

SUMMARY
It is intuitively obvious that the ability of a cell to repair DNA damage is saturable, either by limitation of enzymatic activities, the time allotted to achieve their function, or both. However, very little is known regarding the mechanisms that establish such a threshold. Here we demonstrated that the CUL4A ubiquitin ligase restricts the cellular repair capacity by orchestrating the concerted actions of nucleotide excision repair (NER) and the DNA damage-responsive G1/S checkpoint through selective degradation of the DDB2 and XPC DNA damage sensors and the p21/CIP1/WAF1 checkpoint effector. We generated Cul4a conditional knockout mice and observed that skin-specific Cul4a ablation dramatically increased resistance to UV-induced skin carcinogenesis. Our findings reveal that wild-type cells do not operate at their full DNA repair potential, underscore the critical role of CUL4A in establishing the cellular DNA repair threshold, and highlight the potential augmentation of cellular repair proficiency by pharmacological CUL4A inhibition.
INTRODUCTION
Eukaryotic cells respond to ultraviolet (UV) irradiation by induction of the nucleotide excision repair (NER) pathway, which identifies and removes damaged DNA, as well as activation of the DNA damage checkpoint to halt cell cycle progression, thus allowing time for NER action. NER is the major DNA repair pathway by which cells remove helix-distorting DNA damage caused by UV irradiation and chemical mutagens (Friedberg et al., 2006). It is a multistep process that employs over 30 proteins to carry out the distinct steps of recognizing DNA damage, incising the 5′ and 3′ ends of the lesion to remove damaged DNA, filling in the gap with DNA polymerase, and attaching the newly synthesized DNA to the parental DNA via DNA ligase activity (reviewed in (Friedberg et al., 2006; Sancar, 1996)). NER consists of two pathways with distinct DNA strand specificities: the transcription-coupled repair pathway (TCR) which removes lesions from DNA strands transcribed by RNA polymerase II, and the global genomic repair pathway (GGR) which repairs damage on the non-transcribed strand of expressed genes as well as from inactive chromatin (reviewed in (Hanawalt, 2002)). The NER process has been studied extensively, and the components essential to perform the excision and repair reactions have been defined by in vitro reconstitution using recombinant proteins and damaged DNA templates (Aboussekhra et al., 1995; Araujo et al., 2000; Mu et al., 1996; Mu et al., 1995). However, the regulatory mechanisms governing DNA damage recognition inside the cell are not well understood.
NER factors involved in the GGR pathway of DNA damage recognition include XPA-RPA, XPC-HR23B, and the heterodimeric, damage-specific DNA binding proteins consisting of DDB1 and DDB2 subunits. Among these DNA damage sensors, DDB1-DDB2 exhibit the highest affinity (designated UV-DDB activity) for UV-induced cyclobutane pyrimidine dimers (CPDs) and 6-4 photoproducts (6-4PPs) (Batty et al., 2000). Mutations in DDB2 are responsible for xeroderma pigmentosum complementation group E (XP-E) cases, which are characterized by defects in GGR-mediated removal of damaged DNA and predisposition to skin cancer (Wittschieben and Wood, 2003). Following UV irradiation, DDB1 and DDB2 immediately accumulate on damaged DNA, and are subsequently ubiquitinated and degraded by the CUL4A ubiquitin ligase (Chen et al., 2006; Fitch et al., 2003a; Groisman et al., 2003; Rapic-Otrin et al., 2002). CUL4A is also responsible for the turnover of DDB2 under normal growth conditions (Chen et al., 2001; Nag et al., 2001), which leads to an overall decrease in UV-DDB activity (Chen et al., 2001). However, the physiological role of CUL4A in DNA repair and tumorigenesis remains largely elusive.
The CUL4A ubiquitin ligase is a multimeric complex: the C-terminus of CUL4A interacts with the RING finger protein RBX1/ROC1/Hrt1 (referred to as RBX1 hereafter) to recruit the E2 ubiquitin-conjugating enzyme, and the CUL4A N-terminus interacts with DDB1 (acting as an adaptor), which in turn binds to DDB1, CUL4A associated factors (DCAFs) as specific substrate receptors (Angers et al., 2006; He et al., 2006; Higa et al., 2006; Jin et al., 2006)(reviewed in (Lee and Zhou, 2007)). CUL4B, the other CUL4 family member, shares extensive sequence homology and redundant functions with CUL4A in maintaining cell growth and mediating ubiquitination of certain CUL4 targets (Higa et al., 2003; Hu et al., 2004). However, recent studies also revealed unique functions of CUL4B in assembling atypical ubiquitin ligase complexes to degrade sex steroid hormone receptors (Ohtake et al., 2007). Additionally, CUL4B mutations were identified as the causal genetic defects underlying X-linked mental retardation (Tarpey et al., 2007; Zou et al., 2007).
In addition to NER activation, UV irradiation also elicits the DNA damage checkpoint to halt cell cycle progression and allow time for the stepwise NER reactions. The G1/S checkpoint is triggered upon detection of UV-induced DNA damage by the ATR kinase, which activates the Chk1/2 kinases to phosphorylate and stabilize p53. This leads to transcriptional activation of the cyclin-dependent kinase inhibitor p21 to prevent S phase entry and replication of damaged DNA. While NER and the DNA damage checkpoint are capable of operating independently of each other, little is known about how eukaryotic cells coordinate these two damage response pathways to achieve efficient and timely removal of DNA photolesions and restore genomic integrity.
Here we describe the generation of conditional floxed alleles of Cul4a in mice. While germline-deleted Cul4a mice were viable, healthy, and exhibited no developmental abnormalities, skin-specific deletion of Cul4a resulted in dramatically enhanced resistance to UVB-induced skin carcinogenesis. Consistently, Cul4a deletion led to increased accumulation of both DDB2 and XPC DNA damage sensors, as well as the p21 damage checkpoint effector, resulting in enhanced GGR activity and reinforced UV-responsive G1 DNA damage checkpoint. These studies revealed the coordinated regulation of NER and G1/S checkpoint pathways by the CUL4A ubiquitin ligase, and underscored the benefit of CUL4A inhibition in enhancing the removal of DNA lesions and suppression of tumorigenesis.
RESULTS
Conditional Inactivation of the Cul4a Gene
To investigate the physiological role of CUL4A in DNA repair and tumorigenesis, we generated conditional Cul4a knockout mice using the Cre/lox strategy. Exons 17–19, which span the essential cullin homology domain and neddylation site, were floxed by homologous recombination in embryonic stem (ES) cells (Fig. 1A–C). Homozygous Cul4af/f mice were healthy and phenotypically indistinguishable from their wild-type littermates. Cul4a−/− mice were subsequently generated by interbreeding Cul4af/f mice with EIIA-Cre transgenic mice, a general deleter strain with the adenovirus EIIA promoter that drives Cre-mediated recombination in a wide range of tissues, including the germ cells (Lakso et al., 1996). The absence of full-length CUL4A protein was confirmed by Western blotting in multiple tissues and MEFs (Fig. 1D). A C-terminal truncated CUL4A (designated CUL4A(Δ)) was detectable, but at markedly reduced levels (8%) compared to that of wild-type CUL4A, and failed to interact with RBX1 for recruitment of the E2 ubiquitin-conjugating enzyme (Fig. 1E). Therefore, our Cul4a exon 17–19 knockout strain represents a complete loss-of-function allele.
Figure 1. Generation of floxed and null Cul4a alleles in mice.
