

Abstract
Guggulsterone (GUG), a resin of the Commiphora mukul tree, has been used in ayurvedic medicine for centuries to treat a variety of ailments. Recent studies have suggested that GUG may also possess anticancer effects. In the present study, we show that GUG possesses antitumor-promoting effects in SENCAR mouse skin tumorigenesis model. We first determined the effect of topical application of GUG to mice against 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced conventional markers and other novel markers of skin tumor promotion. We found that topical application of GUG (1.6 μmol per mouse) 30 min prior to TPA (3.2 nmol per mouse) application onto the skin of mice afforded significant inhibition against TPA-mediated increase in skin edema and hyperplasia. Topical application of GUG was also found to result in substantial inhibition against TPA-induced epidermal (i) ornithine decarboxylase (ODC) activity; (ii) ODC, cyclooxygenase-2 and inducible nitric oxide synthase protein expressions; (iii) phosphorylation of extracellular signal-regulated kinase1/2, c-jun N-terminal kinases and p38; (iv) activation of NF-κB/p65 and IKKα/β and (v) phosphorylation and degradation of IκBα. We next assessed the effect of topically applied GUG on TPA-induced skin tumor promotion in 7,12-dimethyl benz[a]anthracene-initiated mice. Compared with non-GUG-pretreated mice, animals pretreated with GUG showed significantly reduced tumor incidence, lower tumor body burden and a significant delay in the latency period for tumor appearance from 5 to 11 weeks. These results provide the first evidence that GUG possesses anti-skin tumor-promoting effects in SENCAR mice and inhibits conventional as well as novel biomarkers of tumor promotion. In summary, GUG could be useful for delaying tumor growth in humans.
Introduction
Guggulsterone (GUG), [4,17(20)-pregnadiene-3,16-dione] (Figure 1A), is a plant polyphenol obtained from the gum resin of the Commiphora mukul tree; it has been used in ayurvedic medicine for centuries to treat a variety of ailments like obesity, diabetes, hyperlipidemia, atherosclerosis and osteoarthritis (1–5). The anti-inflammatory activity of gum guggul is well known (6), and recent studies have suggested that GUG may also possess anticancer effects (7,8) but the molecular mechanisms underlying the anticancer effects of GUG are beginning to emerge (7,8). We considered the possibility that GUG may also possess antitumor-promoting effects.
Fig. 1.
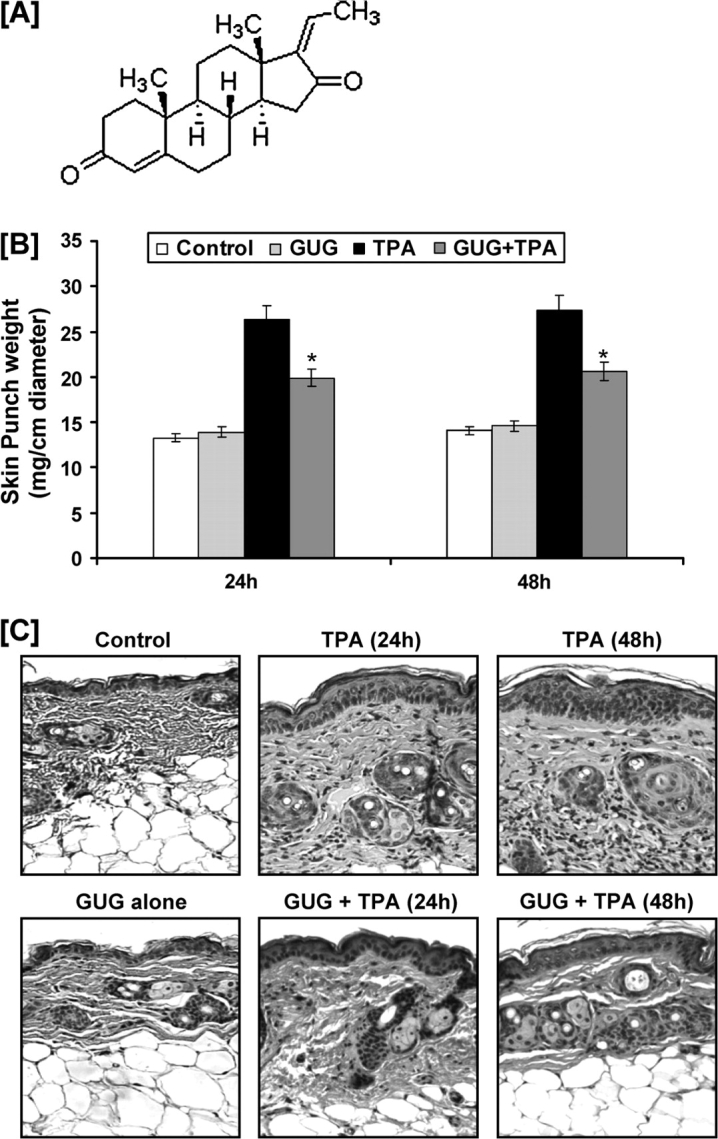
(A) Structure of GUG. Inhibitory effect of GUG on TPA-induced skin edema and hyperplasia in SENCAR mice. (B) Twenty-four and 48 h after TPA treatment, the skin edema was determined by weighing 1 cm diameter punch skin as described in text. At least four determinations were made at different dorsal skin sites per mouse in each group. The data represent the mean ± SE of eight mice (*P < 0.01 versus TPA). (C) Twenty-four and 48 h after treatment, the animals were killed; skin biopsies were processed for hematoxylin and eosin staining. Representative pictures are shown. Sections were photographed using an ×80 objective.
The multistage mouse skin carcinogenesis model, although an artificial one, is an ideal system to study a number of biochemical alterations, changes in cellular functions and histologic changes that take place during the different stages of chemical carcinogenesis (9,10). This system has also served as a useful model for initial screen for cancer chemopreventive effects of most dietary substances (11). Studies have shown that skin application of tumor-promoting agents results in inflammatory responses, such as development of edema, hyperplasia, induction of proinflammatory cytokine interleukin-1α, induction of epidermal ornithine decarboxylase (ODC) and cyclooxygenase-2 (COX-2) protein expression and activity, as well as activation of NF-κB (11–14). Activation of mitogen-activated protein kinases (MAPKs)/NF-κB pathways has been shown to be involved in tumor growth and development (15,16). In the present study, we show that topical application of GUG to SENCAR mice possesses antitumor-promoting effects and these effects are mediated via the ability of GUG to modulate MAPK and NF-κB pathways.
Materials and methods
Materials
GUG, [4,17(20)-pregnadiene-3,16-dione], was purchased from Steraloids (Newport, RI). Extracellular signal-regulated kinase (ERK)1/2 (phospho-p44/42), c-jun N-terminal kinase (JNK) (phospo-p54/46), p38 (phospho-p38), IκBα and IκBα (phospho) antibodies were obtained from New England Biolabs (Beverly, MA). NF-κB/p65 antibody was procured from Geneka Biotechnology (Montreal, Canada). IKKα and ODC, COX-2 and inducible nitric oxide synthase (iNOS) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-mouse or anti-rabbit secondary antibody horseradish peroxidase conjugate was obtained from Amersham Life Science (Arlington Height, IL). 7,12-Dimethyl benz[a]anthracene (DMBA) and 12-O-tetradecanoyl-phorbal-13-acetate (TPA) were purchased from Sigma Chemicals (St Louis, MO). The DC BioRad Protein assay kit was purchased from Bio-Rad Laboratories (Hercules, CA). Novex pre-cast Tris-Glycine gels were obtained from Invitrogen (Carlsbad, CA).
