

Abstract
Secreted frizzled related proteins (sFRPs) have emerged as key regulators of a wide range of developmental and disease processes, with virtually all known functions of mammalian sFRPs attributed to their ability to antagonize Wnt signaling. Recently however, the Xenopus and zebrafish sFRP, Sizzled, was shown to function as an antagonist of Chordin processing by Tolloid-like metalloproteinases, leading to the proposal that sFRPs may function as evolutionarily-conserved antagonists of the chordinase activities of this class of proteinases. Herein, in contrast to this proposal, we show that the mammalian sFRP, sFRP2, does not affect Chordin processing, but instead can serve as a direct enhancer of the procollagen C-proteinase activity of Tolloid-like metalloproteinases. We further show that the level of fibrosis, in which procollagen processing by Tolloid-like proteinases plays a rate-limiting role, is markedly reduced in sFRP2-null mice subjected to myocardial infarction. Importantly, this reduced level of fibrosis is accompanied by significantly improved cardiac function. This study thus uncovers a novel function for sFRP2 and a potential therapeutic application for sFRP2 antagonism in controlling fibrosis in the infarcted heart.
Secreted frizzled related proteins (sFRPs) are a family of secreted proteins that share a cysteine-rich (CR) domain with the extracellular region of the Frizzled Wnt receptors1–5. sFRPs bind Wnt molecules via this CR-domain and inhibit Wnt-mediated signal transduction1–5. In the mouse and human, five family members, sFRP1–5, have been identified and have recently emerged as regulators of a wide–range of developmental and disease processes6–22. All such functions of sFRPs in mice and humans have been attributed to antagonism of Wnt signaling.
However, two studies have proposed that sFRP function can go beyond effects on Wnt signaling23,24. In zebrafish, Xenopus and Drosophila, dorsal-ventral patterning during gastrulation is established by cross-talk between ventral and dorsal centers that spatially and temporally regulate a Bone Morphogenetic Protein (BMP) activity gradient25,26. In vertebrates, the dorsal center secretes BMP antagonists, including Chordin, whereas the ventral center expresses Tolloid (TLD) family metalloproteinases and sFRP family member, Sizzled (Szl). A gradient of BMP activity is set by Chordin which is in turn inactivated via cleavage by TLD-like proteinases. In recent studies of Xenopus and zebrafish dorsal-ventral patterning, Szl was shown to bind TLD-like proteinases, thereby inhibiting Chordin cleavage23,24. Thus, Szl appears to regulate BMP signaling and dorsal-ventral patterning during embryo gastrulation, through its novel activity as a competitive inhibitor of TLD-like proteinases.
In mammals, there are four TLD-like proteinases: BMP1, mTLD, mTLL1 and mTLL227. BMP1 and mTLL1, but not mTLD or mTLL2, are responsible for cleaving Chordin, whereas all four can exhibit some level of procollagen C-proteinase (pCP) activity, which converts procollagen precursors into the major fibrillar components of the extracellular matrix (ECM), with BMP1 showing highest pCP activity27. In fact, the mammalian TLD-like proteinases biosynthetically process a variety of precursors into mature functional proteins involved in ECM formation, and also play roles in activating TGFβ superfamily members myostatin/GDF-8, GDF11, and TGFβ27,28.
Although it has been shown that Szl functions as a competitive inhibitor of TLD-like proteinase cleavage of Chordin in Xenopus and zebrafish, it has not been clear whether this novel function of sFRP proteins is conserved in mammals. Lee, et. al. reported that Szl inhibits Xenopus BMP1 cleavage of a synthetic peptide substrate that resembles a procollagen C-propeptide cleavage site, and that mouse sFRP2 can inhibit Xenopus Chordin cleavage by the Xenopus TLD-like proteinase Xlr23. However, the question of whether mammalian sFRPs, such as sFRP2, actually inhibit cleavage of mammalian physiological substrates by TLD-like proteinases has yet to be explored.
Here we show that sFRP2 does not inhibit cleavage of mammalian Chordin by mammalian BMP1 or mTLL1. Thus, inhibition of Chordin cleavage is not a general property of sFRPs. Rather, sFRP2 is shown to enhance the pCP activity of mammalian BMP1 in vitro, consistent with significantly reduced levels of procollagen processing and collagen deposition in sFRP2-null fibroblast cultures and of fibrosis in sFRP2-null mice subjected to myocardial infarction, as demonstrated here. Thus, our study reveals a novel function of an sFRP, as an enhancer of pCP activity of TLD-like metalloproteinases, in mammals. Furthermore, it suggests the possibility of using sFRP2 antagonists in controlling fibrosis in the infarcted heart.
RESULTS
sFRP2, but not Szl, is an enhancer of BMP1 pCP activity
Recent studies in Xenopus and zebrafish led to the proposal that sFRPs function as evolutionarily-conserved competitive inhibitors of the TLD-like proteinases, with a particularly important role in inhibiting the chordinase activity of these proteinases in development23,24. However, in these studies, the ability of mammalian sFRP2 to inhibit cleavage of mammalian Chordin by mammalian TLD-like proteinase was not shown. Therefore, we reevaluated this notion by studying the effects of a mammalian sFRP, sFRP2, on the processing of various physiological substrates of mammalian TLD-like proteinases. Surprisingly, despite reports that murine sFRP2 inhibits cleavage of Xenopus Chordin by the Xenopus TLD-like proteinase, Xlr23, no obvious differences in the extent of cleavage of murine Chordin by BMP1 or mTLL1 occurred in the presence/absence of sFRP2 (Fig. 1a&b). In contrast, Szl was efficient in inhibiting cleavage of mammalian Chordin by mammalian BMP1 (Fig. 1c), while Szl with the ogon mutation (SzlD92N) showed much less efficient inhibition (Fig. 1d), essentially as reported for Szl and SzlD92N in studies of the inhibition of Xenopus23 and zebrafish24 Chordin cleavage by TLD-like proteinases.
Figure 1.