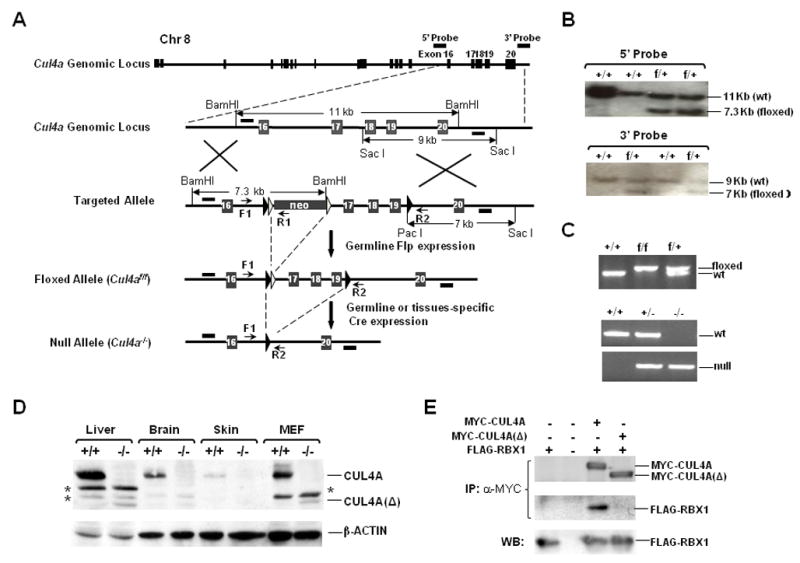
A, Genomic structure of the Cul4a gene and the targeted alleles. LoxP sites (filled arrowheads) were introduced into the intron regions flanking exons 17–19 of the targeting construct for homologous recombination in ES cells. Frt recombination sites (open arrowheads) were engineered for removal of the neomycin selection marker by germline Flp expression following crossbreeding with ACT-FLPe transgenic mice. Arrows, PCR primers (F, R1, R2) for genotyping; bars, 5′ and 3′ probes for Southern blotting. B, Southern blotting of ES clones. DNA was digested with restriction enzymes and hybridized to the probes as indicated in (A). +/+, wild-type; f/+, floxed heterozygous allele. C, PCR genotyping of tail DNA from the indicated wild-type (+), floxed (f) and null Cul4a (−) alleles. D, Western blotting of dissected tissues and MEF cells derived from Cul4a+/+ and Cul4a−/− mice. “*”, non-specific species. E, Deletion of exons 17–19 of Cul4a abrogates the ability of CUL4A(Δ) to recruit RBX1. 293T cells were transiently transfected with the indicated plasmids. Binding of MYC-tagged CUL4A or CUL4A(Δ) to FLAG-tagged RBX1 was assessed by co-immunoprecipitation (α-MYC) and Western blotting with the respective antibodies.
Surprisingly, Cul4a−/− mice were viable and displayed no overt developmental abnormalities throughout their life span. This result is in stark contrast with the embryonic lethality of the published Cul4a exon 1 deletion strain (Li et al., 2002). However, it is noteworthy that the exon 1 targeting construct also abolished expression of the Pcid2 gene that resides on the complementary stand adjacent to Cul4a exon 1. Pcid2 encodes a protein with a PCI domain, which is conserved among the essential subunits of the 26S proteasome, COP9 signalosome, and translation initiation factor 3 complexes (Hofmann and Bucher, 1998). The exon 1 targeting allele by Li et al. inadvertently deleted a 529 base pair region upstream of the first Pcid2 exon, leaving only 4 base pairs upstream of the ATG translation initiation codon (Fig. 2A). Indeed, silencing of Pcid2 expression in primary MEFs by lentiviral Pcid2 shRNA resulted in a rapid loss of viability (Fig. 2B–C). In contrast, growth arrest or cell death was not observed in Cul4a−/−MEFs (Fig. S1A). Therefore, the embryonic lethality phenotype observed by Li et al. is likely due to the coincidental abrogation of the essential Pcid2 gene that resides on the complementary strand adjacent to Cul4a.
Figure 2. The published Cul4a knockout mice by Li et al. inadvertently deleted the essential Pcid2 gene located adjacent to Cul4a on the complementary strand.
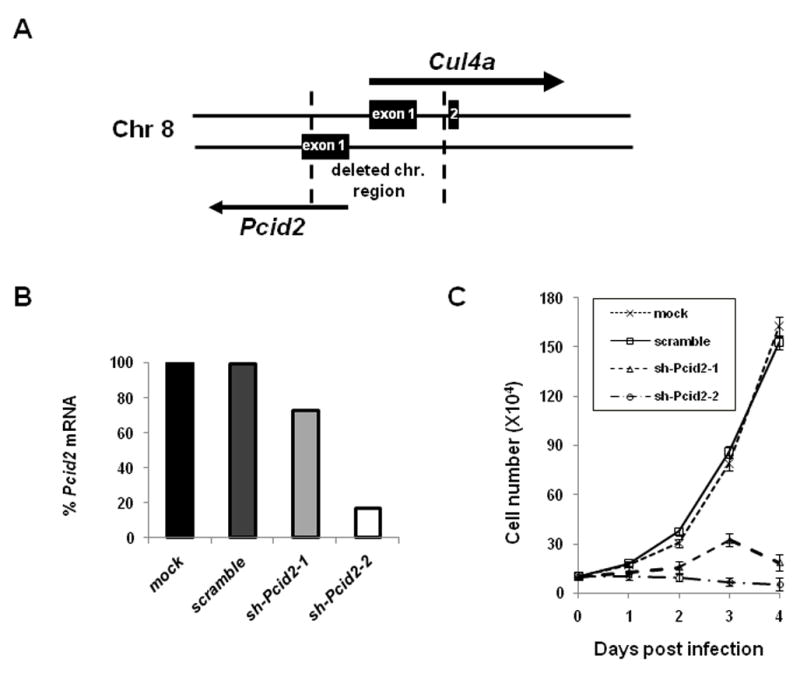
A, Schematic diagram of the Cul4a knockout allele by Li et al. that deletes both Cul4a and Pcid2. The DNA fragment between the broken lines denotes the deleted genomic fragment (Li et al., 2002) that spans both the first exon of Cul4a and the promoter and transcription initiation site of Pcid2. B–C, Silencing of Pcid2 by lentiviral shRNAs resulted in rapid loss of cell viability. Primary wild-type MEF cells were infected with one of two lentiviral shRNAs (shPcid2-1 and shPcid2-2), scrambled shPcid2 (scm-Pcid2), or mocked-infected for 48 hours, and evaluated for Pcid2 mRNA levels by real-time qPCR. Cell growth was evaluated by counting cell numbers at 2, 3, and 4 days following lentiviral infection.
In mammals, the two Cul4 genes are broadly co-expressed and assemble structurally similar ubiquitin ligases. Therefore, CUL4B could complement the loss of CUL4A and thus ensure survival of the Cul4a−/− mice. Accordingly, silencing of Cul4b in Cul4a−/− MEFs led to a dramatic reduction of BrdU incorporation and loss of cell viability (Fig. S1), consistent with what we previously observed with Ddb1−/− MEFs (Cang et al., 2006), and in accordance with the fact that both CUL4 proteins must be inactivated to abolish CUL4 ubiquitin ligase activity (Higa et al., 2003; Hu et al., 2004).
Enhanced DDB2 and XPC stability, UV-DDB and GGR activities in Cul4a−/− cells
Enforced transgenic overexpression of DDB2 conferred increased NER and skin cancer resistance, while Ddb2 null mice were more susceptible to UV-induced skin cancer (Alekseev et al., 2005; Itoh et al., 2004; Yoon et al., 2004). Previous studies identified DDB2 both as a target and a recognition component for other substrates of the CUL4-DDB1 ubiquitin ligase (Angers et al., 2006; Chen et al., 2001; Groisman et al., 2003; He et al., 2006; Higa et al., 2006; Jin et al., 2006; Nag et al., 2001). Indeed, we observed higher steady-state levels of DDB2 protein in the epidermis of Cul4a−/− mice and MEFs, and the half-life of DDB2 was prolonged in primary MEFs derived from E13.5 Cul4a−/− embryos as determined by pulse-chase analysis (Fig. 3A–C). To determine the relative contributions of CUL4A and CUL4B in DDB2 degradation, lentiviral shRNA against mouse Cul4b was generated for infection of early passage (P3) primary Cul4af/f and Cul4a−/− MEFs. shCul4b effectively reduced over 93% of mouse CUL4B, yet had only a marginal effect on DDB2 levels (Fig. 3B, Fig. S2A). In contrast, Cul4a knockout or abrogation of both Cul4a and Cul4b resulted in a 3- to 4.5-fold upregulation of DDB2 protein (Fig. 3B, Fig. S2A). DDB2 mRNA levels remained unaltered or even slightly decreased in response to Cul4a knockout or Cul4b silencing (Fig. S2A).
