

Abstract
Engineering of the methylmalonyl-CoA (mmCoA) metabolite node of the Saccharopolyspora erythraea wild type strain (FL2267) through duplication of the mmCoA mutase (MCM) operon led to a 51% (range 40%-64%, 0.95 CI, N = 152) increase in erythromycin production in a high-performance oil-based fermentation medium. The MCM operon was carried on a 6.8 kb DNA fragment in plasmid pFL2212 which was inserted by homologous recombination into the S. erythraea chromosome. The fragment contained one uncharacterized gene, ORF1; three MCM related genes, mutA, mutB, meaB; and one gntR-family regulatory gene, mutR. Additional strains were constructed containing partial duplications of the MCM operon, as well as a knockout of ORF1, none of these strains showed any significant alteration in their erythromycin production profile. The combined results showed that increased erythromycin production only occurred in strain FL2385 containing a duplication of the entire MCM operon including mutR and a predicted stem-loop structure overlapping the 3′ terminus of the mutR coding sequence.
Keywords: methylmalonyl-CoA mutase (MCM), metabolic engineering, Saccharopolyspora erythraea, erythromycin, precursor, strain improvement
1. Introduction
Microorganisms are used for the commercial fermentative production of many of the world's most important drugs including antibiotics, anticancer agents, and immunosuppressants. A critical aspect of this process is that industrial microorganisms must often be genetically improved for higher production before they can be used commercially (Parekh et al., 2000). The success of a strain improvement program can determine whether a new natural product goes to market or is abandoned (Demain, 2006). For generic drugs, strain improvement determines which manufacturer maintains the greatest market share. Historically, strain improvement has been performed by an empirical mutate-and-screen process and has been performed in close association with process and engineering improvements to optimize large-scale industrial fermentations (Adrio and Demain, 2006).
The emergence of genetic engineering technology in industrial microorganisms in the late 1970's brought with it the potential that rational engineering might be used to improve strains beyond what could be done solely by random mutagenesis (Vinci and Byng, 1999; Baltz, 2001). Presently, although the molecular genetic tools have been developed for most of the important antibiotic-producing organisms and much useful information has been gained describing the genetic control of antibiotic biosynthesis, the application of this new technology to strain improvement of secondary metabolites has met with limited commercial success. Nevertheless, many new innovative approaches towards secondary metabolite strain improvement are now available or under development using a variety of different approaches, some of which show promise for general commercial application to the field (Askenazi et al., 2003; Tang et al., 2000; Murli et al., 2003; Zhang et al., 2002; Lum et al., 2004, Brunker et al., 1998).
More recently, metabolic engineering tools (Bro and Nielsen, 2004) have been introduced including genomic (Kellis et al., 2003), transcriptomic (Blanchard and Hood, 1996; Lander 1999), proteomic (Gavin et al., 2002), and metabolomic (German et al., 2005) technologies, which in combination with genetic engineering tools can further accelerate the development of rational strain improvement strategies. The principal challenge facing the incorporation of these new technologies into secondary metabolite strain improvement programs is to focus their data-generating power onto the most important strain improvement targets.
Our work has been directed towards the identification of the most important erythromycin strain improvement targets for metabolic engineering using a genetic approach. Our improved mutants are generated by a random process, using tagged mutagenesis, rendering the improved mutants amenable to reverse engineering (Reeves et al., 2004, 2006). This process is performed in a step-wise fashion in an industrial organism where strain improvement mutations and corresponding process enhancements are added sequentially. The method of screening directly for the desired phenotype followed by reverse engineering generates high-quality targets that can then be further exploited by “omic” analyses and metabolic engineering (Vemuri and Aristidou, 2005).
The erythromycin fermentation is a classic antibiotic fermentation that has been successfully improved by the traditional mutate-and-screen method in conjunction with corresponding process improvements over the past 50 years. Erythromycin biosynthesis in the mycelial actinomycete, Saccharopolyspora erythraea, has been widely studied as a model system for antibiotic production (Hutchinson et al., 1993; Katz and Donadio, 1995; Leadlay, 1997; McDaniel et al., 2001), and erythromycin and its semi-synthetic derivatives are still widely used in medicine; therefore, improved strains are still highly sought after. An alternative, single-cell erythromycin-producing organism, Aeromicrobium erythreum, is also available for comparative analyses between these two different bacteria (Miller, 1991; Reeves et al., 2004). Both strains have sophisticated methods for genetic engineering, but A. erythreum is easier to genetically manipulate, is more amenable to microfermentation, and is the faster growing of the two organisms.
The genetic analysis of erythromycin strain improvement was begun in a previous study and the first high-quality genetic strain improvement targets were generated and identified as mutB and cobA. In A. erythreum, a mutB knockout, leading to the inactivation of methylmalonyl-CoA mutase (MCM), led directly to a 75% strain improvement effect in a soluble starch carbohydrate-based medium (Reeves et al., 2004). In S. erythraea, a mutB polar mutant grown in the same type of medium caused a 125% strain improvement effect. Further indication that mutB was an important and interesting target in S. erythraea, came from studies performed in a soybean oil-based fermentation medium where a mutB (polar) knockout caused a 65% decrease in erythromycin production (Reeves et al., 2006). A model was presented illustrating how carbon flow at the methylmalonyl-CoA (mmCoA) metabolite node flows in different directions through MCM depending on whether a carbohydrate-based or oil-based fermentation is performed (Reeves et al., 2004; 2006).
The model for MCM activity in oil-based medium led to the current study in which the prediction was tested that if knockouts of the MCM genes cause a decrease in erythromycin production, then overexpression of the MCM genes should lead to an increase in erythromycin production in this medium. Here, we report the results of that study in which the prediction was borne out, and the magnitude of the yield increase was determined through extensive shake flask testing.
2. Materials and methods
2.1. Bacterial strains, culture conditions, chemicals and biochemicals
The bacterial strains and plasmids used in this study are described in Table 1. S. erythraea FL2267, a wild-type strain (not a low producing red variant strain) has been described previously (Reeves et al., 2006). The E. coli strains used in cloning experiments were DH5α-e or the DNA methylation-minus strain MCR-DH5α (Invitrogen, Carlsbad, CA). Spores of S. erythraea strains were produced on E20A agar (Reeves et al., 2002). Thiostrepton, if necessary for plasmid maintenance, was added to E20A agar at a final concentration of 15 μg/ml. Seed cultures used for the preparation of shake flask fermentations were grown in CFM1 medium (Reeves et al., 2006). Fermentations were performed in OFM1 (Reeves et al., 2006) using a 1:20 seed inoculation (1.25 ml in 23.75 ml OFM1) and incubated at 32.5°C for 5 days at 380 rpm. Minimal medium agar (AVMM; Weber and McAlpine, 1992) was supplemented with 50 mM methylmalonic acid (Sigma-Aldrich, St. Louis, MO). Analysis of pigment production in both wild-type and mutant S. erythraea strains was performed on R2T2 agar [R2T agar (Weber et al., 1985) minus peptone] and E20A agar (Reeves et al., 2002). E. coli was routinely cultured on 2×YT agar and grown in Luria-Bertani broth (Sambrook et al., 1989) containing either ampicillin sodium salt at 100 μg/ml or kanamycin sulfate at 50 μg/ml for plasmid maintenance.
Table 1.
