

Abstract
Objective. Antigen-presenting cells (APC) play critical roles in establishing and maintaining peripheral tolerance. This is accomplished in part via expression of negative co-stimulatory molecules such as programmed death ligand-1 (PD-L1) on tolerogenic APC, such as immature myeloid dendritic cells (mDC). Several studies have strongly linked dysfunction of APC, including mDC, to the pathogenesis of SLE. The objective of this study was to determine whether APC expressed PD-L1 protein at normal levels during active lupus.
Methods. Peripheral blood mononuclear cells (PBMC) were isolated from 19 paediatric patients with SLE and from 17 healthy age-matched controls. PBMC from both cohorts were cultured in the absence of exogenously added stimuli, and leucocyte PD-L1 expression was measured by flow cytometry.
Results. Immature mDC and monocytes (Mo) from healthy children expressed little PD-L1 at initial isolation, but spontaneously up-regulated PD-L1 by 24 h. In contrast, both mDC and Mo from patients with active SLE failed to up-regulate PD-L1 over a 5 day time course, expressing this protein only during disease remissions.
Conclusions. These data are the first to link active lupus with reversibly decreased PD-L1 expression on professional APC, suggesting a novel mechanism for loss of peripheral tolerance in SLE.
Keywords: Systemic lupus erythematosus, Human, Antigen-presenting cell, Monocyte, Myeloid dendritic cell, Programmed death ligand-1, Co-stimulation
Introduction
Programmed death ligand-1 (PD-L1; also known as B7-H1/CD274), is a B7 family glycoprotein inducibly expressed on many haematopoetic and parenchymal cells in response to inflammatory stimuli. This molecule regulates immune tolerance by binding to the programmed death-1 (PD-1) receptor on lymphocytes, causing suppression of T-effector function [1–3], and permissiveness of regulatory T-cell function [4]. Recent data suggest that PD-L1 may also suppress T-cell activation by signalling through the B7-1 receptor [5]. Although mRNA for PD-L1 can be found in many healthy human tissues, baseline protein expression appears to be limited to cells of monocytic origin [6, 7]. Both myeloid dendritic cells (mDC) and monocytes (Mo) express PD-L1 protein, and anti-PD-L1 antibody increases the stimulatory capacity of mature and immature DCs for T-effector cells [8, 9], suggesting that PD-L1 may play an important role in the ability of DCs to tolerize T lymphocytes in the periphery. In support of this idea, endogenous or transgene-driven expression of PD-L1 on antigen-presenting DCs leads to diminished T-cell reactivity in vitro and in vivo, as demonstrated in murine models of autoimmunity [10, 11]. The importance of PD-L1 in self-tolerance has also been demonstrated in experimental animals in which blockade or absence of the PD-L1:PD-1 pathway results in various forms of autoimmune disease, including a spontaneous lupus-like glomerulonephritis in C57BL/6 mice [12–16]. However, little is known regarding the role of PD-L1 in human autoimmune disease.
The receptor for PD-L1 is shared by a second ligand, PD-L2, (B7-DC/CD273), which can also inhibit T-cell activation [17], but is less widely expressed and appears to play some non-redundant roles in self-tolerance [15, 18–20]. DNA polymorphisms in the gene for the shared PD-1 receptor have been linked to SLE susceptibility in some populations of adults [21–24] and children [25]; however, T-cell expression of PD-1 protein was not found to differ significantly between SLE patients and controls [26].
In contrast to the PD-1 gene studies, genetic polymorphisms in PD-L1 did not appear to be linked to SLE [27]. However, protein expression of PD-L1 has not been previously studied in lupus APC. We found that both immature mDC and Mo from children with SLE failed to up-regulate PD-L1 normally, and that this deficiency was associated with increased disease activity, suggesting an important role for this negative co-stimulator in the pathogenesis of SLE.
Materials and methods
Patient samples and peripheral blood mononuclear cell isolation
Paediatric donors with and without SLE were recruited under a research protocol approved by the institutional review board of Children's Hospital and Regional Medical Center in Seattle, WA, USA. Peripheral venous blood was collected into heparin- or citrate-containing tubes (Vacutainer, Becton Dickinson, NJ, USA) after written informed consent was obtained from the child and/or parent/guardian. Blood samples were centrifuged and plasma aliquots stored at −80°C. Data on plasma IFN-α levels were kindly provided by Dr Veronika Groh (Fred Hutchinson Cancer Research Center, Seattle, WA, USA), as measured by ELISA (R&D Systems, MN, USA). Peripheral blood mononuclear cells (PBMC) were isolated by density centrifugation over a Ficoll–Paque gradient (Amersham, Uppsala, Sweden), frozen in heat-inactivated A/B human serum (Valley Biomedical, Winchester, MA, USA) with 7% DMSO (Sigma, St. Louis, MO, USA), and stored in liquid nitrogen until use. In preliminary experiments, PBMC samples from four unique donors were split into frozen and fresh aliquots, and evaluated by flow cytometry to confirm a lack of effect of freeze–thaw on expression levels of PD-L1 (P ≥ 0.7).