Figure 3. CUL4A controls the stability of DDB2, XPC and p21.
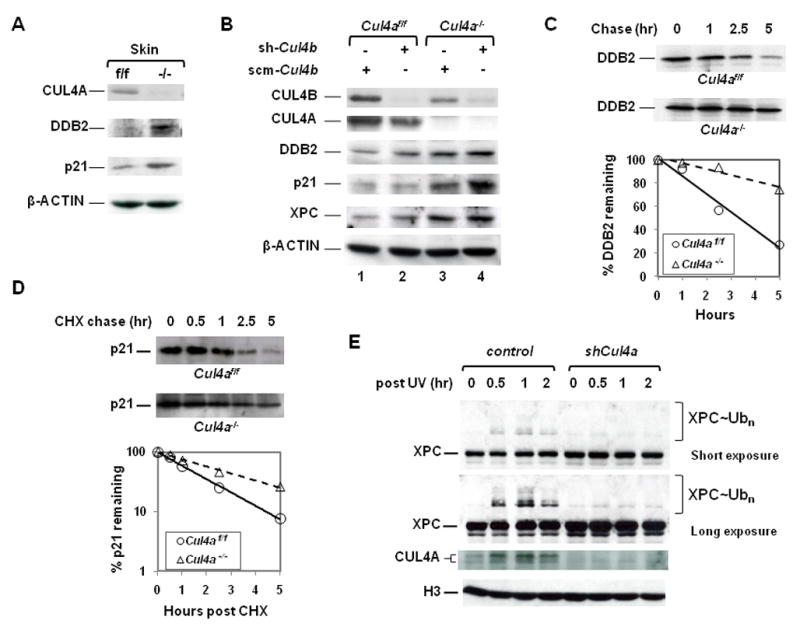
A, Increased DDB2 and p21 levels in Cul4a−/− skin, as determined by Western blotting. B, Accumulation of endogenous DDB2, p21 and XPC protein in primary Cul4a−/−, Cul4bk/d and Cul4a−/− Cul4bk/d MEFs compared to Cul4af/f MEFs. Cul4af/f and Cul4a−/− MEFs were infected with FUGW lentivirus containing either shRNA for mouse Cul4b (sh-Cul4b) or scramble control (scm-Cul4b) and subjected to Western blotting with indicated antibodies. k/d, knockdown. C–D, Pulse-chase and cyclohexamide chase analysis to determine the half-life of endogenous DDB2 and p21 in Cul4a−/− and Cul4af/f MEFs. The percentage of p21 remaining was graphed on a logarithmic scale at the time points indicated. CHX, cyclohexamide. E, CUL4A deficiency impaired XPC ubiquitination post-UV. HCT116 cells were infected with lentiviral shCUL4A or control FUGW for 48 hours, UV irradiated at 10J/m2 and harvested at the indicated time points. The chromatin-bound extracts were prepared and subjected to immunoblotting with the indicated antibodies. Native and neddylated CUL4A species were indicated. H3, histone H3.
XPC is a rate-limiting factor for the DNA damage recognition step of GGR, and also is a direct ubiquitination target of CUL4A-DDB1-DDB2-RBX1 E3 ligase (Friedberg et al., 2006; Sugasawa et al., 2005). Interestingly, under normal conditions, the steady-state levels of XPC protein, but not mRNA, increased 4.4-fold upon Cul4a deletion in primary MEF cells, while Cul4b knockdown had a marginal effect on XPC accumulation (Fig. 3B, compare lanes 1–3, Fig. S2C). Therefore, Cul4a−/− MEFs accumulate not only DDB2, but also the rate-limiting XPC DNA damage sensor for damage recognition and GGR.
Upon UV irradiation, the DDB1-DDB2 complex immediately recognizes damaged DNA and helps recruit the XPC-HR23B complex to DNA damage sites through direct binding to XPC (Fitch et al., 2003b). XPC is then ubiquitinated on DNA by the CUL4A-DDB1-DDB2-RBX1 E3 ligase, and subsequently de-ubiquitinated, rather than undergoing proteasomal-dependent degradation (Sugasawa et al., 2005). The physiological role of XPC ubiquitination on GG-NER has yet to be determined. We have assessed the effect of CUL4A ablation on XPC binding to chromatin and modification by ubiquitin upon UV irradiation. Since the available XPC antibodies were unable to detect ubiquitinated mouse XPC in MEFs, we depleted CUL4A by lentiviral shRNA in human HCT116 cells for these studies. As shown in Fig. 3E, the association of XPC with chromatin increases upon CUL4A silencing by lentiviral shRNA, consistent with higher XPC levels available in the absence of Cul4a (Fig. 3B). Furthermore, CUL4A depletion led to a dramatic inhibition of XPC ubiquitination on chromatin post-UV (Fig. 3E). Collectively, these results underscore a specific role of CUL4A in controlling XPC levels under normal conditions and XPC ubiquitination on chromatin in response to UV irradiation. Moreover, CUL4A is primarily responsible for governing the ubiquitination of DDB2 and XPC, while CUL4B plays a lesser, if any, role in these processes.
Recognition of UV-damaged DNA on chromatin is limited by the cellular pool of available DDB2 (Fitch et al., 2003a; Tang and Chu, 2002). Accumulation of DDB2 in Cul4a−/− MEFs led to increased UV-DDB activity compared to Cul4a f/f MEFs (Fig. 4A), and approximately 20% enhancement of GGR activity for both CPDs and 6-4 PPs, the most common lesions repaired by NER, as measured by ELISA-based GGR assay using anti-CPD and anti-6-4PP antibodies (Fig. 4B). Knockdown of endogenous DDB2 in Cul4a−/− MEFs by SMART pool siRNA (Dharmacon) effectively reduced GGR of CPDs by 50% (Fig. S3), underscoring the critical role of DDB2 upregulation upon Cul4a abrogation in GGR enhancement. In contrast, depletion of Cul4b by shRNA had no discernable effect on GGR efficiency (Fig. 4B). These results demonstrate that CUL4A abrogation permits effective elevation of GGR capacity beyond the threshold attainable in wild-type cells.
Figure 4. Loss of CUL4A enhanced UV-DDB and NER activities, and reinforced the UV-responsive G1/S checkpoint to ensure genomic integrity.