Bacterial strains and plasmids used in this study.
| Plasmid or strain | Description | Reference |
|---|---|---|
| E. coli plasmids | ||
| pFL8 | S. erythraea integration vector. Used to make gene knockouts in the chromosome when modified to contain a DNA insert from the S. erythraea chromosome. (Apr)1 (Thior)2 | Reeves et al. (2002) |
| pFL2212 | S. erythraea integration vector used to insert a duplicate copy of the methylmalonyl-CoA mutase region in the chromosome. Used in the construction of strains FL2385 and FL2421. The total region integrated was 6.791 kb and contained the ORF1, mutA, mutB, meaB, and mutR genes (DNA accession nos. DQ289499 and DQ289500). (Apr) (Thior) | This study |
| pFL3163 | S. erythraea integration vector used to make a knockout in ORF1 by gene replacement of a kanamycin resistance gene (aphI), thus generating strain FL2433. The kanamycin resistance gene cassette was inserted 222 bp into the ORF1 gene in the opposite transcriptional orientation. (Apr) (Thior) (Knr)3 | This study |
| pFL3165 | S. erythraea integration vector used to make a duplication of the methylmalonyl-CoA mutase region minus mutR, thus generating strain FL2435. Performed by religation of the larger fragment of pFL2212 after double digestion with StuI and BamHI, which deleted a 191 bp 3′ end fragment of mutR. (Apr) (Thior) | This study |
| pFL3166 | S. erythraea integration vector used to make a duplication of the ORF1 gene, thus generating strain FL2436. Constructed by religation of pFL2212 after exonuclease III digestion deleted mutR, meaB and 50 bp into the 3′ end of mutB. (Apr) (Thior) | This study |
| S. erythraea strains | ||
| FL2267 | Derivative of S. erythraea ATCC 11635. Wild-type (white) strain. Used as host strain in transformations. | Reeves et al. (2006) |
| FL2281 | Polar knockout strain containing a kanamycin-resistance gene (aphI) inserted into mutB. (Knr) | Reeves et al. (2006) |
| FL2302 | Derivative of FL2267 containing a 126 bp in-frame deletion in mutB. Used as host for generating S. erythraea FL2421. | Reeves et al. (2006) |
| FL2385-1, -2,-3 | Derivative of FL2267 containing integrated pFL2212 by single crossover insertion. Contains duplicate copies of ORF1, mutA, mutB, meaB, and mutR. Used for testing overexpression of MCM region. (Thior) | This study |
| FL2421-1,-2,-3 | Derivative of S. erythraea FL2302 containing an integrated pFL2212 by single crossover insertion. Contains one functional copy of mutB and duplicate functional copies of ORF1, mutA, meaB, and mutR. Used to test overexpression of ORF1 and mutR. (Thior). | This study |
| FL2433-1,-2,-3 | ORF1 knockout strains derived by gene replacement of pFL3163. (Knr) | This study |
| FL2435-1,-2,-3 | Derivative of FL2267 containing integrated pFL3165 by single crossover insertion. Contains duplicate copies of ORF1, mutAB, and meaB. Used for testing overexpression of MCM region except for mutR. (Thior) | This study |
| FL2436-1,-2,-3 | Derivative of FL2267 containing integrated pFL3166 by single crossover insertion. Contains duplicate functional copies of ORF1 and mutA. Used for testing overexpression of ORF1. (Thior) | This study |
| E. coli strains | ||
| DH5α | E. coli host strain for transformations | Invitrogen (Carlsbad, CA) |
denotes that plasmid contains bla, which confers resistance to ampicillin in E. coli;
denotes that plasmid contains tsr, which confers resistance to thiostrepton in S. erythraea;
denotes that plasmid contains aphI, which confers resistance to kanamycin in E. coli and S. erythraea.
2.2. DNA methods
Standard molecular biology techniques were used for cloning all or parts of the MCM region into E. coli plasmids (Sambrook et al., 1989). Restriction enzymes, T4 DNA ligase and Klenow DNA polymerase were obtained from Fermentas (Vilnius, Lithuania). PCR was performed on a Perkin Elmer Geneamp 2400 thermocycler using the 2× PCR Master kit from Fermentas. The final Mg++ concentration in the reaction mixture was 2 mM. PCR products were cloned directly into pGEM-T Easy Vector (Promega, Madison, WI). DNA sequencing reactions, used to confirm all plasmid constructs, were performed using the Big Dye Terminator Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) according to the manufacturer's instructions. All sequencing was performed at SeqWright (Houston, TX) performed on an ABI Prism™ 3730xl sequencer. Sequence chromatograms were analyzed using Sequencher® version 4.6 software (Gene Codes, Ann Arbor, MI).
S. erythraea wild-type and the mutB mutant strain FL2302 were transformed with plasmid DNA using standard protoplast transformation procedures (Weber et al., 1990). Confirmation of plasmid integration was performed by PCR using sequence-specific primers that amplify a 1.04 kb region of the thiostrepton-resistance (tsr) gene contained on pFL8 (Reeves et al., 2004).
2.3. Generation of S. erythraea strain FL2385, which contains a duplicate copy of the MCM operon region
Analysis of the DNA sequence of the MCM operon region revealed two convenient restriction sites, EcoRI and BamHI, for subcloning the entire MCM operon region (Fig. 1) into the S. erythraea integration vector pFL8 (Reeves et al., 2002). Cosmid 6E7 (Reeves et al., 2006) was digested with EcoRI and BamHI, which released the 6.791 kb S. erythraea chromosomal fragment that contained all of mutA, mutB, meaB, mutR and ORF1 (Fig. 1). The EcoRI/BamHI fragment was ligated to similarly digested pFL8 (Reeves et al., 2002) to create plasmid pFL2212. Plasmid pFL2212 and its derivatives were used as the basis for generating strains containing duplicate DNA sequences of all or part of the MCM region in the S. erythraea chromosome. S. erythraea wild-type strain FL2267 was transformed with pFL2212 with selection for single crossover integration by homologous recombination. The recombinant S. erythraea transformants were designated FL2385. Three independent isolates were tested for erythromycin production in shake flask fermentations.
Fig.1.
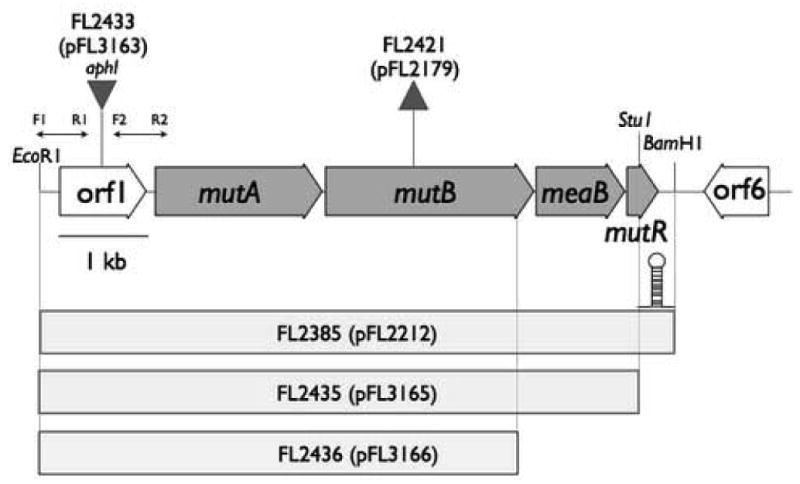
Gene map of the 8.6 kb S. erythraea MCM operon region. The EcoRI/BamHI fragment subcloned for construction of plasmid pFL2212 is 6.8 kb. The three bars beneath the operon map represent the fragments subcloned into pFL8 used for generating full or partial gene duplications through plasmid insertion. Each DNA fragment is labeled with the corresponding strain number and the corresponding insertion plasmid number, in parentheses, used to create the strain. The triangle above ORF1 indicates the location of the kanamycin-resistance gene (aphI) insertion in FL2433. The triangle above mutB represents the in-frame deletion site in S. erythraea FL2302 (Reeves et al., 2006), the host strain used to generate FL2421 by integration of pFL2212. Plasmid numbers are given for each of the two knockout insertions in parentheses. Plasmid pFL2179 was constructed as part of a previous study to generate strain FL2302. The location of a stem-loop structure described in the text and determined from DNA sequence analysis is shown just below the map at the end of the mutR gene. Abbreviations: mutA, encodes the smaller subunit of MCM; mutB, encodes the larger subunit of MCM; meaB, encodes an MCM accessory protein; mutR, gntR-family regulatory gene; 6, ORF6, homology to lipoproteins; F1 and R1, forward and reverse primers, respectively, for PCR fragment 1; F2 and R2, primers for PCR fragment 2 (Materials and Methods).
