

Abstract
To assess the ability of resting state functional magnetic resonance imaging to distinguish known risk factors for AD, we evaluated 17 cognitively normal individuals with a family history of AD and at least one copy of the apolipoprotein e4 allele compared to 12 individuals who were not carriers of the APOE4 gene and did not have a family history of AD. Blood oxygen level dependent fMRI was performed evaluating encoding-associated signal and resting state default mode network signal differences between the two risk groups. Neurocognitive testing revealed that the high risk group performed worse on category fluency testing, but the groups were equivalent on all other cognitive measures. During encoding of novel face-name pairs, there were no regions of encoding-associated BOLD activations that were different in the high risk group. Encoding-associated deactivations were greater in magnitude in the low risk group in the medial and right lateral parietal cortex, similar to findings in AD studies. The resting state DMN analysis demonstrated nine regions in the prefrontal, orbital frontal, temporal and parietal lobes that distinguished the two risk groups. Resting state DMN analysis could distinguish risk groups with an effect size of 3.35, compared to an effect size of 1.39 using encoding-associated fMRI techniques. Imaging of the resting state avoids performance related variability seen in activation fMRI, is less complicated to acquire and standardize, does not require radio-isotopes, and may be more effective at identifying functional pathology associated with AD risk compared to non-resting fMRI techniques.
Keywords: fMRI, Alzheimer's disease, APOE4, default network
INTRODUCTION
Functional imaging has been demonstrated to be able to distinguish people at risk for Alzheimer's disease (AD) prior to any clinical manifestations of neurodegeneration in young and middle aged individuals(Bookheimer et al., 2000; Fleisher et al., 2005; Fleisher et al., 2008; Reiman et al., 1996; Reiman et al., 2004). These studies and others provide evidence that there may be identifiable changes in brain physiology prior to potential clinical manifestations of dementia. Over the past two decades functional magnetic resonance imaging (fMRI) has become a prominent tool for studying blood oxygenation levels associated with various cognitive activities. Most studies have focused on the use of cognitive tasks to explore fMRI blood oxygenation level dependent (BOLD) signal activity, to define functional brain maps relating to anatomical structures, and to define related networks of brain activity. Although this has proven useful for understanding functional brain pathways, it is not clear that these activation techniques will be practical for use as biomarkers to identify individuals predisposed to developing dementia or useful as outcome measures in preventative drug studies. fMRI activation studies unfortunately suffer from intra and inter-subject variability, scanner variability, are dependent on task performance, and often involve lengthy scan times with complex study designs that are hard to standardize, and are difficult to perform in cognitively impaired individuals. fMRI evaluation of the resting state of the brain may be a sufficient and relevant target for studies of pre-clinical dementia.
The amplitude of the hemodynamic response to an external stimulus is dependent on the basal state of the brain(Ances et al., 2008; Brown et al., 2003; Buxton et al., 2004; Davis et al., 1998). Exposure to the same sensory stimulus will generate different fMRI responses depending on whether the basal state is high or low. For example, in individuals with a copy of the apolipoprotien E epsilon 4 allele (APOE4) and a family history of dementia, compared to individuals without these risk factors, differences reported in medial temporal lobe activations during encoding may simply reflect differences in the basal neuro-physiologic state(Fleisher et al., 2008). In fact, when alterations in the resting state are accounted for, the activation states may no longer reveal significant absolute differences. This implies that the resting state may often drive differences reported in activation state fMRI studies of AD and AD risk(Fleisher et al., 2008). One advantage to resting-state fMRI is that it is not dependent on differential task performance such as memory encoding, which is of particular concern when studying neurodegenerative processes and evaluating disease modifying therapies for prevention and treatment of dementia. Due to these issues, in part, there is an increasing interest in development of resting state fMRI as a potential biomarker for preventative drug development in AD.
Echo Planar Imaging of the default mode network (DMN) explores resting state neuronal network dysfunction in AD and has potential to be a sensitive marker for preclinical AD patho-physiology. The DMN represents a network of coordinated low frequency fluctuation (LFF) in specific functional neuronal networks. It is manifested as key brain regions that are elevated in states of relative rest, which are responsible for attention to environmental stimuli, reviewing of past knowledge, and planning of future behaviors(Binder et al., 1999; Raichle et al., 2001). These regions predominantly consist of midline and lateral frontal regions, and medial and lateral parietal regions extending into posterior cingulate/retrosplenial (pC/rsp) cortex(Buckner and Vincent, 2007). These same regions that are activated at rest appear to be suppressed during various cognitive activities, including encoding of new memories(Pihlajamaki et al., 2008; Rombouts et al., 2005; Sorg et al., 2007). For this reason, two methods have been developed utilizing the DMN to identify diseases of cognition and risk for dementia in the BOLD fMRI literature. One method explores task-related deactivations and the other focuses on differences in resting state BOLD networks. These default networks may be particularly effected by the neurodegenerative process of AD(Buckner et al., 2008). With this, several groups have reported both reduced resting state connectivity(Buckner et al., 2005) and alterations in fMRI task-induced deactivation responses in aging (Andrews-Hanna et al., 2007; Lustig et al., 2003) mild cognitive impairment (MCI) (Rombouts et al., 2005) and AD patients (Buckner and Vincent, 2007; Lustig et al., 2003; Persson et al., 2008; Rombouts et al., 2005; Sorg et al., 2007; Wang et al., 2007; Wang et al., 2006) compared to healthy controls.
Abnormalities of the DMN seen with fMRI may signify underlying physiologic defects associated with AD. For instance, there is evidence of a relationship between medial temporal lobe memory networks, frontoparietal attentional networks and the DMN, which appears to be required for successful memory formation(Buckner et al., 2005; Miller et al., 2008; Pihlajamaki et al., 2008). Also, decreased connectivity between the hippocampus, entorhinal cortex and the posterior cingulate cortex in AD has been proposed to represent early changes in functional brain networks in AD(Greicius et al., 2004). Evidence that supports this includes findings that the cortical regions that make up the DMN are similar to areas of early brain atrophy, hypometabolism, decreased perfusion, and fibrillar amyloid deposition in early AD and mild cognitive impairment (MCI)(Buckner et al., 2005; Edison et al., 2007; Forsberg et al., 2007; Jack, Jr. et al., 2008; Johnson et al., 1998; Klunk et al., 2004; Minoshima et al., 1997). In particular, the posterior cingulate and precuneus cortex are regions that have the most prominent deactivations during cognitive tasks and are increased during the resting state(Buckner et al., 2005; Greicius et al., 2004). Very recently data was presented demonstrating that decrease in the DMN in cognitively normal APOE4 carries is associated with increase cortical fibrillar amyloid (Buckner et al., 2009; Hedden et al., 2009). And, differences in DMN signal has been noted in APOE4 carriers as young as 20-35 years old (Filippini et al., 2009). In addition, suppression of encoding-associated fMRI deactivation in the posterior cortical default network is associated with increased amyloid plaque burden in the precuneus/posterior cingulate cortex measured by Pittsburg compound B positron emission tomography (PiB-PET) imaging(Sperling et al., 2008). Further, failure of deactivation of medial posterior DMN during encoding is associated with worse memory performance(Miller et al., 2008) Overall, these findings suggest that “suspending” the default network during working memory is necessary for successful encoding, is impaired in AD, and potentially is associated with underlying amyloid pathology, even prior to clinical dementia in association with the APOE4 allele.
In an effort to further develop tools for early identification of Alzheimer's disease pathology and risk, we studied individuals with high and low risk for developing AD using BOLD fMRI. We compared the ability to distinguish AD risk groups using activation and deactivation fMRI during an encoding task versus resting state BOLD DMN correlation analysis. Resting state data was extracted from the encoding scans which included periods of relative rest. The overall aim of this study was to comparatively evaluate the utility of DMN BOLD fMRI in distinguishing risk for Alzheimer's disease pathology, and as a potential biomarker forpreventative treatment trial.
METHODS
Study population
Twenty-nine healthy right-handed volunteers, fifty to sixty-five years of age, were evaluated. Seventeen had a significant family history of dementia in a first degree relative and at least one copy of the APOE4 gene. Twelve participants had neither a family history of dementia nor a copy of the APOE4 gene. Participants were drawn from normal control participants in the University of San Diego (UCSD) Alzheimer's Disease Research Center, from the UCSD student, staff, and faculty population, as well as the general San Diego community by means of advertisements. The study was conducted according to Good Clinical Practice, the Declaration of Helsinki and U.S. 21 CFR Part 50-Protection of Human Subjects, and Part 56-Institutional Review Boards. Written informed consent for the study was obtained from all of the participants before protocol-specific procedures were performed, including cognitive testing.
All potential participants were screened and excluded for a history of significant head trauma with residual cognitive deficits, other neurological or major psychiatric disorders such as schizophrenia, bipolar disorder, developmental learning disorders, and alcohol or substance abuse. The Geriatric Depression Scale (Yesavage, Brink et al. 1982) was administered to screen for the presence of affective disturbance. Persons with significant cerebrovascular disease (as indexed by modified Rosen ischemic scores greater than 4) were also excluded, as were individuals with unstable diabetes and respiratory disease. All participants underwent careful screening for contraindications for magnetic resonance imaging (i.e., metal in the body, pregnancy, claustrophobia), as well as complete physical and neurological examinations. All participants received APOE4 genotyping using a polymerase chain reaction based method (Saunders, Strittmatter et al. 1993).