SLE disease activity
Clinical and laboratory data were collected for each individual, and all but one of the lupus patients fulfilled the current ACR classification criteria for SLE [28]. As this was a retrospective study, the European Consensus Lupus Activity Measurement (ECLAM) was calculated [29] for all patient samples where information was available (n = 24); ECLAM scores ranged from 0 to 6.5, with a mean ± s.d. of 2.5 ± 2.0. As no patient had documentation of seizures, psychosis, cerebrovascular accident, cranial nerve disorder, visual disturbance, myositis, pleurisy, pericarditis, intestinal vasculitis or peritonitis at the time of blood draw, we used a modified scoring system to group patients with respect to disease activity, consisting of these remaining categories: mucocutaneous disease (rash, alopecia, mucosal ulcers and finger nodules), arthritis, haematuria, thrombocytopenia and hypocomplementaemia. In addition, we used lymphopenia, rather than leucopenia, as a sensitive measure of active paediatric SLE [30]. Several samples were chosen at random and also assayed for PBMC apoptosis and/or plasma levels of IFN-α, as these markers are strongly linked to SLE disease activity [31–33]. PBMC apoptosis was considered to be abnormally high if outside the bounds of the 99.95% CI of control cells (PBMC from seven healthy children tested, data not shown) and plasma IFN-α levels were considered to be abnormal if ≥5 times the upper limit of normal (six healthy children tested, data not shown). As the clinical assessments were gleaned from chart notes written by a panel of different physicians, the objective laboratory data were weighted more heavily in the final determination, with each abnormal laboratory value assigned 2 points, and each abnormal clinical finding assigned 1 point. A total disease activity score of ≥4 points was felt to represent active disease, and called ‘flare’, while a score of <4 was felt to represent inactive disease, and called ‘remission’. This modified scoring system has the limitation that it has not been formally validated; however, there are no validated disease activity scoring systems for paediatric SLE. Moreover, when this modified scale was used to categorize patients into flare and remission groups, the mean ECLAM scores and anti-dsDNA antibody levels were found to be significantly different between the two groups (Table 1), suggesting the potential utility of this approach.
Table 1.
Clinical and laboratory characteristics of SLE patients at the time of blood draw
| Mean value |
Percentage of patients |
Percentage of patients |
||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SLE subgroup | n | Age (yrs) | Disease activity score | ECLAM score | Anti-dsDNA (IU/ml) | Apoptosis (%PBMC) | Mucocu- taneous disease | Arthritis | Haematuria | Thrombocy- topenia | Lymphopenia | Hypocomple- mentemia | High PBMC apoptosis | High plasma IFN-α | IV methyl- prednisolonea | IV cyclophos- phamidea | Daily oral mycophenolate | Daily oral HCQ | Daily oral prednisone | Weekly oral MTX |
| Remission | 14 | 14.7 | 1.6 | 1.8 | 52 | 30 | 43 | 7 | 9 | 0 | 15 | 46 | 0 | 0 | 21 | 7 | 7 | 79 | 57 | 14 |
| Flare | 12 | 13.8 | 5.2 | 3.5 | 312 | 61 | 36 | 36 | 22 | 17 | 50 | 83 | 83 | 29 | 8 | 8 | 0 | 75 | 58 | 25 |
| P-value | NS | <2×10−7 | <0.04 | <0.05 | <0.004 | NS | NS | NS | NS | NS | NS | <0.01 | NS | NS | NS | NS | NS | NS | NS | |
aWithin 3 months.
Cell culture, antibodies and flow cytometry
PBMC were thawed, washed and diluted to 1–2 × 106 cells/ml in culture medium consisting of RPMI 1640 supplemented with l-glutamine (CellGro, Herndon, VA, USA), 10% heat-inactivated A/B human serum, 1% penicillin/streptomycin (CellGro) and 0.1% β-mercaptoethanol. Cells were plated in round-bottom 96-well plates (Corning Costar, Corning, NY, USA) and incubated at 37°C in a humidified cell chamber with 5% CO2. At the time points indicated, PBMC were surface-stained using various fluorochrome- or biotin-conjugated mAbs, including: anti-CD1c, (Miltenyi, Auburn, CA, USA), anti-CD3, anti-PD-L1 (eBioscience, San Diego, CA, USA), anti-CD11b, anti-CD11c, anti-CD14, anti-CD-86, anti-PD-L2 (Pharmingen/BD Biosciences), anti-CD45RO, anti-CD80, anti-CD83 and/or anti-HLA-DR (BioLegend, San Diego, CA, USA), with isotype-matched, fluorochrome-/biotin-labelled irrelevant mAbs as controls. All samples were blocked using 0.5% human serum and anti-FcR antibody (Miltenyi) during staining. After staining, PBMC were fixed using 2% paraformaldehyde in PBS after preliminary experiments indicated no effect of cell fixation on expression levels of PD-L1 or other surface markers (data not shown). Some cultures were stained in parallel with Annexin V and propidium iodide (PI) as per the manufacturer's instructions (both from Becton Dickinson) and apoptosis assessed by enumerating the percent of Annexin V-positive PBMC per culture. Flow cytometry was performed using an LSR II cytometer (Becton Dickinson), and the data were analysed using Flow Jo software (Tree Star, Inc., Ashland, OR, USA).