2.4. Generation of S. erythraea strain FL2435, which contains a duplication of the MCM operon region lacking co-duplication of mutR
To generate a plasmid containing the entire MCM region except for mutR, a double digestion of pFL2212 with the unique cutting restriction enzymes StuI and BamHI was performed. This deleted 191 bp from the 3′ end of mutR. Klenow polymerase was used to fill in the BamHI overhang and the blunt ends were ligated using T4 DNA ligase. This plasmid was designated pFL3165. S. erythraea wild-type strain FL2267 was transformed with pFL3165 with selection for thiostrepton resistance. The S. erythraea strain containing integrated pFL3165 was designated FL2435 (Fig. 1). Three independent FL2435 isolates were tested for erythromycin production in shake flask fermentations.
2.5. Generation of S. erythraea strain FL2421, which contains a duplication of mutR
A strain was constructed to test the effects of duplicating mutR without co-duplication of the MCM genes. This was done by transforming pFL2212 (Fig. 1) into the mutB in-frame deletion strain FL2302 (Reeves et al., 2006) with selection for thiostrepton resistance. FL2302 derivatives containing integrated pFL2212 were obtained by patching individual primary transformants onto E20A agar containing thiostrepton at 15 μg/ml. Thiostrepton-resistant isolates were designated FL2421. Three FL2421 isolates (Fig. 1) were tested in shake flask fermentations for erythromycin production.
2.6. Generation of S. erythraea FL2433, which contains a knockout in ORF1
An ORF1 knockout mutant was generated by replacement of a 375 bp internal fragment of ORF1 with the kanamycin-resistance gene (aphI). PCR primers were designed to amplify two non-contiguous DNA fragments (fragment 1, left; and fragment 2, right; Fig. 1) flanking the 375 bp deletion site in ORF1 with convenient restriction sites engineered at the 5′ ends of the PCR fragments. The primers for fragment 1 were forward primer (F1) containing an engineered EcoRI site 5′-agaattcCAACGGCTGGGAGTACCAC-3′ and the reverse primer (R1), containing an engineered BamHI site, 5′-aggatccGCTTCGCCCGTGCTGCCA-3′. The primers for fragment 2 were: forward (F2), containing an engineered BamHI site, 5′-aggatccGAGAACTGCCGGCTCGGC-3′ and the reverse primer (R2) containing an engineered HindIII site, caagcttGCGTGGTGCTGGCGGACG-3′. Lowercase letters indicate engineered sequences and capital letters indicate S. erythraea sequences. Fragments 1 and 2 were cloned individually into pGEM-T Easy Vector. For isolation of the cloned fragments from pGEM-T Easy Vector, the left fragment (638 bp) was released by digesting with EcoRI and BamHI and the right fragment (764 bp) was released by digesting with BamHI and HindIII. After gel purification of the two cloned PCR products, a four-component ligation reaction was performed by mixing the purified ORF1 fragments with EcoRI-, HindIII-digested pFL8 (Reeves et al., 2002) and aphI previously purified by BamHI digestion of pUC4K (Pharmacia Biochemicals, Piscataway, NJ). Ligation mixtures were electroporated into E. coli DH5α-e with selection for white, kanamycin-resistant colonies after plating on 2×YT agar plates containing kanamycin at 50 μg/ml and X-gal indicator. Several kanamycin-resistant colonies were analyzed further by restriction digestion and sequencing to confirm the cloned fragment. This plasmid was designated pFL3163. S. erythraea protoplasts were transformed with pFL3163 and primary transformants were obtained using a selection for kanamycin resistance. Gene replacement strains containing ORF1 knockouts were identified by patching single transformant colonies onto E20A agar containing kanamycin or thiostrepton. Kanamycin-resistant, thiostrepton-sensitive isolates were analyzed further. In addition, strains containing single crossover integrations of pFL3163 were subjected to an eviction procedure to obtain the ORF1 knockout in a two-step process (Reeves et al., 2002). Evictants were confirmed by replica plate testing on E20A agar containing kanamycin and thiostrepton. The strain containing the knockout in ORF1 was designated FL2433. Three independent FL2433 isolates were tested in shake flask fermentations.
2.7. Generation of S. erythraea FL2436, which contains a duplication of ORF1
To duplicate ORF1 in the S. erythraea wild-type chromosome a derivative of pFL3165 containing a deletion of the mutR, meaB and part of the mutB gene was generated. Plasmid pFL3165 was digested with exonuclease (Fermentas) beginning at the StuI site and continued 1206 bp in the 5′ direction. Sequencing revealed that the endpoint of digestion was 50 bp into the 3′ end of mutB, which would be predicted to inactivate MCM. The plasmid containing a deletion of part of the MCM operon was designated pFL3166. Plasmid pFL3166 was transformed into the S. erythraea wild-type strain with selection for thiostrepton resistance. S. erythraea strains containing integrated pFL3166 were designated FL2436 (Fig. 1). Three independent FL2436 isolates were tested in shake flask fermentations.
2.8. Shake flask fermentations, bioassays and thin-layer chromatography
Shake flask fermentations of S. erythraea wild-type and mutant strains were performed as described in Reeves et al. (2006). Two different shake flask media were used, CFM1 and OFM1. CFM1 is a fermentation medium designed for laboratory use where a soluble medium is desired for convenient analysis of growth and chemical analysis of erythromycin production. CFM1 per liter distilled water: Difco™ soluble starch, 60 g; Bacto™-soytone (Difco™), 20 g; CaCl2•2H2O (Sigma), 0.1 g; Bacto™-yeast extract (Difco™), 1.5 g; MOPS, 26.5 g; pH adjusted to 6.8 with 4N NaOH (Sigma). OFM1 contains insoluble medium components and is meant to closely correlate to an industrial-type fermentation medium. OFM1, per liter distilled water: toasted nutri-soy flour (ADM, Decatur, IL), 22 g; Difco™ soluble starch, 15 g; CaCO3 powder (JT Baker, Phillipsburg, NJ), 3 g; MgSO4.7H2O (JT Baker, Phillipsburg, NJ), 0.5 g; FeSO4•7H2O (JT Baker, Phillipsburg, NJ), 15 mg; Soy oil, 50 ml (ADM, Decatur, IL). Bioassays for the determination of erythromycin concentrations in culture broths were performed as previously described (Reeves et al., 2006). Visualization of the erythromycin profile from culture broth extracts was performed using thin-layer chromatography as previously described (Reeves et al., 2006).