Cognitive testing
All participants received the following neuropsychological test battery within one month prior to imaging: Boston Naming Test(Kaplan, Goodglass et al. 1983), WMS-R Logical Memory Test(Wechsler 1987), Verbal Fluency(Monsch, Bondi et al. 1992), WAIS-R digit span forward and backward(Wechsler 1981), WAIS-R Digit symbol test(Smith 1982), California Verbal Learning Test (CVLT)(Delis, Freeland et al. 1988), Clock Drawing(Mohs, Knopman et al. 1997), Trail Making Test (A and B)(Reitan 1958), and Mini Mental State Exam (MMSE)(Folstein, Folstein et al. 1975). Participants were excluded if they did not fall within age adjusted normal ranges for all cognitive tests, as determined by a licensed neuropsychologist at our center.
Functional MRI Behavioral Task
An associative encoding task was utilized to evaluate interleaved blocks of memorizing pairs of faces and names, and periods of rest. The task was adapted with permission from Dr. Reisa Sperling(Sperling, Bates et al. 2001). This task was chosen due to its ability to activate the medial temporal lobes and deactivate the default mode networks with BOLD fMRI (Pihlajamaki et al., 2008; Sperling et al., 2003a; Sperling et al., 2001). Participants viewed pairs of Novel and previously viewed (Repeated) faces and names in alternating blocks of 40 seconds each, separated by 25 seconds of viewing a central fixation crosshair. Participants were familiarized with the repeated face/name pairs, and tested for recall accuracy prior to active scanning. During the fixation task participants were instructed to lay with their eyes open, stare straight at the crosshair in the middle of the screen, and try to blank their minds of the faces and names previously presented. This task is described in detail in a previous publication(Fleisher et al., 2008) and in an online supplement. Four runs were obtained for each participant in rapid succession, each lasting a total of four minutes and thirty-five seconds. A total of 56 Novel face-name pairs and four Repeated face-name pairs were used over the course of the entire experiment, with 400 total seconds of resting during visual fixation.
Post-scan testing
Immediately following the imaging session, outside the scanner, subjects were tested for their recall of all 60 face-name pairs. The post-scan testing was a cued recall task in which subjects were shown 70 faces individually. They were presented with a choice of three names for each face and instructed to chose the name that was presented during the scanning session. There were ten distractor faces in the post-scan testing that were not presented during the experiment. Percent recall scores were calculated to verify attention to the scanning task and evaluate encoding capabilities in both risk groups.
MRI Acquisition
MRI technique
All scans were performed on a General Electric Signa EXCITE 3.0T short bore, twin speed scanner with a body transmit coil and an 8 channel receive array. High-resolution structural brain images were acquired with a magnetization prepared three-dimensional fast spoiled gradient sequence acquisition (FSPGR: 124 axial slices, 1 mm × 1 mm in-plane resolution, 1.3 mm slice thickness, Field of View = 256 × 256, TR = 7.8 ms, TE = 3.1 ms, flip angle 12°). BOLD data were acquired using echo planar imaging sequences (35 slices, perpendicular to the axis of the Hippocampus, 6mm in plane resolution, 0 spacing, FOV=22, TE=30 ms, TR=2500 ms). Voxel dimensions were 3.4 × 3.4 × 6.0mm.
Physiologic monitoring
During all BOLD scan acquisitions, pulse and respiratory wave forms were collected using a pulse oxymeter and respiratory effort transducer. Data from these instruments were collected at 40 samples per second using a multi-channel data acquisition board (National Instruments). Scanner TTL pulse data (10ms duration, 5 volt pulse per slice acquisition) were recorded at 1 kHz. The TTL pulse data were used to synchronize the physiologic data to the acquired images. Pulse, respiratory, and TTL data were used to calculate physiologic noise regressors to improve neuronal activity related signal identification (Glover, Li et al. 2000; Restom, Behzadi et al. 2006).
Data Analysis
General Post-Scan Image Processing
For each voxel the MR signal associated with physiologic noise from both pulse rate and respiratory rate data were included in a general linear model analysis as regressors to model physiologic fluctuations of the BOLD signal. These physiologic noise component estimates were removed from the data to form corrected echo time series for further analysis of BOLD data. Images were then motion corrected across time points, among all four runs, to the most typical base image in the second run, using a three-dimensional iterated, linearized, weighted least-squares method with Fourier interpolation using the Application for Functional NeuroImaging software (AFNI)(Cox, 1996). Time series were also visually inspected for motion based on the output of a program designed to detect outliers (AFNI-3dToutcount). Time points with isolated head movements not corrected by the registration algorithm were ignored in the statistical analysis. A high (0.01 Hz) and low pass (0.1 Hz) filter was applied to remove signal in nonphysiologic ranges (3dFourier). In addition, all data were smoothed with a 6mm full width at half maximum Gaussian filter. Smoothing in space enhances the signal-to-noise ratio of data (Turner et al., 1998), increasing the validity of statistical tests across groups and compensating for variance in normalization(Ashburner and Friston, 1999). Functional datasets were then resampled to 2mm isotropic voxels.
Task-Related Signal processing
A general linear model (GLM) approach was used for statistical analysis of the individual functional datasets. Data from all four functional runs were concatenated for use in the GLM analysis performed by AFNI's 3dDeconvolve program(Cox, 1996). Stimulus-related MR signal was included in the regression model by convolving the block design stimulus pattern with a gamma density function to create reference vectors using the AFNI Waver program(Cox, 1996). In addition, motion related translation and rotation parameters were used as covariates. Hence, the GLM model included a combination of the following independent variables with MR signal as the dependent variable: reference vectors representing the occurrence of different stimulus types (i.e., Novel face-name pairs, Repeated face-name pairs, or fixation), six motion parameters, a linear trend, and a constant. This procedure produced F statistics representing the strength of associations between stimulus presentation and mean peak signal change for each voxel, averaged across each stimulus block for all four functional acquisitions, for each individual participant. Based on the F statistic, mean peak signal changes that satisfied an alpha level of 0.05 for association with the stimulus were used as the primary outcome variables for activations and deactivations in second level between-group analyses. These mean peak values were converted to percent signal change by dividing mean baseline signal data by the mean peak task-associated values for each scan. The contrast between novel face/name pair encoding blocks and repeated face/name pair encoding blocks was used as the primary outcome measure to isolate BOLD signal associated with novel encoding(Sperling et al., 2001).
Resting state Default Mode Network analysis
An approximate of the resting state was assessed by removing task related signal fluctuations from encoding blocks and evaluating periods of visual fixation. Regression modeling was used to remove all task-associated BOLD activity and isolate fluctuations associated with the resting state throughout all functional datasets. To do this, regression coefficients associated with novel and repeated encoding generated by the GLM used to deconvolve task-associated signal were extracted using AFNI's 3dSynthesize and 3dcalc programs (http://afni.nimh.nih.gov/afni). The default network mode was then assessed by correlating each voxel in individual datasets with low frequency fluctuations within a 12mm spherical seed region placed within the largest area of task-related deactivation in the posterior cingulate/retrospenial region (pC/rsp) (Figure 1C; atlas coordinates: −1, 50, 36 (RAI)). This seed location has been reported in the literature to strongly identify the default mode network with correlation analysis of data extracted from resting portions of other functional tasks(Andrews-Hanna et al., 2007). For each individual's scan, fluctuations in resting BOLD signal during fixation were determined for each voxel and correlated across time to signal fluctuations in the seed region. Whole brain statistical maps were then generated with each voxel containing its representative Pearson r-value. This was done using AFNI's 3dDeconvolve program. Movement parameters included as covariate in the task-associated GLM were also included in this correlation analysis. The resulting correlation maps were converted to z scores using Fisher's r-to-z transformation for further group analysis. This method of correlation analysis of the resting state has been demonstrated on various functional datasets to consistently identify the default mode network(Andrews-Hanna et al., 2007; Buckner et al., 2005; Buckner and Vincent, 2007; Raichle et al., 2001).
Figure 1.

Encoding-associated BOLD signal change. Anatomical image is an average of all 29 subjects. A) Between group differences in encoding-associated percent BOLD signal change from voxel-wise t-tests, showing multiple areas of increased signal in the high risk group compared to the low risk group. B) Regions of encoding-associated increased BOLD signal for each risk group based on within-group t-tests, overlapped on to a single map: yellow = high risk (n=17), pink = low risk (n=12), red = areas of overlapping activations for the low and high risk groups. C) Significant encoding-associated deactivations in the entire cohort (n=29) representing the default mode network. The yellow sphere represents the pC/rsp seed placement for the subsequent resting DMN correlation analysis (Figure 3). D) Regions of encoding-associated decreased BOLD signal for each risk group based on within-group t-tests, overlapped on to a single map: light blue = high risk, dark blue = low risk, purple = areas of overlapping deactivations for the low and high risk groups.
