST6 dictates the ability of prostate cancer cells to proliferate and invade. In highly metastatic PC3 cells, overexpression of CST6 reduced their proliferation and invasive ability whereas downregulation of endogenous CST6 enhanced those abilities (Figure 4 and 5). Furthermore, we show that overexpression of CST6 is sufficient to inhibit growth and invasion of prostate cancer cells in an orthotopic tumor model (Figure 6). Another interesting finding of our study was that overexpression of CST6 significantly reduces the cathepsin B protein and mRNA levels in prostate cancer cells. These results provide support for CST6 functioning as a cathepsin B inhibitor to regulate prostate cancer proliferation and invasion.
In summary, the present study shows that inactivation of CST6 by histone deacetylation is emerging as an important mechanism for promoting prostate cancer cell proliferation and invasion. Moreover, we demonstrate the successful reduction of metastasis by inhibition of cathepsin B by CST6 overexpression in the host microenvironment. These findings provide new and important information on the progression of prostate cancer. These results may yield new strategies for the diagnosis, prevention and treatment of prostate cancer.
MATERIALS & METHODS
Human prostate tissues and immunohistochemistry
Paired human prostate tumor and normal adjacent tissues were obtained from patients undergoing routine therapeutic surgery. Many prostate cancer, PIN, BPH and normal tissues were also obtained as paraffin-embedded, formalin-fixed blocks. For immunohistochemistry, tissue sections were labeled with antibodies against CST6 and cathepsin B (Oncogene, San Diego, CA) and the expression levels graded as negative, weak positive, and strong positive (see Supplementary Information section).
Cell lines and drug treatments
The prostate cancer cell lines RWPE1, LNCaP, DU145, PC3 and MDA-MB-231 were obtained from the American Type Culture Collection and cultured as directed. PC3-M was obtained from the Xenogen Corporation. Total RNA and genomic DNA were isolated from the treated cells using RNA and DNA isolation kits. Cells were treated with TSA, sodium butyrate (SB) and 5-aza as described previously (Pulukuri et al., 2007a) (see Supplementary Information section).
RNA interference and CST6 expression plasmids
Scramble siRNA control and specific siRNA pool for CST6 were synthesized and annealed. Transfection of siRNA pool and control pool in six-well plates was carried out using Lipofectamine 2000 (Invitrogen) as per the manufacturer’s instructions. The siRNA sequences that we used are listed in Supplementary Table S1. The CST6 expression plasmid (pCMV-neo/CST6) was prepared by inserting full-length human CST6 cDNA into a pCMV-neo vector (OriGene, Rockville, MD). The CST6 expression vector and empty vector-transfected cells were selected by growth in the presence of G418 (500 μg/mL) and then analyzed for CST6 expression using RT-PCR and immunoblotting analyses.
Immunoblot, ChIP, RT-PCR and MSP analyses
Immunoblotting, ChIP and RT-PCR analyses were performed as described previously (Pulukuri et al., 2007a). Expression analysis for CST6 mRNA was measured using real-time quantitative PCR with SYBR Green PCR Mastermix (Bio-Rad, Hercules, CA). MSP experiments were performed as described previously (Ai et al., 2006) (see Supplementary Information section).
Proliferation and matrigel invasion assays
Proliferation of PC3 and RWPE1 cells was assessed with the MTT method using a cell proliferation kit (Chemicon, Temecula, CA). Invasion of cells through matrigel was conducted using a Transwell apparatus (Corning Costar) as described previously (Yoon et al., 2001) (see Supplementary Information section).
Orthotopic mouse prostate tumor/metastasis model
Orthotopic implantation was carried out as previously described (Pulukuri and Rao, 2007). PC3 cells stably expressing either empty vector or CST6 expression vector with luciferase reporter were injected into mouse prostate (106 cells per mouse). Prostate tumor growth and subsequent metastasis to lungs were assessed weekly using the in vivo imaging system coupled to Living Image acquisition and analysis software (Xenogen, Alameda, CA) according to the manufacturer’s instructions.
Densitometry
ImageJ software (National Institutes of Health) was used to quantify the mRNA and protein band intensities. Data are represented as relative to the intensity of the indicated loading control.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
This research was supported by National Cancer Institute Grant CA75557, CA92393, CA95058, CA116708, CA138409, N.I.N.D.S. NS47699, NS57529, and NS61835 and Caterpillar, Inc., OSF St. Francis, Inc. Peoria, IL (to J.S.R.). The contents are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
The authors are grateful to Dr. Hnilica of the Department of Pathology at the University of Illinois College of Medicine (Peoria) for kindly providing normal and tumor human prostate tissues. We thank Shellee Abraham for preparing the manuscript and Diana Meister and Sushma Jasti for manuscript review. We also thank Noorjehan Ali and Lavanya Talluri, for technical assistance.