2.9. Statistical analysis
Percent increases and decreases in erythromycin production of MCM region mutants compared to the wild-type strain are given as averages for a range of uncertainty with a confidence interval (CI) of 95%.
3. Results
3.1. Duplication of the complete MCM operon by insertion of plasmid pFL2212 causes a significant increase in erythromycin production
S. erythraea strain FL2385 was constructed by transformation of the wild type strain (FL2267) with plasmid pFL2212 and selection for a single crossover plasmid-integrated transformant. Strain FL2385 therefore contained a duplication of the MCM operon (6.791 kb) between the BamHI and EcoRI sites (Fig. 1) which includes five ORFS: ORF1, mutA, mutB, meaB, and mutR.
Extensive shake flask testing of S. erythraea FL2267 (wild-type) and FL2385 was performed in OFM1 medium. The results of 23 separate experiments and seventy-six independent shake flask cultures measured in duplicate (N=152) by bioassay revealed an average increase in erythromycin production of 51% for FL2385 above the wild-type strain with a range of increase between 40%-64% (Fig. 2A). Fermentation broth extracts were further analyzed by thin-layer chromatography (Fig. 2C) and the results were consistent with the increase in bioactivity measured by bioassay as being due to an increase in production of erythromycin A and not some other bioactive metabolite. Two additional independently derived transformants, FL2385-2 and FL2385-3, were generated and tested in shake flask fermentations. FL2385-2 produced 38% above the wild-type strain with a range of increase between 23% and 53%. FL2385-3 produced 38% above the wild-type strain with a range of increase between 27% and 50% (N=18).
Fig. 2.
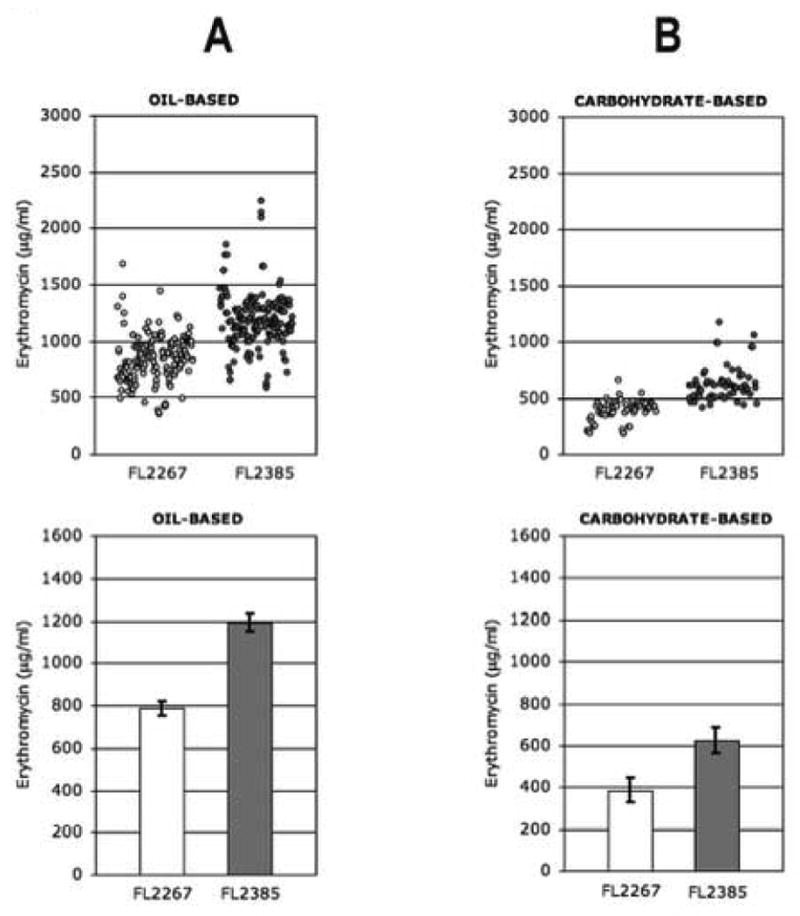
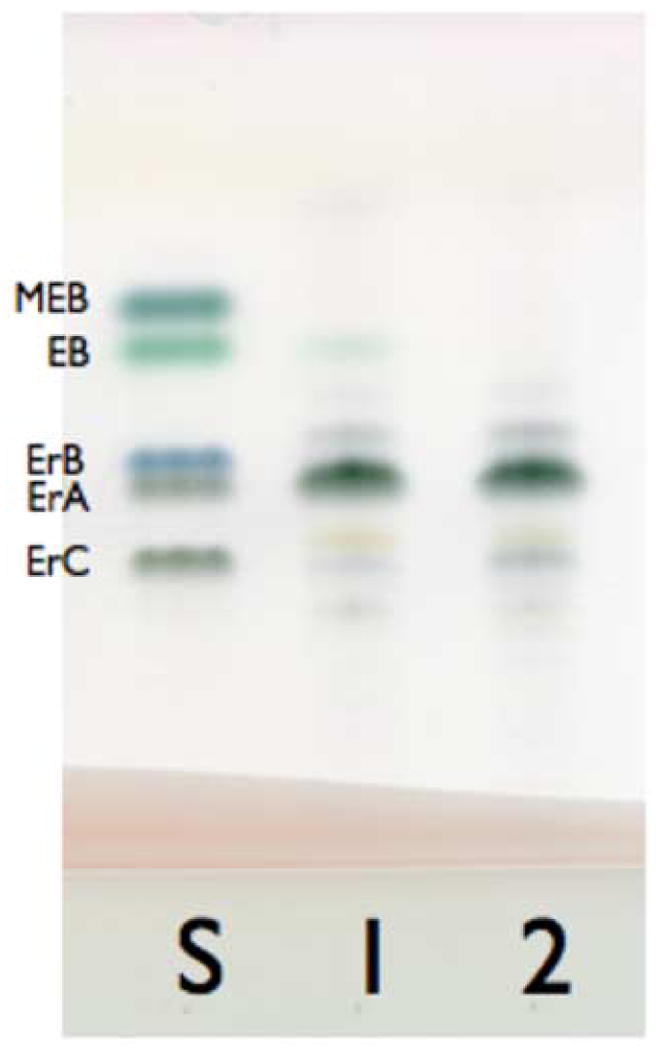
Scatterplot (top) and bar chart (95% CI, bottom) representations of erythromycin production levels of S. erythraea FL2267 (wild-type strain) and FL2385-1 (MCM duplication strain) as determined by bioassay (A) grown in oil-based medium (76 shake flasks, two bioassay discs per flask [N=152]), and (B) grown in carbohydrate-based medium (30 shake flasks, two bioassay discs per flask, [N=60]). In the scatterplots, each circle represents one point of raw data derived from a single bioassay disc (see Material and methods). (C) Thin-layer chromatograph; lane S, erythromycin reference standards; lane 1, ethyl acetate extract of day 5 fermentation broth of the wild type strain (FL2267); lane 2, ethyl acetate extract of day 5 fermentation broth of the MCM duplication strain (FL2385). MEB, 3-alpha-mycarosyl erythronolide B; EB, erythronolide B; ErB, erythromycin B; ErA, erythromycin A; and ErC, erythromycin C.