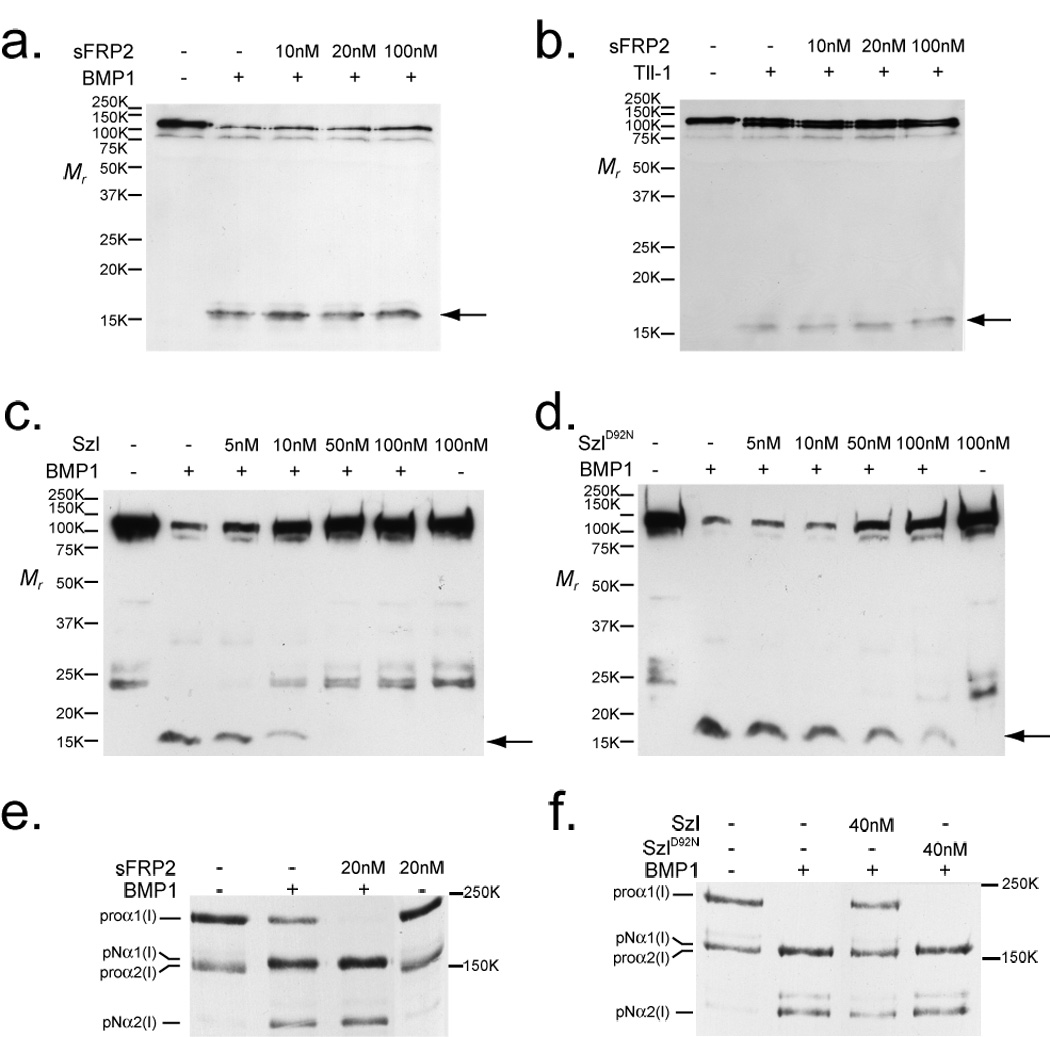
sFRP2 enhances cleavage of procollagen, but not Chordin, by BMP1 and mTLL1. Immunoblots show absence of effects of increasing concentrations of sFRP2 on the cleavage of Chordin by BMP1 (a) or mTLL1 (b), whereas Szl is shown to efficiently inhibit Chordin cleavage by BMP1 (c), with a much reduced inhibitory effect on Chordin cleavage shown by Szl with an ogon-like D92N mutation (d). Chordin cleavage is evidenced by the appearance of a 15-kDa N-terminal fragment (arrow). In contrast to the absence of sFRP2 effects on Chordin cleavage, autofluorography shows that a 2-fold molar excess of sFRP2 enhanced BMP1 pCP activity, such that no unprocessed pro-forms of the two chain classes of type I procollagen (proα1 and proα2) remained, and greater amounts of pN forms (processing intermediates retaining N-propeptides, but from which C-propeptides have been cleaved) for both chain classes were produced, compared to processing with BMP1 alone (e). Szl inhibits, and SzlD92N has no apparent effect, on BMP1 pCP activity (f). Fold-enhancement of pro-α1(I) to pNα1(I) chain processing by sFRP2 ranged from 1.2 – 2.0 fold in six independent experiments (it is 1.4-fold in fig. 1e) and in the four of these experiments in which pro-α2(I) to pNα2(I) chain ratios could be compared, fold-enhancement of pro-α2(I) processing ranged from 1.2 – 2.6 fold (such a comparison could not be made in fig. 1e, in which no uncleaved proα2(V) chains remained after cleavage of procollagen by BMP1 in the presence or absence of sFRP2). For full scans of a–f, see Supplementary Information, Fig. S6.
Due to the lack of sFRP2 effects on the chordinase activity of mammalian TLD-like proteinases, we next assayed for possible effects of mouse sFRP2 on the cleavage of other substrates by TLD-like proteinases. In the course of testing several known substrates for this metalloproteinase family, we found that not only does sFRP2 not have the predicted23 ability to inhibit BMP1 pCP activity, but that it actually enhances this activity (Fig. 1e). In contrast, just as for Chordin, zebrafish Szl was found to inhibit pCP activity (Fig. 1f), with this inhibitory activity similarly suppressed in the ogon-mutant form of Szl (SzlD92N).
These results are consistent with the notion that the non-mammalian sFRP, Szl, inhibits both the chordinase and pCP activities of TLD-like proteinases, whereas the mammalian sFRP, sFRP2, enhances pCP activity, but is ineffective in modulating chordinase activity.
sFRP2 binds BMP1 via its frizzled domain with affinity in the physiological range
To gain insight into the mechanism by which sFRP2 enhances BMP1 pCP activity, we assayed whether sFRP2, like Szl23,24, is capable of binding this class of proteinases. sFRP2 was found to bind BMP1 in a clear-cut manner in pull-down experiments (Fig. 2a), confirming interaction of the two proteins. Surface plasmon resonance (BIAcore) analysis shows the sFRP2-BMP1 interaction to occur with an affinity (KD = 7.13 nM)(Fig. 2b) in the range in which proteins at physiological concentrations are likely to interact, a slightly higher affinity than that previously estimated for the binding of Szl to non-mammalian TLD-like proteinases23.
Figure 2.

sFRP2 binds BMP1 with a KD in the physiological range, predominantly via its Fzl domain. (a) Immunoblots show that His-tagged sFRP2 pulls down FLAG-tagged BMP1, upon precipitation with Ni-NTA agarose. (b) BMP1 binds His-tagged sFRP2 on a BIAcore sensor chip with KD = 7.13 nM. (c) Immunoblots show that the Fc-tagged sFRP2 Fzl domain pulls down FLAG-tagged BMP1 about as effectively as full-length sFRP2-Fc. (d) Fc-tagged Fzl domain of sFRP2 on a Biacore sensor chip binds BMP1 with a KD (KD = 8.68 nM) similar to that of full-length sFRP2-Fc (KD = 6.01 nM), whereas binding of Fc-tagged sFRP2 NTR domain to BMP1 is negligible. (e) Schematic diagram of sFRP2-Fc constructs used for pull-down and BIAcore assays. For full scans of a and c, see Supplementary Information, Fig. S6.
We next determined the sFRP2 protein domain that mediates this interaction. There are two major protein domains in sFRP2: a cysteine-rich frizzled (Fzl) domain and a netrin (NTR) domain (Fig. 2e)1–3,29,30. A pull-down assay clearly showed the sFRP2 Fzl domain sufficient for binding BMP1, whereas little binding was observed for the NTR domain (Fig. 2c). Consistent with the pull-down results, BIAcore analysis found affinity of Fc-tagged sFRP2 Fzl-domain for BMP1 (KD ∼ 8.68 nM) to be in the same physiological range as that of full-length Fc-tagged sFRP2 (KD ∼ 6.01 nM), whereas NTR-domain binding to BMP1 was negligible (Fig. 2d).
These results demonstrate direct and specific interaction of sFRP2 with BMP1 with affinity in the physiological range, suggesting that sFRP2 enhances BMP1 pCP activity by directly interacting with BMP1 via its Fzl-domain.
sFRP2 and Szl bind TLD-like proteinases via non-protease domain sequences
To determine how sFRP2 binding to BMP1 enhances pCP activity of the latter, we evaluated whether sFRP2 directly binds the BMP1 protease domain, which contains the active site. Pull-down assays showed that sFRP2 fails to bind the BMP1 protease domain (BMP1-P), but instead strongly binds a deleted version of BMP1 lacking the protease domain (BMP1-D) (Fig. 3a&e). Similarly, sFRP2 failed to bind the protease domain of another TLD-like proteinase, mTLL1 (mTLL1-P) (Fig. 3b&e). Surprisingly, the same specific mode of interaction was found for Szl, which similarly bound the non-protease domain portions of the TLD-like proteinases (Fig. 3c, d, e), although it has been proposed that Szl was likely to act on the active sites of TLD-like proteinases23. Our results suggest that interaction of sFRP2 with the non-protease domain portions of BMP1 and mTLL1 induces conformational changes of these proteinases, resulting in enhanced pCP, but not chordinase, activity of these enzymes. In contrast, binding of Szl results in suppression of both pCP and chordinase activities of the enzymes.
Figure 3.