Animals and treatment for biomarker studies
Female SENCAR mice (5–6 weeks old) obtained from Charles River Laboratories were housed four per cage and were acclimatized for 1 week before use in both short-term and long-term experiments. Animals were subjected to a 12 h light–dark cycle, housed at 24 ± 2°C and 50 ± 10% relative humidity and fed a Purina chow diet and water ad libitum. For short-term biomarker studies, mice were divided into four groups, shaved on the dorsal side of the skin and treated topically on the shaved area. The mice in the first group received a topical application of 0.2 ml acetone and 0.1 ml dimethyl sulfoxide (DMSO), and those in the second group received 1.6 μmol GUG/0.1 ml DMSO per mouse. The mice in the third group received a topical application of 0.1 ml acetone alone, and those in the fourth group received 0.8 μmol GUG/0.1 ml DMSO per mouse. Thirty minutes after these treatments, the mice in group 3 and group 4 were treated with a single topical application of TPA (3.2 nmol/0.1 ml acetone per mouse). At desired times after these treatments, the mice were killed.
Edema and epidermal hyperplasia
To assess the inhibitory effect of preapplication of GUG on TPA-induced edema, 1 cm diameter punches of skin from vehicle-, GUG-, TPA- or GUG- and TPA-treated animals were removed, made free of fat pads and weighed quickly. After drying for 24 h at 50°C, the skin punches were reweighed, and the loss of water content was determined. The difference in the amount of water gain between the control (vehicle treated) and TPA treated represented the extent of edema induced by TPA, whereas that between the control vehicle and GUG plus TPA represented the inhibitory effect of GUG. For the hyperplasia study, skin was removed, fixed in 10% formalin and embedded in paraffin. Vertical sections (5 μm) were cut, mounted on a glass slide and stained with hematoxylin and eosin.
Preparation of cytosolic and nuclear lysates
Epidermis from the whole skin was separated as described earlier (17) and was homogenized in ice-cold lysis buffer [50 mM Tris–HCl, 150 mM NaCl, 1 mM ethyleneglycol-bis(aminoethylether)-tetraacetic acid, 1 mM ethylenediaminetetraacetic acid (EDTA), 20 mM NaF, 100 mM Na3VO4, 0.5% NP-40, 1% Triton X-100 and 1 mM phenylmethylsulfonyl fluoride (pH 7.4)] with freshly added protease inhibitor cocktail (Protease Inhibitor Cocktail Set III; Calbiochem, La Jolla, CA). The homogenate was then centrifuged at 14 000g for 25 min at 4°C and the supernatant (total cell lysate) was collected, aliquoted and stored at −80°C. For the preparation of nuclear and cytosolic lysates, 0.2 g of the epidermis was suspended in 1 ml of cold buffer [10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH 7.9), 2 mM MgCl2, 10 mM KCI, 1 mM dithiothreitol, 0.1 mM EDTA and 0.1 mM phenylmethylsulfonyl fluoride] with freshly added protease inhibitor cocktail (Protease Inhibitor Cocktail Set III; Calbiochem). After homogenization in a tight-fitting Dounce homogenizer, the homogenates were left on ice for 10 min and then centrifuged at 25000g for 10 min. The supernatant was collected as cytosolic lysate and stored at −80°C. The nuclear pellet was resuspended in 0.1 ml of the buffer containing 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH 7.9), 300 mM NaCI, 50 mM KCI, 0.1 mM EDTA, 1 mM dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride and 10% glycerol with freshly added protease inhibitor cocktail (Protease Inhibitor Cocktail Set III, Calbiochem). The suspension was gently shaken for 20 min at 4°C. After centrifugation at 25000g for 10 min, the nuclear extracts (supernatants) were collected and quickly frozen at −80°C. The protein content in the lysates was measured by DC BioRad assay (Bio-Rad Laboratories) as per the manufacturer’s protocol.
ODC enzyme activity
The epidermis from the dissected skin was separated as described earlier (17) and homogenized at 4°C in a glass-to-glass homogenizer in 10 volumes of ODC buffer [50 mM Tris–HCl buffer (pH 7.5) containing 0.1 mM EDTA, 0.1 mM dithiothreitol, 0.1 mM pyridoxal-5-phosphate, 1 mM 2-mercaptoethanol and 0.1% Tween 80]. The homogenate was centrifuged at 100 000g at 4°C and the supernatant was used for enzyme determination. ODC enzyme activity was determined in epidermal cytosolic fraction by measuring the release of 14CO2 from the D,L-[14C] ornithine by the method described earlier (17). Briefly, 400 μl of the supernatant was added to 0.95 ml of the assay mixture [35 mM sodium phosphate (pH 7.2), 0.2 mM pyridoxal phosphate, 4 mM dithiothreitol, 1 mM EDTA and 0.4 mM L-ornithine containing 0.5 μCi of D,L-[1-14C] ornithine hydrochloride] in 15 ml corex centrifuge tube equipped with rubber stoppers and central well assemblies containing 0.2 ml ethanolamine and methoxyethanol in 2:1(vol/vol) ratio. After incubation at 37°C for 60 min, the reaction was terminated by the addition of 1.0 ml of 2 M citric acid, using a 21G needle per syringe. The incubation was continued for 1 h. Finally, the central well containing the ethanolamine–methoxyethanol mixture to which 14CO2 has been trapped was transferred to a vial containing 10 ml of toluene-based scintillation fluid and 2 ml of ethanol. The radioactivity was measured in a Beckman LS 6000 SC liquid scintillation counter. Enzyme activity was expressed as picomoles CO2 released/h/mg protein.
Western blot analysis
For western analysis, 25–50 μg of the protein was resolved >8–12% polyacrylamide gels and transferred to a nitrocellulose membrane. The blot containing the transferred protein was blocked in blocking buffer for 1 h at room temperature followed by incubation with appropriate monoclonal or polyclonal primary antibody in blocking buffer for 1 h 30 min to overnight at 4°C. This was followed by incubation with anti-mouse, anti-rabbit or anti-sheep secondary antibodies horseradish peroxidase for 1 h 30 min and then washed four times with wash buffer and detected by chemiluminescence (ECL kit, Amersham Life Sciences) and autoradiography using XAR-5 film obtained from Eastman Kodak Co. (Rochester, NY).
Immunostaining for iNOS and COX-2
Mouse skin was fixed in 10% neutralized formalin and embedded in paraffin. Five micormeter sections were cut, deparaffinized in xylol and rehydrated to 70% ethanol and washed in phosphate-buffered saline. For antigen retrieval, sections were heated at 95°C for 30 min in citrate buffer (pH 6.0) and then cooled for 20 min and washed in phosphate-buffered saline. Endogenous peroxidase was quenched by incubation in 0.3% hydrogen peroxide, for 20 min and washed in washing buffer (phosphate-buffered saline plus Tween). Non-specific binding sites were blocked by incubating the sections with goat serum blocking solution for 1 h. Sections were incubated with primary antibody against iNOS and COX-2 overnight at 4°C followed by incubation with specific horseradish peroxidase-labeled secondary antibody for 1 h at room temperature. After washing in wash buffer, the sections were incubated with diaminobenzidene peroxidase substrate solution for 2 min at room temperature and rinsed with distilled water followed by counterstaining with Mayer’s Hematoxylin solution. Sections were rinsed in tap water, dehydrated through 70–100% graded alcohol cleared in xylene and finally mounted in permanent mounting medium.