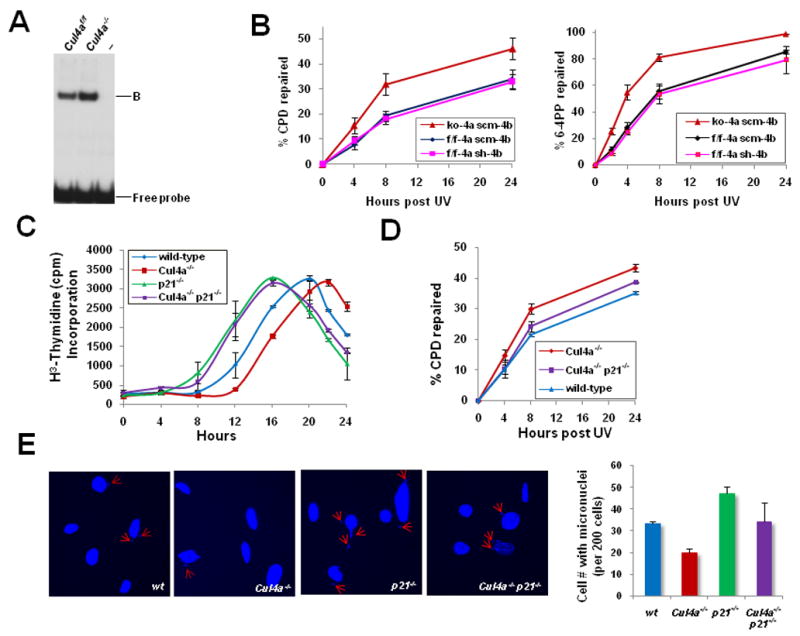
A, UV-DDB activity of Cul4af/f and Cul4a−/− MEFs was determined by electrophoretic mobility shift assay. “B”, DDB-DNA complex. B, GGR activities of CPD and 6-4PP removal were measured in primary Cul4a−/− (ko-4a scm-4b), Cul4bk/d (f/f-4a sh-4b), and control Cul4af/f (f/f-4a scm-4b) MEFs. ko, knockout; k/d, knockdown; scm, scramble. The data represent the mean and standard deviation of three experiments, with each sample point performed in triplicate. C, Primary MEFs of the indicated genotypes were synchronized, UV-irradiated and collected at the indicated time points for [3H]-thymidine incorporation assay. D, GGR for removal of CPDs was measured in primary wild-type, Cul4a−/−, and Cul4a−/− p21−/− MEFs as described in Fig. 4C. E, Micronuclei formation in wild-type, Cul4a−/−, p21−/− and Cul4a−/− p21−/− MEFs, as measured by staining with DAPI at 48 hours post-UV. Error bars represent standard deviations of 3 independent experiments.
Stabilization of p21 and reinforcement of the G1/S DNA damage checkpoint in primary Cul4a−/− MEFs
While the NER and DNA damage checkpoint pathways can operate independently of each other, the concerted action of both pathways is required to ensure efficient and timely removal of DNA lesions. It is currently unclear whether a mechanism exists to coordinate these pathways. We showed recently that p21 was stabilized in response to homozygous deletion of DDB1 (Cang et al., 2006). Indeed, p21 protein, but not mRNA, also accumulated in Cul4a−/− skin and primary MEF cells as a result of increased half-life of p21 upon CUL4A deletion (Fig. 3A, 3B, 3D, Fig. S2B). Silencing of Cul4b by RNAi had little effect, underscoring the predominant role of CUL4A in controlling the stability of not only DDB2 and XPC but also p21 (Fig. 3B). While simultaneous inactivation of both Cul4a and Cul4b resulted in further accumulation of p21, direct comparison of p21 degradation with that of individual Cul4a or Cul4b inactivation could not be made due to the rapid growth arrest of Cul4a−/− Cul4bk/d MEFs (Fig. S1). Consistently, p21 is physically present in the CUL4A-DDB1 complex by co-immunoprecipitation (Fig. S4). During the preparation of this manuscript, several groups published that the CUL4A-DDB1-Cdt2 ubiquitin ligase complex, known to ubiquitinate the DNA replication licensing factor Cdt1, also targeted p21 for degradation (Abbas et al., 2008; Kim et al., 2008; Nishitani et al., 2008). Collectively, these findings corroborate our earlier report of p21 stabilization in Ddb1−/− cells (Cang et al., 2006), and provide the biochemical basis for the increased p21 levels observed in Cul4a−/− cells.
In response to DNA damage, p21 is transcriptionally upregulated through the p53-dependent G1/S DNA damage checkpoint pathway to arrest the cell cycle in G1, thus allowing time for the DNA repair apparatus to remove DNA photolesions (Sancar et al., 2004). p21 is subsequently degraded by both CUL1 and CUL4 ubiquitin ligase complexes, as well as through a ubiquitin-independent pathway (Bendjennat et al., 2003; Lee et al., 2006; Nishitani et al., 2008). To determine whether the posttranslational stabilization of p21 in Cul4a−/− MEFs enforces the p21-dependent DNA damage checkpoint, we measured the kinetics of S phase entry in G0/G1-synchronized primary Cul4a−/− and wild-type MEFs post-UV by [3H]-thymidine incorporation. As shown in Fig. 4C, wild-type MEFs began to exit G1 and incorporate [3H]-thymidine at 12 hours post-UV, and the majority entered S phase at 16–20 hours. However, Cul4a−/− MEFs were delayed by 4–6 hours in S phase entry post-UV. To validate that p21 is the primary downstream target responsible for delaying S phase entry in Cul4a−/− MEFs, Cul4a−/− p21−/− MEFs were generated by crossing Cul4a−/− and p21−/− mice. Indeed, the deletion of p21 effectively abrogated the prolonged G1 arrest associated with CUL4A loss, and resulted in accelerated S phase entry. 18% of Cul4a−/− p21−/− MEFs progressed into S phase at 8 hours post-UV, similar to what was seen in p21−/− MEFs (Fig. 4C). Flow cytometry analysis further confirmed that Cul4a−/− MEFs were delayed at least 6 hours in S phase progression compared to wild-type MEFs, while deletion of p21 effectively eliminated the G1 block in Cul4a−/− MEFs (Fig. S5). Therefore, stabilization of p21 in Cul4a−/− cells enforced the DNA damage-responsive G1 block to prevent premature S phase entry and to allot additional time for the NER machinery to identify and remove DNA photolesions.
Deletion of the p21 gene or knockdown of p21 by lentiviral shRNA in Cul4a−/− MEFs led to an 8–11% reduction of NER activity in CPD removal compared to Cul4a−/− MEFs, underscoring the benefit of p21 upregulation in the removal of UV-induced photolesions (Fig. 4D, data not shown). It is noteworthy that while the GGR enhancement by p21 is modest in the 24-hour repair assay following a single physiological UV dose (10 J/m2), it would be effectively magnified with repeated UV assault, higher UV doses or prolonged irradiation. The advantage of the prolonged G1 arrest in Cul4a−/− MEFs is further underscored by their enhanced ability to maintain genome integrity following UV exposure, as Cul4a−/− MEFs had 40% fewer micronuclei than wild-type MEFs induced by UV irradiation (Fig. 4E). Deletion of p21 abolished the Cul4a−/− MEFs’ ability to prevent perturbation of chromosomal segregation during mitosis. Therefore, stabilization of p21 in Cul4a−/− cells is largely responsible for the enforced DNA damage-responsive G1 block and delayed cell cycle progression into S phase post-UV.
Skin-specific Cul4a knockout mice are resistant to UVB-induced skin carcinogenesis
To evaluate the physiological impact of simultaneous upregulation of NER and the G1/S DNA damage checkpoint upon CUL4A abrogation, we compared skin-specific tamoxifen-inducible Cul4a knockout mice with Cul4af/f control animals in their susceptibility to UV-induced skin carcinogenesis. Cul4a was first deleted in a shaved area of dorsal skin of the Cul4af/f K14-CreERTAM mice by topical administration of tamoxifen, and followed by daily UV-B irradiation. The Cul4af/f control mice started to develop skin tumors at week 27 of UV-B treatment. By week 48, all Cul4af/f mice developed tumors on the shaved dorsal skin area (Fig. 5A). Histological and immunohistochemical analysis confirmed that the tumors in control Cul4af/f mice were mostly squamous cell carcinomas (SCC) derived from epidermal origin (Fig. 5B–F). Strikingly, all but one of the Cul4af/f K14-CreERTAM mice remained SCC-free (Fig. 5A). Non-SCC tumors (e.g. spindle cell neoplasm) also developed and, as expected, a similar incidence was observed in both Cul4af/f and Cul4af/f K14-CreERTAM groups since K14-CreERTAM is not expressed in these cell types (data not shown). The dramatic difference in the onset of SCCs versus non-SCC tumors suggests the specific association of tumor resistance with the loss of CUL4A. Of note, Cul4af/f and Cul4af/f K14-CreERTAM skin displayed similar apoptotic indices, indicating that resistance to SCC is unlikely due to increased susceptibility to cell death in skin-specific Cul4a knockout animals (data not shown). Together, these results provide compelling evidence that the abrogation of CUL4A confers enhanced protection against skin carcinogenesis in response to UV irradiation.
