

Abstract
Huntington's disease (HD) is an autosomal-dominant neurodegenerative disease caused by an expanded polyglutamine tract in the ubiquitously expressed huntingtin protein. Clinically, HD is characterized by motor, cognitive and psychiatric deficits. Striking degeneration of the striatum is observed in HD with the medium spiny neurons (MSNs) being the most severely affected neuronal subtype. Dysfunction of MSNs is marked by characteristic changes in gene expression which precede neuronal death. The ubiquitous expression of the huntingtin protein raises the question as to whether the selective vulnerability of the MSN is cell-autonomous, non-cell-autonomous, or a combination thereof. In particular, growing evidence suggests that abnormalities of the cortex and corticostriatal projections may be primary causes of striatal vulnerability. To examine this issue, we developed transgenic mice that, within the forebrain, selectively express a pathogenic huntingtin species in the MSNs, specifically excluding the neocortex. These mice develop a number of abnormalities characteristic of pan-cellular HD mouse models, including intranuclear inclusion bodies, motor impairment, and changes in striatal gene expression. As this phenotype develops in the presence of normal levels of brain-derived neurotrophic factor and its major striatal receptor, tropomyosin-related kinase B, these data represent the first demonstration of in vivo cell-autonomous transcriptional dysregulation in an HD mouse model. Furthermore, our findings suggest that therapies targeted directly to the striatum may be efficacious at reversing some of the molecular abnormalities present in HD.
INTRODUCTION
Huntington's disease (HD) is an autosomal-dominant, progressive and fatal neurodegenerative disorder caused by an expanded (>35–38) polyglutamine (polyQ) tract in the ubiquitously expressed huntingtin protein. Despite widespread expression of mutant huntingtin (mhtt), selective dysfunction and degeneration of specific neuronal subpopulations characterize HD and lead to motor, cognitive and psychiatric abnormalities. Among these neuronal subtypes, the medium spiny neurons (MSNs) of the striatum demonstrate the most striking vulnerability. As MSNs comprise 95% of total neurons in this nucleus (1), striatal atrophy constitutes a neuropathological hallmark of HD despite the relative sparing of other striatal cell types (2). Notably, however, early and substantial cortical degeneration is also observed in HD patients (3–7), leading to the hypothesis that changes in the cortex may be primarily responsible for dysfunction and death of MSNs.
In HD, the abundant connections between the cortex and striatum serve to link the degenerative processes in these areas and make the delineation of their relative contributions to pathogenesis extremely challenging (8,9). The striatum receives massive glutamatergic innervation (10) and neurotrophic support (11) from the cortex. Abnormalities in either excitotoxic mechanisms and/or trophism, particularly from a decrease in cortical brain-derived neurotrophic factor (BDNF), may contribute to striatal pathology in HD (12–16).
Possibly related to these corticostriatal mechanisms is the transcriptional dysregulation which is likely a critical, early event in HD pathogenesis. Numerous studies have described disrupted transcriptional pathways and altered striatal gene expression profiles in both HD patients and models (17). Microarray studies show that levels of ∼2% of striatal transcripts are altered and that most of these are decreased. These studies also show that the pattern of transcriptional dysregulation observed in human brain is quite accurately reproduced across multiple mouse HD models (18–21). The down-regulation of several specific transcripts, e.g. preproenkephalin (PPE) (22,23), dopamine and cyclic AMP-regulated phosphoprotein, 32 kDa (DARPP-32) (24) and neurotransmitter and neuropeptide receptors, including the D1 and D2 dopamine receptors, the adenosine A2A receptor, and the CB1 cannabanoid receptor (25–29), has been particularly well studied and quantitated. Several mechanisms have been proposed to explain the reductions in steady-state levels of striatal mRNAs in HD. Cell-autonomous mechanisms include altered regulation of histone modifications, abnormal interactions between mhtt and transcription factors and/or directly with DNA, and transcriptional repression due to loss of wild-type huntingtin function (17,30–32). Decreased cortical neurotrophic support is a particularly relevant non-cell-autonomous potential mechanism leading to transcriptional dysregulation as suggested by the similarities of striatal microarray profiles of forebrain-specific BDNF-knockout-mice and human HD (16).
We sought to determine the extent of cell-autonomous striatal pathology and transcriptional dysregulation in a murine model of HD. To this end, we created a line of transgenic mice that, within the forebrain, selectively expresses a cDNA encoding the N-terminal 171 amino acids of human huntingtin with a 98Q polyQ tract expansion (N171-98Q) in the striatal MSNs. Regulatory components of genomic elements from DARPP-32 (i.e. D9) (33) were used to target expression of the pathogenic huntingtin fragment. These mice demonstrate MSN nuclear inclusion formation, a motor phenotype, and striatal gene expression changes similar to those seen in pan-cellular and pan-neuronal HD models. These data provide the first unambiguous evidence of in vivo cell-autonomous MSN transcriptional dysregulation and the development of motor abnormalities in the absence of neocortical transgene expression in an HD model.
RESULTS
DARPP-32 elements target expression of mhtt to the MSNs, resulting in inclusion formation
Genomic fragments from the mouse DARPP-32 gene, hereafter referred to as ‘D9’, are known to direct transgene expression to the MSNs of the striatum (33). In that study, D9 was also noted to direct expression to the choroid plexus, olfactory tubercle, piriform cortex, and in an insertion-site-dependent manner, Purkinje cells. Importantly, these are all areas of endogenous DARPP-32 expression. The identical genomic elements were used to direct expression of a cDNA encoding the N-terminal 171 amino acids of the human huntingtin protein with an expanded polyQ tract length of 98 (D9-N171-98Q; Fig. 1). Western blot analysis of regionally dissected brain tissue from the highest expressing line, DE5, demonstrates high transgene expression in the striatum and cerebellum, and barely detectable expression in the neocortex (Fig. 2A).
Figure 1.

Schematic diagram of the D9-N171-98Q construct used in creation of the DE5 mouse. An IRES sequence followed by cDNA encoding the N-terminal 171 amino acids of huntingtin (N171-98Q) was placed downstream of DARPP-32 genomic elements totaling 9 kb (D9). The use of an IRES sequence was necessary because of the presence of an endogenous start codon (ATG) within D9. The transgene was released from plasmid by digestion with PacI. pA, polyadenosine tail.
Figure 2.

DE5 mice demonstrate striatal-specific transgene expression within the forebrain. (A) Western blotting of line DE5 extracts with 1C2 antibody reveals robust striatal expression and an absence of detectable neocortical expression. Transgene levels are also high in the cerebellum. (B) DE5 striatal mhtt levels are higher than in PrP-N171-82Q mice. The DE5 transgene has a higher molecular weight due to its larger polyQ expansion (98Q versus 82Q). DE5 striatal extract is from 1.5-month-old mouse; PrP-N171-82Q extract is from a 4-month-old mouse. DE5 striatal mhtt levels were unchanged up to 8.5 months of age. NTG, non-transgenic.
The primary goal of the current study is to compare the DE5 line with models ubiquitously expressing mhtt. The comparison with the well-characterized PrP-N171-82Q is particularly relevant, as in this model, the mouse prion promoter directs pan-neuronal and possibly glial expression of an identical mhtt fragment with an 82Q expansion (34,35). These mice die with severe motor and transcriptional abnormalities by 6 months of age (19,34). It is therefore important to note that striatal levels of transgenic protein in DE5 are higher than in PrP-N171-82Q mice at all ages analyzed (Fig. 2B), and a smaller transgene band is evident in both DE5 and PrP-171-82Q, likely an in vivo cleavage product (34). Even more important is the fact that no molecular or behavioral abnormalities are present in mice ubiquitously expressing N171-18Q (non-expanded) (19,34).