Volumetric data processing
Measurements of total intracranial, whole brain, ventricular and hippocampal volumes were calculated using semi-automated segmentation and volumetric output measures from FreeSurfer's ASEG analysis(Fischl, Salat et al. 2002). Whole brain, ventricular, and hippocampal volumes were divided by the total intracranial volumes to adjust for variability due to head size. These normalized volumes were used for between group comparisons to evaluate for significant group differences.
Between group statistics
All demographics, neurocognitive measures and volumetric MRI measures were compared between AD risk groups by chi-square or two sample t-tests, as appropriate, with significance determined as p ≤ 0.05.
All functional and anatomical images were transformed into standard Talairach atlas space(Talairach and Tournoux, 1988) for co-registration and between group voxel-wise analyses. For all between group exploratory analyses a liberal p-threshold <0.05 was used. In addition, to account for type-I errors due to multiple comparisons a cluster thresholding criteria to determine a minimum cluster size necessary to avoid false positive associations was established by a Monte Carlo simulation program (AlphaSim) with an alpha criteria of 0.05(Cox, 1996). This simulation determines the number of adjacent voxels that must be activated so that there is less than a 5% risk of activation occurring by random chance based on the number of voxels being analyses within a predetermined volume.
Task-associated functional datasets were first submitted to two-tailed voxel-wise t-tests between the two different AD risk groups using AFNI's 3dttest program. To further distinguish regions of encoding-associated deactivations versus activations in both risk groups, these datasets were separated into BOLD signal that was decreased due to task presentation and signal that was increased during the task using one sample within group t-tests. Regions of significant within-group encoding-associated deactivations in the low risk group were identified that overlapped with regions of deactivation in the high risk group. These complementary between-group regions of overlapping deactivations were then assessed for between group signal intensity differences with voxel-wise two-tailed t-tests. This was done to assure that only regions with unidirectional significant signal changes were compared between groups. By doing this we avoided comparing regions of deactivation in one group to regions of activation in the other group, potentially making interpretation of default mode network signal changes difficult. In addition, volume of task-associated deactivations between each risk group was compared with two-tailed t-tests. Similarly, regions of overlapping between-group encoding-associated activations were assessed to isolate significant group differences resulting from signal increases in both groups.
Correlation z-score maps for the resting default mode network were compared between groups using voxel-wise two-tailed t-tests. Clusters of correlated DMN activity that differed between risk groups were determined with p-thresholding and cluster size thresholding with an alpha < 0.05 as described above. Mean z-scores from each functional cluster demonstrating significant between-group differences were extracted for each individual.
For both task-associated measures and resting DMN measures, mean voxel values for clusters demonstrating between group differences were subjected to traditional two sample t-tests to calculate specific p-values. Standard deviations and 95% confidence intervals were also calculated using these mean values. In addition, effect sizes for distinguishing differences between the two groups for each of these functional regions of interest (ROI) was calculated using Hedges g scores, accounting for sample sizes(Hedges, 1981). Finally, multivariate effect sizes were calculated using multivariate Hedges g scores to examine potential increased sensitivity when utilizing simultaneous information from all defined ROIs for each fMRI modality (Kline, 2004).
Cognitively normal individuals have decreased memory scores associated with increased amyloid binding on amyloid imaging or attenuation of the DMN on fMRI(Pihlajamaki et al., 2008; Pike et al., 2007). For this reason we performed correlation analyses by group and for the total cohort comparing California verbal learning test scores to mean fMRI measures in regions demonstrating significant group differences.
RESULTS
Demographics and cognitive testing
Participants in the high risk group (n=17) all had a significant family history of dementia in a first degree relative and at least one copy of the APOE4 gene (e3,e4 = 13; e4,e4 = 4). The low risk group (n=12) had no family history of dementia and no copies of the APOE4 gene (e3,e3 = 11; e2,e3 = 1). Ages ranged from 51 years to 65 years (high risk = 58.6 ± 4.1; low risk = 57.6 ± 4.5). These groups did not significantly differ by age, gender or education (Table1). There were no differences in brain volume measurements of whole brain, ventricles, or hippocampi (Table1). There were no group differences in measures of memory (Table 1) or other neurocognitive test scores, except higher scores on category fluency retrieval in the low risk group (low risk = 63.6 ± 6.4 (range:54-74) , high risk = 53.2 ± 8.0 (range:41-68): p<0.001). Category fluency testing consisted of category retrieval of animals, fruits and vegetables in one minute for each category, with total scores calculated by adding the retrieval scores for all three categories. There were no group differences on the post scan recall task of face-name pairs (Table1).
Table 1.
Demographics, Memory Scores, Volumetrics
| High Risk | Low Risk | P value | |
|---|---|---|---|
| Number of Subjects | 17 | 12 | |
| Age (years) | 58.6 ± 4.1 | 57.6 ± 4.5 | 0.53 |
| Sex (%female) | 4M, 13F (76.5%) | 5M, 7F (58.3%) | 0.45 |
| Education (years) | 16.0 ± 2.0 | 16.0 ± 2.4 | 1 |
| MMSE | 29.6 ± 0.71 | 29.8 ± 0.39 | 0.39 |
| CVLT | 49.4 ± 10.0 | 50.3 ± 11.1 | 0.82 |
| Logical memory delayed recalla | 11.7± 4.8 | 12.4 ± 3.9 | 0.67 |
| Category Fluency | 53.2 ± 8.0 | 63.6 ± 6.4 | <0.001 |
| Post MRI Task Recall Score (%) | 65.60 ± 16.2 | 70.0 ± 14.2 | 0.47 |
| Whole brain volumes (% of TIV) | 60.1 ± 2.0 | 60.9 ± 3.4 | 0.43 |
| Ventricular volume (% of TIV) | 1.4 ± 0.52 | 1.5 ± 0.61 | 0.80 |
| Hippocampal volume (% of TIV) | 0.51 ± 0.031 | 0.52 ± 0.043 | 0.54 |
MMSE = Folstein Mini Mental State Exam (Folstein, Folstein et al. 1975).
CVLT = California Verbal Learning Test (Delis, Freeland et al. 1988),
Delayed recall of the WMS-R Logical Memory Test(Wechsler 1987)
TIV = total intracranial volume
Encoding associated comparisons
Whole brain voxel-wise comparison of percent signal change associated with novel encoding demonstrated several regions with relatively higher encoding-associated BOLD signal in the high risk group compared to the low risk group (Figure 1A). This included ten significant clusters of signal that differed between groups in broad regions of the medial and superior frontal lobe, superior marginal gyrus of the parietal lobe, superior temporal gyrus, occipital cortex, and cingulated cortex. Notably, none of these areas represent regions with increased encoding-associated BOLD signal in both groups. In the low risk group, all regions showing between group differences involve areas of deactivation (negative signal change) in response to encoding. The statistical differences between groups is due to relative increased signal in the high risk group with either “less negative” or positive signal change in response to encoding. In other words, these were regions of attenuated deactivation in the high risk group, with no regions of encoding-associated activations (increased signal) in the low risk group. These regions represent deactivations or divergent signal response between groups, and did not include cortical areas typically associated with activation in response to the face-name encoding task such as the hippocampus, pulvinar nucleus of the thalamus, fusiform gyrus and dorsolateral prefrontal cortices(Sperling et al., 2001).
Significant encoding-associated activations were distinguished from areas of deactivations for each risk group using within-group one-sample t-tests to differentiate signal change significantly greater than or less than zero. Figure 1B displays overlapping regions of activations during encoding, demonstrating that the task successfully, and prominently, activates bilateral hippocampus in each risk group, in addition to scattered regions in the frontal lobes, fusiform gyri, and thalami. Task-related deactivations were found in the entire group (n=29) to successfully represent regions consistent with the default mode network, with prominent posterior cingulated/retrospenial, lateral parietal, and medial prefrontal deactivation during encoding (Figure 1C). In addition, both risk groups had consistent independent overlapping areas of deactivation during encoding in these DMN-associated regions (Figure 1D).
Next, regions of activations and deactivations were assessed separately for between-group differences. No overlapping regions of encoding-associated activations (Figure 1B, red regions) were found to have significantly different BOLD signal change between groups. Three distinct regions of overlapping deactivations (Figure 1D, purple regions) were able to distinguish risk groups for Alzheimer's disease. These included DMN regions in the bilateral medial posterior parietal lobes (cuneus/precuneus) and the right lateral parietal lobe (right superior marginal gyrus) (Figure 2A). In all three regions the low risk group had larger encoding-associated deactivations compared to the high risk group, i.e. the group with higher risk for AD had attenuated encoding-associated deactivation in DMN regions (Figure 2B). Mean cluster percent signal change, between-group p-values, and effect sizes for these clusters are presented in Table 2. By calculating an adjusted eta2 we determine that the greatest variability accounted for by group membership for any individual ROI was 17.5%. All three clusters were combined to calculate a multivariate effect size of 1.39, accounting for 25.4% of variance due to risk group membership. Additionally, there were no significant differences in the total volumes of deactivated regions between risk groups.
Figure 2.