Statistical analyses
Populations were compared using a two-tailed t-test and significance assigned where P < 0.05. Due to the fact that some patients had more than one blood draw and were therefore overrepresented in the data set, statistical analyses were repeated using multivariate logistic generalized estimating equations (GEEs), to account for multiple observations in some individuals. Results of GEE analyses confirmed P < 0.05 between populations as identified by t-test.
Results
Patient population
A total of 26 PBMC samples were collected from 19 unique SLE patients ranging in age from 6 to 21 yrs (mean ± s.d. = 14.3 ± 3.7). Clinical and laboratory data for these blood draws are summarized in Table 1. Overall, 12 samples were obtained from patients with active (recurrent or newly diagnosed) SLE and categorized as ‘flare’ samples, while 14 were categorized as ‘remission’ samples, as outlined above. Patient age was not significantly different between the SLE flare (13.8 ± 3.1) and remission (14.7 ± 4.2) groups, and there were no statistically significant differences between the groups with respect to medication usage.
Control PBMC were collected from 15 healthy volunteers ranging in age from 6 to 23 yrs (15.7 ± 5.2); patient age and gender composition were not significantly different between the control and SLE groups. Females comprised 18/19 of the SLE patients and 12/15 of the controls.
Identification and characterization of PD-L1+ cells in primary human PBMC
To test the hypothesis that PD-L1 expression is abnormal on lupus APC, primary human PBMC were cultured for 1 day in the absence of exogenously added stimuli and PD-L1 levels measured using four-color multiparametric flow cytometry. Consistent with prior findings in normal human leucocytes [7], we observed virtually no PD-L1 protein on CD3+ cells, but PD-L1 was expressed on a proportion of CD3− cells from a healthy subject (Fig. 1A). In contrast, there was near-complete absence of PD-L1 on PBMC from a patient with active SLE. Surprisingly, CD3− cells from the same patient during lupus remission had regained the ability to express normal levels of PD-L1. This pattern was reproducible using PBMC from multiple individuals (see below).
Fig. 1.

Primary human PBMC spontaneously up-regulate PD-L1 protein. (A) Cells were cultured for 1 day and live PBMC gated by forward and side scatter. Histograms show mean fluorescence intensity (MFI) of PD-L1 as measured on CD3+ and CD3− cells from a healthy paediatric control (top), from a patient in SLE flare (middle) and from the same patient during SLE remission (bottom). (B) Using control PBMC, PD-L1+ cells were further characterized and found to separate into two groups based on CD14 expression. (C) Using antibodies to phenotypic markers, the PD-L1+ CD14lo and CD14hi cells were identified as immature mDC and Mo, respectively.
To characterize the CD3− cells expressing PD-L1, we assessed levels of several surface markers on normal PBMC and found that the PD-L1+ cells naturally segregated into CD14-low/negative (CD14lo) and CD14-high (CD14hi) populations (Fig. 1B), demonstrating that PD-L1 was primarily expressed by APC of myeloid lineage, consistent with published data [7]. Examination of the CD14lo and CD14hi APC subsets for CD11c, CD11b, CD1c, CD45RO and HLA-DR revealed expression patterns consistent with immature mDC and Mo, respectively (Fig. 1C). Similar to a prior report [34], we did not observe a significant amount of CD83 on these cells, supporting the idea that the mDC in these cultures were phenotypically immature.
APC from patients with active SLE fail to up-regulate PD-L1
To confirm abnormal PD-L1 levels on lupus APC, PBMC were cultured as above and immature mDC and Mo identified by co-staining for CD14 and CD11c (Fig. 2A). As noted, we found that both immature mDC and Mo from children with active SLE failed to up-regulate PD-L1, while APC from children in lupus remission expressed normal or increased levels of this negative co-stimulator (Fig. 2B). As the CD14lo CD11c+ populations in PBMC may have been comprised of a heterogenous mix of differentiating Mo and early mDC, we used the cell surface marker CD1c (BDCA-1) to specifically identify Type I mDC [35]. Gating for CD14lo CD11c+ CD1c+ cells revealed PD-L1 expression consistent with that of the CD14lo CD11c+ population as a whole, confirming the utility of this method for measuring PD-L1 levels on immature mDC (Fig. 2C).
Fig. 2.

PD-L1 protein is deficient on APC during SLE flare, but not during remission. (A) PBMC were cultured for 1 day and APC subsets identified. (B) PD-L1 expression on both mDC and Mo was reduced during SLE flare, but not during remission. (C) CD1c staining demonstrated that PD-L1 expression in CD14lo CD11c+ cells was enriched on Type I immature mDC. (D) PD-L1 levels on mDC (upper graph) and Mo (lower graph) from 15 controls, 12 SLE flare and 14 SLE remission. Circles represent individual PD-L1 values and bars represent the mean MFI (±1 s.d.) for each group. For SLE flare mDC, *P < 6.5 × 10−3 compared with controls; #P < 2.5 × 10−2 compared with remission. For SLE flare Mo, *P < 1.8 × 10−4 compared with controls; #P < 2.4 × 10−6 compared with remission. For SLE remission Mo, *P < 3.4 ×10−3 compared with controls. (E) PD-L1 expression on mDC and Mo was normalized to background levels using the PD-L1 MFI of CD14− CD11c− cells as the denominator for each sample. For SLE flare mDC, *P < 4.1 × 10−3 compared with controls; #P < 2.1 × 10−2 compared with remission. For SLE flare Mo, *P < 2.0 × 10−6 compared with controls; #P < 1.1 × 10−8 compared with remission. (F) SLE flare patients exhibited a lower percentage of PD-L1+ APC (mDC, upper graph and Mo, lower graph). For SLE flare mDC, *P < 4.2 × 10−3 compared with controls. For SLE flare Mo, *P < 1.1 ×10−6 compared with controls; #P < 4.0 ×10−8 compared with remission. (G) Mo PD-L1 values from (E) were graphed for patients with serial blood samples; shaded area denotes the ‘normal range’ of Mo PD-L1 expression observed in healthy controls (mean ± 1 s.d.).