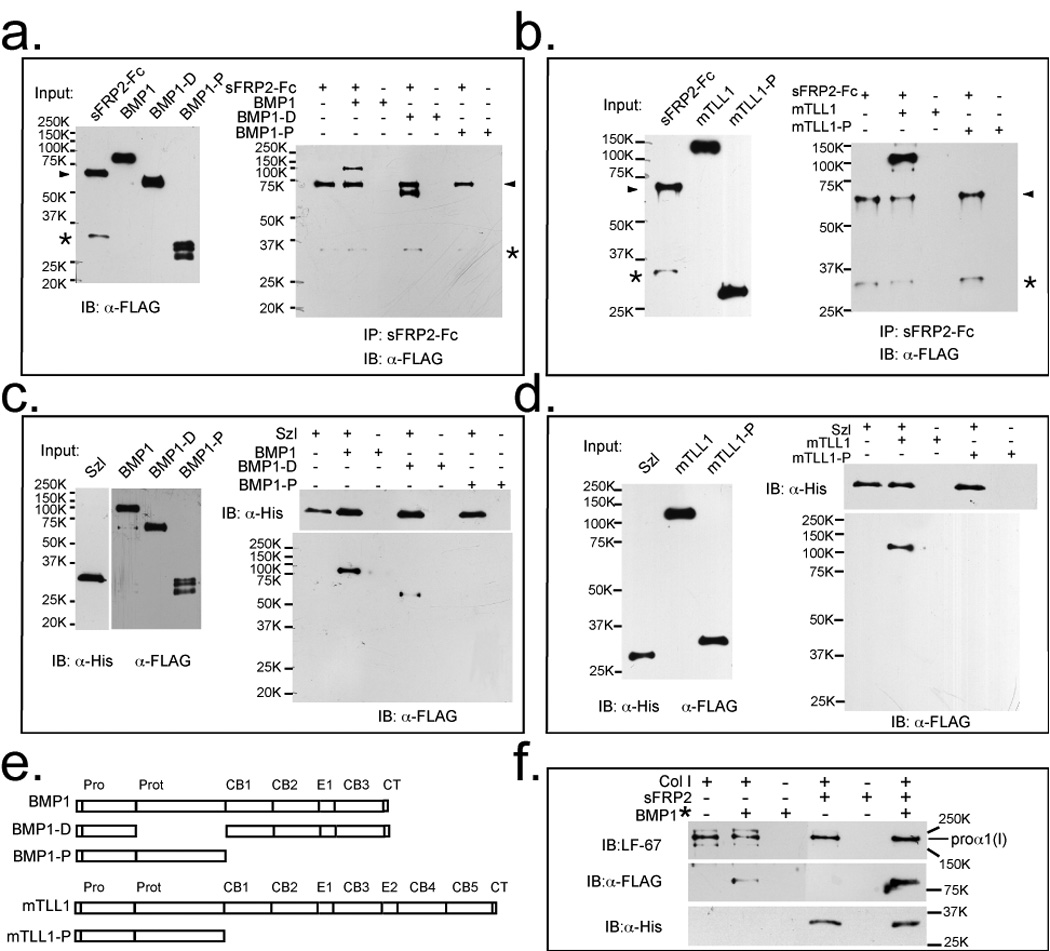
sFRP2 and Szl bind Tolloid-like proteinases via non-protease domain sequences and BMP1/procollagen binding is enhanced by sFRP2. (a) Left, an immunoblot shows input amounts of sFRP2-Fc, FLAG-tagged BMP1 and a deleted form of BMP1 containing all domains except the protease domain (BMP1-D) and isolated BMP1 protease domain (BMP1-P). Right, an immunoblot shows that protein G agarose pull down of sFRP2-Fc pulls down full-length BMP1 and BMP1-D, but not BMP1-P. (b) Left, an immunoblot shows input amounts of sRFP2-Fc and FLAG-tagged mTLL1, or isolated mTLL1 protease domain (mTLL1-P). Right, an immunoblot shows that sFRP2-Fc pulls down full-length mTLL1, but not mTLL1-P. (a and b) arrowheads indicate sFRP2-Fc, and asterisks denote sFRP2 degradation products which bind non-specifically to anti-FLAG antibodies. (c) Left, an immunoblot shows input amounts of His-tagged Szl, and FLAG-tagged BMP1, BMP1-D and BMP1-P. Right, immunoblot shows that Ni-NTA agarose pull down of His-tagged Szl pulls down full-length BMP1 and BMP1-D, but not BMP1-P. (d) Left, an immunoblot shows input amounts of His-tagged Szl, mTLL1, and mTLL1-P. Right, an immunoblot shows that Szl pulls down mTLL1, but not mTLL1-P. (e) Protein domain structures of full-length and truncated forms of BMP1 and mTLL1. Pro, prodomain; Prot, protease domain; CB, CUB domain; E, EGF motif; CT, C-terminal domain. (f) Immunoblots show that procollagen binds sFRP2, and that sFRP2 enhances binding of procollagen to BMP1. Samples were immunoprecipitated with antibody (LF-67) directed against the pro-α1(I) C-telopeptide region. LF-67 is shown to pull down a procollagen/sFRP2-His complex and the presence of sFRP2-His enhances co-immunoprecipitation of procollagen and BMP1*, a form of BMP1 designed to bind but not cleave or release procollagen31 (Quantification of signals on the blots indicate approximately 4-fold enhancement of BMP1* binding by procollagen in the presence of sFRP2). For full scans of a–d and f, see Supplementary Information, Fig. S6.
The interaction of BMP1 proteinase and its substrate, procollagen, is enhanced by sFRP2
We next examined whether sFRP2 might influence the actual binding of procollagen to TLD-like proteinases, in a pull-down assay employing a form of BMP1 designed to bind, but not cleave or release substrates31. Immunoprecipitation of procollagen also pulled down sFRP2, demonstrating that sFRP2 binds procollagen (Fig. 3f). Moreover, co-immunoprecipitation of procollagen and BMP1 was greatly enhanced in the presence of sFRP2 (i.e. the ratio of signal for co-precipitated BMP1 to signal for immunoprecipitated pro-α1(I) chains was ∼4-fold stronger in the presence than in the absence of sFRP2), indicating that sFRP2 enhances BMP1/procollagen binding (Fig. 3f). These results also suggest that sFRP2 enhancement of pCP activity may involve formation of a tripartite complex consisting of sFRP2, BMP1 and procollagen.
Reduced procollagen processing and collagen ECM deposition in sFRP2-null fibroblast cultures
We addressed the question of whether cells actually employ sFRP2 as an enhancer of procollagen processing by TLD-like proteinases by comparing procollagen processing in cultures of wild type and sFRP2-null fibroblasts. sFRP2-null mice were generated by inserting lacZ into the first exon of the Sfrp2 gene (Supplementary Information: Fig. S1) and fibroblasts were isolated from embryos (MEFs) and from adult heart. Both MEFs and heart fibroblasts isolated from wild type mice expressed endogenous sFRP2, whereas sFRP2 expression was, as expected, not detected in sFRP2-null fibroblasts (Supplementary Information: Fig. S2). In conditioned media of wild type MEF and heart fibroblasts, ∼ 63% and 73% of type I procollagen precursor, respectively, was processed to mature collagen (Fig. 4a, b, c, d). In contrast, only 19% and 42% of the precursor was processed to mature collagen in conditioned media from sFRP2-null MEFs and heart fibroblasts, respectively (Fig. 4a, b, c, d). Furthermore, as procollagen C-propeptide cleavage appears to be the rate-limiting step in collagen fibrillogenesis27, it is notable that wild type MEFs were found to deposit 2.5-fold more mature collagen into cell layer-associated ECM than did sFRP2-null MEFs, whereas wild type heart fibroblasts deposited 11-fold more collagen into ECM than did their sFRP2-null counterparts (Fig. 4a, b, e, f). These marked reductions in procollagen processing and in the deposition of collagen into ECM thus support the biochemical data, indicating that sFRP2 is an enhancer of the pCP activity of TLD-like proteinases.
Figure 4.