Figure 5. Skin-specific Cul4a knockout mice are resistant to UV-B-induced skin carcinogenesis.
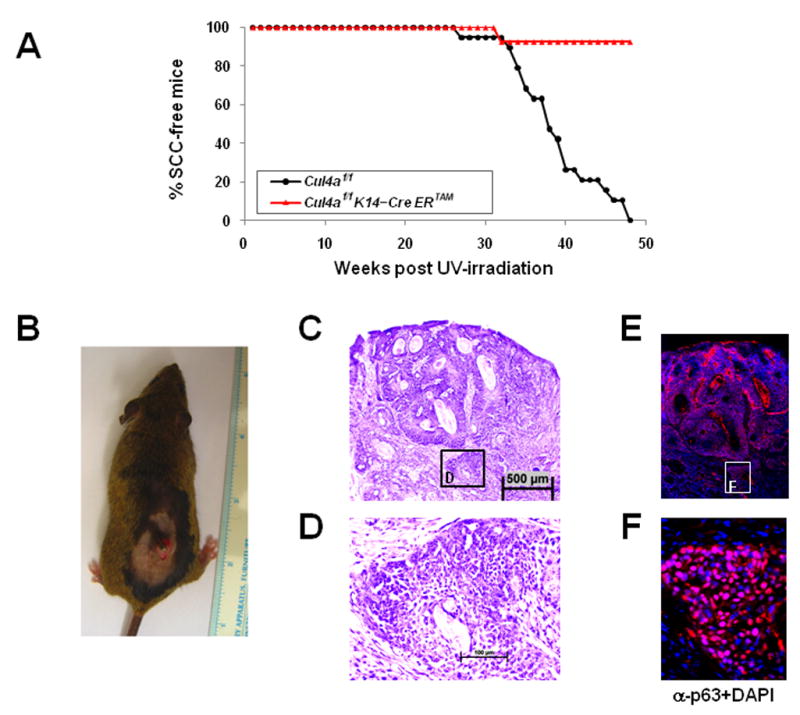
A, Kaplan-Meier curve of the onset of squamous cell carcinomas (SCC) in mice of indicated genotypes after chronic UV-B exposure. Skin-specific Cul4af/f K14-CreERTAM mice (n=13) remained SCC-free, while Cul4af/f (n=19) littermates developed SCC between 27 to 48 weeks after daily UV-B exposure (log-rank test, P<0.00001). B–F, Physical, histopathological and immunohistochemical examination of a UV-B-induced SCC from a representative Cul4af/f mouse. The tumor is a typical SCC, as revealed by H&E staining ((C) and (D)) and positive immunostaining of the basal epidermal marker p63 (counter stained with DAPI in (E) and (F)).
DISCUSSION
Amplification or overexpression of Cul4a has been frequently observed in breast cancer, hepatocarcinomas and other tumor types, consistent with a reduction of UV-DDB activity (Chen et al., 1998; Chen et al., 2001; Yasui et al., 2002). The CUL4A ubiquitin ligase was previously shown to target both soluble and chromatin-bound DDB2 for destruction under normal conditions as well as post-UV irradiation (Chen et al., 2006; Chen et al., 2001; Nag et al., 2001). In this study, we demonstrated that Cul4a null MEFs accumulate both DDB2 and XPC DNA damage sensors and dramatically enhanced GGR activity in CPD and 6-4PP removal. It is noteworthy that CUL4A-DDB1 can also assemble with the Cockayne syndrome protein CSA to form a distinct ubiquitin ligase. Groisman and colleagues showed that in the human cervical carcinoma HeLa cell line, the CUL4A-DDB1-CSA E3 ligase targets a second Cockayne syndrome protein CSB for ubiquitination and degradation several hours after UV irradiation, which may play a role in the resumption of transcription after TCR (Groisman et al., 2006). In contrast, CSB and CSA levels appeared unaltered under the same conditions post-UV in primary wild-type and Cul4a−/− MEF cells (data not shown). Furthermore, UVB-irradiated Cul4af/f and Cul4af/f K14-CreERTAM mice displayed no appreciable signs of apoptosis. Therefore, the enhanced resistance to UV-induced skin carcinogenesis in skin-specific Cul4a knockout mice is attributed primarily to elevated GGR. Future studies should determine a role for the CUL4A-DDB1-CSA-RBX1 ubiquitin ligase in transcription-coupled repair.
DNA damage recognition by DDB1-DDB2 and XPC is dictated by the distorted/kinked DNA structure resulting from CPDs or 6-4PPs DNA adducts, rather than specific DNA sequences. Such structures may also present during normal DNA metabolism. In addition, the DDB1-DDB2 complex was also found capable of binding abasic sites or mismatches, damages that are not repaired by NER (Hanawalt, 2002; Wittschieben et al., 2005). We propose that the inhibitory role of CUL4A serves to restrict the full potential activity of GGR. This is likely a default mechanism to prevent gratuitous NER actions during normal DNA metabolism and interference with other DNA repair pathways (Hanawalt, 2002; Wittschieben et al., 2005). However, under acute UV irradiation conditions in which massive DNA damage is incurred, disengaging the default mechanism that limits NER is likely to augment the NER capacity and reduce mutation frequency. Consistently, skin-specific knockout of Cul4a rendered the animals resistant to UVB-induced skin carcinogenesis, revealing the physiological role of CUL4A in NER and tumorigenesis.
Our Cul4a knockout in skin and MEF cells also revealed the accumulation of the cyclin-dependent kinase inhibitor p21, constituting a second mechanism for enhanced tumor suppression in Cul4a knockouts. p21 is a critical downstream effector of the G1/S DNA damage checkpoint pathway. Following UV irradiation, Cul4a−/− cells harboring high basal levels of p21 sustained an extended arrest period compared to Cul4af/f cells (Fig. 4C, Fig. S5). The enforced UV-responsive G1/S checkpoint is especially beneficial for repairing DNA adducts that induce moderate strand distortions, such as CPDs that are intrinsically difficult for damage sensors to detect. Failure to remove such DNA adducts during NER can give rise to mutations, as error-prone translesion synthesis must be evoked to resolve DNA damage-mediated replication blocks in S phase (Friedberg et al., 2006). Together with the increase in DDB2 and XPC, Cul4a−/− cells are effectively protected against DNA damage by both establishing a higher threshold for repair activity and ensuring that the checkpoints are sufficiently robust to give the cell time to enact repair (Fig. 6). This is validated by the hyper-resistance to UVB-induced squamous cell carcinomas of the skin-specific Cul4af/f K14-CreERTAM mice.
Fig. 6. Proposed role of CUL4A in establishing threshold for DNA repair and tumor suppression.