These findings were reproducible and statistically significant for immature mDC and Mo from multiple individuals (Fig. 2D–F). Compared with control APC, mean PD-L1 expression was more than 3-fold lower on immature mDC and Mo from children in SLE flare, but nearly 2-fold higher on Mo during SLE remission (Fig. 2D). To correct for potential inter-experiment variation, the PD-L1 MFI for each set of APC was normalized to background levels, using the PD-L1 MFI of the CD14− CD11c− cells as the denominator for each sample. However, mDC and Mo from patients in SLE flare remained significantly PD-L1-deficient as compared with both normal and remission APC (Fig. 2E). Not only were PD-L1 protein levels lower on SLE flare APC, but there were also lower percentages of cells expressing PD-L1 (Fig. 2F). Compared with controls, PD-L1 was expressed on nearly 70% fewer SLE flare mDC and nearly 50% fewer Mo. In contrast, the percentages of PD-L1+ APC in lupus remission samples were not significantly different than in controls, consistent with the idea that this negative costimulator may play a role in inhibiting the autoreactive immune response. In support of this concept, serial samples drawn from four patients at different times revealed an inverse correlation between Mo PD-L1 expression and disease activity, with lower levels during SLE flares and higher levels during remissions (Fig. 2G).
PD-L1 deficiency in lupus flare APC is persistent over time and is associated with other immune abnormalities
To rule out the possibility that APC from patients with active SLE were merely delayed in up-regulation of PD-L1, we measured expression of this protein over a 5 day culture period. Normal APC expressed little PD-L1 at initiation of culture, but levels rapidly increased over time, with peak PD-L1 expression in both immature mDC and Mo by days 1–2, and return to baseline by day 5 (data not shown). In contrast, immature mDC and Mo from children with active SLE expressed abnormally low levels of PD-L1 throughout the time course, refuting the idea that the low PD-L1 observed in day 1 cultures was merely due to delayed surface expression of this protein. As in short-term cultures, APC from children in SLE remission exhibited normal or elevated levels of PD-L1, suggesting a potential functional association between PD-L1 expression and disease activity.
In contrast to PD-L1, staining of control PBMC for the related negative co-stimulator, PD-L2, revealed a nearly negligible level of protein expression that did not change over 4 days of culture (data not shown). These findings are in agreement with previous work that showed that purified Mo from healthy adult volunteers expressed virtually no PD-L1 or PD-L2 upon initial isolation, and spontaneously up-regulated only PD-L1 after 24 h of culture [8].
SLE flare APC have increased co-stimulatory potential
To determine whether the defect in lupus flare APC was specific to PD-L1, we measured the level of positive co-stimulatory molecules (a combination of CD80 plus CD86) on PBMC from children with and without SLE. We found that although immature mDC and Mo were clearly PD-L1-deficient during SLE flare, they retained the ability to express CD80/CD86 (Fig. 3), congruent with prior studies that revealed normal or elevated levels of these proteins on mDC and Mo from patients with SLE [36, 37]. Taken together, these observations suggest that the inability of lupus APC to express PD-L1 cannot be attributed to a global decrease in co-stimulatory molecule expression during SLE flare, and that loss of the negative PD-L1 signal is not associated with or compensated for by a decrease in positive co-stimulatory signals.
Fig. 3.
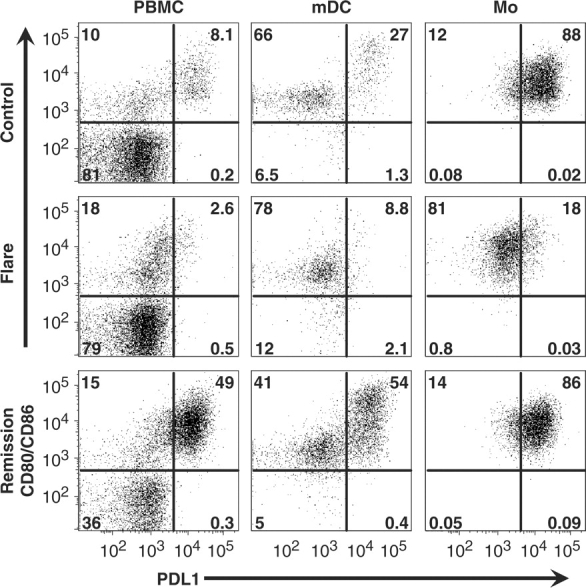
Lupus flare APC express positive co-stimulatory molecules. PBMC were cultured for 1 day and gated for APC as given earlier. Compared with control cells (top row), SLE flare APC (middle row) lacked PD-L1 and the CD80/86hi subset of mDC. In contrast, SLE remission cells (bottom row) expressed both PD-L1 and CD80/86 in a pattern similar to that of controls (numbers in each graph represent the percentage of cells in each quadrant). Results are representative of two separate experiments using PBMC from three healthy controls, two SLE flare and two SLE remission.