Immunocytochemistry with EM48, an antibody preferentially recognizing aggregated huntingtin (36), shows diffuse nuclear staining in DE5 striatal neurons as early as 2 months of age (Fig. 3A). Inclusion bodies are evident by 6 months of age and increase progressively in size and number so that by 22 months, large inclusions are present in virtually every EM48-positive cell in the striatum while cortical expression, particularly in pyramidal projection neurons, remains undetectable (Fig. 3B–E). Of note, inclusions in Purkinje cells are first detected at 8.5 months of age and are less numerous than in the striatum at 22 months of age (Fig. 3F). Thus, within the forebrain, D9-N171-98Q directs expression of high levels of mhtt in the MSNs of the striatum in the absence of neocortical expression and in vivo cell-autonomous mhtt aggregation and inclusion formation occurs in MSNs.
Figure 3.

EM48 immunostaining reveals diffuse nuclear staining and intranuclear inclusion formation in the striatum of DE5 mice. (A) Diffuse nuclear EM48 staining is observed in striatal neurons of DE5 mice by 2 months of age. (B–E) EM48-positive staining and inclusion formation is seen in the striatum of 22-month-old DE5 mice (C) but not in neocortex (E) or age-matched non-transgenic striatum (B) or neocortex (D). (F) Inclusion formation is also observed in DE5 cerebellar Purkinje cells. Scale bars are 30 µm in (A)–(E) and 45 µm in (F).
Forebrain weight is reduced in D9-N171-98Q mice but striatal cellular morphology appears normal
To further characterize the neuropathology of the DE5 mice, we analyzed gross and cellular striatal morphology. DE5 forebrain weight is reduced relative to non-transgenic littermates at 22 months of age (non-transgenic = 318 ± 3.5 mg; DE5 = 305 ± 4.3 mg; weight ± SE; P = 0.028, t-test). Striatal size is not obviously changed with aging, and surface area measurements at the level of the anterior commissure are not significantly different (DE5 left CPu 3.41 ± 0.49 and right CPu 3.46 ± 0.22, P > 0.5, versus control, left CPu 3.37 ± 0.20 and right CPu 3.09 ± 0.36, P > 0.5). Surprisingly, the lateral ventricles progressively decrease in size from 2 to 22 months, perhaps due to encroachment by the septal area (Fig. 4A–D, F–I). MSN cellular morphology appears normal (Fig. 4E and J). Stereological analysis is in progress to determine if more subtle changes in striatal volume or cellularity are evident, and to determine the cause of the decrease in ventricular size. Anti-glial fibrillary acidic protein (GFAP) staining does not reveal reactive gliosis in the striatum (Fig. 5).
Figure 4.

Striatal surface area and cellular morphology appear normal in DE5, but there is a decrease in size of the lateral ventricles. The lateral ventricle progressively decreases in size in DE5 mice (A–C) relative to NTG (non-transgenic) littermates (F–H) from 2 to 11 months of age (scale bar = 300 μm). Striatal area in coronal sections appears unchanged in DE5 (D) versus NTG (I) mice. Nissl-staining reveals apparent normal MSN morphology in DE5 (E) relative to NTG (J) brain (scale bar = 10 µm).
Figure 5.

Reactive gliosis is not observed in DE5 striata. Anti-GFAP staining of the striatum in DE5 mice (A) appears similar to that of non-transgenic littermates (B) at 22 months of age. Scale bar = 100 µm.
D9-N171-98Q mice demonstrate a failure to gain weight, deficit in rotarod performance and hypoactivity
Body mass, performance on an accelerating rotarod apparatus and locomotor activity were measured every 2 months in both male and female D9-N171-98Q (DE5) cohorts up to 16 months of age. DE5 mice of both sexes failed to gain weight beginning at 12 months of age (Fig. 6A). The longitudinal analysis revealed a deficit in rotarod performance in DE5 females relative to their non-transgenic littermates that progressively worsened between 10 and 16 months of age (Fig. 6B). No rotarod deficit was detected in the male cohort and no differences in locomotor activity were observed in either sex (data not shown). Although sex-based differences in phenotype have been reported in HD mouse models and in the human disease (37–39), it was somewhat surprising that the longitudinally tested male cohort failed to demonstrate any significant deficits in rotarod performance. We therefore tested a second larger cohort of males at a single age of 16 months. The observed deficit in this second cohort suggests that motor impairment is not limited to females (Fig. 6C), although sex differences may exist and require further investigation, particularly as to the effects of learning. The second cohort of DE5 male mice also demonstrated hypoactivity in the locomotor apparatus that was most pronounced during the first 5 min, suggesting a decrease in either exploratory behavior or anxiety (Fig. 6D). These data may explain the absence of hypoactivity in mice from the longitudinal studies, which were habituated to the apparatus beginning at 2 months of age. Thus, in the DE5 mice, there is a late onset phenotype characterized by impaired performance on the rotarod, hypoactivity and failure to gain weight.
Figure 6.
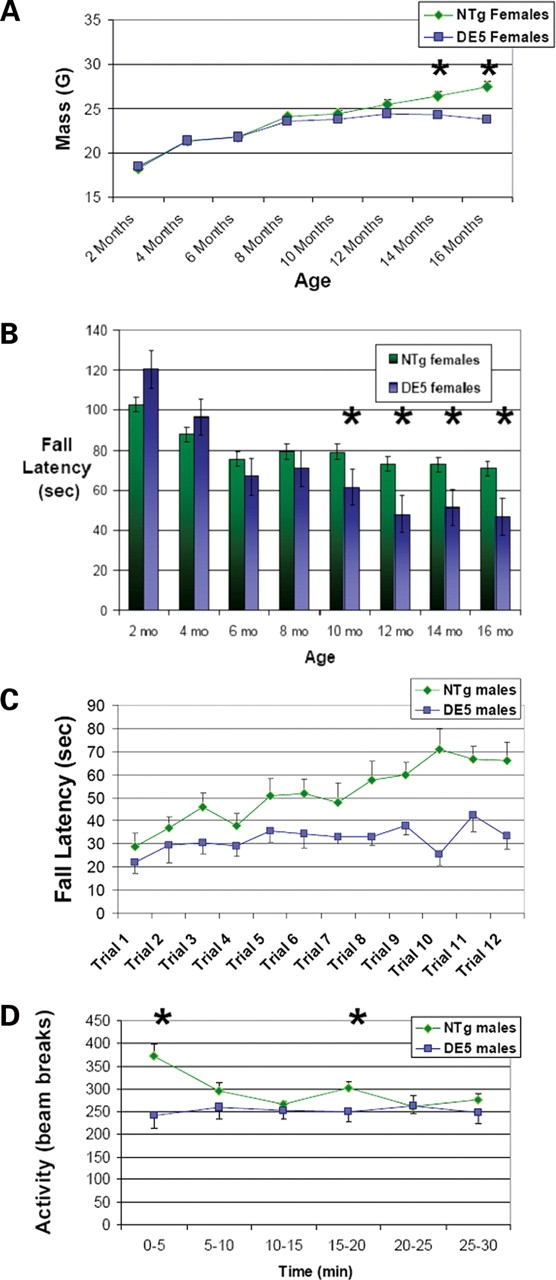
DE5 mice demonstrate failure to gain weight and motor abnormalities. (A) DE5 females (n = 13) demonstrate a failure to gain weight relative to non-transgenic littermates (n = 11) beginning at 14 months of age. DE5 males (n = 7) also failed to gain weight relative to non-transgenics (n = 10) (non-transgenic: 33.8 ± 1.06 g, DE5: 29.57 ± 0.57 g; body mass at 16 months of age ± SEM; P = 0.007, t-test). (B) Longitudinal analysis of female DE5 mice (n = 13) and non-transgenic littermates (n = 11) reveals a progressive deficit in rotarod performance beginning at 10 months of age and continuing through 16 months. Significance was determined by two-way repeated measures ANOVA at each time period. (C) Naïve, 16-month-old male DE5 mice (n = 9) show a marked deficit in rotarod performance relative to non-transgenic littermates (n = 10). (D) Naïve, male DE5 mice (n = 9) are hypoactive relative to non-transgenic littermates (n = 10) tested at 16 months of age. Measurements of activity in 5 min time bins revealed significant hypoactivity from 0 to 5 min and from 15 to 20 min. Total activity > 30 min was also decreased (P = 0.021). Hypoactivity was not detected when animals were tested longitudinally beginning at 2 months. All values are expressed as mean ± SEM. NTg, non-transgenic. *P < 0.05.