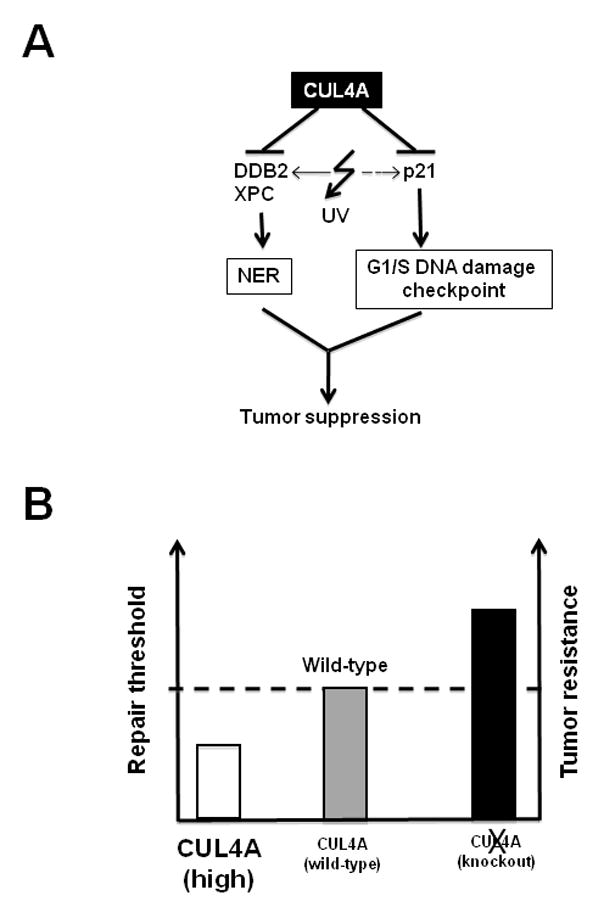
A, CUL4A ubiquitin ligase coordinately suppresses the NER and the G1/S DNA damage checkpoint pathways through targeted degradation of DDB2, XPC, and p21 respectively. B, CUL4A abrogation elevates NER capacity and G1/S DNA damage checkpoint response beyond the threshold attainable in wild-type cells.
The role of p21 in NER has been an issue of debate, as it was reported to either positively or negatively regulate repair, or have no effect on NER (Adimoolam et al., 2001; Bendjennat et al., 2003; Cooper et al., 1999; LaBaer et al., 1997; Maeda et al., 2002; McDonald et al., 1996; Pan et al., 1995; Shivji et al., 1998; Stivala et al., 2001). Given that the threshold levels of p21 dictate whether it stimulates or inhibits cyclin-dependent kinases during the cell cycle, the cellular or physiological conditions likely specify the nature of p21 action (reviewed in (Sherr and Roberts, 1999) and references therein). As such, the seemingly conflicting conclusions on the effect of p21 on NER may result from the use of different in vitro or in vivo experimental systems, cell types, or assay methods to measure NER activities. Our studies with primary (passage 3–4) MEFs derived from Cul4a and/or p21 knockout embryos indicated a positive effect of p21 on NER under these conditions, which is consistent with the increased skin cancer protection we observed in skin-specific Cul4a knockout mice. Whether p21-mediated stimulation of NER is the result of an enforced UV-responsive DNA damage checkpoint, or direct involvement of stepwise NER reactions, or both awaits further investigation.
The CUL4-DDB1 ubiquitin ligase was recently shown to maintain genomic stability through regulation of Cdt1 degradation. Silencing of DDB1 in human cancer cell lines led to DNA re-replication and double strand breaks (Lovejoy et al., 2006), and our acute deletion of DDB1 in primary MEF cells resulted in accumulation of micronuclei and overduplication of centrosome (Cang et al., 2006). In contrast, our primary Cul4a−/− MEFs showed no observable perturbation of DNA replication or cell cycle progression (Fig. S1). This is likely due to the compensatory activity of the CUL4B ubiquitin ligase in mediating Cdt1 turnover (Higa et al., 2003; Hu et al., 2004; Jin et al., 2006; Wertz et al., 2004). Consistently, both germline and skin-specific Cul4a knockout mice remained healthy throughout their lifespan, while poised to markedly elevate GGR activity beyond the repair threshold in wild-type animals. Interestingly, despite the redundant functions shared by CUL4A and 4B in maintaining growth and survival, CUL4B plays little, if any, role in overall NER activity and G1/S checkpoint response (Fig. 3–4). It is noteworthy that the NER machinery is primarily responsible for repairing strand-distorting DNA damage not only induced by UV, but also by chemical carcinogens including polycyclic aromatic hydrocarbons derived from tobacco smoking and environmental pollutants (reviewed in (Luch, 2005)). We propose that pharmacological inhibition of the CUL4A-DDB1 ubiquitin ligase may provide an attractive new strategy for prevention or therapeutic intervention of UV radiation- or chemical carcinogen-induced tumorigenesis.
EXPERIMENTAL PROCEDURES
Cells and Plasmids
Primary MEF, HCT116 and 293T cells were cultured in DMEM medium containing 10% FBS and 1% glutamine. The CUL4A(Δ) mutant (deletion of residues 585–727) was generated by two consecutive PCR reactions and cloned into the pCDNA3 vector. shRNAs for murine Cul4b and Pcid2 as well as scrambled controls were subcloned into the FUGW lentiviral vector and expressed under the U6 promoter. Recombinant shCul4b and shPcid2 lentiviruses were generated for infection of primary Cul4af/f and Cul4a−/− MEF cells (details in supplemental data). Lentiviral shRNA for human CUL4A was described previously (Zhang et al., 2003).
Generation of floxed Cul4a Mice
To generate the floxed Cul4a allele, the 129 genomic library (Invitrogen) was screened with a Cul4a cDNA probe. Five BAC clones were identified, and clone #297L07 was used to generate the final targeting construct. The targeting vector was generated by placing a LoxP site and the pSV-FLP-Cre neomycin resistance cassette 1.1 kb upstream of exon 17 and another LoxP site 300 bps downstream of exon 19. The 5′ and 3′ homologous arms were 3 kb and 5 kb, respectively, and generated by EXL long-range PCR (Stratagene) from the BAC clone. Following electroporation and G418 selection, clones that underwent homologous recombination were identified by Southern blotting (Fig. 1A–B, details in supplemental data). Two positive clones (#379 and #395) after homologous recombination were used to generate chimeric mice by blastocyst injection. The deleted Cul4a allele was created by germline-induced deletion of exons 17 to 19 by crossing floxed Cul4a mice with the EIIA-Cre transgenic line (The Jackson Laboratory). Transmission of the targeted loci was confirmed by Southern blotting and PCR. All mutant animals were backcrossed to C57BL/6J for 12 generations.
Analyses of Cul4a knockout tissues and MEF cells
Proteins were extracted from tissues of Cul4a−/−, Cul4af/f or wild-type littermates using the CelLytic™ MT Mammalian Tissue Lysis/Extraction Reagent (Sigma-Aldrich, St. Louis, MO). Tissue and MEF extracts were immunoblotted with antibodies against DDB2 (Cell Signaling Technology, Danvers, MA), XPC (gift of Dr. Kaoru Sugasawa, RIKEN), p21 (Santa Cruz Biotechnology, Santa Cruz, CA) and β-actin (Sigma, St. Louis, MO). For the damaged DNA binding assay of UV-DDB activity, 2 μg of whole cell extract was incubated with a 32P-labeled DNA probe that was UV-irradiated at 5000 J/m2, and binding was assessed by electrophoretic mobility shift assay (Chen et al., 2001). For micronuclei measurement, MEF cells were irradiated with UV at 10 J/m2, cultured for 48 hours, fixed with 4% paraformaldehyde and subjected to 4′,6-diamidino-2-phenylindole (DAPI) staining. The GGR ELISA assay was performed as previously described (Chen et al., 2006). Primary MEFs were synchronized in G0/G1 by serum starvation for 72 hours, UV-irradiated (10 J/m2) at 3h post-release into serum containing medium, and collected at the indicated time points in Fig. 4C for analysis of cell cycle progression by [3H]-thymidine incorporation assay (Gitig and Koff, 2000).