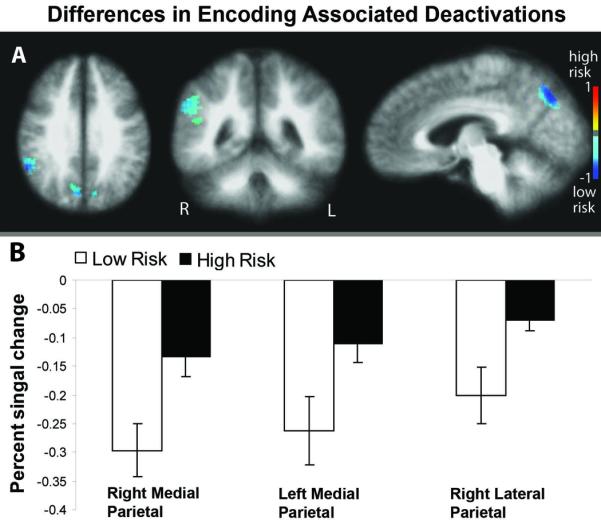
Group differences in overlapping regions of encoding-associated deactivations. A) Voxel-wise t-test maps showing greater deactivations in the low risk group compared to the high risk group. B) Percent signal change during encoding for each risk group in regions showing significant group differences. Error bars represent the standard error of the mean.
Table 2.
Encoding-Associated Deactivation Differences by AD Risk Group
| Cluster Region | Coordinates (RAI) |
Mean % signal change ± SD |
Group difference (%) |
Effect size (95% CI) |
p- values |
|
|---|---|---|---|---|---|---|
| Low Risk | High Risk | |||||
| Right cuneus/precuneus |
−7 71 34 | −0.30 ± 0.16 | −0.13 ± 0.15 | 223.6 | 1.08 (0.26-1.84) |
0.008 |
| Left cuneus/precuneus |
10 73 32 | −0.26 ± 0.21 | −0.11 ± 0.14 | 42.2 | 0.89 (0.1-1.64) |
0.025 |
| Right supramarginal gyrus |
−52 45 30 | −0.20 ± 0.17 | −0.07 ± 0.075 | 289.2 | 1.08 (0.26-1.83) | 0.008 |
Resting state correlation analysis
Correlation analysis of resting-state low frequency BOLD signal fluctuations in the full group (n=29) demonstrated regions of network connectivity with the pC/rsp seed consistent with previously reported DMN correlation analyses in cognitively normal elderly individuals (Figure 3)(Andrews-Hanna et al., 2007). Increased nodal correlation/connectivity was found in the high risk group in the superior, anterior and ventral medial prefrontal cortex, right dorsolateral prefrontal cortex, left postcentral gyrus, middle and superior temporal gyrus, and the right hippocampus, with decreased connectivity in the precuneus and gyrus rectus (Figure 4). A total of nine distinct clusters were found with significant connectivity differences between the two risk groups (Table 3). Mean cluster correlation z-scores, between-group p-values, and effect sizes for each cluster are presented in Table 3, showing that resting state correlation analysis of the DMN provided overall greater effect sizes to separate risk groups than differences in encoding-associated deactivations (Table 2). Adjusted eta2 demonstrate that the greatest variability accounted for by group membership, by any individual ROI, is 36.0% in the bilateral superior prefrontal cortex ROI. All nine connectivity clusters were combined to calculate a multivariate effect size of 3.35, accounting for 62.4% of variance due to risk group membership.
Figure 3.

Z-score maps of the entire cohort (n=29) showing BOLD fluctuations during rest correlated with the posterior cingulate/retrosplenial seed region (Figure 1C), demonstrating the default mode network.
Figure 4.

Resting DMN connectivity with the pC/rsp seed in nine different regions between high risk and low risk individuals. A) Mean z-scores for pC/rsp connectivity for both risk groups in regions showing significant group differences. B) Voxel-wise between-group comparison maps of z-score correlations (−1 to +1) showing increased (hot colors) and decreased (cool colors) connectivity in the high risk group compared to the low risk group for each region. Error bars represent the standard error of the mean.
Table 3.
Resting Default Mode Network Connectivity Differences by AD Risk Group
| Cluster Region | Coordinates (RAI) |
Mean Correlation z-score ± SD |
Z-score group difference (%) |
Effect size (95% CI) |
p-values | |
|---|---|---|---|---|---|---|
| Low Risk | High Risk | |||||
| Bilateral Superior Medial Prefrontal Cortex |
−5 −21 55 | 0.16 ± 0.18 | 0.46 ± 0.19 | 34.7 | 1.62 (0.73-2.41) |
< 0.001 |
| Bilateral VentroMedial Prefrontal cortex |
2 −56 8 | 0.19 ± 0.27 | 0.44 ± 0.16 | 43.6 | 1.18 0.35-1.94 |
0.004 |
| Right Middle/Superior Temporal |
−48 7 −10 | 0.20 ± 0.19 | 0.44 ± 0.16 | 45.0 | 1.4 (0.55-2.19) |
< 0.001 |
| Right Dorsolateral Prefrontal Cortex |
−57 6 29 | 0.091 ± 0.19 | 0.32 ± 0.21 | 28.1 | 1.17 (0.34-1.93) |
0.005 |
| Left AnteroMedial Prefrontal Cortex |
17 −50 33 | 0.12 ± 0.23 | 0.38 ± 0.17 | 31.1 | 1.31 (0.46-2.08) |
0.002 |
| Left Post-Central Parietal |
35 41 60 | 0.13 ± 0.18 | 0.47 ± 0.37 | 26.8 | 1.12 (0.30-1.88) |
0.006 |
| Right Hippocampus |
−27 13 −13 | 0.31± 0.27 | 0.60 ± 0.30 | 51.3 | 1.03 (0.22-1.78) |
0.011 |
| Bilateral Precuneus |
3 74 46 | 0.75 ± 0.17 | 0.46 ± 0.20 | −61.8 | 1.51 (0.64-2.30) |
< 0.001 |
| Bilat Gyrus Rectus | −3 −33 −21 | 0.25 ± 0.13 | 0.076 ± 0.091 |
−30.8 | 1.55 (0.67-2.34) |
< 0.001 |
Planned corrections with cognitive test scores
Higher CVLT scores were associated with larger encoding-associated deactivation in the right lateral parietal lobe in the full cohort (n=29: Pearson r=−0.51, p=0.005), as well as in each risk group (high risk: r=−0.59, p=0.012; low risk: r=−0.58, p=0.048). In the high risk group only there was a trend association between increased CVLT scores and increased DMN connectivity z-scores in the precuneus (r=0.46, p=0.061).
Post-hoc Analysis
Recognizing the group difference in category fluency scores, we sought to evaluate the relationship between total category fluency scores and fMRI measures that differed between the two risk groups. Pearson r scores were calculated to measure the relationship between category fluency scores and encoding-associated BOLD deactivations (ROIs from Figure 2) as well as DMN resting connectivity scores (ROIs from Figure 4) in clusters that were different between groups. There were no significant correlations between category fluency and encoding-associated deactivations in any of the three reported ROIs. Category fluency had a negative correlation with resting connectivity scores in three of the reported ROIs: right middle/superior temporal gyrus (r=−0.44, p=0.017), right dorsolatertal prefrontal cortex (r=−0.39, p=0.036), and the right hippocampus (r=−0.4, p=0.031). Statistical trends were seen in the bilateral superior medial prefrontal cortex (r=−0.32, p=0.087), and the left anterior superior prefrontal cortex (r=−0.35, p=0.061). Positive correlations were seen with connectivity scores in the bilateral gyrus rectus (r=0.45, p=0.008) and the precuneus region (r=0.42, p=0.022) (Figure 5).
Figure 5.

Scatterplots showing the relationship between resting state connectivity z-scores and category fluency scores. Z-scores increase with decreasing category fluency scores in the right temporal and dorso-lateral pre-prefrontal cortex, with trends in medial superior frontal lobes. Connectivity with the pC/rsp decrease with decreasing category fluency scores in the precuneus and rectus gyrus.
To account for confounding effects of performance in category fluency between groups on differences seen in the DMN, we performed analysis of co-variance (ANCOVA) on mean cluster values for each ROI that demonstrated a correlation between category fluency and DMN z-scores (ROIs noted above). For the seven functional ROIs assessed, when category fluency scores were controlled for, between-group z-scores remained significantly different with a p<0.05 for all ROIs except for the right hippocampus (p=0.11).
DISCUSSION
This study supports recent reports that that resting state BOLD fMRI can identify differences based on risk for AD In comparison with encoding-associated BOLD activations and deactivations, evaluation of network connectivity of low frequency fluctuations more readily detected variability associated with AD risk and provided superior effect sizes for distinguishing risk groups. Medial and dorsolateral prefrontal cortex and temporal lobe structures showed increased DMN connectivity, with decreased connectivity in the precuneus and medial orbital frontal cortex, areas that have been consistently demonstrated to have reduced DMN activity in AD(Greicius et al., 2004; Wang et al., 2006). Worse performance in the high risk group on category fluency testing was associated with increased temporal and prefrontal cortex resting connectivity, while associated with decreased connectivity in the precuneus and orbital frontal lobes. In addition, memory scores correlated with the degree of parietal deactivation during encoding. Overall our findings suggest that abnormalities in resting state networks may represent early functional impairment associated with risk for AD. Resting state BOLD fMRI is a viable candidate for a biomarker of preclinical dementia, and potentially a valuable tool in dementia prediction, prognosis, and preventative treatment development in AD.