FL2385 (FL2385-1) was also tested extensively in carbohydrate-based medium CFM1 (Reeves et al., 2006). Thirty independent shake flask fermentations were performed and their erythromycin yields were measured in duplicate (N= 60) by bioassay. The results showed an average increase of 63% above the parent with a range of increase between 43%-85% (Fig. 2B). FL2385-2 and FL2385-3 were also tested in CFM1 medium, though less extensively than FL2385-1, and showed a similar reproducible increase in erythromycin production.
3.2. The strain improvement effect is not seen without including mutR in the duplicated region
A strain containing a duplication of the MCM operon region, without co-duplication of mutR, was constructed. The resulting strain, S. erythraea FL2435, contained two copies of ORF1, mutA, mutB, and meaB but only one copy of mutR. Due to the deletion of the 3′ end of mutR in plasmid pFL3165, the new strain, FL2435, was also lacking a stem-loop structure at the end of the duplicated region (Fig. 1). Three independently derived isolates of FL2435 were initially tested in shake flask fermentations and behaved similarly; one strain was extensively further tested (Fig. 3A). The results showed no statistically significant difference in the erythromycin production levels between FL2435 and the wild-type strain, showing that inclusion of mutR in the duplicated region is essential for the strain improvement effect observed in FL2385.
Fig. 3.
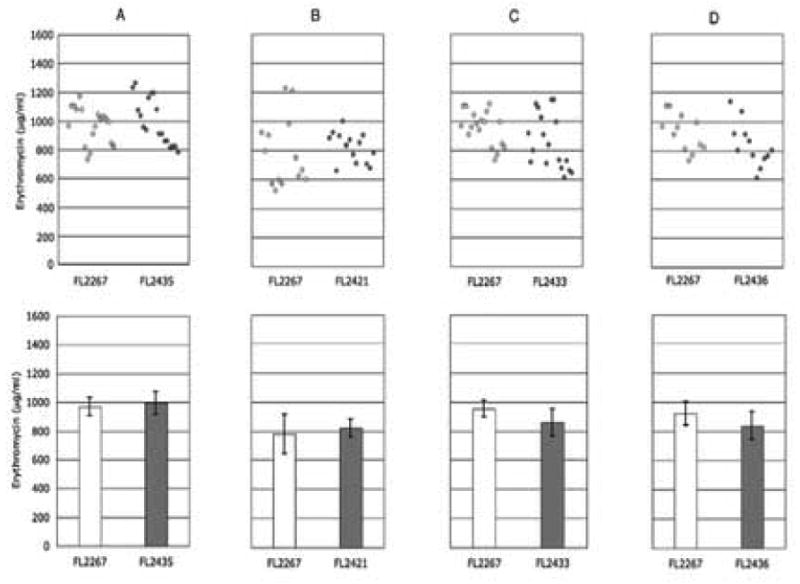
Scatterplot (top) and bar chart (bottom) representations of shake flask fermentations comparing S. erythraea wild-type FL2267 with S. erythraea strains: (A) FL2435, (B) FL2421, (C) FL2433, and (D) FL2436. Each graph represents the results of three independent shake flask fermentations. The bar chart graphs show the average erythromycin yield (N=18) with error bars (95% CI).
3.3. Duplication of mutR alone does not produce a strain improvement effect
To test whether the strain improvement effect in strain FL2385 was solely due to the presence of a duplicate copy of mutR or whether it required co-duplication of the MCM complex genes, a third strain, FL2421, was constructed and tested in shake flask fermentations. This strain contains an integrated pFL2212, as in FL2385, but the host was the non-polar mutB knockout mutant FL2302 (Reeves et al., 2006). The chromosomal copy of mutB is inactivated in FL2302, therefore, integration of an unmodified copy of the MCM region containing mutR would effectively test duplication of only mutR on erythromycin production. Three independently-derived FL2421 strains were initially tested in shake flask fermentations and showed similar results. Extensive further testing of FL2421-1 showed no statistically significant differences between itself and the wild-type strain (Fig. 3B). These results suggest that the strain improvement effect observed in strain FL2385 is not merely the result of duplication of the mutR regulatory gene.
3.4. ORF1 is not involved in erythromycin production
The 3′ end of ORF1 is separated from the predicted start codon of mutA by 87 bp, suggesting that they are coded within two separate transcriptional units (Fig. 1, DNA accession no. AY117133). The kanamycin resistance gene (aphI) was inserted into ORF1 in the opposite transcriptional orientation using plasmid pFL3163 to create S. erythraea strain FL2433. The knockout strain displayed normal morphological and pigment characteristics, unlike the mutB knockout strains FL2302 and FL2281 (Reeves et al., 2006). Furthermore, the ORF1 knockout strains had a wild-type erythromycin production phenotype (Fig. 3C). These results are consistent with ORF1 being in a separate transcriptional unit from the MCM genes. If ORF1 were in the same transcriptional unit as the MCM genes, the ORF1 knockout strain should have displayed the defective sporulation and pigmentation phenotypes characteristic of the MCM (mutB) knockout strains (Fig. 4). The results indicate that ORF1 plays no role in the function of MCM and no role in erythromycin production. Furthermore, ORF1 has no DNA or amino acid sequence homologs in the Genbank database to help assess its possible function.
Fig. 4.

Complementation of the pigmentation defect in mutB knockout strain FL2302 using pFL2212. Two independent isolates of FL2421 were plated alongside S. erythraea wild-type and the two mutB knockout strains FL2302 (in-frame mutB deletion) and FL2281 (polar knockout) on R2T2 agar media. Pigmentation defects are observed clearly on R2T2 where both mutB knockout strains lack diffusible reddish-brown pigment compared to the wild-type strain (FL2267), which makes abundant pigment. Integration of plasmid pFL2212 into strain FL2302 to create S. erythraea strain FL2421 causes full restoration of reddish-brown pigment production. Not shown here, but also investigated was the red-variant strain FL1347 (Reeves et al., 2002). The mutB knockout in FL1347 also led to a block in red pigment production on R2T2 agar.
Another strain, FL2436, was constructed to contain two copies of ORF1. This was accomplished by exonuclease III digestion of pFL2212 into the 3′ end of mutB (see Materials and methods) and transformation of this truncated plasmid (pFL3166) into wild type S. erythraea FL2267. Shake flask fermentations of FL2436 strains showed that erythromycin production was not statistically different between the two strains (Figs. 3D).
3.5. Complementation of mutB morphological, pigmentation and growth phenotypes
In a previous report, S. erythraea mutB mutants, FL2302 and FL2281, were described as having visible pleiotropic defects in both pigmentation and sporulation (Reeves et al., 2006). In addition, these mutants were unable to grow on methylmalonic acid as sole carbon source. In this study we showed that the mutant phenotypes associated with mutB knockouts could be complemented through integration of plasmid pFL2212 into the mutB deletion strain FL2302 (S. erythraea FL2421; Figs. 1 and 4). S. erythraea FL2421 showed complete restoration of reddish-brown pigment production when compared to the wild-type strain on R2T2 agar plates (Fig. 4). It can be concluded, therefore, that the genes of the MCM region carried on plasmid pFL2212, and derivatives of pFL2212, are expressed upon integration into the chromosome of the S. erythraea strains described in this study.