Mouse UVB irradiation and skin carcinogenesis
Eight- to twelve-week-old littermate mice topically treated with tamoxifen/DMSO solution to induce deletion of the CUL4A allele. In three independent experiments, a total of thirteen CUL4A knockout mice and nineteen Cul4af/f littermates were irradiated at an initial dose of 2,500 J/m2 per day and gradually increased to a maximal dose of 3,500 J/m2 per day. Individual mice were irradiated until skin tumors appeared or for a maximum of 48 weeks, corresponding to the week at which all Cul4af/f control mice developed skin tumors. Statistical significance was measured using the log-rank test. The tumors were further analyzed by H&E and immunohistochemistry staining (details in supplemental data).
Supplementary Material
Acknowledgments
We thank John Petrini, Selina Chen-Kiang, and Jennifer Lee for helpful discussions/critical reading of the manuscript, the reviewers for suggesting key experiments on XPC, E. Fuchs, S. Dymecki, B. Hempstead, N. Sugasawa, A. Sancar, and T. Mori for mouse strains and reagents, and J. Zhou for technical support. P.Z. is a Leukemia and Lymphoma Society Scholar. This work was supported by NIH grants CA098210 and CA118085 (P.Z.), and ES014482 (L.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbas T, Sivaprasad U, Terai K, Amador V, Pagano M, Dutta A. PCNA-dependent regulation of p21 ubiquitylation and degradation via the CRL4Cdt2 ubiquitin ligase complex. Genes Dev. 2008;22:2496–2506. doi: 10.1101/gad.1676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protic M, Hubscher U, Egly JM, Wood RD. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell. 1995;80:859–868. doi: 10.1016/0092-8674(95)90289-9. [DOI] [PubMed] [Google Scholar]
- Adimoolam S, Lin CX, Ford JM. The p53-regulated cyclin-dependent kinase inhibitor, p21 (cip1, waf1, sdi1), is not required for global genomic and transcription-coupled nucleotide excision repair of UV-induced DNA photoproducts. J Biol Chem. 2001;276:25813–25822. doi: 10.1074/jbc.M102240200. [DOI] [PubMed] [Google Scholar]
- Alekseev S, Kool H, Rebel H, Fousteri M, Moser J, Backendorf C, de Gruijl FR, Vrieling H, Mullenders LH. Enhanced DDB2 expression protects mice from carcinogenic effects of chronic UV-B irradiation. Cancer Res. 2005;65:10298–10306. doi: 10.1158/0008-5472.CAN-05-2295. [DOI] [PubMed] [Google Scholar]
- Angers S, Li T, Yi X, MacCoss MJ, Moon RT, Zheng N. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature. 2006;443:590–593. doi: 10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- Araujo SJ, Tirode F, Coin F, Pospiech H, Syvaoja JE, Stucki M, Hubscher U, Egly JM, Wood RD. Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Dev. 2000;14:349–359. [PMC free article] [PubMed] [Google Scholar]
- Batty D, Rapic’-Otrin V, Levine AS, Wood RD. Stable binding of human XPC complex to irradiated DNA confers strong discrimination for damaged sites. J Mol Biol. 2000;300:275–290. doi: 10.1006/jmbi.2000.3857. [DOI] [PubMed] [Google Scholar]
- Bendjennat M, Boulaire J, Jascur T, Brickner H, Barbier V, Sarasin A, Fotedar A, Fotedar R. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell. 2003;114:599–610. doi: 10.1016/j.cell.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Cang Y, Zhang J, Nicholas SA, Bastien J, Li B, Zhou P, Goff SP. Deletion of DDB1 in mouse brain and lens leads to p53-dependent elimination of proliferating cells. Cell. 2006;127:929–940. doi: 10.1016/j.cell.2006.09.045. [DOI] [PubMed] [Google Scholar]
- Chen LC, Manjeshwar S, Lu Y, Moore D, Ljung BM, Kuo WL, Dairkee SH, Wernick M, Collins C, Smith HS. The human homologue for the Caenorhabditis elegans cul-4 gene is amplified and overexpressed in primary breast cancers. Cancer Res. 1998;58:3677–3683. [PubMed] [Google Scholar]
- Chen X, Zhang J, Lee J, Lin PS, Ford JM, Zheng N, Zhou P. A kinase-independent function of c-Abl in promoting proteolytic destruction of damaged DNA binding proteins. Mol Cell. 2006;22:489–499. doi: 10.1016/j.molcel.2006.04.021. [DOI] [PubMed] [Google Scholar]
- Chen X, Zhang Y, Douglas L, Zhou P. UV-damaged DNA-binding Proteins Are Targets of CUL-4A-mediated Ubiquitination and Degradation. J Biol Chem. 2001;276:48175–48182. doi: 10.1074/jbc.M106808200. [DOI] [PubMed] [Google Scholar]
- Cooper MP, Balajee AS, Bohr VA. The C-terminal domain of p21 inhibits nucleotide excision repair In vitro and In vivo. Mol Biol Cell. 1999;10:2119–2129. doi: 10.1091/mbc.10.7.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch ME, Cross IV, Turner SJ, Adimoolam S, Lin CX, Williams KG, Ford JM. The DDB2 nucleotide excision repair gene product p48 enhances global genomic repair in p53 deficient human fibroblasts. DNA Repair (Amst) 2003a;2:819–826. doi: 10.1016/s1568-7864(03)00066-1. [DOI] [PubMed] [Google Scholar]
- Fitch ME, Nakajima S, Yasui A, Ford JM. In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. J Biol Chem. 2003b;278:46906–46910. doi: 10.1074/jbc.M307254200. [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede WW, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2. Washington, D.C: ASM press; 2006. [Google Scholar]
- Gitig DM, Koff A. Cdk pathway: cyclin-dependent kinases and cyclin-dependent kinase inhibitors. Methods Mol Biol. 2000;142:109–123. doi: 10.1385/1-59259-053-5:109. [DOI] [PubMed] [Google Scholar]
- Groisman R, Kuraoka I, Chevallier O, Gaye N, Magnaldo T, Tanaka K, Kisselev AF, Harel-Bellan A, Nakatani Y. CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes Dev. 2006;20:1429–1434. doi: 10.1101/gad.378206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–367. doi: 10.1016/s0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- Hanawalt PC. Subpathways of nucleotide excision repair and their regulation. Oncogene. 2002;21:8949–8956. doi: 10.1038/sj.onc.1206096. [DOI] [PubMed] [Google Scholar]
- He YJ, McCall CM, Hu J, Zeng Y, Xiong Y. DDB1 functions as a linker to recruit receptor WD40 proteins to CUL4-ROC1 ubiquitin ligases. Genes Dev. 2006;20:2949–2954. doi: 10.1101/gad.1483206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higa LA, Mihaylov IS, Banks DP, Zheng J, Zhang H. Radiation-mediated proteolysis of CDT1 by CUL4-ROC1 and CSN complexes constitutes a new checkpoint. Nat Cell Biol. 2003;5:1008–1015. doi: 10.1038/ncb1061. [DOI] [PubMed] [Google Scholar]
- Higa LA, Wu M, Ye T, Kobayashi R, Sun H, Zhang H. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat Cell Biol. 2006;8:1277–1283. doi: 10.1038/ncb1490. [DOI] [PubMed] [Google Scholar]
- Hofmann K, Bucher P. The PCI domain: a common theme in three multiprotein complexes. Trends Biochem Sci. 1998;23:204–205. doi: 10.1016/s0968-0004(98)01217-1. [DOI] [PubMed] [Google Scholar]
- Hu J, McCall CM, Ohta T, Xiong Y. Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat Cell Biol . 2004;6:1003–1009. doi: 10.1038/ncb1172. Epub 2004 Sep 1026. [DOI] [PubMed] [Google Scholar]