Skin tumorigenesis
Female SENCAR mice were used in DMBA- and TPA-induced, two-stage skin tumorigenesis protocol. The dorsal side of the skin was shaved using electric clippers, and the mice with hair cycles in the resting phase were used for tumor studies. In each group, 20 animals were used. Tumor induction was initiated by a single topical application of 50 nmol DMBA in 200 μl acetone, and 1 week later, the tumor growth was promoted with twice-weekly topical applications of 3.2 nmol TPA in 200 μl acetone. Treatment with TPA alone or with GUG plus TPA was repeated twice weekly up to the termination of the experiments at 20 weeks. Animals in both the groups were watched for any apparent signs of toxicity, such as weight loss or mortality during the entire period of study. Skin tumor formation was recorded weekly, and tumors >1 mm in diameter were included in the cumulative number if they persisted for 2 weeks or more.
Microscopy and photography
Images from immunostaining experiments were obtained using a Zeiss Axioplot microscope (Thornwood, NY) and Kodak Ektachrome 160T film (Rochester, NY). These images were scanned (SprintScan; Polaroid, Cambridge, MA) and formatted as Tag Image File Format images in Adobe Photoshop 6.0 software to make the composite figures.
Statistical analysis
A two-tailed Student’s t-test was used to assess the statistical significance between the TPA-treated and GUG plus TPA-treated groups. A P value <0.05 was considered statistically significant. In tumorigenesis experiments, the statistical significance of difference in terms of tumor incidence and multiplicity between TPA and GUG plus TPA groups was evaluated by the Wilcoxon rank-sum test and chi-square analysis. An advantage of Wilcoxon rank-sum test is that its validity does not depend on any assumption about the shape of the distribution of tumor multiplicities.
Results
Inhibitory effect of GUG on TPA-induced cutaneous edema
Studies from our laboratory and others have shown that TPA application to mouse skin results in cutaneous edema (17,18). In the present study, we evaluated the protective effects of topical application of GUG in TPA-mediated cutaneous edema in SENCAR mice. We tested two doses 0.8 and 1.6 μmol of GUG per animal in our preliminary studies. Since 0.8 μmol GUG did not exhibit any significant effect on primary biomarkers of tumor promotion (data not shown), therefore we selected dose of 1.6 μmol of GUG for further studies. The SENCAR mice were topically treated with GUG (1.6 μmol per mouse) and 30 min later were topically treated with TPA (3.2 nmol per mouse). As determined by the weight of 1 cm diameter punch of the dorsal skin, application of TPA to SENCAR mouse skin resulted in significant development of skin edema at 24 and 48 h post-TPA treatment compared with control and GUG-treated groups (Figure 1B). The skin application of GUG 30 min prior to that of TPA application showed significant protection against TPA-induced skin edema measured at 24 (51%; P < 0.01) and 48 (49%; P < 0.01) h posttreatment. We found that topical application of GUG alone to mice did not result in increase in skin edema at 24 and 48 h posttreatment (Figure 1B).
Inhibitory effect of GUG on TPA-induced epidermal hyperplasia
The effect of topical application of GUG on TPA-mediated induction of epidermal hyperplasia was then assessed. As shown in Figure 1C, topical application of TPA resulted in an increase in epidermal hyperplasia at 24 and 48 h after treatment when compared with control-treated animals. The topical application of GUG, however, prior to that of TPA application to mouse skin resulted in inhibition in the induction of epidermal hyperplasia (Figure 1C). GUG alone did not induce any epidermal hyperplasia as the histology of these animals was comparable with that of control mice (Figure 1C).
Inhibitory effect of GUG on TPA-induced ODC activity
In order to determine the effect of GUG against the TPA-induced ODC activity in SENCAR mice, groups of animals were treated topically with GUG (1.6 μmol per animal) 30 min prior to topical application of TPA (3.2 nmol per animal). TPA was applied in 0.2 ml acetone and GUG was applied in 0.1 ml DMSO. As shown in Figure 2A, pretreatment of animals with GUG resulted in inhibition of the TPA-caused induction of epidermal ODC activity. GUG at a dose of 1.6 μmol per animal caused a 53% inhibition (P < 0.005) in the epidermal ODC activity in mice treated with TPA. Topical application of GUG alone (1.6 μmol per animal) was without any effect on basal epidermal ODC activity.
Fig. 2.

Inhibitory effect of GUG on TPA-induced epidermal ODC activity and protein expression in SENCAR mice. (A) The animals were killed at 6 h after TPA treatment, epidermal cytosolic fraction was prepared and ODC activity was determined. The data are shown as ODC activity (pmol/h/mg protein) and represent the mean ± SE of eight mice; each assay was performed in duplicate (*P < 0.01 versus TPA). (B) At different time after treatment, the animals were killed, epidermal lysates were prepared and protein expression was determined as described in text. Equal loading was confirmed by stripping the immunoblot and reprobing it for β-actin. The immunoblots shown here are representative of three independent experiments with similar results. The values above the figures represent relative density of the band normalized to β-actin.
Inhibitory effect of GUG on TPA-induced epidermal ODC protein expression
Next, we assessed the effect of skin application of GUG on TPA-caused enhanced expression of ODC protein in the epidermis. Western blotting revealed that at 24 h posttreatment of TPA, there was maximum expression of epidermal ODC protein expression and it gradually declined with the passage of time, (48 h post-TPA treatment) (Figure 2B). Treatment of TPA caused a 3- to 4-fold increase in epidermal ODC protein level as compared with acetone-treated control, whereas pretreatment of animals with GUG resulted in a significant inhibition against TPA-caused induction of epidermal ODC protein expression at all time points investigated (Figure 2B). Densitometric analysis of these blots indicated that, under experimental conditions used, the inhibition varied from 45 to 50% in GUG-pretreated animals.
Inhibitory effect of GUG on TPA-induced epidermal COX-2 and iNOS protein expressions
COX-2 and iNOS are well-established biomarkers of inflammation and tumor promotion. We next assessed the effect of skin application of GUG on TPA-induced epidermal iNOS and COX-2 protein expression. We found that topical application of TPA to SENCAR mice resulted in an increase in epidermal COX-2 protein expression, which was maximum (5.1-fold) at 6 h post-TPA treatment when compared with the control (Figure 3A). The TPA-caused induction in the expression level of epidermal COX-2 gradually declined with time, that is, at 24 and 48 h post-TPA treatment. However, at all time points, the expression of COX-2 in mouse skin following TPA application remained higher than corresponding control group.
Fig. 3.