Gene expression analysis reveals cell-autonomous reductions in striatal mRNA levels
As noted, an early molecular hallmark in human HD and in its mouse models is transcriptional dysregulation in MSNs. Quantitative reverse transcription–polymerase chain reaction (RT–PCR) on striatal mRNA was utilized to measure levels of numerous transcripts in DE5 mice, chosen from amongst those frequently found to be abnormal in other HD mouse models and/or relevant to cortical regulation of striatal function. The earliest significant decreases in mRNA levels are observed in PPE (P = 0.003) and endogenous DARPP-32 (P = 0.046) at 8.5 months (data not shown). Although a trend is evident at 4.5 months, the differences at that age are not statistically significant. By 13.5 months of age, there are significant reductions in steady-state mRNA levels of PPE (P = 0.002), prodynorphin (PDYN, P = 0.006), endogenous DARPP-32 (P < 0.001), the CB1 cannabinoid receptor (P = 0.035), the A2A adenosine receptor (A2AAR, P < 0.001), the dopamine D1 receptor (DRD1, P < 0.001), and the dopamine D2 receptor (DRD2, P < 0.001) (Fig. 7). mRNA levels of the NMDA (N-methyl-D-aspartic acid) receptor subunits NR2A and NR2B are unchanged, whereas NR1 mRNA levels are slightly decreased (P = 0.035). Striatal mRNA levels of calbindin-28K (Calb1) are also unchanged. Importantly, BDNF mRNA levels in the cortex, and those of its main receptor, tropomyosin-related kinase B (trkB), in the striatum, are equal in DE5 mice and controls (Figs 7 and 8A). Transgene mRNA levels are also relatively decreased from 4.5 to 13.5 months in a pattern that mirrors the decrease in endogenous DARPP-32 levels (Fig. 8B). As D9 is derived from DARPP-32, this is an anticipated finding, but importantly, indicates that the regulatory elements that confer cell-specificity also confer down-regulation by mhtt. Finally, analysis of cerebellar mRNA levels demonstrates a decrease in Calb1 mRNA (P = 0.040) representing a contrast to the lack of change observed in the striatum. The apparent decrease in DARPP-32 levels did not achieve significance (P = 0.065) (Fig. 8C). In summary, select and region-specific changes in MSN mRNA levels occur in the striatum in D9-N171-98Q mice.
Figure 7.

DE5 mice demonstrate characteristic striatal gene expression changes. Quantitative RT–PCR revealed select reductions in striatal transcripts in 13.5 mo DE5 mice (n = 3) relative to non-transgenic littermates (n = 3). PPE, preproenkephalin; PDYN, prodynorphin; Calb1, calbindin-28K; D32, endogenous DARPP-32; CB1, cannabinoid receptor CB1; A2AAR, A2A adenosine receptor; DRD1, dopamine D1 receptor; DRD2, dopamine D2 receptor; NR1, NR1 NMDA receptor subunit; NR2A, NR2A NMDA receptor subunit; NR2B, NR2B NMDA receptor subunit. Graph shows relative mRNA expression using β-actin mRNA as loading control. Values are expressed as mean ± SEM. *P < 0.05.
Figure 8.
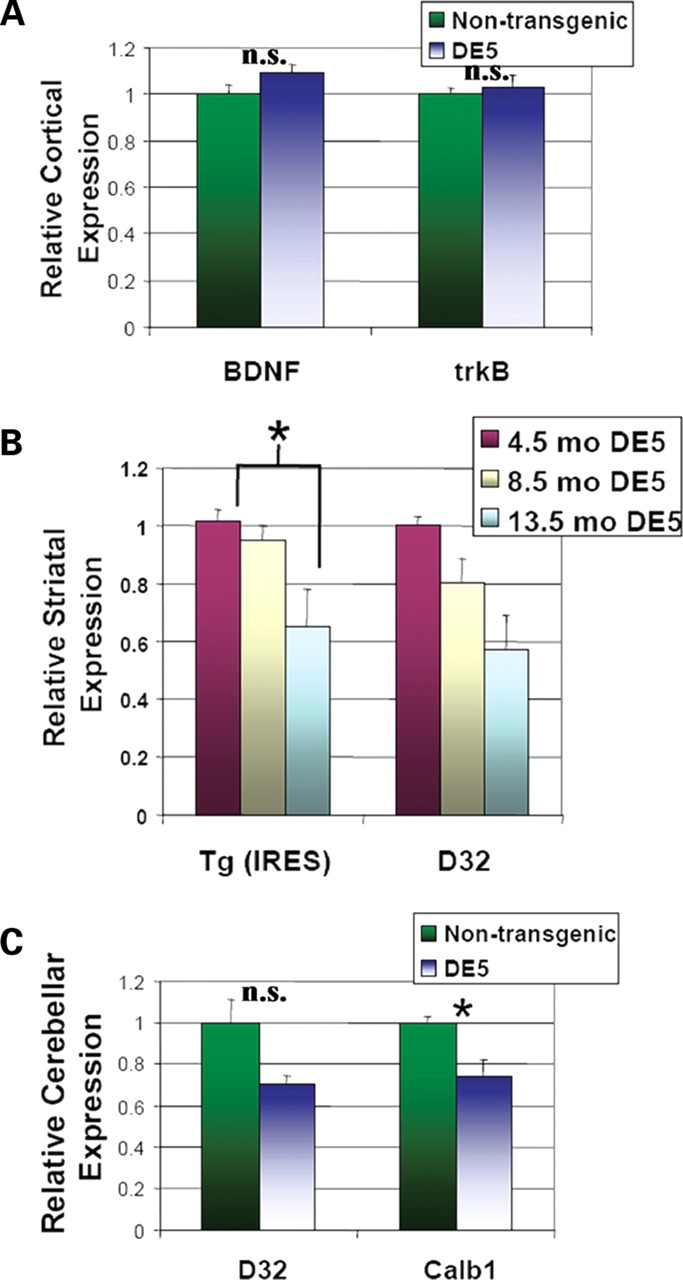
Striatal transcriptional changes in DE5 mice are cell-autonomous and region-specific. (A) BDNF and trkB mRNA levels in the cortex are unchanged. (B) Reductions in transgene (Tg) mRNA levels mirror decreases in endogenous DARPP-32 levels between 4.5 mo (n = 3), 8.5 mo (n = 3) and 13.5 mo (n = 3) DE5 mice. Primers derived against the IRES sequence were used to measure transgene levels. (C) Calb1 mRNA levels are decreased in cerebellum of DE5 mice. Graphs show relative mRNA expression as determined by quantitative RT–PCR using β-actin mRNA as loading control. Values are expressed as mean ± SEM. *P < 0.05.