Another implication of this result is that the reddish-brown pigment produced by S. erythraea on R2T2 agar requires a functional MCM reaction to be produced. A red pigment, flaviolin, derived from malonyl-CoA, has been previously reported to be produced by a S. erythraea red variant strain (Cortes et al., 2002). Results from our study showed that both the red variant strain FL1347 (Reeves et al., 2002) and the wild-type strain FL2267 (also known as the white or gray strain) were blocked in reddish-brown pigment production by the mutB knockout on R2T2 agar, much like the rppA mutation of Cortes et al. (2002) led to a similar block in reddish-brown pigment production. Interestingly, Cortes et al. (2002) tested for a strain improvement effect in their rppA mutants but found none. Our mutation in MCM on the other hand does block pigment production and does have a positive effect on erythromycin production in a carbohydrate-based medium (Reeves et al., 2006) where pigment production appears to be the greatest. One explanation for this result is that carbon needed for red pigment formation could come from the mmCoA pool that feeds into erythromycin, and that blocking pigment formation at the MCM gateway diverts carbon that would normally go into pigment production into the erythromycin pathway. Blocking pigment production at a step after the MCM reaction and therefore after succinyl-CoA formation, such as at the rppA step, does not have the same benefit simply because once carbon has exited from the mmCoA pool, it may no longer benefit erythromycin production. It may be that malonyl-CoA accumulates in rppA mutants but apparently it does not efficiently cycle back to the mmCoA pool in carbohydrate-based medium.
4. Discussion
The importance of the mmCoA metabolite node in erythromycin strain improvement was uncovered in a previous study using a reverse engineering approach (Reeves et al., 2004). The current study was begun to determine whether we could manipulate the mmCoA metabolite node in an industrially relevant oil-based medium in a way to increase production of erythromycin. The results presented in this report show that increased production of erythromycin is possible through the duplication of the MCM operon which includes mutA, mutB, meaB, and mutR. The best modified strain showed an increase in erythromycin production of 51% (range 40-64% for FL2385-1) above the wild type S. erythraea strain.
Previously, mmCoA had been shown to be the limiting factor for erythromycin production in the wild-type strain and that knockouts of the mutB gene, coding for the larger subunit of the MCM, led to significant decreases in erythromycin production in oil-based fermentation medium (Reeves et al., 2006). This led to the conclusion that the net flow of the MCM reaction under oil-based growth conditions was from succinyl-CoA to mmCoA (Reeves et al., 2006). This model logically suggested that overexpression of the MCM genes should lead to an increase in production of erythromycin in the oil-based medium, and the results in this report are consistent with this prediction.
A model, incorporating the results described in this report, is presented (Fig. 5). In oil-based medium, where the MCM acts as a feeder route into the mmCoA metabolite pool, strain FL2385 is postulated to receive additional carbon flow for erythromycin production from the overexpression of MCM complex genes (indicated by an enlarged arrow from succinyl-CoA [S] to mmCoA [M]).
Fig. 5.
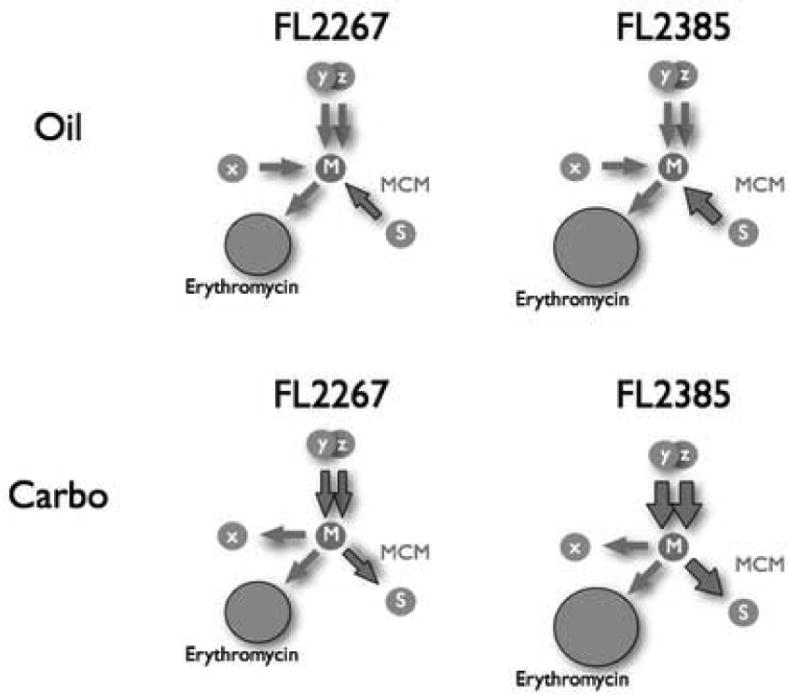
Metabolic pathway model showing feed routes leading into the methylmalonyl-CoA pool when S. erythraea wild-type, FL2267, and MCM duplication strain, FL2385, are grown in an oil-based medium or a carbohydrate-based medium. Top row, oil-based medium: a boost in the carbon flow from succinyl-CoA (metabolite S) to mmCoA (metabolite M) due to a duplication of the MCM operon is shown as a thick gray arrow in FL2385 compared to FL2267. This increase in flow into the mmCoA pool is the explanation currently favored for the increase in erythromycin production seen in FL2385. Other metabolites: x, unknown metabolite connected to the mmCoA pool via an unknown enzyme activity; y, z, additional metabolites connected to the mmCoA pool. Bottom row, carboyhydrate-based medium: a boost in carbon flow from mmCoA to succinyl-CoA is shown due to the duplication of the the MCM operon. Also shown is a boost in carbon flow from two feeder pathways involving metabolites, “y” and “z”, into the mmCoA pool due to postulated regulatory effects of mutR. The postulated increased carbon flow into the mmCoA pool from the two feeder pathways in strain FL2385 more than compensates for the loss of carbon via the MCM reaction such that there is an overall net gain in carbon flow into the mmCoA pool and therefore a net gain in erythromycin production.
A similar model and concept for strain improvement was demonstrated for the cephamycin C producer but it was arrived at from a much different approach (Malmberg and Hu, 1993). In cephamycin C strain improvement, a theoretical kinetic analysis was used to predict the rate-limiting factor for cephamycin C biosynthesis to be alpha-aminoadipic acid (Malmberg and Hu, 1991). This led to the construction of highly improved strains (two- to five-fold improvements were obtained) through duplication of lat, the gene that codes for lysine e-aminotransferase. Overexpression of the LAT gene in cephamycin C biosynthesis, and overexpression of the MCM genes in erythromycin biosynthesis have similar effects on strain improvement presumably because both apparently increase the pool size of the rate-limiting metabolite for biosynthesis of their respective antibiotics. Together, these two studies present experimental verification of two strain improvement concepts that may have general application: first, that strain improvement can be achieved through increasing carbon flow towards the metabolite node of the limiting factor, and second, that the limiting factor is a metabolite that occupies a boundary position between primary and secondary metabolism. Another remarkably similar strain improvement strategy involving gene duplication by plasmid insertion into a Streptomyces genome has been recently reported by Li et al. regarding nikkomycin biosynthesis where a strain improvement effect of 1.8 -fold was achieved using this method (2005). The results from these three highly related studies suggests that a new effective general method for strain improvement could involve the creation and screening of libraries of integrated transformants containing duplicated segments of the chromosome. Our results suggest that larger fragments would be preferrable, for example, in the range of 10 kb to accommodate larger operons such as the MCM operon. No knowledge of the genes involved would be required in advance, but reverse engineering and follow-up studies would reveal the genetic basis of the strain improvement effect, in much the same way as gene knockout libraries are now screened and reverse engineered (Reeves et al., 2004 and Alper et al., 2005).