Reference List
- Abrahamson M. Cystatins. Methods Enzymol. 1994;244:685–700. doi: 10.1016/0076-6879(94)44051-4. [DOI] [PubMed] [Google Scholar]
- Ai L, Kim WJ, Kim TY, Fields CR, Massoll NA, Robertson KD, et al. Epigenetic silencing of the tumor suppressor cystatin M occurs during breast cancer progression. Cancer Res. 2006;66:7899–7909. doi: 10.1158/0008-5472.CAN-06-0576. [DOI] [PubMed] [Google Scholar]
- Alvarez-Fernandez M, Barrett AJ, Gerhartz B, Dando PM, Ni J, Abrahamson M. Inhibition of mammalian legumain by some cystatins is due to a novel second reactive site. J Biol Chem. 1999;274:19195–19203. doi: 10.1074/jbc.274.27.19195. [DOI] [PubMed] [Google Scholar]
- Bogenrieder T, Herlyn M. Axis of evil: molecular mechanisms of cancer metastasis. Oncogene. 2003;22:6524–6536. doi: 10.1038/sj.onc.1206757. [DOI] [PubMed] [Google Scholar]
- Brunner N, Pyke C, Hansen CH, Romer J, Grondahl-Hansen J, Dano K. Urokinase plasminogen activator (uPA) and its type 1 inhibitor (PAI-1): regulators of proteolysis during cancer invasion and prognostic parameters in breast cancer. Cancer Treat Res. 1994;71:299–309. doi: 10.1007/978-1-4615-2592-9_16. [DOI] [PubMed] [Google Scholar]
- Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet. 1999;21:103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- DeClerck YA, Imren S. Protease inhibitors: role and potential therapeutic use in human cancer. Eur J Cancer. 1994;30A:2170–2180. doi: 10.1016/0959-8049(94)00460-m. [DOI] [PubMed] [Google Scholar]
- Esteller M, Corn PG, Baylin SB, Herman JG. A gene hypermethylation profile of human cancer. Cancer Res. 2001;61:3225–3229. [PubMed] [Google Scholar]
- Hake SB, Xiao A, Allis CD. Linking the epigenetic ‘language’ of covalent histone modifications to cancer. Br J Cancer. 2004;90:761–769. doi: 10.1038/sj.bjc.6601575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppler D, Sloane BF. Cathepsin B: multiple enzyme forms from a single gene and their relation to cancer. Enzyme Protein. 1996;49:94–105. doi: 10.1159/000468619. [DOI] [PubMed] [Google Scholar]
- Kim TY, Zhong S, Fields CR, Kim JH, Robertson KD. Epigenomic profiling reveals novel and frequent targets of aberrant DNA methylation-mediated silencing in malignant glioma. Cancer Res. 2006;66:7490–7501. doi: 10.1158/0008-5472.CAN-05-4552. [DOI] [PubMed] [Google Scholar]
- Konduri SD, Yanamandra N, Siddique K, Joseph A, Dinh DH, Olivero WC, et al. Modulation of cystatin C expression impairs the invasive and tumorigenic potential of human glioblastoma cells. Oncogene. 2002;21:8705–8712. doi: 10.1038/sj.onc.1205949. [DOI] [PubMed] [Google Scholar]
- Krueger S, Kellner U, Buehling F, Roessner A. Cathepsin L antisense oligonucleotides in a human osteosarcoma cell line: effects on the invasive phenotype. Cancer Gene Ther. 2001;8:522–528. doi: 10.1038/sj.cgt.7700341. [DOI] [PubMed] [Google Scholar]
- Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001a;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- Marks PA, Rifkind RA, Richon VM, Breslow R. Inhibitors of histone deacetylase are potentially effective anticancer agents. Clin Cancer Res. 2001b;7:759–760. [PubMed] [Google Scholar]
- Ni J, Abrahamson M, Zhang M, Fernandez MA, Grubb A, Su J, et al. Cystatin E is a novel human cysteine proteinase inhibitor with structural resemblance to family 2 cystatins. J Biol Chem. 1997;272:10853–10858. doi: 10.1074/jbc.272.16.10853. [DOI] [PubMed] [Google Scholar]