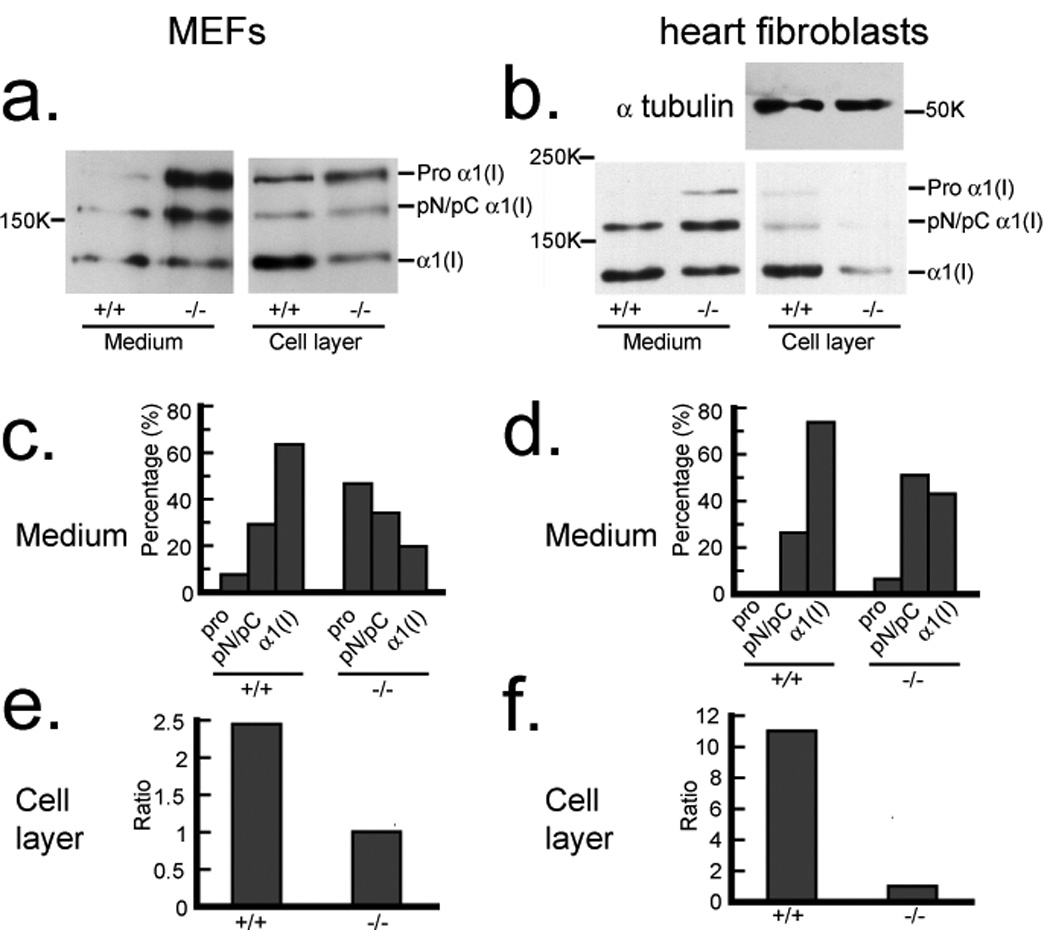
Reduced processing of type I procollagen and deposition of collagen into ECM by Sfrp2−/− fibroblasts. Western blot analysis with antibody against the proα1(I) C-telopeptide domain (a small globular region retained on mature α chains, between the triple-helical domain and BMP1 cleavage site) demonstrates decreased processing of procollagen in conditioned media and decreased deposition of collagen into the ECM associated with cell layers of Sfrp2−/− MEF cultures (a) and heart fibroblasts (b) compared to wild-type (sFRP2+/+) cells. Due to the dramatic difference in collagen levels in the cell layer samples of wild-type and Sfrp2−/− heart fibroblasts, samples were re-probed with anti-α tubulin antibody to demonstrate equal loading in the two lanes. Densitometric quantification is shown of the relative amounts of the various collagen forms in conditioned media of MEFs (c) and heart fibroblasts (d). Pro forms are precursors that retain both N- and C-propeptides; α1(I) forms are mature chains, capable of forming fibrils, from which both N- and C-propeptides have been proteolytically removed; pN forms are processing intermediates of proα1(I) chains from which the C- but not N-propeptide has been removed by BMP1-like proteinases; pC forms are processing intermediates from which the N- but not C-propeptide has been removed by non-BMP1-like proteinases. pC and pN forms were not separated by SDS-PAGE in these experiments. Densitometric quantification is shown of the relative amounts of mature α1(I) chains incorporated into cell layer-associated ECM of wild type (+/+) and sFRP2-null (−/−) MEFs (e) and heart fibroblasts (f). For full scans of a and b, see Supplementary Information, Fig. S6.
sFRP2 expression is upregulated in the fibrotic phase of myocardial infarction
Procollagen processing by TLD-like proteinases is an important rate-limiting step in collagen fibrillogenesis, and thus in the pathological process of fibrosis27, which occurs in many fatal disease conditions, including myocardial infarction, the leading cause of death in men and women32,33. We therefore examined sFRP2 expression in a mouse myocardial infarction model (Fig. 5a&b). Following coronary artery ligation, insufficient oxygen supply leads to damage and death of cardiomyocytes due to apoptosis and necrosis32,33. sFRP2 expression was not detected during this initial phase (Fig. 5a). This first phase is followed by fibrosis, in which dead myocytes are removed and replaced by collagenous ECM that forms the fibrotic scar tissue. We detected 20- to 30-fold up-regulation of sFRP2 RNA transcript levels at day 4 following coronary artery ligation (Fig. 5b), coinciding with appearance of extraneous sirius-red positive collagen fibers, indicating the beginning of fibrosis (Fig. 5a). The sFRP2 expressing cells exhibit a fibroblastic-appearance and scattered distribution. By day 7, we detected ∼ 80- to 100-fold up-regulation of sFRP2 transcript levels (Fig. 5b), and the sirius-red-positive fibrotic area had expanded, replacing dead myocytes (Fig. 5a). The sFRP2-expressing fibroblastic-cells were distributed widely throughout the fibrotic area. At day 14 following ligation, the sFRP2 transcript level was somewhat reduced, but a ∼ 30- to 40-fold increase was still detected (Fig. 5b). Upregulation of BMP1 expression followed the same temporal expression pattern as that of sFRP2 in infarcted heart (Fig. 5b), whereas mTLL1, another TLD-like proteinase capable of procollagen processing, exhibited little expression in infarcted heart (Fig. 5b). Temporal correspondence of sFRP2 and BMP1 expression patterns, together with the specific expression of sFRP2 in fibroblastic cells in fibrotic areas, is consistent with the conclusion that sFRP2 enhancement of BMP1-mediated procollagen processing plays an important role in the fibrotic component of myocardial infarction.
Figure 5.

Induction of Sfrp2 and Bmp1 during the fibrosis phase in the infarcted heart. (a) β-galactosidase staining of the infarcted sFRP2lacZ/+ left-ventricle at days 1, 4, 7, and 14 following coronary artery ligation. Higher magnifications of the boxed areas in the top panels are in bottom panels. At day one, the sirius-red counterstaining (orange) identifies normal collagens in the interstitial space. At this stage, no sFRP2 expression was detected. At day four, the slightly wider areas are stained by sirius-read (orange), which is accompanied by scattered distribution of β-galactosidase-stained sFRP2-expressing fibroblastic cells (blue). By day seven, extensive collagenous fibrosis stained by sirius-red (orange) is detected. Within the fibrotic area, many fibroblastic cells express sFRP2 (blue). At day 14, more extensive sirius-red staining, denoting collagenous scar tissue that has replaced dead myocytes, is clearly detected, and many fibroblastic cells are seen to express sFRP2 (blue). Upregulation of sFRP2 expression in the infarcted area was also confirmed by in situ hybridization staining with a riboprobe for sFRP2 (Supplementary Information: Fig. S3). Scale bars are shown in each panel. (b) Quantitative RT-PCR analysis of sFRP2, BMP1 and mTLL1. Whole ventricle tissue RNA was isolated at days four, seven and 14 following coronary artery ligation. The amounts of sFRP2, BMP1 and mTLL1 were compared to those of non-operated normal ventricle tissue and fold-increases are shown. Duplicate PCR reactions for each sample were carried out and the standard deviations were calculated from the results obtained from two independent experiments. Marked increases of sFRP2 and BMP1 transcripts were detected by day four and their transcript levels continued to increase until day seven. By day 14, their transcript levels decreased, but still remained relatively elevated. The mTLL1 transcript level increase was much less that those of sFRP2 and BMP1. The absolute amounts of sFRP2 transcripts at day 0, day4, day7, day14 were quantified and determined as 6.8×102, 1.5×104, 6.5×104, 1.3×104 molecules per 20ng total RNA, respectively. Expression analysis of all Wnt ligands and Wnt antagonists genes indicates that the expression of Sfrp2 gene exhibits the highest upregulation within the fibrotic area of infarcted hearts (Supplementary Information: Figs. S3&S4).