Consistent with a prior report [38], we also observed that although Mo populations were fairly homogenous with respect to expression of CD80/CD86, immature mDC segregated into CD80/CD86lo and CD80/CD86hi-expressing groups, suggesting differing abilities for T-cell stimulation (Fig. 3). Moreover, in control and SLE remission PBMC, the majority of PD-L1 protein was expressed by CD80/CD86hi mDC, suggesting that T-cell stimulation by these most potent APC is normally held in check by this negative regulator. Surprisingly, the CD80/CD86hi subset of mDC was markedly lacking in SLE flare, although the reasons for this are currently unclear.
Discussion
To our knowledge, this is the first report of abnormal PD-L1 expression on APC from patients with active SLE. The finding of decreased PD-L1 protein during active SLE has significant implications for conversion of APC to a pathological state. Although immature mDC and Mo from children with active SLE failed to up-regulate PD-L1, both cell types retained the ability to express several other markers, including CD80/CD86, at the APC surface. As CD80/CD86-mediated T-effector signalling is normally countered by PD-L1 [1, 5], lupus APC could potentially have an abnormally high capacity for positive T-cell co-stimulation during SLE flare. A hyperstimulatory role for lupus APC is supported by data showing that mDC and Mo from patients with SLE have an increased ability to activate allogeneic T-cells [32, 37, 39].
Not only do DCs depend upon PD-L1 signalling to diminish T-cell stimulation, but negative co-stimulation by PD-L1 is more effective in immature DCs than in mature DCs [40], suggesting a mechanism for the immunogenic presentation of autoantigens in SLE. Immature mDC ingest apoptotic bodies and cross-present Ags to cytotoxic T-cells [41], and lack of PD-1 signalling in vivo results in DC-mediated CD8+ T-cell priming rather than tolerization [42]. Therefore, our data may provide a partial explanation for the self-reactivity observed in lupus patients, whereby PD-L1-deficient immature mDC present apoptosis-related antigens in a pro-inflammatory context.
While examining CD80/CD86 expression, we also noted that the CD80/CD86hi subgroup of mDC was diminished during SLE flare. This is intriguing, as SLE PBMC proliferate poorly in autologous mixed leucocyte reactions (aMLRs) [43], and it has recently been suggested that CD80/CD86hi mDC are integral for T-cell proliferation during aMLRs [38]. The reason behind the loss of these cells in active SLE is unclear, however, and may be related to increased apoptosis or to tissue sequestration—it has been reported that patients with active Class III and IV lupus nephritis have significantly fewer circulating mDC along with a concomitant increase of immature mDC in renal tissues [44]. It would be interesting to determine whether these renal mDC retain the ability to express PD-L1.
In addition to potentially stimulating autoreactive T effector cells, PD-L1-deficient APC may promote abnormal function and/or development of regulatory T lymphocytes (Treg). It has been demonstrated that PD-L1 signalling is necessary for the suppressive activity of classic CD4+ CD25+ Treg in an animal model of GVHD [4], and that anti-CD3-stimulated naïve CD4+ T cells could be induced to become Tr1-type regulatory cells if co-stimulated with PD-L1-Ig [45]. Although decreased Treg number and function have been reported in human SLE [46, 47] it remains to be determined whether PD-L1 plays any role in Treg-related deficiencies.
The decreased PD-L1 levels we observed on APC from patients with active SLE were not likely a result of medication effects, as the use of immunosuppressive agents was comparable between flare and remission groups (Table 1). Three of the children with active SLE and low PD-L1 had been newly diagnosed and had never received any immunosuppression. Additionally, all four of the subjects who provided serial samples (Fig. 2G) were on minimally varying medication regimens at the time of their blood draws. Similarly, a prior study of SLE patients revealed no correlation between the use of immunosuppressive agents in vivo and changes in cell surface markers on peripheral blood DC, as well as no significant effect of chloroquine, steroids, 6-mercaptopurine or mycophenolate mofetil on markers of Mo differentiation and maturation in vitro [39].
In the course of these studies, we did attempt to identify the cause of low PD-L1 on SLE APC. We had previously observed that lupus T cells spontaneously secreted high levels of Th2-type cytokines (data not shown), and therefore we examined the effect of recombinant cytokines and soluble anti-cytokine antibodies on APC PD-L1 levels. However, in preliminary studies, none of the agents tested (including IL-2, IL-4, IL-12, IL-17, TNF-α, IFN-α, anti-IL-4, anti-IL-12 or anti-IFN-γ) resulted in down-modulation of PD-L1 (data not shown). These cytokines were functionally active, as they could alter the expression of other cell surface markers; however, this occurred without loss of PD-L1.