Transgenic expression of striatal DARPP-32 does not cause motor, transcriptional, or morphological abnormalities
In DE5 mice, expression of transgenic DARPP-32 protein from D9, as detected by an increase in total DARPP-32 levels, is present up to 3 months of age in striatum (Fig. 9B) and in cerebellum at all ages analyzed (data not shown). Analysis of striatal DARPP-32 protein expression in DE5 mice was performed on several groups of young mice from 3 weeks to 3 months of age and results varied from 0 to a 70% increase in DARPP-32 protein levels in DE5 relative to non-transgenic littermates. Based on these data, controls in all studies include non-transgenic littermates, as presented earlier, and in addition, we studied a second D9 transgenic mouse line, F6, with comparable striatal DARPP-32 expression (Fig. 9). This latter line was developed in an attempt to create a full-length model similar to D9-N171-98Q, in which D9 was used to direct expression of a cDNA encoding full-length human huntingtin with an expanded polyQ tract of 98 downstream of the internal ribosomal entry site (IRES) (D9-FLhtt-98Q). In the highest-expressing line, F6, striatal huntingtin expression was only 15% of the endogenous level (data not shown). Despite the low expression of transgenic huntingtin, we longitudinally followed cohorts in the same experimental paradigms as for DE5. F6 mice display normal brain morphology without a decrease in ventricular size (Fig. 9D) and do not develop transcriptional abnormalities (Fig. 9E), nor do they demonstrate any motor deficits up to 18 months of age in rotarod testing (non-transgenic: 91.6 ± 10.0 s, F6: 94.4 ± 8.0 s; average fall latency ± SEM; P = 0.829, two-way repeated measures ANOVA). Immunostaining for huntingtin in F6 was undetectable (data not shown). Thus, transgenic huntingtin expression in F6 is too low to draw conclusions about the cell-autonomous effects of full-length mhtt. Importantly, however, as striatal DARPP-32 expression is comparable with DE5 (Fig. 9B and C), a lack of phenotype in F6 allows for the exclusion of effects of transient striatal DARPP-32 over-expression.
Figure 9.

A second D9 transgenic mouse line (F6) controls for effects of transient striatal DARPP-32 over-expression in DE5. (A) Quantitative RT–PCR on striatal tissue demonstrates equivalent levels of transgene mRNA (IRES) between DE5 mice (n = 3) and F6 mice (F6 het, n = 3). Predictably, levels in homozygous F6 mice (F6 hom, n = 3) are approximately twice as high. (B and C) Representative western blots demonstrating higher DARPP-32 striatal expression in 2 mo DE5 mice (P = 0.03) (B) and 2 mo F6 homozygotes (P = 0.003) (C) relative to age-matched NTG (non-transgenic) littermates. Over-expression of DARPP-32 was not apparent after 8.5 months of age in striatum (data not shown). (D) 14-month-old F6 mice possess normal gross morphology with open lateral ventricles (scale bar = 300 µm). (E) mRNA levels are unchanged in F6 in 14.5-month-old F6 heterozygote mice relative to non-transgenic mice. All values are expressed as mean ± SEM. *P < 0.05.
DISCUSSION
Numerous pan-cellular and pan-neuronal mouse models have been created in an attempt to recapitulate HD (40,41). A challenge in research into the mechanisms of cell death and dysfunction in HD is to determine whether the pathophysiology of these abnormalities is cell-autonomous, i.e. ‘suicide’ by MSNs, or non-cell-autonomous, i.e. ‘murder’ of MSNs by cortical cells, or a combination thereof. Although in vitro results do not always translate to in vivo, cell culture studies support intracellular, cell-autonomous toxicity of mhtt fragments, as evidenced by enhanced cell death dependent on polyQ length (42–44), and great effort is being expended on the identification of cell-autonomous therapies. Recent in vivo studies, however, suggest a requirement for pathological cell-to-cell interactions for the development of most of the mouse HD phenotype (8,9). Ours is the first report of a transgenic HD model documenting motor and transcriptional abnormalities in the absence of neocortical transgene expression.
Yang and colleagues utilized a Cre-recombinase-mediated strategy to express mhtt (exon 1) 103Q in MSNs and a subset of cortical interneurons (RosaHD/Dlx-5/6-Cre) (9). Transgenic product accumulated in striatal nuclei and an electrophysiological abnormality was detected, but inclusions and a motor phenotype did not develop. Importantly, the D9-N171-98Q mouse and the RosaHD/Dlx-5/6-Cre differ in the length of the expressed mhtt fragment (N171 versus exon 1), pattern of extrastriatal expression, and timing of expression. Shorter mhtt fragments are usually more toxic, and therefore the results perhaps suggest lower striatal mhtt expression in the RosaHD model relative to the D9-N171-98Q. The relationship between expression level and phenotypic severity is well established (34,45).
Striatal-specific expression has also been achieved by lentiviral-mediated delivery of mhtt in rats and primates (45,46). Most of the observations are related to neuropathology, including inclusion formation and cell death, although putaminal injections in primates lead to abnormal movement. These data are overall in support of in vivo mhtt toxicity in MSNs in the absence of neocortical expression.
D9-N171-98Q mice exhibit a motor phenotype characterized by impaired rotarod performance and hypoactivity, both features common to many pan-neuronal HD mouse models. However, relative to the PrP-N171-82Q mouse, the D9-N171-98Q mouse exhibits a more protracted disease course and lacks several other reported behavioral abnormalities (34). In particular, the PrP-N171-82Q mouse also displays tremors, an abnormal gait and frequent hindlimb clasping. Furthermore, deficits in rotarod performance are evident by 3 months of age and premature death occurs ∼6 months of age. Thus, despite higher striatal expression and greater polyQ length, the D9-N171-98Q mouse only recapitulates some of the features of its most comparable pan-neuronal model, the PrP-N171-82Q mouse. Possible contributors to these differences in phenotype and its severity certainly include the presence of neocortical expression in PrP mice, as well as systemic abnormalities and background strain.
In several HD models, contributions to motor abnormalities from Purkinje cell dysfunction cannot be ruled out, but motor dysfunction is notably present in an HD mouse model lacking cerebellar transgene expression (47). Hindlimb clasping, present in several mouse HD models and in naturally occurring cerebellar-specific mutant mice (41,48,49) is absent in D9-N171-98Q suggesting that this abnormality may include cortical dysfunction when originating from the forebrain. Finally, the D9-N171-98Q phenotype may be moderated by background strain. In the YAC128 mouse, the phenotype is less severe on a C57BL/6 background (50).
In general, D9-N171-98Q mice appear healthy, with normal breeding and lifespan, but there is a failure to gain weight followed by a small weight loss after 12 months of age. Proposed mechanisms of weight loss in HD and its models include hypothalamic dysfunction and/or a global defect in mitochondrial function (51,52). The reduced body weight in D9-N171-98Q mice in the presence of a restricted pattern of transgene expression suggests other causes. Interestingly, both the dorsal and ventral striatum are involved in the regulation of appetite and the level of the enkephalin neuropeptide may play a specific role (53).