It is difficult to ascribe specific pathophysiologic deficits associated with the APOE4 gene directly to differences in low frequency BOLD fluctuation correlations, such as those demonstrated here. And, some authors have cautioned against over interpretation of the resting state in terms of brain dysfunction(Morcom and Fletcher, 2007). Yet, there is strong evidence of a link to pre-clinical AD pathology and APOE4 carrier status. Apolipoprotein (APOE) e4 is a common susceptibility gene for Alzheimer's Disease (AD) and plays a key role in coordinating the mobilization and redistribution of cholesterol, phospholipids and fatty acids. In addition to AD, it has been associated with poorer outcomes in traumatic brain injury(Teasdale et al., 1997), age related cognitive impairment(Deary et al., 2002) and cardiovascular disease(Lenzen et al., 1986). It is implicated in mechanisms of neuronal development, brain plasticity and repair functions (Mahley, 1988; Mahley and Rall, Jr., 2000). There are also many studies that demonstrate a role of the APOE4 gene directly on amyloid processing and toxicity. There is evidence that it promotes formation of the beta-pleated sheet conformation of beta-amyloid (abeta) peptides into amyloid fibers, and inhibits the neurotoxic effect of abeta in an allele-specific manner (E2 > or = E3 > E4)(Jordan et al., 1998; Ma et al., 1996; Strittmatter et al., 1993). And, the APOE4 gene appears to modulate amyloid beta toxicity to vascular endothelium (Folin et al., 2006). More recently, in vivo cortical amyloid imaging shows a strong relationship between fibrillar amyloid deposition and APOE4 carrier status(Reiman et al., 2009). This relationship has also been shown with APOE4 status and CSF Aβ42 levels(Li et al., 2007). Relevant to our findings, it was recently demonstrated that there is a relationship between suppression of the DMN and increased cortical PET PiB amyloid binding uptake in cognitively normal individuals(Hedden et al., 2009; Sperling et al., 2008). And, a recent study showed increased DMN BOLD signal and encoding associated hippocampal signal in young APOE4 carriers (ages 20-35), indicating that resting state fMRI may be a sensitive technique for identifying patterns associated with risk for AD many decades prior to development of AD pathology or clinical symptoms(Filippini et al., 2009). In combination with our finding of the strength of DMN for identifying AD risk, these studies demonstrate that resting state fMRI may represent a biomarker for identifying pre-clinical Alzhemer's disease. Yet to determine this, further pathological correlates and longitudinal studies must be performed to assess the true value of DMN imaging as a predictor and biomarker of dementia of the Alzheimer's type.
Encoding-associated differences in those at increased risk for AD have been well reported in the BOLD fMRI literature (Bondi et al., 2005; Bondi et al., 2004; Bookheimer et al., 2000; Fleisher et al., 2005; Fleisher et al., 2008) The specific task used in our study has previously been reported to distinguish AD and MCI from normal controls (Dickerson et al., 2005; Sperling et al., 2003b), as well as identify cognitively normal individuals at increased risk for AD(Fleisher et al., 2008). In association with the APOE4 genotype, the literature has been somewhat variable, with some studies showing increased BOLD signal in the medial temporal lobe (MTL) during encoding associated with AD risk(Bookheimer et al., 2000; Fleisher et al., 2005), while others have seen decreased or mixed BOLD response to encoding (Bondi et al., 2005; Fleisher et al., 2008; Johnson et al., 2006; Trivedi et al., 2006), and some not finding encoding-associated MTL differences at all(Han et al., 2006). In the current study we first performed standard between-group voxel-wise t-tests evaluating all encoding-associated signal. We did not find differences in MTL structures, which reflects the inconsistency in the literature and perhaps a high level of variability and poor sensitivity of activation fMRI tasks in this population. The group differences we identified were all due to encoding-associated deactivations in the low risk group with relatively increased BOLD signal changes in the high risk group. No differences in activations were found in regions typically associated with this associative encoding task(Sperling et al., 2001).
Differences in encoding-associated deactivations were seen in DMN areas including the postero-medial and right lateral parietal lobes. In these regions the high risk group demonstrated attenuated deactivations compared to the low risk group, similar to differences seen in MCI and AD compared to elderly normal controls(Pihlajamaki et al., 2008; Rombouts et al., 2005). Adequate suppression of these posterior attentional networks appears to be important for successful encoding(Miller et al., 2008; Pihlajamaki et al., 2008). Memory scores are associated with PiB amyloid burden in the parietal and prefrontal cortex of cognitively normal elderly(Pike et al., 2007). Similarly, we found correlations between degree of deactivations and memory scores in the lateral parietal lobe, with statistical trends in the precuneus of the high risk group, suggesting underlying early functional pathology associated with known AD risk factors. This is further supported by the observation that the posterior parietal lobe (including the precuneus region) is one area of early fibrillar amyloid deposition in AD, MCI and even asymptomatic elderly based on pathology and amyloid imaging studies (Braak and Braak, 1991; Braak and Braak, 1997; Buckner et al., 2005; Forsberg et al., 2007; Fripp et al., 2008; Klunk et al., 2004; Mintun et al., 2006; Pike et al., 2007; Thal et al., 2002).
Comparison of resting-state DMN connectivity with the posterior cingulate/retrospenial region demonstrated larger and more widespread areas associated with risk for AD than encoding-associated deactivation analysis. In addition, effect sizes for distinguishing the two risk groups were substantially larger using this technique, with a combined ROI effect size more than twice as large, more than doubling the variance explained by AD risk group membership. Two of the nine regions demonstrating differences based on AD risk showed decreased DMN connectivity in the high risk group. These regions, the precuneus and the gyrus rectus in the medial orbitofrontal cortex, are areas of early amyloid pathology noted in AD(Braak and Braak, 1991; Braak and Braak, 1997; Buckner et al., 2005; Klunk et al., 2004; Thal et al., 2002), MCI (Forsberg et al., 2007; Pike et al., 2007), and elderly individuals with asymptomatic fibrillar amyloid deposition(Mintun et al., 2006; Pike et al., 2007). They also represent regions associated with successful memory performance(Miller et al., 2008; Stuss et al., 1982). Areas of increased connectivity were noted in regions of the medial prefrontal cortex (both superior and middle), right dorso-lateral prefrontal cortex, the right medial and lateral temporal lobe, including the hippocampus, and the left lateral parietal lobe. These are regions previously demonstrated to have decreased connectivity in resting DMN studies of AD, and may represent a disruption of temporal-frontal-parietal functional network connectivity(Greicius et al., 2004; Sorg et al., 2007; Wang et al., 2006). In addition to key roles in memory acquisition, retrieval and working memory(Nyberg et al., 1995; Stuss and Benson, 1984; Wilson et al., 1993), these cortical networks are important for sustained attention during relative periods of cognitive rest(Binder et al., 1999; Corbetta and Shulman, 2002; Raichle et al., 2001; Stuss et al., 1995). Reorganization of these functional networks may explain differences seen in DMN connectivity in high risk individuals.
Our finding of increased DMN connectivity with the posterior parietal seed region in various frontal and temporal cortex areas in high risk individuals is somewhat counterintuitive if we associate decreased DMN activity with impairment in AD. Why would risk for AD connote increased DMN connectivity in similar regions? One possibility is that prefrontal and temporal attentional and working memory networks are necessarily compensating for strained novel encoding abilities associated with risk for AD. For instance, increased fMRI activity in the prefrontal cortex is associated with degree of cognitive task difficulty(Braver et al., 1997; Grady et al., 1996; Grady et al., 2003; Maril et al., 2001), giving evidence of prefrontal recruitment based on cognitive effort. Successful encoding appears to require coordination of neural networks involving the hippocampus, prefrontal cortex, and medial parietal regions; and low performing individuals demonstrate increased prefrontal activation to achieve successful encoding(Miller et al., 2008). Even in early AD, compared to controls, there is increased prefrontal and temporal BOLD activity during semantic and episodic memory tasks associated with successful performance(Grady et al., 2003). This compensatory recruitment of prefrontal cortex may be due to its role in mediating organizational and executive functions (D'Esposito et al., 1995; Fuster, 2000; Stuss and Benson, 1984). This is also supported by the observation that prefrontal recruitment is related to task complexity across various cognitive tasks including working memory, semantic memory and perceptual tasks (Braver et al., 1997; Grady et al., 1996; Maril et al., 2001). In addition, compensatory recruitment has been reported in frontal and temporal regions associated with the APOE4 gene in normal elderly, proposed to be mediated by increased frontal-executive function(Bondi et al., 2005; Bookheimer et al., 2000; Han et al., 2006; Han and Bondi, 2008).