In addition to cytokine dysregulation, SLE leucocytes undergo apoptosis at an increased rate, and we did note an inverse correlation between PD-L1 expression and PBMC apoptosis (Table 1). Following this lead, we have preliminary data demonstrating that in vitro treatment of PBMC with polycaspase inhibitors not only reduced leucocyte apoptosis, but increased the expression of PD-L1 on mDC and Mo in all cultures (data not shown). These findings suggest a role for caspase activity in the normal regulation of PD-L1 and provide a potential explanation for the loss of this negative co-stimulator on APC from patients with active SLE. In support of this idea, it has been reported that caspase-3 is directly responsible for the decreased CD3ζ-chain expression on the surface of SLE T cells [48].
Our findings complement what is already known regarding PD-L1 expression in human disease; levels of PD-L1 are increased on circulating APC from patients with chronic HIV, hepatitis B or hepatitis C infection [49–51], and decreased on DC from patients with multiple sclerosis [34]. As preliminary studies in our laboratory have also indicated abnormally low levels of PD-L1 on APC from patients with some other types of active autoimmune disease (data not shown), we propose that diminished expression of PD-L1 on circulating APC may be a hallmark of active multi-organ autoimmunity, while elevated levels of PD-L1 on circulating APC may be indicative of chronic infection. If verified in larger samples, this distinction may be medically useful, as it is often unclear whether clinical deterioration in SLE patients represents disease flare or infection.
In summary, our findings link active SLE with the inability of peripheral blood APC to express PD-L1, suggesting that PD-L1 may be functionally important in the maintenance of immune tolerance in SLE. Lack of this protein on the surface of immature mDC also suggests a mechanism for the propensity of the immune system to target apoptosis-associated molecules in SLE, as immature mDC typically ingest and present these self-antigens. Given the inverse correlation between PD-L1 and SLE disease activity, future investigations may reveal a role for PD-L1 fusion proteins or other molecules capable of ligating PD-1 in the treatment of SLE or other autoimmune diseases. Larger studies may determine whether intermittent measurements of PD-L1 on circulating APC could provide an additional tool for monitoring the clinical course of SLE.

Acknowledgements
The authors would like to thank Dr Keith Elkon, Dr Troy Torgerson and Dr Yufeng Peng for helpful discussion and critical review of the manuscript, Dr Veronika Groh for plasma IFN-α levels, Dr Do Peterson for statistical analyses and Ms. Sharon Goodwin for collection of clinical samples.
Funding: This work was supported in part by NIH grant T32 AR007108 to the University of Washington Division of Rheumatology, by NIH grant M01-RR-00037 to the University of Washington General Clinical Research Center, and by funding from Children's Hospital and Regional Medical Center in Seattle, WA, USA.
Disclosure statement: The authors/institution have applied for a patent based on the contents of this manuscript.
References
- 1.Freeman GJ, Long AJ, Iwai Y, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keir ME, Liang SC, Guleria I, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203:883–95. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–45. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 4.Kitazawa Y, Fujino M, Wang Q, et al. Involvement of the programmed death-1/programmed death-1 ligand pathway in CD4+CD25+ regulatory T-cell activity to suppress alloimmune responses. Transplantation. 2007;83:774–82. doi: 10.1097/01.tp.0000256293.90270.e8. [DOI] [PubMed] [Google Scholar]
- 5.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–22. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong H, Strome SE, Salomao DR, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 7.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–9. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 8.Brown JA, Dorfman DM, Ma FR, et al. Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol. 2003;170:1257–66. doi: 10.4049/jimmunol.170.3.1257. [DOI] [PubMed] [Google Scholar]
- 9.Kim HK, Guan H, Zu G, et al. High-level expression of B7-H1 molecules by dendritic cells suppresses the function of activated T cells and desensitizes allergen-primed animals. J Leukoc Biol. 2006;79:686–95. doi: 10.1189/jlb.0805436. [DOI] [PubMed] [Google Scholar]
- 10.He FR, Zhu HF, Huang H, et al. Programmed death-1 ligands-transfected dendritic cells loaded with glutamic acid decarboxylase 65 (GAD65) inhibit both the alloresponse and the GAD65-reactive lymphocyte response. Clin Exp Immunol. 2008;151:86–93. doi: 10.1111/j.1365-2249.2007.03546.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hirata S, Senju S, Matsuyoshi H, Fukuma D, Uemura Y, Nishimura Y. Prevention of experimental autoimmune encephalomyelitis by transfer of embryonic stem cell-derived dendritic cells expressing myelin oligodendrocyte glycoprotein peptide along with TRAIL or programmed death-1 ligand. J Immunol. 2005;174:1888–97. doi: 10.4049/jimmunol.174.4.1888. [DOI] [PubMed] [Google Scholar]