Reduced fibrosis and improved cardiac function of the infarcted sFRP2-null heart
We next examined the extent of fibrosis in the sFRP2-null heart after coronary artery ligation (Fig. 6). Age-matched sFRP2-null and wild type mice were subjected to this surgical procedure and areas of fibrosis in the left-ventricle were measured by staining collagen fibers with sirius-red. By day 14 after coronary artery ligation, ∼25 – 35% of the left-ventricle of wild type heart was stained by sirius-red, indicating severe fibrosis (Fig. 6a&b). In contrast, only ∼15– 20% of the left-ventricle of sFRP2-null heart was stained by sirius-red (Fig. 6a&b). As an independent readout of fibrosis, we measured amounts of hydroxyproline which reflects the total amount of mature fully-processed collagen deposited into tissue fibrils34,35. In sham operated control wild type and sFRP2-null hearts, ∼2 – 5 µg hydroxyproline was present per mg dry weight left-ventricle (Fig. 6c). In the infarcted wild type heart, ∼10 – 16 µg hydroxyproline was detected per mg dry weight left-ventricle (Fig. 6c). In contrast, only 3 – 8 µg hyroxyproline per dry weight left ventricle was detected in sFRP2-null heart (Fig. 6c). This reduced fibrosis and mature collagen content of infarcted sFRP2-null heart is consistent with the role of sFRP2 in enhancing BMP1 procollagen processing, and thus collagen deposition, indicated by biochemical and cell culture studies (Fig. 1&Fig. 4).
Figure 6.

Reduced fibrosis in the infarcted sFRP2-deficient heart. (a) Sirius-red stained cross sectioned infarcted left ventricle of wild-type (WT) and sFRP2-deficient (Sfrp2lacZ/lacZ) hearts at day 14 after coronary artery ligation. The infarcted wild-type ventricle shows dilatation and extensive sirius-red stained fibrosis (orange). The infarcted Sfrp2lacZ/lacZ ventricle shows much less dilation, and the sirius-red stained area (orange) is reduced. Scale bars are shown in each panel. (b) Quantification of the sirius-red stained fibrotic area. Sirius-red stained areas, determined using AxioVision image analysis software (Carl Zeiss), were compared to whole ventricular areas and are shown as percentages. A representative section from each of a total of nine (n=9) wild type and five (n=5) sFRP2 null hearts was evaluated and the standard deviations were calculated. The sirius-red stained areas in wild type and Sfrp2lacZ/lacZ left ventricles were ∼25 – 35% and ∼15 – 20% of the entire ventricle areas, respectively. (c) Quantification of total hydroxyproline (HP) amounts. Samples from four independent experiments (n=4) for each group were assayed and shown here. The standard deviations and P values are shown. In sham operated control wild type and sFRP2-null hearts, approximately 2 – 5µg of hydroxyproline was present per mg dry weight left-ventricle. In the infarcted wild type heart, approximately 10 – 16µg of hydroxyproline was detected per mg dry weight left-ventricle. In contrast, only 3 – 8µg of hydroxyproline per dry weight left ventricle was detected in the sFRP2-null heart.
The functional consequence of reduced fibrosis in sFRP2-null hearts was studied by cardiac echocardiography monitoring in both wild type and sFRP2-null mice subsequent to coronary artery ligation. At day 7 following coronary artery ligation, both sFRP2-null and wild type control hearts exhibited a wide range of ejection fraction (EF) and no significant differences were detected between them (Fig. 7a&c). By day 14 following ligation, wild type heart EFs dropped consistently to 10 – 20%, markedly lower than EFs of sham-operated control hearts (Fig. 7b – d). In contrast, EFs of sFRP2-null hearts remained in the 25 – 65% range, with some of them even staying at 60 – 65%, a range essentially indistinguishable from that of sham-operated controls (Fig. 7b – d). Time-lapse cinematography revealed relatively active hearts beating in the infarcted sFRP2-null mice (Supplementary Information: movie 2). In contrast, infarcted wild type hearts showed very little beating, due to severe dilation of the ventricular wall resulting from extensive myocardial cell death and fibrosis (Supplementary Information: movie 1).
Figure 7.

Improved cardiac function of the infarcted sFRP2-deficient heart. (a) Ejection fraction (EF) of wild-type (WT) and sFRP2-deficient (Sfrp2lacZ/lacZ) hearts at day seven following coronary artery ligation. Each dot represents an individual mouse (i.e., WT sham: n=3; WT MI: n=7; Sfrp2lacZ/lacZ sham: n=3; Sfrp2lacZ/lacZ MI: n=6). At day seven, the infarcted (MI) hearts of both wild type (WT) and sFRP2-deficient (Sfrp2lacZ/lacZ ) mice showed wide ranges of EF (10% – 55%) and no significant (NS) difference. The EF of sham operated mice showed a normal range of EF. (b) EF of wild-type (WT) and sFRP2-deficient (sFRP2lacZ/lacZ) hearts at day 14 following coronary artery ligation. Each dot represents an individual mouse (i.e., WT sham: n=3; WT MI: n=9; Sfrp2lacZ/lacZ sham: n=3; Sfrp2lacZ/lacZ MI: n=7). At day 14, The infarcted (MI) hearts of all wild type (WT) mice showed consistently low EF (10 – 20%). In contrast, the infarcted (MI) hearts of sFRP2-deficient (Sfrp2lacZ/lacZ) mice showed higher EFs (25 – 60%) and two of the null mice showed EFs (60 –70%) equivalent to those of sham operated mice. The EF of sham operated mice showed a normal range of EF. (c) Change of EFs over time. The EFs of the infarcted wild-type (WT) hearts continued to decline over time, indicating functional failure. In contrast, the EFs of the infarcted sFRP2-deficient (sFRP2lacZ/lacZ) hearts in general remained approximately the same or in some cases slightly improved over time. The data shown in a&b are used to generate this plot. (d) M-mode images of representative wild-type (WT) and sFRP2-deficient (sFRP2lacZ/lacZ) hearts at day 14 following coronary artery ligation. The wild-type heart showed virtually no pumping. In contrast, the sFRP2-deficient heart showed some pumping activity. The time-lapse movie of each heart can be viewed in the (supplementary information Movies 1 & 2).
DISCUSSION
sFRPs have long been considered endogenous antagonists of Wnt signaling36. However, recent studies in Xenopus and zebrafish have questioned this notion and shown that at least one sFRP, Szl, functions as a competitive inhibitor of Chordin cleavage by directly binding TLD-like proteinases23,24. This novel, non-Wnt signaling function of Szl appears to play a key role in dorsal-ventral patterning of early embryos23,24. Furthermore, it was reported that mammalian sFRP2 can inhibit Xenopus Chordin cleavage by the Xenopus TLD-like proteinase, Xlr23. Based on this result, it was proposed that ability to block the Chordinase activity of TLD-like proteinases is shared by all sFRPs and is evolutionarily conserved23.
Here, we present biochemical evidence that mammalian sFRP2 does not act as an inhibitor of the chordinase activity of mammalian TLD-like proteinases BMP1 and mTLL1 (Fig. 1a&b), but that it instead functions as an enhancer of the procollagen pCP activity of such proteinases (Fig. 1e). The conclusion that sFRP2 is a physiological enhancer of procollagen processing is further substantiated by the marked reduction of type I procollagen processing by sFRP2-null fibroblasts (Fig. 4) and of fibrosis in infarcted sFRP2-null hearts (Fig. 6).
What is the mechanism underlying this novel pCP enhancer function of sFRP2? In previous studies23,24, it was shown that Szl directly binds TLD-like proteinases via its frizzled-domain, thus mediating the inhibition of chordinase activity. In one of the studies23, it was further proposed that Szl inhibits chordinase activity by acting directly on the active site within the protease domain of TLD-like proteinases. This proposal was primarily based on competitive enzyme kinetics assays23, but direct interaction of Szl with the protease domains of TLD-like proteinases was not demonstrated.
In our present studies we show that sFRP2, like Szl, has specific and physiological interactions with both BMP1 and mTLL1 via its frizzled domain (Fig. 2). However, contrary to the previous proposal, our study shows that both Szl and sFRP2 fail to bind the protease domain of BMP1 or mTLL1, instead binding the non-protease portions of these two proteinases (Fig. 3). Based on these results, we propose an alternative mechanism: Szl and sFRP2 exert their differing effects on TLD-like proteinases via binding domains other than the active-site containing protease domain. Szl likely inhibits chordinase activity by competing with Chordin for binding to non-protease domain sites of TLD-like proteinases, or perhaps such binding by Szl induces conformational changes in the proteinases that preclude Chordin binding. Similarly, sFRP2 binding to non-protease domain sites likely enhances pCP activity by facilitating the interaction between procollagen substrates and TLD-like proteinases, which appears to involve formation of a tripartite complex comprising sFRP2, proteinase, and procollagen substrate (Fig. 3f). Interestingly, this latter mechanism of pCP enhancement by sFRP2 is similar to that of the other family of pCP enhancers, PCOLCEs/PCPEs, which have been shown to enhance pCP activity via binding both procollagen substrates and the non-protease domains of TLD-like proteinases37–40. Similarly, the mechanism involving Szl inhibition of chordinase activity via interactions with non-protease domain sequences is consistent with the previous finding that the chordinase activity of a given TLD-like proteinase can in large part be determined by its non-protease protein domains41.