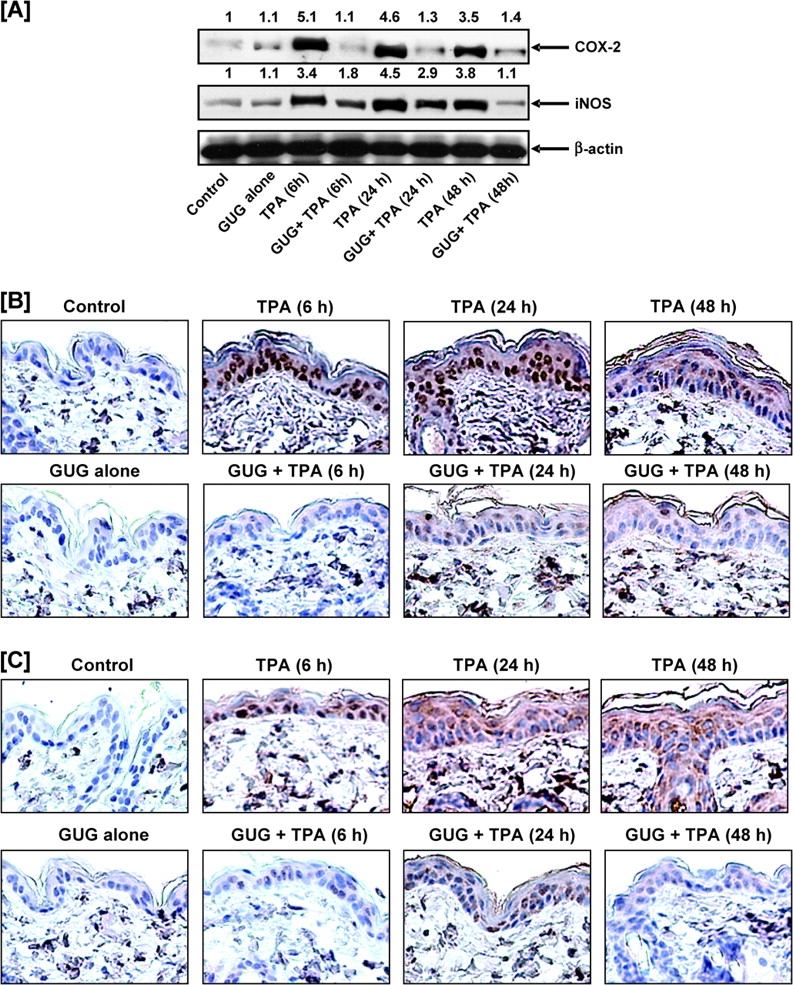
Inhibitory effect of GUG on TPA-induced epidermal COX-2 and iNOS protein expression in SENCAR mice. (A) At different time after treatment, the animals were killed, epidermal lysates were prepared and protein expression was determined as described in text. Equal loading was confirmed by stripping the immunoblot and reprobing it for β-actin. The immunoblots shown here are representative of three independent experiments with similar results. The values above the figures represent relative density of the band. Immunostaining of (B) COX-2 and (C) iNOS. At different time after treatment, the animals were killed, the skin punch biopsies were fixed in 10% neutralized formalin and embedded in paraffin. Five micrometer sections were cut, deparaffinized in xylol and rehydrated to 70% ethanol and washed in phosphate-buffered saline and immunostaining of COX-2 and iNOS was performed as detailed in text. A representative picture from three independent immunostaining is shown. Scale bar = 50 μm.
We further observed that topical application of TPA to SENCAR mice resulted in a significant increase in the expression of epidermal iNOS protein (Figure 3A). The expression of iNOS was observed to reach its peak at 24 h post-TPA treatment. Topical application of TPA alone caused 3- to 4-fold increase in iNOS protein expression in mouse skin as compared with vehicle-treated controls at 6 and 24 h posttreatment; however, pretreatment of GUG to the skin caused inhibition against TPA-caused increases of iNOS protein expression. Densitometric analysis of blots revealed that mice pretreated with GUG (1.6 μmol per animal) showed 68% inhibition against TPA-induced epidermal iNOS protein expression at 48 h posttreatment (Figure 3A). The application of GUG alone at the dose of 1.6 μmol did not produce any change in epidermal COX-2 and iNOS protein expression when compared with vehicle-treated control animals. Immunostaining analysis further validated these observations. As evident from Figure 3B and C, there was a significant increase in protein expressions of COX-2 and iNOS in the epidermis of TPA-treated mice as compared with the untreated controls. In contrast, GUG-treated mice showed a significant decrease in the expression level of both proteins at the selected time points. This suggests that GUG has the ability to suppress the tumor promoter activity of TPA in the murine skin.
Inhibitory effect of GUG on TPA-mediated phosphorylation of MAPKs
To determine whether TPA could induce activation of MAPKs under in vivo condition in SENCAR mice, western blot analysis was performed using phospho-specific MAPKs antibodies. In the present study, as evident from western blot analysis, we found that topical application of TPA resulted in an increased phosphorylation of ERK1/2, JNK1/2 and p38 (Figure 4A). There was no effect on the total amount of ERK1/2, JNK1/2 and p38 proteins after TPA or GUG treatment (Figure 4A). We found that topical application of TPA resulted in an increased phosphorylation of ERK1/2 (p44 and p42) at 6 h post-TPA application that gradually decreases. The effect of TPA application was more pronounced on phosphorylation of p38 at 24 h and JNK at 6 h for given post-TPA application; then it gradually subsides but was still higher when compared with control. Topical application of GUG prior to TPA application resulted in inhibition of TPA-mediated phosphorylation of MAPKs protein (Figure 4A).
Fig. 4.
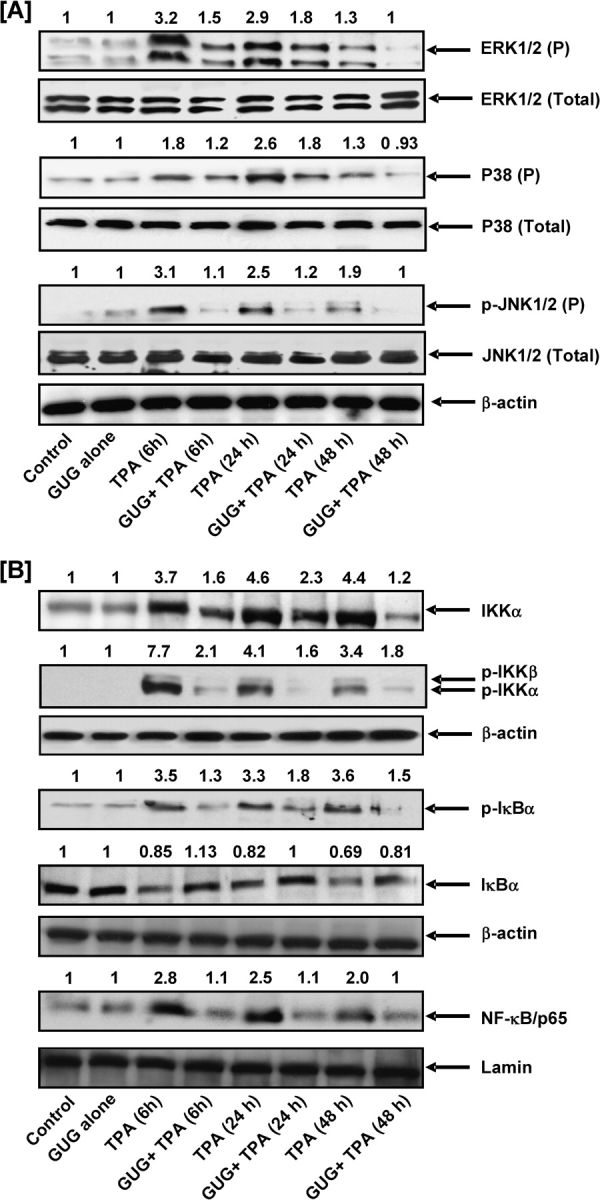
Inhibitory effect of GUG on TPA-induced phosphorylation of MAPKs, activation of NF-κB and IKKα/β, phosphorylation and degradation of IκBα in SENCAR mice. (A and B) At different time after treatment, the animals were killed, epidermal cytosolic and nuclear lysates were prepared and protein expression was determined as described in text. Equal loading was confirmed by stripping the immunoblot and reprobing it for β-actin. The immunoblots shown here are representative of three independent experiments with similar results. The values above the figures represent relative density of the band normalized to β-actin.
