

Abstract
Polycystin-1 (PC1), the product of the PKD1 gene mutated in the majority of autosomal dominant polycystic kidney disease (ADPKD) cases, undergoes a cleavage resulting in the intracellular release of its C-terminal tail (CTT). Here, we demonstrate that the PC1 CTT co-localizes with and binds to β-catenin in the nucleus. This interaction requires a nuclear localization motif present in the PC1 CTT as well as the N-terminal portion of β-catenin. The PC1 CTT inhibits the ability of both β-catenin and Wnt ligands to activate T-cell factor (TCF) -dependent gene transcription, a major effector of the canonical Wnt signaling pathway. The PC1 CTT may produce this effect by reducing the apparent affinity of the interaction between β-catenin and the TCF protein. DNA microarray analysis reveals that the canonical Wnt signaling pathway is activated in ADPKD patient cysts. Our results suggest a novel mechanism through which PC1 cleavage may impact upon Wnt-dependent signaling and thereby modulate both developmental processes and cystogenesis.
INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is a common hereditary disorder associated with the progressive replacement of normal renal tubular architecture with a profusion of fluid-filled cysts that ultimately leads to massive kidney enlargement and compromised kidney function (1,2). Important insights into the pathogenesis of ADPKD were obtained through the positional cloning of two genes, PKD1 and PKD2, (that encode the polycystin proteins 1 and 2, respectively), which together are responsible for more than 95% of all clinical cases of the disease. Detailed reconstruction and dissection studies have shown that renal cysts develop initially as saccular outpouchings arising from any nephron segment. Most of these dilated tubules eventually become cysts as a result of cellular proliferation and fluid secretion, and detach from their tubule of origin when enlarged beyond a few millimeters in diameter (3). Recent studies have provided important insights into the molecular basis of cystogenesis in ADPKD (1,4,5). Monoclonal expansion of individual epithelial cells that have undergone a second hit mutagenesis event, resulting in biallelic inactivation of either PKD1 or PKD2, appears to be a major mechanism for the initiation of focal cyst formation (6). The polycystins may thus exert a growth suppressive influence, and the loss of heterozygosity at a PKD locus abrogates this activity, resulting in proliferation of affected cells that ultimately lead to cyst formation. Despite intense investigation, the physiological functions of the polycystins, as well as the mechanisms through which their absence or dysfunction induces cystic disease, remain to be precisely defined.
Polycystin-1 (PC1) is a very large integral membrane protein with a molecular mass exceeding 460 kDa. Structurally, it is predicted to traverse the membrane 11 times and includes a massive extracellular amino terminal domain and a short intracellular carboxy terminal tail. The N-terminal portion of the PC1 molecule may participate in establishing cell–cell and cell–matrix interactions. By virtue of its apparent capacity to regulate numerous signal transduction cascades, including the Wnt (7), activator protein-1 (8–10) and nuclear factor of activated T-cells pathways (11), the C-terminus may act as a conduit that transmits extracellular information across the plasma membrane. Thus, the pathogenesis of ADPKD may involve perturbations of numerous developmentally important signaling pathways.
Wnts are a family of secreted glycoproteins involved in embryonic induction, cell polarity determination and cell proliferation and differentiation (12). The canonical Wnt signaling pathway functions to regulate the cellular level of β-catenin, which is a multifunctional polypeptide that plays essential roles in two cellular processes: calcium-dependent cellular adhesion and T-cell factor/lymphoid-enhancing factor (TCF/LEF)-dependent transcriptional activation (12,13). Compelling evidence directly supporting the involvement of the Wnt/β-catenin signaling cascade in ADPKD pathogenesis has been provided by in vivo studies performed using transgenic mice harboring mutations that result in the accumulation of β-catenin (14–16). Targeted expression of a stabilized form of β-catenin to renal epithelial cells gives rise to animals with a cystic phenotype remarkably similar to that seen in ADPKD (15). This link between ADPKD and β-catenin is substantiated at the molecular level by the presence of a multiprotein complex comprised of PC1 and β-catenin at the plasma membrane and at sites of cell–cell contact (17,18). Recent evidence also indicates that PC1 may play a role in regulating the stability of a pool of the β-catenin protein (19).
In the current study, we used a systems biology approach to discover growth-modulating gene pathways for human PKD1 renal cysts. Using the gene set enrichment analysis (GSEA), a novel bioinformatics algorithm for discovering differentially expressed gene pathways between two biological states, we found strong evidence that the canonical Wnt/β-catenin signaling pathway is activated in human PKD1 cysts of different sizes. Studies performed at both the whole animal and cellular level have recently demonstrated a novel mode of PC1 regulation whereby the full-length protein undergoes a regulated proteolytic event that results in the cleavage and release of its C-terminal tail (CTT) (20,21). The physiological regulation of this proteolytic event appears to be achieved, at least in part, through mechanical stimuli that may be sensed at the level of the primary cilium located on the luminal surface of renal epithelial cells. Here we show that a soluble CTT fragment of PC1 similar to that which results from PC1 cleavage forms a complex with β-catenin and profoundly inhibits canonical Wnt signaling. Taken together, these data suggest that loss of the PC1 CTT removes an important restraint on the Wnt signaling pathway, which in turn could underlie the development of renal cysts.