Gene expression changes in striatal MSNs represent an early marker of neuronal dysfunction in HD, although the causal relationship with motor abnormalities is unknown. The striatal microarray profile of the aforementioned forebrain-specific BDNF-knockout-mice (Emx-BDNFKO) has been shown to be statistically more similar to human HD than that of several mouse HD models (16). Striatal BDNF is critical for the maturation and maintenance of MSNs in vitro and in vivo (15,54–56). Wild-type, but not mutant, huntingtin promotes the transcription and vesicular transport of BDNF (13,14,31), and BDNF protein is reduced in the cortex and striatum of HD patients and mouse models (13,57–59). The primary striatal receptor for BDNF, trkB, may also be decreased in the presence of mhtt, further compromising BDNF-derived trophic support (58,61). Our results clearly demonstrate that striatal transcriptional dysregulation occurs in the absence of alterations in cortical BDNF and/or striatal trkB levels.
Of the transcripts that we measured, the D9-N171-98Q mouse recapitulates several, but not all, of the changes observed in human HD, pan-cellular mouse models and the Emx-BDNFKO mouse. In particular, calbindin mRNA is unchanged in D9-171-98Q mice, whereas mRNA levels of A2AAR and PDYN are decreased in the D9-171-98Q mouse and several other mouse models of HD (19,29), but are normal in the Emx-BDNFKO mouse (16), suggesting that a decrease in BDNF does not play a key role in the reduction of these two transcripts. Two striking similarities between D9-N171-98Q and Emx-BDNFKO are decreases in PPE and the D2 dopamine receptor, markers of the subpopulation of MSNs that is first affected in HD (62). The high level of vulnerability of this subpopulation may arise from the convergence of cell-autonomous and BDNF-related pathogenic mechanisms. Microarray analysis of D9-N171-98Q will provide a more comprehensive comparison with human HD, ubiquitous mhtt mouse models, and BDNF-null models. These comparisons may uncover other MSN-specific differences in expression, potentially revealing novel pathogenic mechanisms and therapeutic opportunities.
In addition to offering mechanistic insights into a number of the molecular abnormalities observed in HD, our findings have important therapeutic implications. The ability to reverse neuropathology and motor dysfunction in an HD mouse model by blocking expression of mhtt (47) predicts potential efficacy of therapies targeting mhtt expression, e.g. intrabodies and RNA interference (63,64). Our demonstration of cell-autonomous MSN dysfunction further suggests that delivery of these therapies directly to the striatum may be sufficient to reverse key molecular abnormalities.
MATERIALS AND METHODS
Vector design
A huntingtin cDNA encoding the first 171 amino acids with a CAG repeat length of 98 was removed from the plasmid pC3HD NXQ150 GFP (provided by Marcy MacDonald, Harvard Medical School, Boston, MA) with EcoRI and XhoI. This cDNA fragment and a poly linker encoding a stop codon were inserted into pBluescriptIIKS+ (Stratagene, La Jolla, CA, USA) at EcoRI and HindIII sites to create pBKSHttStop. An IRES was removed from pIRES2-EGFP (Clontech, Palo Alto, CA, USA) with BamHI and NcoI and was inserted into pBKSHttStop upstream of the huntingtin cDNA at BamHI and NcoI sites to create pBKSIresHttStop. A polyA sequence was removed from pTracerCMV (Invitrogen, Carlsbad, CA, USA) at PvuII and Sau3AI sites and inserted downstream of the stop codon in pBKSIresHtt98Qstop to create pIresHttstpBGHpA. Finally, a NotI to SalI fragment was excised from pIresHTstpBGHpA and inserted into D9 in place of the IRES and lacZ marker gene (33). The linearized 11.7kb transgenic vector was excised from D9 with PacI.
Transgenesis and genotyping
The 11.7 kb D9-N171-98Q fragment was separated on 0.9% agarose gel, purified by electrophoretic elution, treated by phenol–chloroform mixture (1:1), precipitated, dissolved in injection buffer, and injected into fertilized eggs of C57BL/6J mice at the transgenic facility of The Rockefeller University. Transgenic animals were identified by performing PCR on genomic DNA purified from tail biopsies using the REDExtract-N-Amp Tissue PCR kit (Sigma, St Louis, MO, USA). The primers amplified downstream of the CAG repeat across the exon 2–3 junction: sense 5′-CCgCTgCACCgACCAAAgAA-3′; and antisense 5′-gCATTCgTCAgCCACCATCC-3′. Additionally, tail biopsies were sent to Laragen, Inc. (Los Angeles, CA, USA) for genotyping by PCR as well as determination of the CAG repeat length. Mice were maintained on a C57BL/6J background.
Protein expression analysis
Protein levels were measured by western blot analysis of protein extracted from regionally dissected brain tissue. Soluble protein was extracted from tissue in 20 mm Hepes (pH 7.6), 150 mm NaCl, 0.5 mm EDTA, and 0.5% Triton X-100, supplemented with 1X complete protease inhibitor cocktail (Roche Diagnostics GmbH, Mannheim, Germany), 1 mm PMSF, 50 mm NaF and 1 mm Na-orthovanadate. Protein levels in the extracts were determined using the Bio-Rad DC Protein Assay. Antibodies used included 1C2 antibody (1:1000, MAB1574, Chemicon, Temecula, CA, USA), anti-DARPP-32 (1:1000, AB1656, Chemicon), anti-β-actin (1:1000, Sigma) and anti-α-tubulin (1:2000, DM1A, Sigma). Blots were developed using Amersham (Piscataway, NJ, USA) enhanced chemiluminescence western blot detection reagents. Densitometry was performed using LabWorks™ Image Acquisition and Analysis Software from BioImaging Systems. Statistical analyses of densitometry results were performed using a Student's t-test and statistical significance was achieved at P < 0.05.
Behavioral analysis
Motor performance in male and female cohorts was assessed using a Rotamex Rotarod (Columbus Instruments, Columbus, OH, USA) beginning at 2 months of age and continuing every other month up to 16 months of age. In a second male cohort, the first test was performed on naïve animals at 16 months of age. The first test consisted of three trials per day for 4 days (12 trials total). A trial involved placing a mouse on a slowly rotating rod that began at 8 rpm and increased 1 rpm every 8 s until the mouse fell off the rod or gripped the rod and rotated around with it. Mice received an hour of rest between trials. In subsequent months, only 3 days of testing were performed (9 total trials). For all testing periods, only the last 6 trials were used, with the initial trials serving as training. Data were analyzed by two-way repeated measures ANOVA (SigmaStat software). For the animals tested only at 16 months, the initial velocity of the rod was set to 4 rpm and 12 trials were performed >4 days. All 12 trials were analyzed by two-way repeated measures ANOVA.
Locomotor activity level was measured as previously described (65). Briefly, animals were placed in one of eight identical transparent Plexiglas boxes equipped with MicroMax Animal Activity Monitors (Accuscan Instruments, Inc., Columbus, OH, USA) for 30 min. The monitors use infrared light emitters and detectors to detect animal presence. Total activity is measured as the total number of photo beam breaks over the allotted time. Testing was performed on the same animals that underwent rotarod testing. A Student's t-test was used to compare activity levels of transgenic and non-transgenic animals at each time bin and for total activity, and statistical significance was achieved at P < 0.05.