Inhibitory effect of GUG on TPA-induced activation of NF-κB and IKKα and phosphorylation and degradation of IκBα protein expression
Studies have shown that one of the critical events in NF-κB activation is its dissociation with subsequent degradation of inhibitory protein IκBα via phosphorylation and ubiquitination (19–21). Activation and nuclear translocation of NF-κB is preceded by the phosphorylation and proteolytic degradation of IκBα (20,22). To determine whether the inhibitory effect of GUG was attributable to an effect on IκB degradation, we examined the cytoplasmic level of IκBα protein expression by western blot analysis. We found that TPA application to mouse skin resulted in the degradation of IκBα protein expression at 6, 12 and 24 h after treatment. However, topical application of GUG 30 min prior to TPA application resulted in inhibition of TPA-induced degradation of IκBα protein (Figure 4B). We next assessed whether TPA application affects the phosphorylation of IκBα protein. As shown by western blot, TPA induced a marked increase in the phosphorylation level of IκBα protein at Ser (23) after treatment, which was inhibited by topical application of GUG prior to TPA application (Figure 4B). Studies have shown that IKKα activity is necessary for IκBα protein phosphorylation/degradation (24,25). To determine whether inhibition of TPA-induced IKKα activation by GUG is attributable to suppression of IκBα phosphorylation/degradation, we also measured IKKα protein level. We found that TPA application resulted in the activation of IKKα protein that in turn phosphorylated and degraded IκBα protein. Topical application of GUG prior to TPA application inhibited TPA-induced increase expression of IKKα and phosphorylation of IKKα/β (Figure 4B). Next, we investigated whether topical application of GUG inhibited TPA-induced activation and nuclear translocation of NF-κB/p65, the functionally active subunit of NF-κB in mouse skin. In the nuclear fraction, we found that TPA application onto the skin of SENCAR mice resulted in the activation and nuclear translocation of NF-κB/p65. However, topical application of GUG prior to TPA application inhibited TPA-induced NF-κB/p65 activation and nuclear translocation (Figure 4B).
Inhibitory effect of GUG on TPA-induced skin tumor promotion
We next assessed the effect of skin application of GUG on TPA-induced skin tumor promotion in DMBA-initiated SENCAR mice. As shown by data in Figure 5, topical application of GUG prior to that of TPA in DMBA-initiated SENCAR mice skin resulted in an inhibition of skin tumorigenesis. This inhibition was evident when tumor data were considered as the percentage of mice with tumors (Figure 5A), the number of tumors per group (Figure 5B) and the number of tumors per mouse (Figure 5C). The animals pretreated with GUG showed substantially reduced tumor incidence and lower tumor body burden when assessed as total number of tumors per group, percent of mice with tumors and number of tumors per animal as compared with animals that did not receive GUG (Figure 5A). In TPA-treated group, 100% of the mice developed tumors at 14 weeks on test, whereas at this time in GUG-treated group, only 30% of the mice exhibited tumors (Figure 5A). Skin application of GUG prior to TPA application resulted in a delay in the latency period from 5 to 11 weeks and afforded protection when tumor data were considered in terms of tumor incidence and tumor multiplicity throughout the treatment period (P < 0.05, chi-square test). At the termination of the experiment at 20 weeks, 50% of the mice were tumor free in the group that received skin application of GUG prior to each TPA application. At the termination of the experiment at 20 weeks on test, compared with a total of 212 tumors in TPA-treated group of animals, only 66 tumors in GUG-treated group were recorded (Figure 5B). Compared with the non-GUG-treated group, such decrease in the total number of tumors in the GUG-treated group corresponded to 69% inhibition. When these tumor data were considered in terms of number of tumors per mice, at the termination of the experiment at 20 weeks on test, compared with 10.6 tumors per mouse in TPA-treated group of animals, only 3.3 tumors per mouse in GUG-treated group were recorded (Figure 5C). Compared with the non-GUG-treated group, such decrease in the number of tumor per mouse in the GUG-treated group corresponded to 69% inhibition.
Fig. 5.
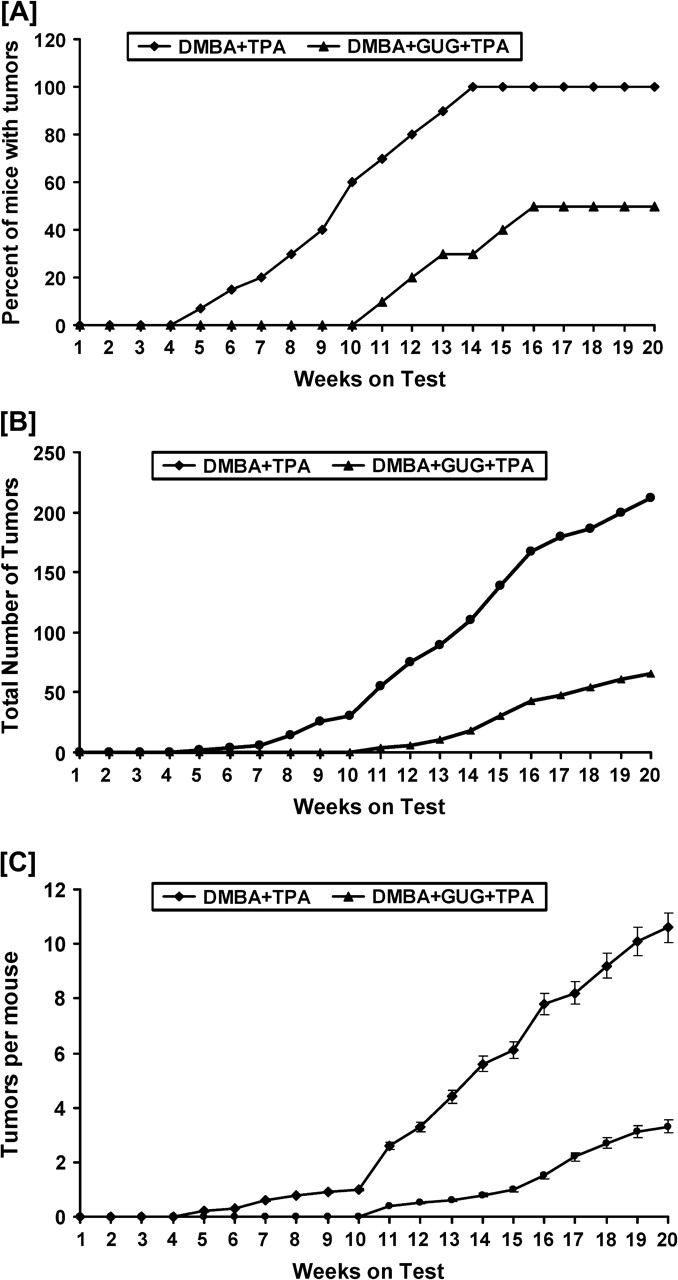
Inhibitory effect of GUG on DMBA-initiated and TPA-promoted tumor formation in SENCAR mice. In each group, 20 animals were used. Tumorigenesis was initiated in the animals by a single topical application of 50 nmol DMBA in 0.2 ml vehicle on the dorsal shaved skin, and 1 week later, the tumor growth was promoted with twice-weekly applications of 3.2 nmol TPA in 0.2 ml vehicle. To assess its anti-skin tumor-promoting effect, GUG at a dose of 0.5 mg per animal was applied topically 30 min prior to each TPA application in different groups. Treatment with TPA alone or GUG plus TPA was repeated twice weekly up to the termination of the experiments at 20 weeks. Animals in all the groups were watched for any apparent signs of toxicity, such as weight loss or mortality during the entire period of study. Skin tumor formation was recorded weekly, and tumors >1 mm in diameter were included in the cumulative number only if they persisted for 2 weeks or more. The tumor data are represented as (A) the percentage of mice with tumors, (B) the number of tumors per group and (C) the number of tumors per mouse. The data were analyzed by Wilcoxon rank-sum test and chi-square analysis.