RESULTS
Hierarchical cluster analysis
We probed DNA microarrays to profile the expression of ∼47 400 transcripts and variants in 13 renal cysts of different size, 5 minimally cystic tissue (MCT) samples and 3 normal renal cortical tissue samples. Cysts were derived from ADPKD patients carrying germline mutations in PKD1. Using the top 200, 500, 1000 or 2000 most variable genes across all samples in unsupervised hierarchical cluster analysis, we found all cyst samples consistently clustered as a single group, while the MCT and normal renal cortical tissue samples clustered as a second group (Supplementary Material, Fig. S1). These results suggest that the gene expression pattern is very similar between renal cysts of different size, and between MCT and normal renal cortical tissue.
Gene set enrichment analysis
To identify potential gene pathways that modulate renal cyst growth, we performed GSEA and tested 440 gene sets in 13 cysts against 5 MCT samples. Due to its small sample size (n = 3), we did not include the normal renal cortical tissue samples in this analysis. Using a NOM P-value ≤ 0.01, we identified that 25 gene sets were up-regulated and 24 gene sets were down-regulated (Supplementary Material, Table S1). From the up-regulated gene sets, GSEA suggests that renal cysts displayed a rich signature of gene pathways that regulate proliferation (up-regulation of ribosomal proteins, cancer-related transcription factors, genes up-regulated in hepatoma tissue of Myc transgenic mice, MYC direct target genes, proliferation-related genes, PPARA pathway, tumor suppressor genes and tenascin-C target genes), cell cycle (PML nuclear bodies regulated transcription, Ras, Rac and Rho on G1/S transition signaling, cell cycle arrest, p53 up-regulated genes and cancer-related genes involved in the cell cycle), Ca2+ signaling (Ca2+-mediated synaptic vesicle endocytosis, calcineurin pathway, control of skeletal myogenesis by HDAC & calcium/CaMK), hypoxic responses (HIF pathway) and tissue fibrosis (EMT target gene). In contrast, most of the down-regulated genes sets were involved in amino acid, urea cycle, fatty acid and ATP metabolism.
Of interest, four up-regulated pathways contained either components of the canonical Wnt/β-catenin signaling cascade or their nuclear target genes, suggesting that this pathway was activated in the cyst samples. Specifically, both the canonical Wnt/β-catenin signaling pathway (NES = 1.57, NOM P-value = 0.006) and its direct target genes (NES = 1.86, NOM P-value < 0.001) were enriched in the cysts compared to MCT. Additionally, the pathways for Myc transgenic mice (NES = 1.86, NOM P-value < 0.001) and MYC target genes (NES = 1.69, NOM P-value = 0.004) were similarly enriched in the cyst samples. Leading-edge analysis from the GSEA further showed that 28/72 component genes and 21/41 direct target genes of the canonical Wnt/β -catenin signaling pathway were enriched in the renal cysts (Supplementary Material, Table S2). The hierarchical clustering of these enriched component genes and direct target genes is shown in Figure 1A and B. All of them were also shown to be differentially expressed in the renal cysts by significance analysis of microarrays (SAM), using a false discovery rate ≤0.5%.
Figure 1.