- 12.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–51. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 13.Latchman YE, Liang SC, Wu Y, et al. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci USA. 2004;101:10691–6. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang SC, Latchman YE, Buhlmann JE, et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. 2003;33:2706–16. doi: 10.1002/eji.200324228. [DOI] [PubMed] [Google Scholar]
- 15.Martin-Orozco N, Wang YH, Yagita H, Dong C. Cutting edge: programmed death (PD) ligand-1/PD-1 interaction is required for CD8+ T cell tolerance to tissue antigens. J Immunol. 2006;177:8291–5. doi: 10.4049/jimmunol.177.12.8291. [DOI] [PubMed] [Google Scholar]
- 16.Fife BT, Guleria I, Gubbels Bupp M, et al. Insulin-induced remission in new-onset NOD mice is maintained by the PD-1-PD-L1 pathway. J Exp Med. 2006;203:2737–47. doi: 10.1084/jem.20061577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Latchman Y, Wood CR, Chernova T, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–8. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 18.Zhu B, Guleria I, Khosroshahi A, et al. Differential role of programmed death-ligand 1 and programmed death-ligand 2 in regulating the susceptibility and chronic progression of experimental autoimmune encephalomyelitis. J Immunol. 2006;176:3480–9. doi: 10.4049/jimmunol.176.6.3480. [DOI] [PubMed] [Google Scholar]
- 19.Fukushima A, Yamaguchi T, Azuma M, Yagita H, Ueno H. Involvement of programmed death-ligand 2 (PD-L2) in the development of experimental allergic conjunctivitis in mice. Br J Ophthalmol. 2006;90:1040–5. doi: 10.1136/bjo.2006.091314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carter LL, Leach MW, Azoitei ML, et al. PD-1/PD-L1, but not PD-1/PD-L2, interactions regulate the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2007;182:124–34. doi: 10.1016/j.jneuroim.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 21.Thorburn CM, Prokunina-Olsson L, Sterba KA, et al. Association of PDCD1 genetic variation with risk and clinical manifestations of systemic lupus erythematosus in a multiethnic cohort. Genes Immun. 2007;8:279–87. doi: 10.1038/sj.gene.6364383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prokunina L, Castillejo-Lopez C, Oberg F, et al. A regulatory polymorphism in PDCD1 is associated with susceptibility to systemic lupus erythematosus in humans. Nat Genet. 2002;32:666–9. doi: 10.1038/ng1020. [DOI] [PubMed] [Google Scholar]
- 23.Ferreiros-Vidal I, Gomez-Reino JJ, Barros F, et al. Association of PDCD1 with susceptibility to systemic lupus erythematosus: evidence of population-specific effects. Arthritis Rheum. 2004;50:2590–7. doi: 10.1002/art.20436. [DOI] [PubMed] [Google Scholar]
- 24.Wang SC, Chen YJ, Ou TT, et al. Programmed death-1 gene polymorphisms in patients with systemic lupus erythematosus in Taiwan. J Clin Immunol. 2006;26:506–11. doi: 10.1007/s10875-006-9048-9. [DOI] [PubMed] [Google Scholar]
- 25.Velazquez-Cruz R, Orozco L, Espinosa-Rosales F, et al. Association of PDCD1 polymorphisms with childhood-onset systemic lupus erythematosus. Eur J Hum Genet. 2007;15:336–41. doi: 10.1038/sj.ejhg.5201767. [DOI] [PubMed] [Google Scholar]
- 26.Tsutsumi Y, Jie X, Ihara K, et al. Phenotypic and genetic analyses of T-cell-mediated immunoregulation in patients with type 1 diabetes. Diabet Med. 2006;23:1145–50. doi: 10.1111/j.1464-5491.2006.01951.x. [DOI] [PubMed] [Google Scholar]
- 27.Wang SC, Lin CH, Ou TT, et al. Ligands for programmed cell death 1 gene in patients with systemic lupus erythematosus. J Rheumatol. 2007;34:721–5. [PubMed] [Google Scholar]
- 28.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40:1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 29.Mosca M, Bencivelli W, Vitali C, Carrai P, Neri R, Bombardieri S. The validity of the ECLAM index for the retrospective evaluation of disease activity in systemic lupus erythematosus. Lupus. 2000;9:445–50. doi: 10.1191/096120300678828640. [DOI] [PubMed] [Google Scholar]
- 30.Yu HH, Wang LC, Lee JH, Lee CC, Yang YH, Chiang BL. Lymphopenia is associated with neuropsychiatric manifestations and disease activity in paediatric systemic lupus erythematosus patients. Rheumatology. 2007;46:1492–4. doi: 10.1093/rheumatology/kem182. [DOI] [PubMed] [Google Scholar]
- 31.Chen DY, Hsieh TY, Hsieh CW, Lin FJ, Lan JL. Increased apoptosis of peripheral blood lymphocytes and its association with interleukin-18 in patients with active untreated adult-onset Still's disease. Arthritis Rheum. 2007;57:1530–8. doi: 10.1002/art.23088. [DOI] [PubMed] [Google Scholar]