The rational strain improvement mechanism presented here is for the S. erythraea wild type strain, as opposed to the much lower producing red-variant strain. This is the strain closest in phenotype to most high producing industrial strains. The mechanism works for this strain when it is grown in its most productive fermentation medium, an oil-based medium. Even though the results presented in this report are for the publicly available wild-type strain, the strain improvement mechanisms could also improve production of higher-producing commercial strains, if mmCoA is also the limiting factor for erythromycin production in those strains. No information has been reported, to our knowledge, regarding factors that are limiting production of erythromycin in the commercial strains. Nevertheless it would be logical to assume that mmCoA is the limiting factor in commercial strains if they accumulate erythromycin A as the primary end product, and if they show no evidence of a pathway bottleneck, for example, by the accumulation of a pathway intermediate.
Interestingly, the strain improvement effect did not result from the simple duplication of the MCM complex genes themselves, mutA, mutB, and meaB (Padovani and Banerjee, 2006); but also required duplication of the regulatory gene mutR. This is understandable considering mutR is likely to be transcribed together with the other three genes as part of the MCM operon and also likely to be involved in the regulation of expression of the MCM operon (Fig. 1). Another possible explanation for the need to include mutR in the gene duplication comes from inspection of the DNA sequence of mutR and the region downstream of the MCM operon. A 12 basepair stem-loop secondary structure with only 2 mismatches and a loop of 8 nucleotides is located at the very end of mutR overlapping the TAG stop codon for mutR (Fig. 1). This structure could play a role in transcription termination or transcript stability, and its exclusion from the duplicated region in strain FL2435 could explain why the yield improvement effect was not seen in this strain (Pulido and Jimenez, 1987). Other mechanisms that could affect regulation of the MCM operon or stability of its mRNA transcript (Bralley et al., 2006) could be involved and certainly more experimentation will be required to clarify the role of mutR or other factors on the strain improvement effect.
Interestingly, improved erythromycin production was also seen in strain FL2385 fermentations in carbohydrate-based medium. This seemingly contradicted the model where the MCM reaction is shown to operate from mmCoA towards succinyl-CoA in carbohydrate medium (Reeves et al., 2006). The model predicted that overexpression of the MCM genes would accelerate the draining of the mmCoA metabolite pool and cause a drop in erythromycin production in carbohydrate-based medium. However, the duplication of the MCM operon in strain FL2385 may have had effects on other metabolic pathways besides MCM due to the unknown targets of the mutR regulatory protein. For example, if mutR duplication also up-regulated carbon flow through the two other feeder pathways in the model (fig 5) or down-regulated carbon flow through a drain pathway (not diagrammed), then even if the MCM reaction was also up-regulated in strain FL2385, as predicted, the net metabolic effect would be to increase, not decrease, production of erythromycin in carbohydrate medium. Further study of the mmCoA node to better understand the true strain improvement mechanism in carbohydrate medium could prove helpful for the development of additional rational strain improvement strategies.
At some point in the strain improvement process of the wild type strain the mmCoA metabolite pool may be increased to the point where mmCoA ceases to be the limiting factor affecting erythromycin production. At that point, a new limiting factor may appear. In order to increase production of erythromycin further, efforts would have to be refocused on those metabolic pathways or regulatory circuits that can be manipulated to overcome the scarcity of the new limiting factor. In this way, rational strain improvement may in some cases become a step-wise process of increasing the supply of a series of limiting factors. The amount of limiting factor may be increased by making appropriate genetic and physiological manipulations at the proper metabolic or regulatory node for each stage of the strain improvement process.
Future experimental directions will involve optimizing the overexpression of the MCM operon with the hope that further increases in erythromycin yields can still be achieved. Additionally, other genes and pathways connected to the mmCoA metabolite node should be investigated and manipulated to determine their effects on erythromycin production.
Acknowledgments
We gratefully acknowledge the National Institute of General Medical Sciences for financial support (Small Business Innovation Research awards R44GM58943 and R44GM063278); and the John Innes Centre-Streptomyces club for technical and material support. We also thank Robert Snell for technical support and helpful discussions and Roy Wesley for administrative support and helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adrio JL, Demain AL. Genetic improvement of processes yielding microbial products. FEMS Microbiol Rev. 2006;30:187–214. doi: 10.1111/j.1574-6976.2005.00009.x. [DOI] [PubMed] [Google Scholar]
- Alper H, Miyaoku K, Stephanopoulos G. Construction of lycopene-overproducing E. coli strains by combining systematic and combinatorial gene knockout targets. Nat Biotechnol. 2005;23:612–6. doi: 10.1038/nbt1083. [DOI] [PubMed] [Google Scholar]
- Askenazi M, Driggers EM, Holtzman DA, Norman TC, Iverson S, Zimmer DP, Boers ME, Blomquist PR, Martinez EJ, Monreal AW, Feibelman TP, Mayorga ME, Maxon ME, Sykes K, Tobin JV, Cordero E, Salama SR, Trueheart J, Royer JC, Madden KT. Integrating transcriptional and metabolite profiles to direct the engineering of lovastatin-producing fungal strains. Nature Biotechnol. 2003;21:150–156. doi: 10.1038/nbt781. [DOI] [PubMed] [Google Scholar]
- Baltz RH. Genetic methods and strategies for secondary metabolite yield improvement in actinomycetes. Antonie Van Leeuwenhoek. 2001;79:251–9. doi: 10.1023/a:1012020918624. [DOI] [PubMed] [Google Scholar]
- Blanchard AP, Hood L. Sequence to array: probing the genome's secrets. Nat Biotechnol. 1996;14:1649. doi: 10.1038/nbt1296-1649. [DOI] [PubMed] [Google Scholar]
- Bralley P, Gust B, Chang S, Chater KF, Jones GH. RNA 3′-tail synthesis in Streptomyces: in vitro and in vivo activities of RNase PH, the SCO3896 gene product and polynucleo-tide phosphorylase. Microbiology. 2006;152:627–636. doi: 10.1099/mic.0.28363-0. [DOI] [PubMed] [Google Scholar]
- Bro C, Nielsen J. Impact of ‘ome’ analyses on inverse metabolic engineering. Metab Eng. 2004;6:204–211. doi: 10.1016/j.ymben.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Brunker P, Minas W, Kallio PT, Bailey JE. Genetic engineering of an industrial strain of Saccharopolyspora erythraea for stable expression of the Vitreoscilla haemoglobin gene (vhb) Microbiology. 1998 Sep;144(Pt 9):2441–8. doi: 10.1099/00221287-144-9-2441. [DOI] [PubMed] [Google Scholar]