Histology
Immunocytochemistry was performed as previously described (33). Briefly, brains were fixed by transcardial perfusion with 4% paraformaldehyde in PBS followed by overnight immersion fixation in the same fixative and then stored in PBS at 4°C. Brains were sectioned coronally on a vibratome (Leica Microsystems, Wetzlar, Germany) at 40 µm. The following primary antibodies were used for immunohistochemistry:rabbit polyclonal EM48 (1:1000, generously provided by Xiao-Jiang Li, Emory University School of Medicine, Atlanta, GA) (36), anti-NeuN (1:500, MAB377, Millipore, Billerica, MA, USA) and anti-GFAP (1:100, 3670, Cell Signaling Technology, Beverly, MA, USA). Immunostaining was performed with biotinylated secondary antibodies, the ABC Vectastain kit (Vector Laboratories, Burlingame, CA, USA) and the DAB Reagent Set (KPL, Gaithersburg, MD, USA). Coronal brain sections were also mounted on glass slides and stained for nissl substance with a 0.1% cresyl violet acetate solution. Measurement of striatal surface area was calculated with the aid of MicroBrightField's Stereo Investigator 7.00. Contours were traced around the regions of caudate putamen, anterior commissure anterior, or anterior commissure posterior in matched, coronal plane sections. Contour perimeter and area were obtained for each structure and sorted by hemisphere for each section and animal. Results are reported in arbitrary units.
Gene expression analysis
RNA was isolated from regionally dissected brain tissue and treated with Amplification Grade DNaseI (Invitrogen) with the TRIzol Plus RNA Purification Kit (Invitrogen) according to the manufacturer's protocol. cDNA was synthesized with the SuperScript III First-Strand Synthesis System for RT–PCR (Invitrogen). Quantitative PCR was then performed using Taqman Gene Expression Assays on a 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) according to the manufacturer's protocol. Custom primers were designed against the IRES sequence to detect transgene (forward primer: ggCCAAAAgCCACgTgTATAAgATA, reverse primer: CCACAACTATCCAACTCACAACgT, reporter: TTgTgCCgCCTTTgCA). Custom primers specific for endogenous DARPP-32 amplified a region of 3′-UTR not present in D9 (forward primer: gCTgCTTgCTgCgTTTTgT, reverse primer: CCCTAAggACAgATAgTAAgTgCATT, reporter: CTggCACCAAAgCACA). Relative quantification (RQ) of gene expression was performed by the comparative CT method using β-actin as the endogenous reference for each sample. All reactions were performed in triplicate from at least three individual mice in 20 µl reaction volumes. Primary data analysis was performed using system software from Applied Biosystems to obtain RQ values. Statistical analysis of RQ values were performed using a Student's t-test and statistical significance was achieved at P < 0.05. Assay IDs: trkB, Mm00435422_m1; PPE, Mm01212875_m1; PDYN, Mn00457572_m1; Calb1, Mm00486645_m1; CB1, Mm00432621_m1; A2AAR, Mm00802075_m1; DRD1, Mm01353211_m; DRD2, Mm00438541_m1; NR1, Mm00433800_m1; NR2A, Mm00433802_m1; NR2B, Mm00433820_m1; BDNF, Mm00432069_m1; β-actin, Part# 435935E.
FUNDING
This work was supported by NIH NS0045942 and NS052452 and the High Q Foundation.
ACKNOWLEDGEMENTS
J. Pelta-Heller, E. Sluzas, C. Thomas and C. Marshall provided technical support. We thank Drs M. MacDonald (Harvard Medical School), M. Hayden (University of British Columbia), and G. Lawless (University of California at Los Angeles) for huntingtin plasmids, and X.-J. Li (Emory University School of Medicine) for polyclonal EM48 antibody and PrP-N171-82Q tissue.
Conflict of Interest statement. The authors have no conflict of interest to declare.
REFERENCES
- 1.Ouimet C.C., Langley-Gullion K.C., Greengard P. Quantitative immunocytochemistry of DARPP-32-expressing neurons in the rat caudatoputamen. Brain Res. 1998;808:8–12. doi: 10.1016/s0006-8993(98)00724-0. [DOI] [PubMed] [Google Scholar]
- 2.Vonsattel J.P., DiFiglia M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Rosas H.D., Koroshetz W.J., Chen Y.I., Skeuse C., Vangel M., Cudkowicz M.E., Caplan K., Marek K., Seidman L.J., Makris N., et al. Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology. 2003;60:1615–1620. doi: 10.1212/01.wnl.0000065888.88988.6e. [DOI] [PubMed] [Google Scholar]
- 4.Rosas H.D., Liu A.K., Hersch S., Glessner M., Ferrante R.J., Salat D.H., van der Kouwe A., Jenkins B.G., Dale A.M., Fischl B. Regional and progressive thinning of the cortical ribbon in Huntington's disease. Neurology. 2002;58:695–701. doi: 10.1212/wnl.58.5.695. [DOI] [PubMed] [Google Scholar]
- 5.Rosas H.D., Salat D.H., Lee S.Y., Zaleta A.K., Pappu V., Fischl B., Greve D., Hevelone N., Hersch S.M. Cerebral cortex and the clinical expression of Huntington's disease: complexity and heterogeneity. Brain. 2008;131:1057–1068. doi: 10.1093/brain/awn025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halliday G.M., McRitchie D.A., Macdonald V., Double K.L., Trent R.J., McCusker E. Regional specificity of brain atrophy in Huntington's disease. Exp. Neurol. 1998;154:663–672. doi: 10.1006/exnr.1998.6919. [DOI] [PubMed] [Google Scholar]
- 7.Macdonald V., Halliday G. Pyramidal cell loss in motor cortices in Huntington's disease. Neurobiol. Dis. 2002;10:378–386. doi: 10.1006/nbdi.2002.0528. [DOI] [PubMed] [Google Scholar]
- 8.Gu X., Li C., Wei W., Lo V., Gong S., Li S.H., Iwasato T., Itohara S., Li X.J., Mody I., et al. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant huntingtin contribute to cortical pathogenesis in HD mice. Neuron. 2005;46:433–444. doi: 10.1016/j.neuron.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 9.Gu X., Andre V.M., Cepeda C., Li S.H., Li X.J., Levine M.S., Yang X.W. Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington's disease. Mol. Neurodegener. 2007;2:8. doi: 10.1186/1750-1326-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cepeda C., Wu N., Andre V.M., Cummings D.M., Levine M.S. The corticostriatal pathway in Huntington's disease. Prog. Neurobiol. 2007;81:253–271. doi: 10.1016/j.pneurobio.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altar C.A., Cai N., Bliven T., Juhasz M., Conner J.M., Acheson A.L., Lindsay R.M., Wiegand S.J. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- 12.Cepeda C., Hurst R.S., Calvert C.R., Hernandez-Echeagaray E., Nguyen O.K., Jocoy E., Christian L.J., Ariano M.A., Levine M.S. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington's disease. J. Neurosci. 2003;23:961–969. doi: 10.1523/JNEUROSCI.23-03-00961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zuccato C., Ciammola A., Rigamonti D., Leavitt B.R., Goffredo D., Conti L., MacDonald M.E., Friedlander R.M., Silani V., Hayden M.R., et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 14.Gauthier L.R., Charrin B.C., Borrell-Pages M., Dompierre J.P., Rangone H., Cordelieres F.P., De Mey J., MacDonald M.E., Lessmann V., Humbert S., et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 15.Baquet Z.C., Gorski J.A., Jones K.R. Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J. Neurosci. 2004;24:4250–4258. doi: 10.1523/JNEUROSCI.3920-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Strand A.D., Baquet Z.C., Aragaki A.K., Holmans P., Yang L., Cleren C., Beal M.F., Jones L., Kooperberg C., Olson J.M., et al. Expression profiling of Huntington's disease models suggests that brain-derived neurotrophic factor depletion plays a major role in striatal degeneration. J. Neurosci. 2007;27:11758–11768. doi: 10.1523/JNEUROSCI.2461-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cha J.H. Transcriptional signatures in Huntington's disease. Prog. Neurobiol. 2007;83:228–248. doi: 10.1016/j.pneurobio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luthi-Carter R., Strand A., Peters N.L., Solano S.M., Hollingsworth Z.R., Menon A.S., Frey A.S., Spektor B.S., Penney E.B., Schilling G., et al. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum. Mol. Genet. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- 19.Luthi-Carter R., Strand A.D., Hanson S.A., Kooperberg C., Schilling G., La Spada A.R., Merry D.E., Young A.B., Ross C.A., Borchelt D.R., et al. Polyglutamine and transcription: gene expression changes shared by DRPLA and Huntington's disease mouse models reveal context-independent effects. Hum. Mol. Genet. 2002;11:1927–1937. doi: 10.1093/hmg/11.17.1927. [DOI] [PubMed] [Google Scholar]
- 20.Hodges A., Strand A.D., Aragaki A.K., Kuhn A., Sengstag T., Hughes G., Elliston L.A., Hartog C., Goldstein D.R., Thu D., et al. Regional and cellular gene expression changes in human Huntington's disease brain. Hum. Mol. Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- 21.Kuhn A., Goldstein D.R., Hodges A., Strand A.D., Sengstag T., Kooperberg C., Becanovic K., Pouladi M.A., Sathasivam K., Cha J.H., et al. Mutant huntingtin's effects on striatal gene expression in mice recapitulate changes observed in human Huntington's disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum. Mol. Genet. 2007;16:1845–1861. doi: 10.1093/hmg/ddm133. [DOI] [PubMed] [Google Scholar]
- 22.Augood S.J., Faull R.L., Love D.R., Emson P.C. Reduction in enkephalin and substance P messenger RNA in the striatum of early grade Huntington's disease: a detailed cellular in situ hybridization study. Neuroscience. 1996;72:1023–1036. doi: 10.1016/0306-4522(95)00595-1. [DOI] [PubMed] [Google Scholar]
- 23.Menalled L., Zanjani H., MacKenzie L., Koppel A., Carpenter E., Zeitlin S., Chesselet M.F. Decrease in striatal enkephalin mRNA in mouse models of Huntington's disease. Exp. Neurol. 2000;162:328–342. doi: 10.1006/exnr.1999.7327. [DOI] [PubMed] [Google Scholar]
- 24.Bibb J.A., Yan Z., Svenningsson P., Snyder G.L., Pieribone V.A., Horiuchi A., Nairn A.C., Messer A., Greengard P. Severe deficiencies in dopamine signaling in presymptomatic Huntington's disease mice. Proc. Natl Acad. Sci. USA. 2000;97:6809–6814. doi: 10.1073/pnas.120166397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Denovan-Wright E.M., Robertson H.A. Cannabinoid receptor messenger RNA levels decrease in a subset of neurons of the lateral striatum, cortex and hippocampus of transgenic Huntington's disease mice. Neuroscience. 2000;98:705–713. doi: 10.1016/s0306-4522(00)00157-3. [DOI] [PubMed] [Google Scholar]
- 26.McCaw E.A., Hu H., Gomez G.T., Hebb A.L., Kelly M.E., Denovan-Wright E.M. Structure, expression and regulation of the cannabinoid receptor gene (CB1) in Huntington's disease transgenic mice. Eur. J. Biochem. 2004;271:4909–4920. doi: 10.1111/j.1432-1033.2004.04460.x. [DOI] [PubMed] [Google Scholar]
- 27.Augood S.J., Faull R.L., Emson P.C. Dopamine D1 and D2 receptor gene expression in the striatum in Huntington's disease. Ann. Neurol. 1997;42:215–221. doi: 10.1002/ana.410420213. [DOI] [PubMed] [Google Scholar]
- 28.Cha J.H., Kosinski C.M., Kerner J.A., Alsdorf S.A., Mangiarini L., Davies S.W., Penney J.B., Bates G.P., Young A.B. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human Huntington disease gene. Proc. Natl Acad. Sci. USA. 1998;95:6480–6485. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tarditi A., Camurri A., Varani K., Borea P.A., Woodman B., Bates G., Cattaneo E., Abbracchio M.P. Early and transient alteration of adenosine A2A receptor signaling in a mouse model of Huntington disease. Neurobiol. Dis. 2006;23:44–53. doi: 10.1016/j.nbd.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 30.Sadri-Vakili G., Cha J.H. Mechanisms of disease: histone modifications in Huntington's disease. Nat. Clin. Pract. Neurol. 2006;2:330–338. doi: 10.1038/ncpneuro0199. [DOI] [PubMed] [Google Scholar]
- 31.Zuccato C., Tartari M., Crotti A., Goffredo D., Valenza M., Conti L., Cataudella T., Leavitt B.R., Hayden M.R., Timmusk T., et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]
- 32.Zuccato C., Belyaev N., Conforti P., Ooi L., Tartari M., Papadimou E., MacDonald M., Fossale E., Zeitlin S., Buckley N., et al. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington's disease. J. Neurosci. 2007;27:6972–6983. doi: 10.1523/JNEUROSCI.4278-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bogush A.I., McCarthy L.E., Tian C., Olm V., Gieringer T., Ivkovic S., Ehrlich M.E. DARPP-32 genomic fragments drive cre expression in postnatal striatum. Genesis. 2005;42:37–46. doi: 10.1002/gene.20118. [DOI] [PubMed] [Google Scholar]
- 34.Schilling G., Becher M.W., Sharp A.H., Jinnah H.A., Duan K., Kotzuk J.A., Slunt H.H., Ratovitski T., Cooper J.K., Jenkins N.A., et al. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum. Mol. Genet. 1999;8:397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 35.Wang J., Xu G., Slunt H.H., Gonzales V., Coonfield M., Fromholt D., Copeland N.G., Jenkins N.A., Borchelt D.R. Coincident thresholds of mutant protein for paralytic disease and protein aggregation caused by restrictively expressed superoxide dismutase cDNA. Neurobiol. Dis. 2005;20:943–952. doi: 10.1016/j.nbd.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 36.Gutekunst C.A., Li S.H., Yi H., Mulroy J.S., Kuemmerle S., Jones R., Rye D., Ferrante R.J., Hersch S.M., Li X.J. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J. Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roos R.A., Vegter-van der Vlis M., Hermans J., Elshove H.M., Moll A.C., van de Kamp J.J., Bruyn G.W. Age at onset in Huntington's disease: effect of line of inheritance and patient's sex. J. Med. Genet. 1991;28:515–519. doi: 10.1136/jmg.28.8.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kovtun I.V., Therneau T.M., McMurray C.T. Gender of the embryo contributes to CAG instability in transgenic mice containing a Huntington's disease gene. Hum. Mol. Genet. 2000;9:2767–2775. doi: 10.1093/hmg/9.18.2767. [DOI] [PubMed] [Google Scholar]