Discussion
Cancer chemoprevention has become an important area of cancer research, which, in addition to providing a practical approach to identifying potentially useful inhibitors of cancer development, also affords excellent opportunities to study the mechanisms of carcinogenesis (26,27). One excitement of chemoprevention is that agents can be targeted for intervention either at the initiation, promotion or progression stage of multistage carcinogenesis. The intervention of cancer at the promotion stage appears to be the most appropriate and practical. The major reason for this relates to the fact that tumor promotion is a reversible event at least in early stages and requires repeated and prolonged exposure of a promoting agent (28). For this reason, it is important to identify mechanism-based effective novel antitumor-promoting agents. It is appreciated that those agents, which have the ability to intervene at more than one critical pathway in the carcinogenic process, will have greater advantage over other single-target agents. This study was designed to show the chemopreventive potential of GUG by using carcinogenesis-associated biochemical endpoints in a mouse skin tumorigenesis model. The topical application of TPA to mouse skin or its treatment in certain epidermal cells is known to result in a number of biochemical alterations, changes in cellular functions and histologic changes leading to skin tumor promotion (13,24,29). Our data clearly demonstrate that topical application of GUG prior to TPA application affords significant inhibition of TPA-induced skin edema and hyperplasia (Figure 1B and C).
ODC, the first and the rate-limiting enzyme in the biosynthesis of polyamines, plays an important role in the regulation of cell proliferation and development of cancer (30). Studies with the mouse skin model have shown an excellent correlation between the induction of ODC activity and the tumor-promoting ability of a variety of substances (31,32). Several lines of evidence indicate that aberrations in ODC regulation and subsequent polyamine accumulation are intimately associated with neoplastic transformation (33,34). Elevated levels of ODC gene products are consistently detected in transformed cell lines, virtually all animal tumors and in certain tissues predisposed to tumorigenesis (33). Agents that block induction of ODC can prevent tumor formation; therefore, its inhibition was shown to be a promising tool for screening inhibitors of tumorigenesis (35,36). In the present study, topical application of GUG prior to that of TPA resulted in a significant inhibition of TPA-mediated induction of epidermal ODC activity (Figure 2A). It is reasonable to believe that GUG application inhibited the action of the tumor promoter and/or the enzymatic pathways that regulates the ODC induction rather than interacting directly with the enzyme. In addition, our data obtained from western blot analysis demonstrate that prior application of GUG to that of TPA showed an inhibitory effect of GUG against TPA-induced increases in the levels of epidermal ODC protein in the mouse skin (Figure 2B). The magnitude of the inhibitory effect of topical application of GUG on TPA-induced increases in ODC protein expression seems to be similar to that for inhibition of TPA-induced increases in ODC enzyme activity.
Tumor promotion is closely linked to inflammation and oxidative stress, and it is probable that compounds that have anti-inflammatory and antioxidative properties act as antitumor promoters as well (37). COX-2 isoform and iNOS are important enzymes involved in mediating the inflammatory process (38,39). COX-2 and iNOS have been reported to play an important role in cutaneous inflammation, cell proliferation and skin tumor promotion (40,41). There is considerable body of compelling evidence that inhibition of COX-2 and iNOS expression or activity is important for not only alleviating inflammation but also for the prevention of cancer (41). Previous studies have demonstrated that GUG inhibits cytokine-induced COX-2 expression and NF-κB activation (42). In this study, we showed the inhibitory effects of GUG against TPA-caused induction of epidermal COX-2 and iNOS protein expression in SENCAR mice (Figure 3). These inhibitory effects also correlate with the inhibitory effect of GUG against TPA-caused induction of skin edema (Figure 1B) and hyperplasia (Figure 1C). These inhibitory effects of GUG against TPA-mediated responses in the mouse skin suggest that the primary effect of GUG may be against inflammatory responses, which may then result in the inhibition of tumor promotion.
MAPKs constitute a superfamily of proteins that include ERK1/2, JNK1/2 and p38 kinase (15,27). The involvement of MAPKs pathway in tumor proliferation is well documented. Activation of the MAPKs pathway occurs in response to integrin-mediated cellular adhesion to the extracellular matrix, which plays a critical role in both tumor metastasis and angiogenesis (43,44). In the present study, employing western blot analysis, we found that topical application of TPA resulted in a marked increase in the phosphorylated form of ERK1/2, JNK1/2 and p38 protein expression. Importantly, topical application of GUG prior to TPA application was found to inhibit TPA-mediated phosphorylation of MAPKs (Figure 4A). Several studies have shown that JNK pathway plays a major role in cellular function, such as cell proliferation and transformation (45), whereas the ERK pathway suppresses apoptosis and enhances cell survival or tumorigenesis (46).
Studies have shown that ERK1/2 and p38 are involved in the transcriptional activation of NF-κB (47,48). NF-κB has emerged as one of the most promising molecular targets in the prevention of cancer. We next investigated the effect of GUG on the pattern of NF-κB activation and its nuclear translocation by TPA in SENCAR mice skin. NF-κB resides in the inactive state in the cytoplasm as a heterotrimer consisting of p50, p65 and IκBα subunits. An IκBα kinase, IKKα, phosphorylates serine residues in IκBα at position 32 and 36 (49). Upon phosphorylation and subsequent degradation of IκBα, NF-κB activates and translocates to the nucleus, where it binds to DNA and activates the transcription of various genes (49,50). Several lines of evidence suggest that proteins from the NF-κB and IκB families are involved in carcinogenesis. Studies have also shown that NF-κB activity affects cell survival and determines the sensitivity of cancer cells to cytotoxic agents as well as ionizing radiation (51). Shishodia et al. (42) have shown that GUG inhibits tumor necrosis factor-induced IKK activity and also suppresses inducible and constitutive NF-κB activation without directly affecting binding of NF-κB to DNA. In the present study, we have demonstrated that topical application of TPA to mouse skin resulted in activation and nuclear translocation of NF-κB/p65 (Figure 4B). We also found that TPA application to mouse skin resulted in an increased expression of IKKα and phosphorylation and degradation of IκBα protein (Figure 4B). Interestingly, we observed that topical application of GUG prior to TPA application to mouse skin inhibited TPA-induced NF-κB/p65 and IKKα activation and phosphorylation and degradation of IκBα protein (Figure 4B). Because GUG inhibited IκBα phosphorylation and degradation, this study suggests that the effect of GUG on NF-κB/p65 is through inhibition of phosphorylation and subsequent proteolysis of IκBα. Since GUG contains two α,β-unsaturated carbonyl moiety, it seems that the compound may cause cysteine thiol modification in key molecule such as IKK of the NF-κB-signaling pathway (52).
The results in Figure 5 show the protective effects of skin application of GUG on TPA-caused tumor promotion in DMBA-initiated SENCAR mouse skin. The preapplication of GUG to that of TPA showed substantially reduced tumor incidence and lower tumor body burden when assessed as total number of tumors per group, percent of mice with tumors and number of tumors per animal, as compared with animals that did not receive GUG (Figure 5). These chemopreventive and antitumor promotion observations in murine skin by GUG can be explained by the biochemical mechanisms examined in the present study.
In summary, our results suggest that topical application of GUG prior to TPA application to SENCAR mice resulted in a significant decrease in skin edema, hyperplasia, epidermal ODC activity and protein expression of ODC, COX-2 and iNOS classical markers of inflammation and tumor promotion. In addition, our results also suggest that topical application of GUG prior to TPA application also resulted in inhibition of phosphorylation of MAPKs, activation of NF-κB/p65 and IKKα/β and degradation and phosphorylation of IκBα. Our data clearly demonstrate that GUG could be a potent antitumor-promoting agent because it inhibits several biomarkers of TPA-induced tumor promotion in an in vivo animal model. One might envision the use of chemopreventive agents such as GUG in an emollient or patch for chemoprevention or treatment of skin cancer.