It is not clear, however, how compensatory brain functions during cognitive tasks are related to increased functional connectivity in the DMN during rest. Yet, it is conceivable that the same type of compensatory recruitment seen during challenging cognitive function mirrors redistributed network connectivity during the resting state. As the DMN is believed to function in baseline attention to environmental stimuli, reviewing of past knowledge, and planning of future behaviors(Binder et al., 1999; Raichle et al., 2001), impaired posterior parietal DMN function may result in recruitment of increased anterior connectivity to support these functions. Recall that our study also demonstrated decreased posterior parietal resting connectivity and attenuated encoding-associated deactivation in the setting of this increased prefrontal and temporal functional connectivity during the resting state.
The two risk cohorts in this study differed on scores assessing semantic memory recall processing based on category fluency testing. Increased resting DMN connectivity z-scores in the precuneus and medial orbital frontal cortex were associated with increased category fluency performance. There was also increased resting state connectivity in the ventral and superior prefrontal cortex, and medial and lateral right temporal cortex in association with lower scores in category fluency. These relationships did not account for risk group differences seen in resting DMN connectivity, except perhaps for the right hippocampus. Category fluency testing is impaired with damage to the prefrontal cortex or lateral temporal lobes (Henry et al., 2004), structures important in key functions of semantic knowledge(Baldo and Shimamura, 1998; Levy et al., 2004). In addition, category fluency testing shows early and prominent decline in AD(Henry et al., 2004; Monsch et al., 1992; Monsch et al., 1994), and preclinical AD within two years of diagnosis(Mickes et al., 2007). In APOE4 studies there are no clear reports of category fluency abnormalities distinguishing carriers from non carriers(Bondi et al., 1999). Note that all of our participant's category fluency scores were within normal range for their age and education. The association we found between increased temporal and frontal DMN connectivity and difficulty with semantic memory is consistent with the idea of functional reorganization of fronto-temporal networks in individuals predisposed to developing AD. And, fMRI studies of verbal fluency demonstrate involvement of medial temporal lobe, inferior frontal and retrosplenial cortex(Pihlajamaki et al., 2000). Coupled with our findings of attenuated deactivation in the parietal cortex associated with worse episodic memory performance, these findings suggest preclinical functional relevance of DMN connectivity differences in our population of cognitively normal individuals at high risk for AD.
There are several limitations to this study that should be noted. Most notably, the primary results in this study pertain to resting state low frequency fluctuation in the DMN; however, this was not a true resting state analysis. In order to do a direct comparison with activation state fMRI, all resting data was extracted from interleaved resting blocks and from baseline signal underlying an encoding paradigm. We cannot claim that the encoding paradigm did not influence our findings in this extrapolated resting state analysis. In addition, since there were some differences seen in encoding-related BOLD signal, it is not clear if extraction of this signal may have differentially affected the resting state baseline signal. Yet, task-related BOLD signal is a difference contrast between the baseline signal and the signal change associated with the task. Therefore, when removing the signal related to the task, the basal signal should be left intact. Yet this is difficult to verify. However, similar analyses have been reported that demonstrate that the DMN can be extracted from various cognitive paradigms in both block designs and event-related fMRI(Andrews-Hanna et al., 2007; Buckner et al., 2005; Buckner and Vincent, 2007; Fair et al., 2007; Raichle et al., 2001), suggesting minimal effects of the task on the resting state analysis. Another limitation to consider is, despite being a voxel-wise analysis, this study was not done entirely without a priori knowledge in that we used a predetermined retrosplenial seed region to perform a voxel-wise correlation analysis. Although this technique has been well validated(Andrews-Hanna et al., 2007), an approach without a priori hypotheses regarding expected brain changes, such as independent component analysis(Greicius et al., 2004), would validate our results, which we intend to perform. In addition to these limitations, this study was performed in a relatively small cohort, and replication of this work in larger cohorts with a true resting acquisition will be necessary for validation.
Use of resting state BOLD fMRI may be a more sensitive marker of preclinical functional reorganization in those at risk for AD, potentially representing underlying early AD pathology, as compared to activation and deactivation task-associated fMRI. It has distinct advantages in that the resting state does not suffer from variability related to task performance and may be easier to standardize across study sites. Yet, the resting state of the brain is not possible to standardize, as we cannot control a participant's mental state during during visual fixation or with eyes closed. It is also not dependent on radio-isotopes and may serve as a valuable marker for early AD pathology. However, we must emphasize that our data merely demonstrates an association with known risk factors for AD, and does not establish that those with altered network connectivity will progress to AD. Therefore, we do not imply that this data is useful for predicting subsequent AD, and we have not presented any data demonstrating a known relationship to AD pathology. Prospective longitudinal studies and association with AD pathology are required to address the potential use of these fMRI techniques as biomarkers for predicting future dementia. Cross-sectional studies such as this one are a first step in establishing true biomarkers for preclinical AD which are critical for development of preventative treatments and clinical screening tools and for prediction of impending dementia.
Supplementary Material
Acknowledgements
This research was supported by grant k23 AG24062 from the National Institute on Aging, National Institutes of Health
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Ances BM, Leontiev O, Perthen JE, Liang C, Lansing AE, Buxton RB. Regional differences in the coupling of cerebral blood flow and oxygen metabolism changes in response to activation: implications for BOLD-fMRI. Neuroimage. 2008;39:1510–1521. doi: 10.1016/j.neuroimage.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews-Hanna JR, Snyder AZ, Vincent JL, Lustig C, Head D, Raichle ME, Buckner RL. Disruption of large-scale brain systems in advanced aging. Neuron. 2007;56:924–935. doi: 10.1016/j.neuron.2007.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner J, Friston KJ. Nonlinear spatial normalization using basis functions. Hum Brain Mapp. 1999;7:254–266. doi: 10.1002/(SICI)1097-0193(1999)7:4<254::AID-HBM4>3.0.CO;2-G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldo JV, Shimamura AP. Letter and category fluency in patients with frontal lobe lesions. Neuropsychology. 1998;12:259–267. doi: 10.1037//0894-4105.12.2.259. [DOI] [PubMed] [Google Scholar]
- Binder JR, Frost JA, Hammeke TA, Bellgowan PS, Rao SM, Cox RW. Conceptual processing during the conscious resting state. A functional MRI study. J.Cogn Neurosci. 1999;11:80–95. doi: 10.1162/089892999563265. [DOI] [PubMed] [Google Scholar]
- Bondi MW, Houston WS, Eyler LT, Brown GG. FMRI Evidence of Compensatory mechanisms in Older adults at Genetic Risk for Alzheimer's Disease. Neurology. 2005;64:501–508. doi: 10.1212/01.WNL.0000150885.00929.7E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondi MW, Salmon DP, Galasko D, Thomas RG, Thal LJ. Neuropsychological function and apolipoprotein E genotype in the preclinical detection of Alzheimer's disease. Psychol Aging. 1999;14:295–303. doi: 10.1037//0882-7974.14.2.295. [DOI] [PubMed] [Google Scholar]
- Bondi MW, Houston WS, Eyler LT, Brown GG. Differential BOLD Brain Response to Verbal Paired-Associate Learning by APOE Genotype in Nondemented Older Adults: A Functional MRI Study. Neurobiology of Aging. 2004;25:506. doi: 10.1016/j.neurobiolaging.2005.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookheimer SY, Strojwas MH, Cohen MS, Saunders AM, Pericak-Vance MA, Mazziotta JC, Small GW. Patterns of brain activation in people at risk for Alzheimer's disease. N Engl J Med. 2000;343:450–456. doi: 10.1056/NEJM200008173430701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- Braver TS, Cohen JD, Nystrom LE, Jonides J, Smith EE, Noll DC. A parametric study of prefrontal cortex involvement in human working memory. Neuroimage. 1997;5:49–62. doi: 10.1006/nimg.1996.0247. [DOI] [PubMed] [Google Scholar]