Genetic studies of zebrafish identified the ogon mutant in which a missense Asp>Asn mutation in the Szl Fzl-domain at position 92 (D92N) results in production of functionally null Szl protein42,43. In previous studies23,24, functional significance of Fzl-domain mediated interaction of Szl with TLD-like proteinases was further implied by showing that the D92N ogon-mutation in the Fzl domain abrogates inhibition of chordinase activity by Szl in vitro and in vivo23. However, seemingly corresponding mutation of Asp105 to Asn (D105N) in the Fzl-domain of mammalian sFRP2 had no effects on pCP enhancer activity (data not shown).
Excessive collagen fiber formation results in fibrosis in various life-threatening human diseases. Removal of procollagen C-terminal propeptides by TLD-like proteinases is a critical and rate-limiting biochemical step in this process27. Myocardial infarction is one disease in which fibrosis reflects the severity of the prognosis. Our study shows significant co-induction of sFRP2 and BMP1 expression during fibrosis (Fig. 5 and Supplementary Information: Figs. S3&S4) and, importantly, markedly reduced fibrosis (Fig. 6) in the infarcted sFRP2-null heart. These in vivo results are consistent with our biochemical results indicating that sFRP2 is an enhancer of BMP1 pCP activity (Fig 1&Fig 4). Based on these results, we propose that the co-induction of BMP1 and sFRP2, demonstrated herein to occur during fibrosis, and the consequently enhanced procollagen processing, are key drivers of the formation and deposition of the extensive collagen fibrils that constitute severe fibrosis.
The possibility that sFRP2 might function as an autocrine factor to either enhance proliferation of, or suppress the death of, sFPR2-expressing fibroblastic cells, appears to be excluded. In this scenario, the total number of sFRP2-expressing fibroblastic cells would be reduced in infarcted sFRP2-null heart, thereby reducing the extent of fibrosis, since collagens are deposited by these fibroblastic cells44,45. Analysis of the number of formerly sFRP2-expressing fibroblastic cells after coronary artery ligation did not, however, provide evidence for such a reduction of fibroblastic cells (Supplementary Information: Fig. S5.), arguing against a role for sFRP2 as a proliferative or survival factor for cardiac fibroblastic cells involved in fibrosis.
Cardiac fibrosis is known to have adverse effects on left-ventricular (LV) function44–46, thus forming a therapeutic basis for use of antifibrotic agents to inhibit or reverse such adverse effects44–46. Our study shows that reduced fibrosis in the infarcted sFRP2-null heart is accompanied by markedly improved performance compared to infarcted wild type heart (Fig. 7), suggesting that antagonism of sFRP2 may prove to be an effective therapeutic approach towards controlling fibrosis, and improving cardiac function in myocardial infarction, which accounts for ∼13% of deaths worldwide and is the leading cause of death in developed countries.
In summary, we provide in vitro and in vivo evidence supporting the concept of sFRP2 as an endogenous enhancer of procollagen C-proteinase activity. Based on our finding, urgent reevaluation of previously reported roles for sFRPs in various processes presumed to be dependent on the inhibition of Wnt signaling is needed. Furthermore, our biochemical studies suggest that pCP enhancement mediated via formation of a tripartite complex consisting of a TLD-like proteinase, a procollagen (i.e. substrate) and a pCP enhancer may be a common mechanism underlying the function of two distinct classes of pCP enhancers, sFRP2 and PCOLCEs/PCPEs.
METHODS
Biochemical and cell biology methods
Recombinant murine Chordin, and human BMP1 and mTLL1 were prepared, and cleavage assays performed as described previously47. Mutant BMP1 (BMP1*) designed to bind but not cleave or release substrates, due to an E214A substitution at the active site has been described31, as has a FLAG-tagged BMP1 consisting only of pro- and protease domains40. FLAG-tagged BMP1 lacking the protease domain, and C-terminal His-tagged Xenopus wild type Szl and SzlD92N were prepared as detailed in Supplementary Information.
For Chordin cleavage, 15 ng BMP1 or mTLL1 was incubated with 100 ng Chordin 16 h at 37 °C with/without mouse sFRP2 (R&D), Szl, or SzlD92N, in amounts noted in the figure, in 50 mM Tris-HCl, 150 mM NaCl, 5 mM CaCl2 (buffer A). Subsequent to SDS-PAGE on 10% acrylamide gels, Western blotting was with a previously described anti-Chordin N-terminus antibody48. For procollagen processing, 400 ng 3H-labeled type I procollagen, prepared as described47, was incubated in buffer A with 15 ng BMP1 at 37°C with/without 20 nM sFRP2, or 40 nM Szl or SzlD92N. 20 nM sFRP2, or 40 nM Szl or SzlD92N, in the absence of proteinase, were used as controls. Reactions were stopped after 16 h at 37 °C by adding SDS-sample buffer. Proteins were separated on 5% SDS-PAGE gels, followed by treatment with EN3HANCE (PerkinElmer) and autofluorography.
Various immunoprecipitations and co-immunoprecipitations are described in detail in Supplementary Information.
Surface plasmon resonance measurements were performed in a BIAcore 2000 system, as described in detail in Supplementary Information.
To compare collagen processing and deposition in cell-associated matrix, wild type and sFRP2-null fibroblasts were cultured in DMEM, 10% FBS, and treated 24 h with 50 µg/ml ascorbate. Cells were washed twice with PBS, and incubated 15 min in serum-free DMEM at 37 °C. Cells were then washed once with PBS, followed by addition of serum-free DMEM containing 50 µg/ml ascorbate, 40 µg/ml soybean trypsin inhibitor. Conditioned media were harvested after 24 h and cell layers washed twice with ice-cold PBS, and scraped into hot SDS-sample buffer. Western blotting was performed with anti-α1(I) C-telopeptide polyclonal antibody LF67, as described49. Blots were scanned and quantified with Image J 1.36b software.
Generation of sFRP2 null mice
The sFRP2 null mice were generated by standard gene-targeting method as detailed in the Supplementary Information and Fig. S1.
Isolation and culture of fibroblasts
Fibroblasts were isolated and cultured by standard methods as detailed in Supplementary Information.
Quantitative RT-PCR (qRT-PCR)
Total RNA was isolated by RNeasy Midi kit (QIAGEN) according to the manufacturer’s protocol. For RNA isolation from heart tissues, homogenate was passed through a cell strainer (100 µm) to prevent the column from clogging. cDNA was synthesized using the SuperScript first-strand synthesis system (Invitrogen). qRT-PCR was performed using the QuantiTect SYBR Green PCR kit (QIAGEN) and M×4000 multiplex Quantitative PCR system (Stratagene). The sequences of the primers are indicated in Supplementary Information.
GAPDH qRT-PCR was used as an internal control for, and to normalize, each gene-specific qRT-PCR. The absolute amounts of sFRP2 transcripts were determined by using the standard curve of known amounts of sFRP2 cDNA in the quantitative RT-PCR reactions.
Myocardial infarction
Male mice on a CD1 background, ranging in age from 7–12 wks, underwent coronary artery ligation to produce myocardial infarction. Mice were anesthetized by intraperitoneal injection of 1.25% Avertin (2,2,2-tribromoethanol, 20 µl/g body weight) and intubated with a 20-gauge intravenous catheter. Animals were ventilated with a volume-controlled respirator (Harvard Apparatus Co., Holliston, Massachusetts, USA) with a 200 µl/cycle at a respiratory rate of 110 cycles/min. After thoracotomy, ligation of the left coronary artery was performed with an 8–0 nylon suture 1–2 mm below the tip of the left auricle. Occlusion was confirmed by the change of color (pallor) of the anterior wall of the left ventricle. The chest cavity and the skin were closed with 5–0 silk sutures. The animals were extubated and allowed to recover from surgery on a warm plate for 1 hr. Sham-operated mice underwent the same procedure without tying the suture, but passing it below the coronary artery.