- Itoh T, Cado D, Kamide R, Linn S. DDB2 gene disruption leads to skin tumors and resistance to apoptosis after exposure to ultraviolet light but not a chemical carcinogen. Proc Natl Acad Sci U S A. 2004;101:2052–2057. doi: 10.1073/pnas.0306551101. Epub 2004 Feb 2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Arias EE, Chen J, Harper JW, Walter JC. A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell. 2006;23:709–721. doi: 10.1016/j.molcel.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Kim Y, Starostina NG, Kipreos ET. The CRL4Cdt2 ubiquitin ligase targets the degradation of p21Cip1 to control replication licensing. Genes Dev. 2008;22:2507–2519. doi: 10.1101/gad.1703708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Zeng SX, Lu H. UV Induces p21 rapid turnover independently of ubiquitin and Skp2. J Biol Chem. 2006;281:26876–26883. doi: 10.1074/jbc.M605366200. [DOI] [PubMed] [Google Scholar]
- Lee J, Zhou P. DCAFs, the Missing Link of the CUL4-DDB1 Ubiquitin Ligase. Mol Cell. 2007;26:775–780. doi: 10.1016/j.molcel.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Li B, Ruiz JC, Chun KT. CUL-4A Is Critical for Early Embryonic Development. Mol Cell Biol. 2002;22:4997–5005. doi: 10.1128/MCB.22.14.4997-5005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovejoy CA, Lock K, Yenamandra A, Cortez D. DDB1 maintains genome integrity through regulation of Cdt1. Mol Cell Biol. 2006;26:7977–7990. doi: 10.1128/MCB.00819-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luch A. Nature and nurture - lessons from chemical carcinogenesis. Nat Rev Cancer. 2005;5:113–125. doi: 10.1038/nrc1546. [DOI] [PubMed] [Google Scholar]
- Maeda T, Chong MT, Espino RA, Chua PP, Cao JQ, Chomey EG, Luong L, Tron VA. Role of p21(Waf-1) in regulating the G1 and G2/M checkpoints in ultraviolet-irradiated keratinocytes. J Invest Dermatol. 2002;119:513–521. doi: 10.1046/j.1523-1747.2002.01828.x. [DOI] [PubMed] [Google Scholar]
- McDonald ER, 3rd, Wu GS, Waldman T, El-Deiry WS. Repair Defect in p21 WAF1/CIP1 −/− human cancer cells. Cancer Res. 1996;56:2250–2255. [PubMed] [Google Scholar]
- Mu D, Hsu DS, Sancar A. Reaction mechanism of human DNA repair excision nuclease. J Biol Chem. 1996;271:8285–8294. doi: 10.1074/jbc.271.14.8285. [DOI] [PubMed] [Google Scholar]
- Mu D, Park CH, Matsunaga T, Hsu DS, Reardon JT, Sancar A. Reconstitution of human DNA repair excision nuclease in a highly defined system. J Biol Chem. 1995;270:2415–2418. doi: 10.1074/jbc.270.6.2415. [DOI] [PubMed] [Google Scholar]
- Nag A, Bondar T, Shiv S, Raychaudhuri P. The xeroderma pigmentosum group e gene product ddb2 is a specific target of cullin 4a in mammalian cells. Mol Cell Biol. 2001;21:6738–6747. doi: 10.1128/MCB.21.20.6738-6747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitani H, Shiomi Y, Iida H, Michishita M, Takami T, Tsurimoto T. CDK inhibitor p21 is degraded by a PCNA coupled Cul4-DDB1Cdt2 pathway during S phase and after UV irradiation. J Biol Chem. 2008;283:29045–29052. doi: 10.1074/jbc.M806045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H, Takahashi S, Kouzmenko A, Nohara K, Chiba T, et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature. 2007;446:562–566. doi: 10.1038/nature05683. [DOI] [PubMed] [Google Scholar]
- Pan ZQ, Reardon JT, Li L, Flores-Rozas H, Legerski R, Sancar A, Hurwitz J. Inhibition of nucleotide excision repair by the cyclin-dependent kinase inhibitor p21. J Biol Chem. 1995;270:22008–22016. doi: 10.1074/jbc.270.37.22008. [DOI] [PubMed] [Google Scholar]
- Rapic-Otrin V, McLenigan MP, Bisi DC, Gonzalez M, Levine AS. Sequential binding of UV DNA damage binding factor and degradation of the p48 subunit as early events after UV irradiation. Nucleic Acids Res. 2002;30:2588–2598. doi: 10.1093/nar/30.11.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancar A. DNA excision repair. Annu Rev Biochem. 1996;65:43–81. doi: 10.1146/annurev.bi.65.070196.000355. [DOI] [PubMed] [Google Scholar]
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Shivji MK, Ferrari E, Ball K, Hubscher U, Wood RD. Resistance of human nucleotide excision repair synthesis in vitro to p21Cdn1. Oncogene. 1998;17:2827–2838. doi: 10.1038/sj.onc.1202352. [DOI] [PubMed] [Google Scholar]
- Stivala LA, Riva F, Cazzalini O, Savio M, Prosperi E. p21(waf1/cip1)-null human fibroblasts are deficient in nucleotide excision repair downstream the recruitment of PCNA to DNA repair sites. Oncogene. 2001;20:563–570. doi: 10.1038/sj.onc.1204132. [DOI] [PubMed] [Google Scholar]
- Sugasawa K, Okuda Y, Saijo M, Nishi R, Matsuda N, Chu G, Mori T, Iwai S, Tanaka K, Hanaoka F. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell. 2005;121:387–400. doi: 10.1016/j.cell.2005.02.035. [DOI] [PubMed] [Google Scholar]
- Tang J, Chu G. Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein. DNA Repair (Amst) 2002;1:601–616. doi: 10.1016/s1568-7864(02)00052-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpey PS, Raymond FL, O’Meara S, Edkins S, Teague J, Butler A, Dicks E, Stevens C, Tofts C, Avis T, et al. Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am J Hum Genet. 2007;80:345–352. doi: 10.1086/511134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wertz IE, O’Rourke KM, Zhang Z, Dornan D, Arnott D, Deshaies RJ, Dixit VM. Human De-Etiolated-1 Regulates c-Jun by Assembling a CUL4A Ubiquitin Ligase. Science. 2004;303:1371–1374. doi: 10.1126/science.1093549. [DOI] [PubMed] [Google Scholar]
- Wittschieben BB, Wood RD. DDB complexities. DNA Repair (Amst) 2003;2:1065–1069. doi: 10.1016/s1568-7864(03)00113-7. [DOI] [PubMed] [Google Scholar]
- Wittschieben BO, Iwai S, Wood RD. DDB1–DDB2 (xeroderma pigmentosum group E) protein complex recognizes a cyclobutane pyrimidine dimer, mismatches, apurinic/apyrimidinic sites, and compound lesions in DNA. J Biol Chem. 2005;280:39982–39989. doi: 10.1074/jbc.M507854200. [DOI] [PubMed] [Google Scholar]
- Yasui K, Arii S, Zhao C, Imoto I, Ueda M, Nagai H, Emi M, Inazawa J. TFDP1, CUL4A, and CDC16 identified as targets for amplification at 13q34 in hepatocellular carcinomas. Hepatology. 2002;35:1476–1484. doi: 10.1053/jhep.2002.33683. [DOI] [PubMed] [Google Scholar]
- Yoon T, Chakrabortty A, Franks R, Valli T, Kiyokawa H, Raychaudhuri P. Tumor-prone phenotype of the DDB2-deficient mice. Oncogene. 2004;24:469–478. doi: 10.1038/sj.onc.1208211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Morrone G, Zhang J, Chen X, Lu X, Ma L, Moore M, Zhou P. CUL-4A stimulates ubiquitylation and degradation of the HOXA9 homeodomain protein. Embo J. 2003;22:6057–6067. doi: 10.1093/emboj/cdg577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Liu Q, Chen B, Zhang X, Guo C, Zhou H, Li J, Gao G, Guo Y, Yan C, et al. Mutation in CUL4B, which encodes a member of cullin-RING ubiquitin ligase complex, causes X-linked mental retardation. Am J Hum Genet. 2007;80:561–566. doi: 10.1086/512489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.