Funding
United States Public Health Service (R01 CA 78809, R01 CA 101039, R01 CA 120451, R21 AT 002429).
Acknowledgments
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- COX-2
cyclooxygenase
- DMBA
7,12-dimethyl benz[a]anthracene
- DMSO
dimethyl sulfoxide
- EDTA
ethylenediaminetetraacetic acid
- ERK
extracellular signal-regulated kinase
- GUG
guggulsterone
- iNOS
inducible nitric oxide synthase
- JNK
c-jun N-terminal kinase
- MAPK
mitogen-activated protein kinases
- ODC
ornithine decarboxylase
- TPA
12-O-tetradecanoylphorbol-13-acetate
References
- 1.Gujral ML, et al. Antiarthritic and anti-inflammatory activity of gum guggul (Balsamodendron mukul Hook) Indian J. Physiol. Pharmacol. 1960;4:267–273. [PubMed] [Google Scholar]
- 2.Sharma JN, et al. Comparison of the anti-inflammatory activity of Commiphora mukul (an indigenous drug) with those of phenylbutazone and ibuprofen in experimental arthritis induced by mycobacterial adjuvant. Arzneimittelforschung. 1977;27:1455–1457. [PubMed] [Google Scholar]
- 3.Sinal CJ, et al. Guggulsterone: an old approach to a new problem. Trends Endocrinol. Metab. 2002;13:275–276. doi: 10.1016/s1043-2760(02)00640-9. [DOI] [PubMed] [Google Scholar]
- 4.Urizar NL, et al. GUGULIPID: a natural cholesterol-lowering agent. Annu. Rev. Nutr. 2003;23:303–313. doi: 10.1146/annurev.nutr.23.011702.073102. [DOI] [PubMed] [Google Scholar]
- 5.Tripathi YB, et al. Thyroid stimulatory action of (Z)-guggulsterone: mechanism of action. Planta. Med. 1988;4:271–277. doi: 10.1055/s-2006-962431. [DOI] [PubMed] [Google Scholar]
- 6.Singh BB, et al. The effectiveness of Commiphora mukul for osteoarthritis of the knee: an outcomes study. Altern. Ther. Health Med. 2003;9:74–79. [PubMed] [Google Scholar]
- 7.Singh SV, et al. Guggulsterone-induced apoptosis in human prostate cancer cells is caused by reactive oxygen intermediate dependent activation of c-Jun NH2-terminal kinase. Cancer Res. 2007;67:7439–7449. doi: 10.1158/0008-5472.CAN-07-0120. [DOI] [PubMed] [Google Scholar]
- 8.Shishodia S, et al. Guggulsterone inhibits tumor cell proliferation, induces S-phase arrest, and promotes apoptosis through activation of c-Jun N-terminal kinase, suppression of Akt pathway, and downregulation of antiapoptotic gene products. Biochem. Pharmacol. 2007;74:118–130. doi: 10.1016/j.bcp.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slaga TJ, et al. Studies on the mechanisms involved in multistage carcinogenesis in mouse skin. J. Cell. Biochem. 1982;18:99–119. doi: 10.1002/jcb.1982.240180109. [DOI] [PubMed] [Google Scholar]
- 10.Katiyar SK, et al. Inhibition of phorbol ester tumor promoter 12-O-tetradecanoylphorbol-13-acetate-caused inflammatory responses in SENCAR mouse skin by black tea polyphenols. Carcinogenesis. 1997;18:1911–1916. doi: 10.1093/carcin/18.10.1911. [DOI] [PubMed] [Google Scholar]
- 11.Katiyar SK, et al. Inhibition of tumor promotion in SENCAR mouse skin by ethanol extract of Zingiber officinale rhizome. Cancer Res. 1996;56:1023–1030. [PubMed] [Google Scholar]
- 12.Katiyar SK, et al. Inhibition of 12-O-tetradecanoylphorbol-13-acetate and other skin tumor-promoter-caused induction of epidermal interleukin-1 alpha mRNA and protein expression in SENCAR mice by green tea polyphenols. J. Invest. Dermatol. 1995;105:394–398. doi: 10.1111/1523-1747.ep12321030. [DOI] [PubMed] [Google Scholar]
- 13.Chun KS, et al. Effects of yakuchinone A and yakuchinone B on the phorbol ester-induced expression of COX-2 and iNOS and activation of NF-κB in mouse skin. J. Environ. Pathol. Toxicol. Oncol. 2002;21:131–139. [PubMed] [Google Scholar]
- 14.Seo HJ, et al. Inhibitory effects of the standardized extract (DA-9601) of Artemisia asiatica Nakai on phorbol ester-induced ornithine decarboxylase activity, papilloma formation, cyclooxygenase-2 expression, inducible nitric oxide synthase expression and nuclear transcription factor kappa B activation in mouse skin. Int. J. Cancer. 2002;100:456–462. doi: 10.1002/ijc.10489. [DOI] [PubMed] [Google Scholar]
- 15.Afaq F, et al. Suppression of UVB-induced phosphorylation of mitogen-activated protein kinases and nuclear factor kappa B by green tea polyphenol in SKH-1 hairless mice. Oncogene. 2003;22:9254–9264. doi: 10.1038/sj.onc.1207035. [DOI] [PubMed] [Google Scholar]
- 16.Shishodia S, et al. Ursolic acid inhibits nuclear factor-kappaB activation induced by carcinogenic agents through suppression of IkappaBalpha kinase and p65 phosphorylation: correlation with down-regulation of cyclooxygenase 2, matrix metalloproteinase 9, and cyclin D1. Cancer Res. 2003;63:4375–4383. [PubMed] [Google Scholar]
- 17.Afaq F, et al. Anthocyanin- and hydrolyzable tannin-rich pomegranate fruit extract modulates MAPK and NF-κB pathways and inhibits skin tumorigenesis in CD-1 mice. Int. J. Cancer. 2005;113:423–433. doi: 10.1002/ijc.20587. [DOI] [PubMed] [Google Scholar]
- 18.Saleem M, et al. Lupeol modulates NF-κB and PI3K/Akt pathways and inhibits skin cancer in CD-1 mice. Oncogene. 2004;23:5203–5214. doi: 10.1038/sj.onc.1207641. [DOI] [PubMed] [Google Scholar]
- 19.Takada Y, et al. Flavopiridol inhibits NF-κB activation induced by various carcinogens and inflammatory agents through inhibition of Ikappa Balpha kinase and p65 phosphorylation: abrogation of cyclin D1, cyclooxygenase-2 and matrix metalloprotease-9. J. Biol. Chem. 2004;279:4750–4759. doi: 10.1074/jbc.M304546200. [DOI] [PubMed] [Google Scholar]
- 20.Bharti AC, et al. Chemopreventive agents induce suppression of nuclear factor-kappaB leading to chemosensitization. Ann. N. Y. Acad. Sci. 2002;973:392–395. doi: 10.1111/j.1749-6632.2002.tb04671.x. [DOI] [PubMed] [Google Scholar]
- 21.Israel A. A role for phosphorylation and degradation in the control of NF-kappa B activity. Trends Genet. 1995;11:203–205. doi: 10.1016/s0168-9525(00)89045-9. [DOI] [PubMed] [Google Scholar]
- 22.Afaq F, et al. Inhibition of ultraviolet B-mediated activation of nuclear factor kappaB in normal human epidermal keratinocytes by green tea constituent (-)-epigallocatechin-3-gallate. Oncogene. 2003;22:1035–1044. doi: 10.1038/sj.onc.1206206. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka T, et al. Tannins and related compounds: XLI, isolation and characterization of novel ellagitannins, puicacorteins A, B, C and D, and punigluconin from the bark of Punicum granatum. L. Chem. Pharm. Bull. 1986;34:656–663. [Google Scholar]