Histology and in situ hybridization
Hematoxylin-Eosin (H-E) staining, sirius-red staining, the detection of β-galactosidase activity and in situ hybridization analysis were performed as detailed in Supplementary Information.
Hydroxyproline assay
Hydroxyproline content of heart tissues was measured as previously described34,35 with some modifications. Left-ventricle was freshly harvested from each heart and vacuum-dried. Each vacuum-dried tissue was weighed, minced and hydrolyzed in vacuum-sealed 6 M HCl (1ml) at 116°C overnight. Hydrolyzed samples were dried in a speed-vac overnight. The dried pellet was dissolved in water and re-dried in a speed-vac overnight. The dried pellet was re-dissolved in water and centrifuged 5 min. To each supernantant, 40 µl phenolphthalein was added as pH indicator, and KOH solution was added until the solution color became pale pink. Neutralized samples were centrifuged for 5 min and the supernatant was used for the hydroxyproline measurement. To each diluted (typically 20-fold) sample (2 ml), 1 ml of 0.05 M chloramine-T (0.285 g/5 ml dH2O, 7.5 ml Methyl Cellosolve, 12.5 ml Na-citrate/Na-acetate buffer) was added and incubated 20 min. at room temperature. Chrolamine-T was then destroyed with 1 ml 3.15 M perchloric acid (7.56 ml 60% perchloric acid, 16.44 ml dH2O). After 5 min at room temperature, 1 ml 20% p-dimethylaminobenzaldehyde solution (dissolved in Methyl Cellosolve) was added and each sample was vortexed, and incubated 20 min at 60°C. Hydroxyproline content was determined by measuring the absorbance of each sample at 557 nm. A standard curve was generated by using hydroxyproline solutions (0, 2, 4, 6, 8, 10 µg/2 ml).
Echocardiography
Echocardiography was performed on wild type and sFRP2-null infarcted and sham-operated mice at days 1, 7 and 14 using a VisualSonic Vevo 770 ultrasound imager equipped with a 40 MHz RSV-704 scan head. In each instance mice were anesthetized with 1% isoflurane and parasternal short-axis and long-axis B-mode and M-mode views were recorded. The short-axis view was used to measure anteroposterior internal diameter, anterior wall thickness and posterior wall thickness at end-diastole and end-systole at the mid-papillary level. The long-axis view was used to monitor outflow through the aortic root by pulsed Doppler to measure peak velocity of blood through the aortic valve. Left ventricular systolic function was assessed by ejection fraction: %EF =[(EDv - ESv)/EDv ] × 100. The observer was unaware of the genotypes and treatment.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Kuangfu Hsiao, Patricia Cobo, and Guy Hoffman for technical assistance, and Dr. Tony Brown (Cornell) for providing information regarding qPCR primers for Wnt genes. K.K. and M.L. would like to acknowledge Dr. Rosemary Kraemer’s teaching of technical procedures for the mouse myocardial infarction model. Work in the laboratory of A.K. is supported by funding from the German Research Foundation (DFG) for the Cluster of Excellence REBIRTH (from Regenerative Biology of Reconstructive Therapy), and by the European Union FP6 contract “Heart Repair” (LSHM-CT-2005-018630). C.T.B. is an Established Investigator of the American Heart Association and is supported by the Snart Cardiovascular Fund. This work was supported in part by grants from NIH (C.T.B, D.S.G, T.N.S).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare that they have no competing interests.
REFERENCES
- 1.Rattner A, et al. A family of secreted proteins contains homology to the cysteine-rich ligand-binding domain of frizzled receptors. Proc Natl Acad Sci U S A. 1997;94:2859–2863. doi: 10.1073/pnas.94.7.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mayr T, et al. Fritz: a secreted frizzled-related protein that inhibits Wnt activity. Mech Dev. 1997;63:109–125. doi: 10.1016/s0925-4773(97)00035-x. [DOI] [PubMed] [Google Scholar]
- 3.Finch PW, et al. Purification and molecular cloning of a secreted, Frizzled-related antagonist of Wnt action. Proc Natl Acad Sci U S A. 1997;94:6770–6775. doi: 10.1073/pnas.94.13.6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melkonyan HS, et al. SARPs: a family of secreted apoptosis-related proteins. Proc Natl Acad Sci U S A. 1997;94:13636–13641. doi: 10.1073/pnas.94.25.13636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salic AN, Kroll KL, Evans LM, Kirschner MW. Sizzled: a secreted Xwnt8 antagonist expressed in the ventral marginal zone of Xenopus embryos. Development. 1997;124:4739–4748. doi: 10.1242/dev.124.23.4739. [DOI] [PubMed] [Google Scholar]
- 6.Bodine PV, et al. The Wnt antagonist secreted frizzled-related protein-1 is a negative regulator of trabecular bone formation in adult mice. Mol Endocrinol. 2004;18:1222–1237. doi: 10.1210/me.2003-0498. [DOI] [PubMed] [Google Scholar]
- 7.Barandon L, et al. Involvement of FrzA/sFRP-1 and the Wnt/frizzled pathway in ischemic preconditioning. Circ Res. 2005;96:1299–1306. doi: 10.1161/01.RES.0000171895.06914.2c. [DOI] [PubMed] [Google Scholar]
- 8.Satoh W, Gotoh T, Tsunematsu Y, Aizawa S, Shimono A. Sfrp1 and Sfrp2 regulate anteroposterior axis elongation and somite segmentation during mouse embryogenesis. Development. 2006;133:989–999. doi: 10.1242/dev.02274. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki H, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36:417–422. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- 10.Zou H, et al. Aberrant methylation of secreted frizzled-related protein genes in esophageal adenocarcinoma and Barrett's esophagus. Int J Cancer. 2005;116:584–591. doi: 10.1002/ijc.21045. [DOI] [PubMed] [Google Scholar]
- 11.Lee JL, Lin CT, Chueh LL, Chang CJ. Autocrine/paracrine secreted Frizzled-related protein 2 induces cellular resistance to apoptosis: a possible mechanism of mammary tumorigenesis. J Biol Chem. 2004;279:14602–14609. doi: 10.1074/jbc.M309008200. [DOI] [PubMed] [Google Scholar]
- 12.Zi X, et al. Expression of Frzb/secreted Frizzled-related protein 3, a secreted Wnt antagonist, in human androgen-independent prostate cancer PC-3 cells suppresses tumor growth and cellular invasiveness. Cancer Res. 2005;65:9762–9770. doi: 10.1158/0008-5472.CAN-05-0103. [DOI] [PubMed] [Google Scholar]
- 13.Dufourcq P, et al. FrzA, a secreted frizzled related protein, induced angiogenic response. Circulation. 2002;106:3097–3103. doi: 10.1161/01.cir.0000039342.85015.5c. [DOI] [PubMed] [Google Scholar]
- 14.Oshima T, et al. Myeloma cells suppress bone formation by secreting a soluble Wnt inhibitor, sFRP-2. Blood. 2005 doi: 10.1182/blood-2004-12-4940. [DOI] [PubMed] [Google Scholar]
- 15.Kim AS, Anderson SA, Rubenstein JL, Lowenstein DH, Pleasure SJ. Pax-6 regulates expression of SFRP-2 and Wnt-7b in the developing CNS. J Neurosci. 2001;21:RC132. doi: 10.1523/JNEUROSCI.21-05-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barandon L, et al. Reduction of infarct size and prevention of cardiac rupture in transgenic mice overexpressing FrzA. Circulation. 2003;108:2282–2289. doi: 10.1161/01.CIR.0000093186.22847.4C. [DOI] [PubMed] [Google Scholar]
- 17.Roth W, et al. Secreted Frizzled-related proteins inhibit motility and promote growth of human malignant glioma cells. Oncogene. 2000;19:4210–4220. doi: 10.1038/sj.onc.1203783. [DOI] [PubMed] [Google Scholar]