- Chistoserdova LV, Lidstrom ME. Molecular characterization of a chromosomal region involved in the oxidation of acetyl-CoA to glyoxylate in the isocitrate-lyase-negative methylotroph Methylobacterium extorquens AM1. Microbiology. 1996;142:1459–1468. doi: 10.1099/13500872-142-6-1459. [DOI] [PubMed] [Google Scholar]
- Cortes J, Velasco J, Foster G, Blackaby AP, Rudd BA, Wilkinson B. Identification and cloning of a type III polyketide synthase required for diffusible pigment biosynthesis in Saccharopolyspora erythraea. Mol Microbiol. 2002;44:1213–1224. doi: 10.1046/j.1365-2958.2002.02975.x. [DOI] [PubMed] [Google Scholar]
- Demain AL. From natural products discovery to commercialization: a success story. J Ind Microbiol Biotechnol. 2006;33:486–495. doi: 10.1007/s10295-005-0076-x. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, Remor M, Hofert C, Schelder M, Brajenovic M, Ruffner H, Merino A, Klein K, Hudak M, Dickson D, Rudi T, Gnau V, Bauch A, Bastuck S, Huhse B, Leutwein C, Heurtier MA, Copley RR, Edelmann A, Querfurth E, Rybin V, Drewes G, Raida M, Bouwmeester T, Bork P, Seraphin B, Kuster B, Neubauer G, Superti-Furga G. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–7. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- German JB, Hammock BD, Watkins SM. Metabolomics: building on a century of biochemistry to guide human health. Metabolomics. 2005;1:3–9. doi: 10.1007/s11306-005-1102-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson CR, Decker H, Madduri K, Otten SL, Tang L. Genetic control of polyketide biosynthesis in the genus Streptomyces. Antonie Van Leeuwenhoek. 1993;64:165–176. doi: 10.1007/BF00873025. [DOI] [PubMed] [Google Scholar]
- Katz L, Donadio S. Macrolides. Biotechnol. 1995;28:385–420. doi: 10.1016/b978-0-7506-9095-9.50023-x. [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–54. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Lander ES. Array of hope. Nat Genet. 1999;21:3–4. doi: 10.1038/4427. [DOI] [PubMed] [Google Scholar]
- Leadlay PF. Combinatorial approaches to polyketide biosynthesis. Curr Opin Chem Biol. 1997;1:162–168. doi: 10.1016/s1367-5931(97)80005-1. [DOI] [PubMed] [Google Scholar]
- Li Y, Ling H, Li W, Tan H. Improvement of nikkomycin production by enhanced copy of sanU and sanV in Streptomyces ansochromogenes and characterization of a novel glutamate mutase encoded by sanU and sanV. Metab Eng. 2005;7:165–173. doi: 10.1016/j.ymben.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Lum AM, Huang J, Hutchinson CR, Kao CM. Reverse engineering of industrial pharmaceutical-producing actinomycete strains using DNA microarrays. Metab Eng. 2004;6:186–96. doi: 10.1016/j.ymben.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Malmberg LH, Hu WS. Kinetic analysis of cephalosporin biosynthesis in Streptomyces clavuligerus. Biotechnol Bioeng. 1991;38:941–947. doi: 10.1002/bit.260380815. [DOI] [PubMed] [Google Scholar]
- Malmberg LH, Hu WS, Sherman DH. Precursor flux control through targeted chromosomal insertion of the lysine epsilon-aminotransferase (lat) gene in cephamycin C biosynthesis. J Bacteriol. 1993;175:6916–6924. doi: 10.1128/jb.175.21.6916-6924.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel R, Licari P, Khosla C. Process development and metabolic engineering for the overproduction of natural and unnatural polyketides. Adv Biochem Eng Biotechnol. 2001;73:31–52. doi: 10.1007/3-540-45300-8_3. [DOI] [PubMed] [Google Scholar]
- Miller ES. Cloning vectors, mutagenesis, and gene disruption (ermR) for the erythromycin-producing bacterium Aeromicrobium erythreum. Appl Environ Microbiol. 1991;57:2758–2761. doi: 10.1128/aem.57.9.2758-2761.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murli S, Kennedy J, Dayem LC, Carney JR, Kealey JT. Metabolic engineering of Escherichia coli for improved 6-deoxyerythronolide B production. J Ind Microbiol Biotechnol. 2003;8:500–509. doi: 10.1007/s10295-003-0073-x. [DOI] [PubMed] [Google Scholar]
- Padovani D, Banerjee R. Assembly and protection of the radical enzyme, methylmalonyl-CoA mutase, by its chaperone. Biochemistry. 2006;45:9300–9306. doi: 10.1021/bi0604532. [DOI] [PubMed] [Google Scholar]
- Parekh S, Vinci VA, Strobel RJ. Improvement of microbial strains and fermentation processes. Appl Microbiol Biotechnol. 2000;54:287–301. doi: 10.1007/s002530000403. [DOI] [PubMed] [Google Scholar]
- Pulido D, Jimenez A. Optimization of gene expression in Streptomyces lividans by a transcription terminator. Nucleic Acids Res. 1987;15:4227–40. doi: 10.1093/nar/15.10.4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves AR, Weber G, Cernota WH, Weber JM. Analysis of an 8.1-kb DNA fragment contiguous with the erythromycin gene cluster of Saccharopolyspora erythraea in the eryCI-flanking region. Antimicrob Agents Chemother. 2002;46:3892–3899. doi: 10.1128/AAC.46.12.3892-3899.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves AR, Cernota WH, Brikun IA, Wesley RK, Weber JM. Engineering precursor flow for increased erythromycin production in Aeromicrobium erythreum. Metab Eng. 2004;6:300–312. doi: 10.1016/j.ymben.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Reeves AR, Brikun IA, Cernota WH, Leach BI, Gonzalez MC, Weber JM. Effects of methylmalonyl-CoA mutase gene knockouts on erythromycin production in carbohydrate-based and oil-based fermentations of Saccharopolyspora erythraea. J Ind Microbiol Biotechnol. 2006;33:600–609. doi: 10.1007/s10295-006-0094-3. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manua. 2nd. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1989. [Google Scholar]
- Tang L, Shah S, Chung L, Carney J, Katz L, Khosla C, Julien B. Cloning and heterologous expression of the epothilone gene cluster. Science. 2000;287:640–2. doi: 10.1126/science.287.5453.640. [DOI] [PubMed] [Google Scholar]
- Vemuri GN, Aristidou AA. Metabolic engineering in the -omics era: elucidating and modulating regulatory networks. Microbiol and Molec Biol Rev. 2005;69:197–216. doi: 10.1128/MMBR.69.2.197-216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinci VA, Byng G. In: Manual of Industrial Microbiology and Biotechnology. 2. Demain AL, Davies JE, Atlas RM, Cohen G, Hershberger CL, Hu WS, Sherman DH, Willson RC, Wu JHD, editors. American Society for Microbiology; Washington DC: 1999. pp. 103–113. [Google Scholar]
- Weber JM, Wierman CK, Hutchinson CR. Genetic Analysis of erythromycin production in Streptomyces erythreus. J Bacteriol. 1985;164:425–433. doi: 10.1128/jb.164.1.425-433.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JM, McAlpine JB. Erythromycin derivatives. U.S. Patent, 5,141,926 1992
- Weber JM, Leung JO, Maine GT, Potenz RH, Paulus TJ, DeWitt JP. Organization of a cluster of erythromycin genes in Saccharopolyspora erythraea. J Bacteriol. 1990;172:2372–2383. doi: 10.1128/jb.172.5.2372-2383.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YX, Perry K, Vinci VA, Powell K, Stemmer WP, del Cardayre SB. Genome shuffling leads to rapid phenotypic improvement in bacteria. Nature. 2002;415:644–646. doi: 10.1038/415644a. [DOI] [PubMed] [Google Scholar]