- 24.Baldwin AS., Jr The NF-kappa B and I kappa B proteins: new discoveries and insights. Annu. Rev. Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- 25.Maniatis T. Catalysis by a multiprotein IkappaB kinase complex. Science. 1997;278:818–819. doi: 10.1126/science.278.5339.818. [DOI] [PubMed] [Google Scholar]
- 26.Bickers DR, et al. Novel approaches to chemoprevention of skin cancer. J. Dermatol. 2000;27:691–695. doi: 10.1111/j.1346-8138.2000.tb02259.x. [DOI] [PubMed] [Google Scholar]
- 27.Ding M, et al. Inhibition of AP-1 and neoplastic transformation by fresh apple peel extract. J. Biol. Chem. 2004;279:10670–10676. doi: 10.1074/jbc.M311465200. [DOI] [PubMed] [Google Scholar]
- 28.DiGiovanni J. Modification of multistage carcinogenesis. In: Ito N, Sugano H, editors. Modification of Tumor Development in Rodents: Progress in Experimental Tumor Research. Vol. 33. Basel, Switzerland: Karger; 1991. pp. 192–229. [DOI] [PubMed] [Google Scholar]
- 29.Katiyar SK, et al. Protective effects of silymarin against photocarcinogenesis in a mouse skin model. J. Natl Cancer Inst. 1997;89:556–566. doi: 10.1093/jnci/89.8.556. [DOI] [PubMed] [Google Scholar]
- 30.Thomas T, et al. Polyamine metabolism and cancer. J. Cell. Mol. Med. 2003;7:113–126. doi: 10.1111/j.1582-4934.2003.tb00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Einspahr JG, et al. Skin cancer chemoprevention: strategies to save our skin. Recent Results Cancer Res. 2003;163:151–164. doi: 10.1007/978-3-642-55647-0_14. [DOI] [PubMed] [Google Scholar]
- 32.Ahmad N, et al. A definitive role of ornithine decarboxylase in photocarcinogenesis. Am. J. Pathol. 2001;159:885–892. doi: 10.1016/S0002-9440(10)61764-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Auvinen M. Cell transformation, invasion, and angiogenesis: a regulatory role for ornithine decarboxylase and polyamines? J. Natl Cancer Inst. 1997;89:533–537. doi: 10.1093/jnci/89.8.533. [DOI] [PubMed] [Google Scholar]
- 34.Mohan RR, et al. Overexpression of ornithine decarboxylase in prostate cancer and prostatic fluid in humans. Clin. Cancer Res. 1999;5:143–147. [PubMed] [Google Scholar]
- 35.Verma AK, et al. Correlation of the inhibition by retinoids of tumor promoter-induced mouse epidermal ornithine decarboxylase activity and of skin tumor promotion. Cancer Res. 1979;39:419–425. [PubMed] [Google Scholar]
- 36.Nakadate T, et al. Inhibition of teleocidin-caused epidermal ornithine decarboxylase induction by phospholipase A2-, cyclooxygenase- and lipoxygenase-inhibitors. Jpn. J. Pharmacol. 1985;37:253–258. doi: 10.1254/jjp.37.253. [DOI] [PubMed] [Google Scholar]
- 37.Bhimani RS, et al. Inhibition of oxidative stress in HeLa cells by chemopreventive agents. Cancer Res. 1993;53:4528–4533. [PubMed] [Google Scholar]
- 38.Herschman HR. Regulation of prostaglandin synthase-1 and prostaglandin synthase-2. Cancer Metastasis Rev. 1994;13:241–256. doi: 10.1007/BF00666095. [DOI] [PubMed] [Google Scholar]
- 39.Smith WL, et al. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and -2. J. Biol. Chem. 1996;271:33157–33160. doi: 10.1074/jbc.271.52.33157. [DOI] [PubMed] [Google Scholar]
- 40.Furstenberger G, et al. In: Arachidonic Acid Metabolism and Tumor Promotion. Fischer SM, Slaga TJ, editors. Boston, MA: Martinus Nijhoff Publishing; 1985. pp. 49–72. [Google Scholar]
- 41.Kim DJ, et al. Chemoprevention of colon cancer by Korean food plant components. Mutat. Res. 2003;523–524:99–107. doi: 10.1016/s0027-5107(02)00325-1. [DOI] [PubMed] [Google Scholar]
- 42.Shishodia S, et al. Guggulsterone inhibits NF-κB and IkappaBalpha kinase activation, suppresses expression of anti-apoptotic gene products, and enhances apoptosis. J. Biol. Chem. 2004;279:47148–47158. doi: 10.1074/jbc.M408093200. [DOI] [PubMed] [Google Scholar]
- 43.Chen W, et al. UVB irradiation-induced activator protein-1 activation correlates with increased c-fos gene expression in a human keratinocyte cell line. J. Biol. Chem. 1998;273:32176–32181. doi: 10.1074/jbc.273.48.32176. [DOI] [PubMed] [Google Scholar]
- 44.Zhu WH, et al. Regulation of angiogenesis by vascular endothelial growth factor and angiopoietin-1 in the rat aorta model: distinct temporal patterns of intracellular signaling correlate with induction of angiogenic sprouting. Am. J. Pathol. 2002;161:823–830. doi: 10.1016/S0002-9440(10)64242-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Potapova O, et al. c-Jun N-terminal kinase is essential for growth of human T98G glioblastoma cells. J. Biol. Chem. 2000;275:24767–24775. doi: 10.1074/jbc.M904591199. [DOI] [PubMed] [Google Scholar]
- 46.Huang C, et al. Shortage of mitogen-activated protein kinase is responsible for resistance to AP-1 transactivation and transformation in mouse JB6 cells. Proc. Natl Acad. Sci. USA. 1998;95:156–161. doi: 10.1073/pnas.95.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adderley SR, et al. Oxidative damage of cardiomyocytes is limited by extracellular regulated kinases 1/2-mediated induction of cyclooxygenase-2. J. Biol. Chem. 1999;274:5038–5046. doi: 10.1074/jbc.274.8.5038. [DOI] [PubMed] [Google Scholar]
- 48.Carter AB, et al. The p38 mitogen-activated protein kinase is required for NF-κB-dependent gene expression: the role of TATA-binding protein (TBP) J. Biol. Chem. 1999;274:30858–30863. doi: 10.1074/jbc.274.43.30858. [DOI] [PubMed] [Google Scholar]
- 49.Karin M, et al. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000;18:621–663. doi: 10.1146/annurev.immunol.18.1.621. [DOI] [PubMed] [Google Scholar]
- 50.Garg A, et al. Nuclear transcription factor-kappaB as a target for cancer drug development. Leukemia. 2002;16:1053–1068. doi: 10.1038/sj.leu.2402482. [DOI] [PubMed] [Google Scholar]
- 51.Epinat JC, et al. Diverse agents act at multiple levels to inhibit the Rel/NF-κB signal transduction pathway. Oncogene. 1999;18:6896–6909. doi: 10.1038/sj.onc.1203218. [DOI] [PubMed] [Google Scholar]
- 52.Na HK, et al. Transcriptional regulation via cysteine thiol modification: a novel molecular strategy for chemoprevention and cytoprotection. Mol. Carcinog. 2006;45:368–380. doi: 10.1002/mc.20225. [DOI] [PubMed] [Google Scholar]