- 18.Lee CS, Buttitta LA, May NR, Kispert A, Fan CM. SHH-N upregulates Sfrp2 to mediate its competitive interaction with WNT1 and WNT4 in the somitic mesoderm. Development. 2000;127:109–118. doi: 10.1242/dev.127.1.109. [DOI] [PubMed] [Google Scholar]
- 19.Kim BM, Buchner G, Miletich I, Sharpe PT, Shivdasani RA. The stomach mesenchymal transcription factor Barx1 specifies gastric epithelial identity through inhibition of transient Wnt signaling. Dev Cell. 2005;8:611–622. doi: 10.1016/j.devcel.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 20.Polesskaya A, Seale P, Rudnicki MA. Wnt signaling induces the myogenic specification of resident CD45+ adult stem cells during muscle regeneration. Cell. 2003;113:841–852. doi: 10.1016/s0092-8674(03)00437-9. [DOI] [PubMed] [Google Scholar]
- 21.Lei Q, et al. Wnt signaling inhibitors regulate the transcriptional response to morphogenetic Shh-Gli signaling in the neural tube. Dev Cell. 2006;11:325–337. doi: 10.1016/j.devcel.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 22.Mirotsou M, et al. Secreted frizzled related protein 2 (Sfrp2) is the key Akt-mesenchymal stem cell-released paracrine factor mediating myocardial survival and repair. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0610024104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee HX, Ambrosio AL, Reversade B, De Robertis EM. Embryonic dorsal-ventral signaling: secreted frizzled-related proteins as inhibitors of tolloid proteinases. Cell. 2006;124:147–159. doi: 10.1016/j.cell.2005.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muraoka O, et al. Sizzled controls dorso-ventral polarity by repressing cleavage of the Chordin protein. Nat Cell Biol. 2006;8:329–338. doi: 10.1038/ncb1379. [DOI] [PubMed] [Google Scholar]
- 25.De Robertis EM, Kuroda H. Dorsal-ventral patterning and neural induction in Xenopus embryos. Annu Rev Cell Dev Biol. 2004;20:285–308. doi: 10.1146/annurev.cellbio.20.011403.154124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schier AF, Talbot WS. Molecular genetics of axis formation in zebrafish. Annu Rev Genet. 2005;39:561–613. doi: 10.1146/annurev.genet.37.110801.143752. [DOI] [PubMed] [Google Scholar]
- 27.Hopkins DR, Keles S, Greenspan DS. The bone morphogenetic protein 1/Tolloid-like metalloproteinases. Matrix Biol. 2007 doi: 10.1016/j.matbio.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ge G, Greenspan DS. BMP1 controls TGFbeta1 activation via cleavage of latent TGFbeta-binding protein. J Cell Biol. 2006;175:111–120. doi: 10.1083/jcb.200606058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci. 2003;116:2627–2634. doi: 10.1242/jcs.00623. [DOI] [PubMed] [Google Scholar]
- 30.Banyai L, Patthy L. The NTR module: domains of netrins, secreted frizzled related proteins, and type I procollagen C-proteinase enhancer protein are homologous with tissue inhibitors of metalloproteases. Protein Sci. 1999;8:1636–1642. doi: 10.1110/ps.8.8.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ge G, Fernandez CA, Moses MA, Greenspan DS. Bone morphogenetic protein 1 processes prolactin to a 17-kDa antiangiogenic factor. Proc Natl Acad Sci U S A. 2007;104:10010–10015. doi: 10.1073/pnas.0704179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cleutjens JP, Blankesteijn WM, Daemen MJ, Smits JF. The infarcted myocardium: simply dead tissue, or a lively target for therapeutic interventions. Cardiovasc Res. 1999;44:232–241. doi: 10.1016/s0008-6363(99)00212-6. [DOI] [PubMed] [Google Scholar]
- 33.Swynghedauw B. Molecular mechanisms of myocardial remodeling. Physiol Rev. 1999;79:215–262. doi: 10.1152/physrev.1999.79.1.215. [DOI] [PubMed] [Google Scholar]
- 34.Woessner JF., Jr The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch Biochem Biophys. 1961;93:440–447. doi: 10.1016/0003-9861(61)90291-0. [DOI] [PubMed] [Google Scholar]
- 35.Berg RA. Determination of 3-and 4-hydroxyproline. Methods Enzymol. 1982;82(Pt A):372–398. doi: 10.1016/0076-6879(82)82074-0. [DOI] [PubMed] [Google Scholar]
- 36.Wodarz A, Nusse R. Mechanisms of Wnt signaling in development. Annu Rev Cell Dev Biol. 1998;14:59–88. doi: 10.1146/annurev.cellbio.14.1.59. [DOI] [PubMed] [Google Scholar]
- 37.Kessler E, Adar R. Type I procollagen C-proteinase from mouse fibroblasts. Purification and demonstration of a 55-kDa enhancer glycoprotein. Eur J Biochem. 1989;186:115–121. doi: 10.1111/j.1432-1033.1989.tb15184.x. [DOI] [PubMed] [Google Scholar]
- 38.Takahara K, et al. Type I procollagen COOH-terminal proteinase enhancer protein: identification, primary structure, and chromosomal localization of the cognate human gene (PCOLCE) J Biol Chem. 1994;269:26280–26285. [PubMed] [Google Scholar]
- 39.Moali C, et al. Substrate-specific modulation of a multisubstrate proteinase. C-terminal processing of fibrillar procollagens is the only BMP-1-dependent activity to be enhanced by PCPE-1. J Biol Chem. 2005;280:24188–24194. doi: 10.1074/jbc.M501486200. [DOI] [PubMed] [Google Scholar]
- 40.Ge G, Zhang Y, Steiglitz BM, Greenspan DS. Mammalian tolloid-like 1 binds procollagen C-proteinase enhancer protein 1 and differs from bone morphogenetic protein 1 in the functional roles of homologous protein domains. J Biol Chem. 2006;281:10786–10798. doi: 10.1074/jbc.M511111200. [DOI] [PubMed] [Google Scholar]
- 41.Garrigue-Antar L, Francois V, Kadler KE. Deletion of epidermal growth factor-like domains converts mammalian tolloid into a chordinase and effective procollagen C-proteinase. J Biol Chem. 2004;279:49835–49841. doi: 10.1074/jbc.M408134200. [DOI] [PubMed] [Google Scholar]
- 42.Martyn U, Schulte-Merker S. The ventralized ogon mutant phenotype is caused by a mutation in the zebrafish homologue of Sizzled, a secreted Frizzled-related protein. Dev Biol. 2003;260:58–67. doi: 10.1016/s0012-1606(03)00221-5. [DOI] [PubMed] [Google Scholar]
- 43.Yabe T, et al. Ogon/Secreted Frizzled functions as a negative feedback regulator of Bmp signaling. Development. 2003;130:2705–2716. doi: 10.1242/dev.00506. [DOI] [PubMed] [Google Scholar]
- 44.Sun Y, Weber KT. Infarct scar: a dynamic tissue. Cardiovasc Res. 2000;46:250–256. doi: 10.1016/s0008-6363(00)00032-8. [DOI] [PubMed] [Google Scholar]
- 45.Jugdutt BI. Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation. 2003;108:1395–1403. doi: 10.1161/01.CIR.0000085658.98621.49. [DOI] [PubMed] [Google Scholar]
- 46.Jugdutt BI. Remodeling of the myocardium and potential targets in the collagen degradation and synthesis pathways. Curr Drug Targets Cardiovasc Haematol Disord. 2003;3:1–30. doi: 10.2174/1568006033337276. [DOI] [PubMed] [Google Scholar]
- 47.Scott IC, et al. Mammalian BMP-1/Tolloid-related metalloproteinases, including novel family member mammalian Tolloid-like 2, have differential enzymatic activities and distributions of expression relevant to patterning and skeletogenesis. Dev Biol. 1999;213:283–300. doi: 10.1006/dbio.1999.9383. [DOI] [PubMed] [Google Scholar]
- 48.Pappano WN, Steiglitz BM, Scott IC, Keene DR, Greenspan DS. Use of Bmp1/Tll1 doubly homozygous null mice and proteomics to identify and validate in vivo substrates of bone morphogenetic protein 1/tolloid-like metalloproteinases. Mol Cell Biol. 2003;23:4428–4438. doi: 10.1128/MCB.23.13.4428-4438.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fisher LW, Stubbs JT, 3rd, Young MF. Antisera and cDNA probes to human and certain animal model bone matrix noncollagenous proteins. Acta Orthop Scand Suppl. 1995;266:61–65. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.