- Brown GG, Eyler Zorrilla LT, Georgy B, Kindermann SS, Wong EC, Buxton RB. BOLD and perfusion response to finger-thumb apposition after acetazolamide administration: differential relationship to global perfusion. J Cereb Blood Flow Metab. 2003;23:829–837. doi: 10.1097/01.WCB.0000071887.63724.B2. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Andrews-Hanna JR, Schacter DL. The Brain's Default Network: Anatomy, Function, and Relevance to Disease. Ann N y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011. [DOI] [PubMed] [Google Scholar]
- Buckner RL, Sepulcre J, Talukdar T, Krienen FM, Liu H, Hedden T, ndrews-Hanna JR, Sperling RA, Johnson KA. Cortical hubs revealed by intrinsic functional connectivity: mapping, assessment of stability, and relation to Alzheimer's disease. J.Neurosci. 2009;29:1860–1873. doi: 10.1523/JNEUROSCI.5062-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, Mintun MA. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Vincent JL. Unrest at rest: default activity and spontaneous network correlations. Neuroimage. 2007;37:1091–1096. doi: 10.1016/j.neuroimage.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Buxton RB, Uludag K, Dubowitz DJ, Liu TT. Modeling the hemodynamic response to brain activation. Neuroimage. 2004;23(Suppl 1):S220–S233. doi: 10.1016/j.neuroimage.2004.07.013. [DOI] [PubMed] [Google Scholar]
- Corbetta M, Shulman GL. Control of goal-directed and stimulus-driven attention in the brain. Nat.Rev.Neurosci. 2002;3:201–215. doi: 10.1038/nrn755. [DOI] [PubMed] [Google Scholar]
- Cox RW. AFNI: software for analysis and visualization of functional magnetic resonance neuroimages. Comput Biomed Res. 1996;29:162–173. doi: 10.1006/cbmr.1996.0014. [DOI] [PubMed] [Google Scholar]
- D'Esposito M, Detre JA, Alsop DC, Shin RK, Atlas S, Grossman M. The neural basis of the central executive system of working memory. Nature. 1995;378:279–281. doi: 10.1038/378279a0. [DOI] [PubMed] [Google Scholar]
- Davis TL, Kwong KK, Weisskoff RM, Rosen BR. Calibrated functional MRI: mapping the dynamics of oxidative metabolism. Proc Natl Acad Sci U S A. 1998;95:1834–1839. doi: 10.1073/pnas.95.4.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deary IJ, Whiteman MC, Pattie A, Starr JM, Hayward C, Wright AF, Carothers A, Whalley LJ. Cognitive change and the APOE epsilon 4 allele. Nature. 2002;418:932. doi: 10.1038/418932a. [DOI] [PubMed] [Google Scholar]
- Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, Bertram L, Mullin K, Tanzi RE, Blacker D, Albert MS, Sperling RA. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology. 2005;65:404–411. doi: 10.1212/01.wnl.0000171450.97464.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edison P, Archer HA, Hinz R, Hammers A, Pavese N, Tai YF, Hotton G, Cutler D, Fox N, Kennedy A, Rossor M, Brooks DJ. Amyloid, hypometabolism, and cognition in Alzheimer disease: an [11C]PIB and [18F]FDG PET study. Neurology. 2007;68:501–508. doi: 10.1212/01.wnl.0000244749.20056.d4. [DOI] [PubMed] [Google Scholar]
- Fair DA, Schlaggar BL, Cohen AL, Miezin FM, Dosenbach NU, Wenger KK, Fox MD, Snyder AZ, Raichle ME, Petersen SE. A method for using blocked and event-related fMRI data to study “resting state” functional connectivity. Neuroimage. 2007;35:396–405. doi: 10.1016/j.neuroimage.2006.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippini N, MacIntosh BJ, Hough MG, Goodwin GM, Frisoni GB, Smith SM, Matthews PM, Beckmann CF, Mackay CE. Distinct patterns of brain activity in young carriers of the APOE-{varepsilon}4 allele. Proc.Natl.Acad.Sci.U.S.A. 2009;106:7209–7214. doi: 10.1073/pnas.0811879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher AS, Houston WS, Eyler LT, Frye S, Jenkins C, Thal LJ, Bondi MW. Identification of Alzheimer disease risk by functional magnetic resonance imaging. Arch Neurol. 2005;62:1881–1888. doi: 10.1001/archneur.62.12.1881. [DOI] [PubMed] [Google Scholar]
- Fleisher AS, Podraza KM, Bangen KJ, Taylor C, Sherzai A, Sidhar K, Liu TT, Dale AM, Buxton RB. Cerebral perfusion and oxygenation differences in Alzheimer's disease risk. Neurobiol Aging. 2008;4:4. doi: 10.1016/j.neurobiolaging.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folin M, Baiguera S, Guidolin D, Di Liddo R, Grandi C, De Carlo E, Nussdorfer GG, Parnigotto PP. Apolipoprotein-E modulates the cytotoxic effect of beta-amyloid on rat brain endothelium in an isoform-dependent specific manner. Int J Mol Med. 2006;17:821–826. [PubMed] [Google Scholar]
- Forsberg A, Engler H, Almkvist O, Blomquist G, Hagman G, Wall A, Ringheim A, Langstrom B, Nordberg A. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.03.029. [DOI] [PubMed] [Google Scholar]
- Fripp J, Bourgeat P, Acosta O, Raniga P, Modat M, Pike KE, Jones G, O'Keefe G, Masters CL, Ames D, Ellis KA, Maruff P, Currie J, Villemagne VL, Rowe CC, Salvado O, Ourselin S. Appearance modeling of (11)C PiB PET images: Characterizing amyloid deposition in Alzheimer's disease, mild cognitive impairment and healthy aging. Neuroimage. 2008 doi: 10.1016/j.neuroimage.2008.07.053. [DOI] [PubMed] [Google Scholar]
- Fuster JM. Executive frontal functions. Exp.Brain Res. 2000;133:66–70. doi: 10.1007/s002210000401. [DOI] [PubMed] [Google Scholar]
- Grady CL, Horwitz B, Pietrini P, Mentis MJ, ungerleider LG, Rapoport SI, Haxby JV. Effect of task difficulty on cerebral blood flow during perceptual matching of faces. Human Brain Mapping. 1996;4:227–239. doi: 10.1002/(SICI)1097-0193(1996)4:4<227::AID-HBM1>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Grady CL, McIntosh AR, Beig S, Keightley ML, Burian H, Black SE. Evidence from functional neuroimaging of a compensatory prefrontal network in Alzheimer's disease. J Neurosci. 2003;23:986–993. doi: 10.1523/JNEUROSCI.23-03-00986.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greicius MD, Srivastava G, Reiss AL, Menon V. Default-mode network activity distinguishes Alzheimer's disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci U S A. 2004;101:4637–4642. doi: 10.1073/pnas.0308627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SD, Bondi MW. Revision of the apolipoprotein E compensatory mechanism recruitment hypothesis. Alzheimers.Dement. 2008;4:251–254. doi: 10.1016/j.jalz.2008.02.006. [DOI] [PubMed] [Google Scholar]
- Han SD, Houston WS, Jak AJ, Eyler LT, Nagel BJ, Fleisher AS, Brown GG, Corey-Bloom J, Salmon DP, Thal LJ, Bondi MW. Verbal paired-associate learning by APOE genotype in non-demented older adults: fMRI evidence of a right hemispheric compensatory response. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2005.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedden T, van Dijk K, Becker A, Mehta A, Sperling RA, Buckner RL, Johnson K. Relationship of amyloid deposition to default netwrok functional connectivity in non-demented adults; Human Amyloid Imaging conference; Seattle, WA: 2009. [Google Scholar]
- Hedges LV. Distribution theory for Glass's estimator of effect size and related estimators. Journal of Educational Statistics. 1981;6:107–128. [Google Scholar]
- Henry JD, Crawford JR, Phillips LH. Verbal fluency performance in dementia of the Alzheimer's type: a meta-analysis. Neuropsychologia. 2004;42:1212–1222. doi: 10.1016/j.neuropsychologia.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr., Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, Knopman DS, Boeve BF, Klunk WE, Mathis CA, Petersen RC. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131:665–680. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Jones K, Holman BL, Becker JA, Spiers PA, Satlin A, Albert MS. Preclinical prediction of Alzheimer's disease using SPECT. Neurology. 1998;50:1563–1571. doi: 10.1212/wnl.50.6.1563. [DOI] [PubMed] [Google Scholar]
- Johnson SC, Schmitz TW, Trivedi MA, Ries ML, Torgerson BM, Carlsson CM, Asthana S, Hermann BP, Sager MA. The influence of Alzheimer disease family history and apolipoprotein E epsilon4 on mesial temporal lobe activation. J Neurosci. 2006;26:6069–6076. doi: 10.1523/JNEUROSCI.0959-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan J, Galindo MF, Miller RJ, Reardon CA, Getz GS, LaDu MJ. Isoform-specific effect of apolipoprotein E on cell survival and beta-amyloid-induced toxicity in rat hippocampal pyramidal neuronal cultures. J Neurosci. 1998;18:195–204. doi: 10.1523/JNEUROSCI.18-01-00195.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline RB. Supplemental chapter on multivariate effect size estimation. 2004 Retrieved October 29, 2008 from http://www.apa.org/books/resoources/kline.