- Pulukuri SM, Gorantla B, Rao JS. Inhibition of histone deacetylase activity promotes invasion of human cancer cells through activation of urokinase plasminogen activator (uPA) J Biol Chem. 2007a;282:35594–35603. doi: 10.1074/jbc.M705867200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Pulukuri SM, Patibandla S, Patel J, Estes N, Rao JS. Epigenetic inactivation of the tissue inhibitor of metalloproteinase-2 (TIMP-2) gene in human prostate tumors. Oncogene. 2007b;26:5229–5237. doi: 10.1038/sj.onc.1210329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulukuri SM, Rao JS. Small interfering RNA directed reversal of urokinase plasminogen activator demethylation inhibits prostate tumor growth and metastasis. Cancer Res. 2007;67:6637–6646. doi: 10.1158/0008-5472.CAN-07-0751. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Qiu J, Ai L, Ramachandran C, Yao B, Gopalakrishnan S, Fields CR, et al. Invasion suppressor cystatin E/M (CST6): high-level cell type-specific expression in normal brain and epigenetic silencing in gliomas. Lab Invest. 2008;88:910–925. doi: 10.1038/labinvest.2008.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci USA. 2000;97:10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roshy S, Sloane BF, Moin K. Pericellular cathepsin B and malignant progression. Cancer Metastasis Rev. 2003;22:271–286. doi: 10.1023/a:1023007717757. [DOI] [PubMed] [Google Scholar]
- Sambucetti LC, Fischer DD, Zabludoff S, Kwon PO, Chamberlin H, Trogani N, et al. Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. J Biol Chem. 1999;274:34940–34947. doi: 10.1074/jbc.274.49.34940. [DOI] [PubMed] [Google Scholar]
- Schagdarsurengin U, Pfeifer GP, Dammann R. Frequent epigenetic inactivation of cystatin M in breast carcinoma. Oncogene. 2007;26:3089–3094. doi: 10.1038/sj.onc.1210107. [DOI] [PubMed] [Google Scholar]
- Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64:9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- Shridhar R, Zhang J, Song J, Booth BA, Kevil CG, Sotiropoulou G, et al. Cystatin M suppresses the malignant phenotype of human MDA-MB-435S cells. Oncogene. 2004;23:2206–2215. doi: 10.1038/sj.onc.1207340. [DOI] [PubMed] [Google Scholar]
- Sotiropoulou G, Anisowicz A, Sager R. Identification, cloning, and characterization of cystatin M, a novel cysteine proteinase inhibitor, down-regulated in breast cancer. J Biol Chem. 1997;272:903–910. doi: 10.1074/jbc.272.2.903. [DOI] [PubMed] [Google Scholar]
- Turk B, Turk D, Turk V. Lysosomal cysteine proteases: more than scavengers. Biochim Biophys Acta. 2000;1477:98–111. doi: 10.1016/s0167-4838(99)00263-0. [DOI] [PubMed] [Google Scholar]
- Turk V, Bode W. The cystatins: protein inhibitors of cysteine proteinases. FEBS Lett. 1991;285:213–219. doi: 10.1016/0014-5793(91)80804-c. [DOI] [PubMed] [Google Scholar]
- Veena MS, Lee G, Keppler D, Mendonca MS, Redpath JL, Stanbridge EJ, et al. Inactivation of the cystatin E/M tumor suppressor gene in cervical cancer. Genes Chromosomes Cancer. 2008;47:740–754. doi: 10.1002/gcc.20576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamandra N, Gumidyala KV, Waldron KG, Gujrati M, Olivero WC, Dinh DH, et al. Blockade of cathepsin B expression in human glioblastoma cells is associated with suppression of angiogenesis. Oncogene. 2004;23:2224–2230. doi: 10.1038/sj.onc.1207338. [DOI] [PubMed] [Google Scholar]
- Yoon SO, Kim MM, Chung AS. Inhibitory effect of selenite on invasion of HT1080 tumor cells. J Biol Chem. 2001;276:20085–20092. doi: 10.1074/jbc.M101143200. [DOI] [PubMed] [Google Scholar]
- Zhang J, Shridhar R, Dai Q, Song J, Barlow SC, Yin L, et al. Cystatin m: a novel candidate tumor suppressor gene for breast cancer. Cancer Res. 2004;64:6957–6964. doi: 10.1158/0008-5472.CAN-04-0819. [DOI] [PubMed] [Google Scholar]
- Zhong S, Fields CR, Su N, Pan YX, Robertson KD. Pharmacologic inhibition of epigenetic modifications, coupled with gene expression profiling, reveals novel targets of aberrant DNA methylation and histone deacetylation in lung cancer. Oncogene. 2007;26:2621–2634. doi: 10.1038/sj.onc.1210041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.