- 39.Dorner J.L., Miller B.R., Barton S.J., Brock T.J., Rebec G.V. Sex differences in behavior and striatal ascorbate release in the 140 CAG knock-in mouse model of Huntington's disease. Behav. Brain Res. 2007;178:90–97. doi: 10.1016/j.bbr.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levine M.S., Cepeda C., Hickey M.A., Fleming S.M., Chesselet M.F. Genetic mouse models of Huntington's and parkinson's diseases: illuminating but imperfect. Trends Neurosci. 2004;27:691–697. doi: 10.1016/j.tins.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 41.Rubinsztein D.C. Lessons from animal models of Huntington's disease. Trends Genet. 2002;18:202–209. doi: 10.1016/s0168-9525(01)02625-7. [DOI] [PubMed] [Google Scholar]
- 42.Hackam A.S., Singaraja R., Wellington C.L., Metzler M., McCutcheon K., Zhang T., Kalchman M., Hayden M.R. The influence of huntingtin protein size on nuclear localization and cellular toxicity. J. Cell Biol. 1998;141:1097–1105. doi: 10.1083/jcb.141.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cooper J.K., Schilling G., Peters M.F., Herring W.J., Sharp A.H., Kaminsky Z., Masone J., Khan F.A., Delanoy M., Borchelt D.R., et al. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum. Mol. Genet. 1998;7:783–790. doi: 10.1093/hmg/7.5.783. [DOI] [PubMed] [Google Scholar]
- 44.Saudou F., Finkbeiner S., Devys D., Greenberg M.E. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 45.de Almeida L.P., Ross C.A., Zala D., Aebischer P., Deglon N. Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces a selective neuropathology modulated by polyglutamine repeat size, huntingtin expression levels, and protein length. J. Neurosci. 2002;22:3473–3483. doi: 10.1523/JNEUROSCI.22-09-03473.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Palfi S., Brouillet E., Jarraya B., Bloch J., Jan C., Shin M., Conde F., Li X.J., Aebischer P., Hantraye P., et al. Expression of mutated huntingtin fragment in the putamen is sufficient to produce abnormal movement in non-human primates. Mol. Ther. 2007;15:1444–1451. doi: 10.1038/sj.mt.6300185. [DOI] [PubMed] [Google Scholar]
- 47.Yamamoto A., Lucas J.J., Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 48.Lalonde R. Motor abnormalities in staggerer mutant mice. Exp. Brain Res. 1987;68:417–420. doi: 10.1007/BF00248807. [DOI] [PubMed] [Google Scholar]
- 49.Lalonde R. Motor abnormalities in weaver mutant mice. Exp. Brain Res. 1987;65:479–481. doi: 10.1007/BF00236322. [DOI] [PubMed] [Google Scholar]
- 50.van Raamsdonk J.M., Metzler M., Slow E., Pearson J., Schwab C., Carroll J., Graham R.K., Leavitt B.R., Hayden M.R. Phenotypic abnormalities in the YAC128 mouse model of Huntington disease are penetrant on multiple genetic backgrounds and modulated by strain. Neurobiol. Dis. 2007;26:189–200. doi: 10.1016/j.nbd.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 51.Petersen A., Bjorkqvist M. Hypothalamic-endocrine aspects in Huntington's disease. Eur. J. Neurosci. 2006;24:961–967. doi: 10.1111/j.1460-9568.2006.04985.x. [DOI] [PubMed] [Google Scholar]
- 52.Weydt P., Pineda V.V., Torrence A.E., Libby R.T., Satterfield T.F., Lazarowski E.R., Gilbert M.L., Morton G.J., Bammler T.K., Strand A.D., et al. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell. Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 53.Kelley A.E., Baldo B.A., Pratt W.E., Will M.J. Corticostriatal-hypothalamic circuitry and food motivation: integration of energy, action and reward. Physiol. Behav. 2005;86:773–795. doi: 10.1016/j.physbeh.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 54.Ivkovic S., Polonskaia O., Farinas I., Ehrlich M.E. Brain-derived neurotrophic factor regulates maturation of the DARPP-32 phenotype in striatal medium spiny neurons: studies in vivo and in vitro. Neuroscience. 1997;79:509–516. doi: 10.1016/s0306-4522(96)00684-7. [DOI] [PubMed] [Google Scholar]
- 55.Ivkovic S., Ehrlich M.E. Expression of the striatal DARPP-32/ARPP-21 phenotype in GABAergic neurons requires neurotrophins in vivo and in vitro. J. Neurosci. 1999;19:5409–5419. doi: 10.1523/JNEUROSCI.19-13-05409.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jones K.R., Farinas I., Backus C., Reichardt L.F. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ferrer I., Goutan E., Marin C., Rey M.J., Ribalta T. Brain-derived neurotrophic factor in Huntington disease. Brain Res. 2000;866:257–261. doi: 10.1016/s0006-8993(00)02237-x. [DOI] [PubMed] [Google Scholar]
- 58.Zuccato C., Marullo M., Conforti P., Macdonald M.E., Tartari M., Cattaneo E. Systematic assessment of BDNF and its receptor levels in human cortices affected by Huntington's disease. Brain Pathol. 2007;18:225–238. doi: 10.1111/j.1750-3639.2007.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zuccato C., Liber D., Ramos C., Tarditi A., Rigamonti D., Tartari M., Valenza M., Cattaneo E. Progressive loss of BDNF in a mouse model of Huntington's disease and rescue by BDNF delivery. Pharmacol. Res. 2005;52:133–139. doi: 10.1016/j.phrs.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 60.Duan W., Guo Z., Jiang H., Ware M., Li X.J., Mattson M.P. Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc. Natl Acad. Sci. USA. 2003;100:2911–2916. doi: 10.1073/pnas.0536856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gines S., Bosch M., Marco S., Gavalda N., Diaz-Hernandez M., Lucas J.J., Canals J.M., Alberch J. Reduced expression of the TrkB receptor in Huntington's disease mouse models and in human brain. Eur. J. Neurosci. 2006;23:649–658. doi: 10.1111/j.1460-9568.2006.04590.x. [DOI] [PubMed] [Google Scholar]
- 62.Reiner A., Albin R.L., Anderson K.D., D'Amato C.J., Penney J.B., Young A.B. Differential loss of striatal projection neurons in Huntington disease. Proc. Natl Acad. Sci. USA. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Harper S.Q., Staber P.D., He X., Eliason S.L., Martins I.H., Mao Q., Yang L., Kotin R.M., Paulson H.L., Davidson B.L. RNA interference improves motor and neuropathological abnormalities in a Huntington's disease mouse model. Proc. Natl Acad. Sci. USA. 2005;102:5820–5825. doi: 10.1073/pnas.0501507102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miller T.W., Zhou C., Gines S., MacDonald M.E., Mazarakis N.D., Bates G.P., Huston J.S., Messer A. A human single-chain fv intrabody preferentially targets amino-terminal huntingtin's fragments in striatal models of Huntington's disease. Neurobiol. Dis. 2005;19:47–56. doi: 10.1016/j.nbd.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 65.Niculescu M., Ehrlich M.E., Unterwald E.M. Age-specific behavioral responses to psychostimulants in mice. Pharmacol. Biochem. Behav. 2005;82:280–288. doi: 10.1016/j.pbb.2005.08.014. [DOI] [PubMed] [Google Scholar]