- 32.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–3. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 33.Feng X, Wu H, Grossman JM, et al. Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum. 2006;54:2951–62. doi: 10.1002/art.22044. [DOI] [PubMed] [Google Scholar]
- 34.Karni A, Abraham M, Monsonego A, et al. Innate immunity in multiple sclerosis: myeloid dendritic cells in secondary progressive multiple sclerosis are activated and drive a proinflammatory immune response. J Immunol. 2006;177:4196–202. doi: 10.4049/jimmunol.177.6.4196. [DOI] [PubMed] [Google Scholar]
- 35.Robak E, Smolewski P, Wozniacka A, Sysa-Jedrzejowska A, Stepien H, Robak T. Relationship between peripheral blood dendritic cells and cytokines involved in the pathogenesis of systemic lupus erythematosus. Eur Cytokine Netw. 2004;15:222–30. [PubMed] [Google Scholar]
- 36.Liu MF, Li JS, Weng TH, Lei HY. Differential expression and modulation of costimulatory molecules CD80 and CD86 on monocytes from patients with systemic lupus erythematosus. Scand J Immunol. 1999;49:82–7. doi: 10.1046/j.1365-3083.1999.00452.x. [DOI] [PubMed] [Google Scholar]
- 37.Decker P, Kotter I, Klein R, Berner B, Rammensee HG. Monocyte-derived dendritic cells over-express CD86 in patients with systemic lupus erythematosus. Rheumatology. 2006;45:1087–95. doi: 10.1093/rheumatology/kel061. [DOI] [PubMed] [Google Scholar]
- 38.Scheinecker C, Machold KP, Majdic O, Hocker P, Knapp W, Smolen JS. Initiation of the autologous mixed lymphocyte reaction requires the expression of costimulatory molecules B7-1 and B7-2 on human peripheral blood dendritic cells. J Immunol. 1998;161:3966–73. [PubMed] [Google Scholar]
- 39.Ding D, Mehta H, McCune WJ, Kaplan MJ. Aberrant phenotype and function of myeloid dendritic cells in systemic lupus erythematosus. J Immunol. 2006;177:5878–89. doi: 10.4049/jimmunol.177.9.5878. [DOI] [PubMed] [Google Scholar]
- 40.Chen C, Qu QX, Huang JA, et al. Expression of programmed-death receptor ligands 1 and 2 may contribute to the poor stimulatory potential of murine immature dendritic cells. Immunobiology. 2007;212:159–65. doi: 10.1016/j.imbio.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 41.Albert ML, Pearce SF, Francisco LM, et al. Immature dendritic cells phagocytose apoptotic cells via alphavbeta5 and CD36, and cross-present antigens to cytotoxic T lymphocytes. J Exp Med. 1998;188:1359–68. doi: 10.1084/jem.188.7.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Probst HC, McCoy K, Okazaki T, Honjo T, van den Broek M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat Immunol. 2005;6:280–6. doi: 10.1038/ni1165. [DOI] [PubMed] [Google Scholar]
- 43.Sakane T, Steinberg AD, Green I. Failure of autologous mixed lymphocyte reactions between T and non-T cells in patients with systemic lupus erythematosus. Proc Natl Acad Sci USA. 1978;75:3464–8. doi: 10.1073/pnas.75.7.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fiore N, Castellano G, Blasi A, et al. Immature myeloid and plasmacytoid dendritic cells infiltrate renal tubulointerstitium in patients with lupus nephritis. Mol Immunol. 2008;45:259–65. doi: 10.1016/j.molimm.2007.04.029. [DOI] [PubMed] [Google Scholar]
- 45.Ding Q, Lu L, Wang B, et al. B7H1-Ig fusion protein activates the CD4+ IFN-gamma receptor+ type 1 T regulatory subset through IFN-gamma-secreting Th1 cells. J Immunol. 2006;177:3606–14. doi: 10.4049/jimmunol.177.6.3606. [DOI] [PubMed] [Google Scholar]
- 46.Valencia X, Yarboro C, Illei G, Lipsky PE. Deficient CD4+CD25 high T regulatory cell function in patients with active systemic lupus erythematosus. J Immunol. 2007;178:2579–88. doi: 10.4049/jimmunol.178.4.2579. [DOI] [PubMed] [Google Scholar]
- 47.Alvarado-Sanchez B, Hernandez-Castro B, Portales-Perez D, et al. Regulatory T cells in patients with systemic lupus erythematosus. J Autoimmun. 2006;27:110–8. doi: 10.1016/j.jaut.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 48.Krishnan S, Kiang JG, Fisher CU, et al. Increased caspase-3 expression and activity contribute to reduced CD3zeta expression in systemic lupus erythematosus T cells. J Immunol. 2005;175:3417–23. doi: 10.4049/jimmunol.175.5.3417. [DOI] [PubMed] [Google Scholar]
- 49.Geng L, Jiang G, Fang Y, et al. B7-H1 expression is upregulated in peripheral blood CD14+ monocytes of patients with chronic hepatitis B virus infection, which correlates with higher serum IL-10 levels. J Viral Hepat. 2006;13:725–33. doi: 10.1111/j.1365-2893.2006.00746.x. [DOI] [PubMed] [Google Scholar]
- 50.Trabattoni D, Saresella M, Biasin M, et al. B7-H1 is up-regulated in HIV infection and is a novel surrogate marker of disease progression. Blood. 2003;101:2514–20. doi: 10.1182/blood-2002-10-3065. [DOI] [PubMed] [Google Scholar]
- 51.Golden-Mason L, Palmer B, Klarquist J, Mengshol JA, Castelblanco N, Rosen HR. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J Virol. 2007;81:9249–58. doi: 10.1128/JVI.00409-07. [DOI] [PMC free article] [PubMed] [Google Scholar]