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Lenzen HJ, Assmann G, Buchwalsky R, Schulte H. Association of apolipoprotein E polymorphism, low-density lipoprotein cholesterol, and coronary artery disease. Clin.Chem. 1986;32:778–781. [PubMed] [Google Scholar]
- Levy DA, Bayley PJ, Squire LR. The anatomy of semantic knowledge: medial vs. lateral temporal lobe. Proc.Natl.Acad.Sci.U.S.A. 2004;101:6710–6715. doi: 10.1073/pnas.0401679101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Sokal I, Quinn JF, Leverenz JB, Brodey M, Schellenberg GD, Kaye JA, Raskind MA, Zhang J, Peskind ER, Montine TJ. CSF tau/Abeta42 ratio for increased risk of mild cognitive impairment: a follow-up study. Neurology. 2007;69:631–639. doi: 10.1212/01.wnl.0000267428.62582.aa. [DOI] [PubMed] [Google Scholar]
- Lustig C, Snyder AZ, Bhakta M, O'Brien KC, McAvoy M, Raichle ME, Morris JC, Buckner RL. Functional deactivations: change with age and dementia of the Alzheimer type. Proc Natl Acad Sci U S A. 2003;100:14504–14509. doi: 10.1073/pnas.2235925100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Brewer HB, Jr., Potter H. Alzheimer A beta neurotoxicity: promotion by antichymotrypsin, ApoE4; inhibition by A beta-related peptides. Neurobiol Aging. 1996;17:773–780. doi: 10.1016/0197-4580(96)00112-1. [DOI] [PubMed] [Google Scholar]
- Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- Mahley RW, Rall SC., Jr. Apolipoprotein E: far more than a lipid transport protein. Annu Rev Genomics Hum Genet. 2000;1:507–537. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- Maril A, Wagner AD, Schacter DL. On the tip of the tongue: an event-related fMRI study of semantic retrieval failure and cognitive conflict. Neuron. 2001;31:653–660. doi: 10.1016/s0896-6273(01)00396-8. [DOI] [PubMed] [Google Scholar]
- Mickes L, Wixted JT, Fennema-Notestine C, Galasko D, Bondi MW, Thal LJ. Progressive impairment on neuropsychological tasks in a longitudinal study of preclinical Alzheimer's disease. Neuropsychology. 2007;21:696–705. doi: 10.1037/0894-4105.21.6.696. [DOI] [PubMed] [Google Scholar]
- Miller SL, Celone K, DePeau K, Diamond E, Dickerson BC, Rentz D, Pihlajamaki M, Sperling RA. Age-related memory impairment associated with loss of parietal deactivation but preserved hippocampal activation. Proc Natl Acad Sci U S A. 2008;105:2181–2186. doi: 10.1073/pnas.0706818105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer's disease. Ann.Neurol. 1997;42:85–94. doi: 10.1002/ana.410420114. [DOI] [PubMed] [Google Scholar]
- Mintun MA, Larossa GN, Sheline YI, Dence CS, Lee SY, Mach RH, Klunk WE, Mathis CA, DeKosky ST, Morris JC. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- Monsch AU, Bondi MW, Butter N, Paulsen JS, Salmon DP, Brugger P, Swenson MR. A comparison of category and letter fluency in alzheimer's disease and huntington's disease. Neuropsychology. 1994;8:25–30. [Google Scholar]
- Monsch AU, Bondi MW, Butters N, Salmon DP, Katzman R, Thal LJ. Comparisons of verbal fluency tasks in the detection of dementia of the Alzheimer type. Arch Neurol. 1992;49:1253–1258. doi: 10.1001/archneur.1992.00530360051017. [DOI] [PubMed] [Google Scholar]
- Morcom AM, Fletcher PC. Does the brain have a baseline? Why we should be resisting a rest. Neuroimage. 2007;37:1073–1082. doi: 10.1016/j.neuroimage.2007.06.019. [DOI] [PubMed] [Google Scholar]
- Nyberg L, Tulving E, Habib R, Nilsson LG, Kapur S, Houle S, Cabeza R, McIntosh AR. Functional brain maps of retrieval mode and recovery of episodic information. Neuroreport. 1995;7:249–252. [PubMed] [Google Scholar]
- Persson J, Lind J, Larsson A, Ingvar M, Sleegers K, Van BC, Adolfsson R, Nilsson LG, Nyberg L. Altered deactivation in individuals with genetic risk for Alzheimer's disease. Neuropsychologia. 2008;46:1679–1687. doi: 10.1016/j.neuropsychologia.2008.01.026. [DOI] [PubMed] [Google Scholar]
- Pihlajamaki M, DePeau KM, Blacker D, Sperling RA. Impaired medial temporal repetition suppression is related to failure of parietal deactivation in Alzheimer disease. Am J Geriatr Psychiatry. 2008;16:283–292. doi: 10.1097/JGP.0b013e318162a0a9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihlajamaki M, Tanila H, Hanninen T, Kononen M, Laakso M, Partanen K, Soininen H, Aronen HJ. Verbal fluency activates the left medial temporal lobe: a functional magnetic resonance imaging study. Ann.Neurol. 2000;47:470–476. [PubMed] [Google Scholar]
- Pike KE, Savage G, Villemagne VL, Ng S, Moss SA, Maruff P, Mathis CA, Klunk WE, Masters CL, Rowe CC. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer's disease. Brain. 2007;130:2837–2844. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. A default mode of brain function. Proc.Natl.Acad.Sci.U.S.A. 2001;98:676–682. doi: 10.1073/pnas.98.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, Thibodeau SN, Osborne D. Preclinical evidence of Alzheimer's disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med. 1996;334:752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc Natl Acad Sci U S A. 2004;101:284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, Ayutyanont N, Keppler J, Reeder SA, Langbaum JB, Alexander GE, Klunk WE, Mathis CA, Price JC, Aizenstein HJ, DeKosky ST, Caselli RJ. Fibrillar amyloid-{beta} burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc.Natl.Acad.Sci.U.S.A. 2009 doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rombouts SA, Barkhof F, Goekoop R, Stam CJ, Scheltens P. Altered resting state networks in mild cognitive impairment and mild Alzheimer's disease: an fMRI study. Hum Brain Mapp. 2005;26:231–239. doi: 10.1002/hbm.20160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg C, Riedl V, Muhlau M, Calhoun VD, Eichele T, Laer L, Drzezga A, Forstl H, Kurz A, Zimmer C, Wohlschlager AM. Selective changes of resting-state networks in individuals at risk for Alzheimer's disease. Proc Natl Acad Sci U S A. 2007;104:18760–18765. doi: 10.1073/pnas.0708803104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R, Chua E, Cocchiarella A, Rand-Giovannetti E, Poldrack R, Schacter DL, Albert M. Putting names to faces: successful encoding of associative memories activates the anterior hippocampal formation. Neuroimage. 2003a;20:1400–1410. doi: 10.1016/S1053-8119(03)00391-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Bates JF, Cocchiarella AJ, Schacter DL, Rosen BR, Albert MS. Encoding novel face-name associations: a functional MRI study. Hum Brain Mapp. 2001;14:129–139. doi: 10.1002/hbm.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Dickerson B, Bates JF, Chua EF, Cocchiarella AJ, Rentz DM, Rosen BR, Schacter DL, Albert MS. fMRI studies of associative encoding in young and elderly controls and mild Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2003b;74:44–50. doi: 10.1136/jnnp.74.1.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, O'Keefe J, O'Brien J, Deluca A, Pihlajamaki M, Rentz D, Marshall GA, Eng E, Rastegar S, Carmasin JS, Becker JA, Johnson KA. Cortical amyloid deposition assoiciated with impaired default network activity in non-demented older individuals; Human Amyloid Imagin Conference; Chicago, IL; 2008. p. 17. [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuss DT, Benson DF. Neuropsychological studies of the frontal lobes. Psychol.Bull. 1984;95:3–28. [PubMed] [Google Scholar]
- Stuss DT, Kaplan EF, Benson DF, Weir WS, Chiulli S, Sarazin FF. Evidence for the involvement of orbitofrontal cortex in memory functions: an interference effect. J Comp Physiol Psychol. 1982;96:913–925. doi: 10.1037/0735-7036.96.6.913. [DOI] [PubMed] [Google Scholar]
- Stuss DT, Shallice T, Alexander MP, Picton TW. A multidisciplinary approach to anterior attentional functions. Ann.N.Y.Acad.Sci. 1995;769:191–211. doi: 10.1111/j.1749-6632.1995.tb38140.x. [DOI] [PubMed] [Google Scholar]
- Talairach J, Tournoux P. Co-Planar stereotaxic atlas of the human brain. Thieme Medical; New York: 1988. [Google Scholar]
- Teasdale GM, Nicoll JA, Murray G, Fiddes M. Association of apolipoprotein E polymorphism with outcome after head injury. Lancet. 1997;350:1069–1071. doi: 10.1016/S0140-6736(97)04318-3. [DOI] [PubMed] [Google Scholar]
- Thal DR, Rub U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- Trivedi MA, Schmitz TW, Ries ML, Torgerson BM, Sager MA, Hermann BP, Asthana S, Johnson SC. Reduced hippocampal activation during episodic encoding in middle-aged individuals at genetic risk of Alzheimer's disease: a cross-sectional study. BMC Med. 2006;4:1. doi: 10.1186/1741-7015-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner R, Howseman A, Rees GE, Josephs O, Friston K. Functional magnetic resonance imaging of the human brain: data acquisition and analysis. Experimental Brain Research. 1998;123:5–12. doi: 10.1007/s002210050538. [DOI] [PubMed] [Google Scholar]
- Wang K, Liang M, Wang L, Tian L, Zhang X, Li K, Jiang T. Altered functional connectivity in early Alzheimer's disease: a resting-state fMRI study. Hum Brain Mapp. 2007;28:967–978. doi: 10.1002/hbm.20324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zang Y, He Y, Liang M, Zhang X, Tian L, Wu T, Jiang T, Li K. Changes in hippocampal connectivity in the early stages of Alzheimer's disease: evidence from resting state fMRI. Neuroimage. 2006;31:496–504. doi: 10.1016/j.neuroimage.2005.12.033. [DOI] [PubMed] [Google Scholar]
- Wilson FA, Scalaidhe SP, Goldman-Rakic PS. Dissociation of object and spatial processing domains in primate prefrontal cortex. Science. 1993;260:1955–1958. doi: 10.1126/science.8316836. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.