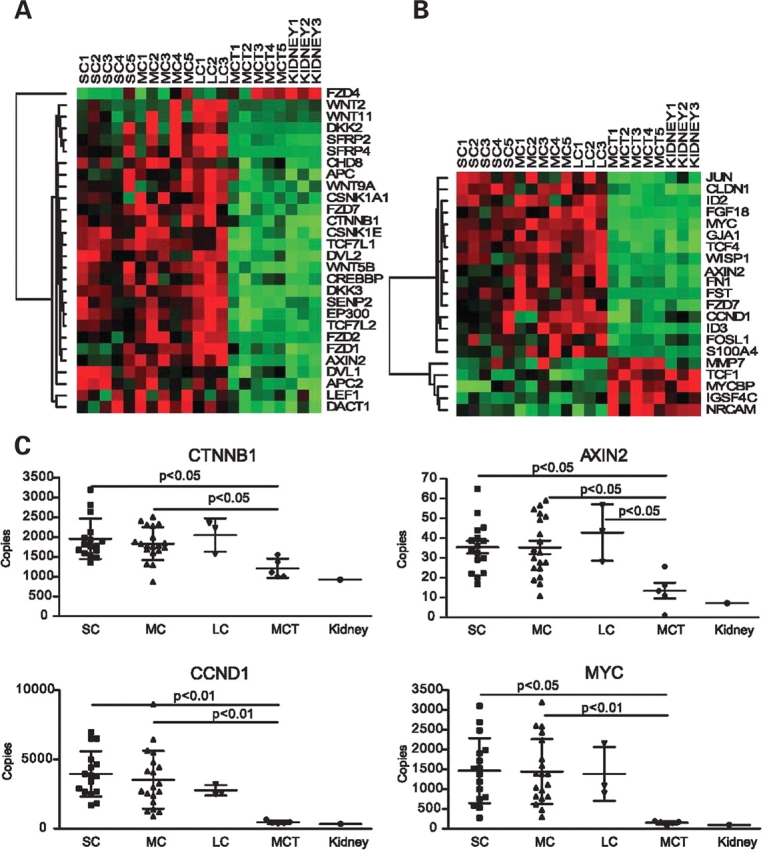
Gene expression profiles based on 28 component (A) and 21 target genes (B) suggest activation of the Wnt/β-catenin signaling pathway in PKD1 renal cysts. All the genes listed in the two panels (A and B) were differentially expressed between the cysts and MCT samples with a false discovery rate of ≤0.05%. (C) Real-time RT–PCR analysis of β-catenin (CTNNB1), AXIN2, cyclin D1 (CCND1) and MYC in an expanded sample set (SC = 16; MC = 18; LC = 3; MCT = 5). Corresponding expression values of these genes from normal renal cortical tissue (pooled from three normal subjects) were also shown to illustrate the similarity to MCT.
Validation of microarray results by qPCR
Using real-time RT–PCR in an expanded sample set of cysts and MCT derived from the same PKD1 kidneys, we independently confirmed the microarray results on seven Wnt/β-catenin component and target genes. We found that the expression of β-catenin (CTNNB1) and its nuclear target genes (AXIN 2, CCND1 and MYC) were increased while GSK-3β expression level did not differ between the cyst and MCT samples (see Fig. 1C for selected examples). Additionally, the expression level of secreted frizzle-related protein 4 (SFRP4), an antagonist of the canonical Wnt/β-catenin pathway (22), was also increased in the cyst samples.
The PC1 C-terminal tail causes nuclear accumulation of β-catenin
Heterologous expression studies employing various anchoring strategies to induce membrane-targeted overexpression of the CTT of PC1 have yielded conflicting results with respect to the effect of this manipulation on the Wnt signaling cascade (7,9,21). Since the CTT of PC1 has recently been described to undergo inducible release from the full-length protein (20,21), in the current study we investigated the relationship between the PC1 CTT and β-catenin. First, we examined whether the PC1 CTT could bring about a change in the subcellular localization of β-catenin. CHO cells co-transfected with constructs encoding the C-terminal 200 amino acids of PC1 (p200) and β-catenin flag-tagged at the C-terminus were processed for immunofluorescence analysis (Fig. 2). As a control, we made use of p200Δ, a cDNA construct encoding a mutant form of the PC1 CTT that lacks a 21 amino acid putative nuclear localization motif (20). Expression of the PC1 CTT constructs was detected using an antibody directed against the Xpress epitope tag, which is appended to these proteins’ N-termini. We chose to use CHO cells for these experiments since they permit us to examine changes in β-catenin expression independent of the possible effects of E-cadherin, which is not expressed endogenously (as is also the case for β-catenin) at appreciable levels in this cell type. In CHO cells transiently transfected with β-catenin, we observed β-catenin localization throughout the entire cell body (Fig. 2A). When β-catenin was co-transfected with p200, however, nuclear localization of p200 (Fig. 2D) was associated with a readily visible redistribution of β-catenin (Fig. 2C) to the same compartment (Fig. 2E). In contrast, expression of p200Δ, which was confined largely to the cytosol (Fig. 2G), was without any appreciable effect on the pattern of β-catenin distribution (Fig. 2F). Upon subcellular fractionation and subsequent immunoblotting for β-catenin expression, we found a slight decrease of β-catenin in the non-nuclear fraction and a readily visible increase in the amount of β-catenin recognized in the nuclear fraction in material prepared from cells that express p200 (Fig. 2I). p200 was also found predominantly in the nuclear fraction, whereas p200Δ was found in the non-nuclear fraction. This nuclear fraction represents a substantial enrichment of the nuclear compartment, since the nuclear marker histone deacetylase-1 was detected exclusively in this fraction. The lack of effect of p200Δ on β-catenin enrichment in the nucleus was also confirmed by these cell fractionation experiments. Cell fractionation experiments performed using HEK 293T cells show that p200 also localizes to the nucleus in this cell type (Supplementary Material, Fig. S2).
Figure 2.
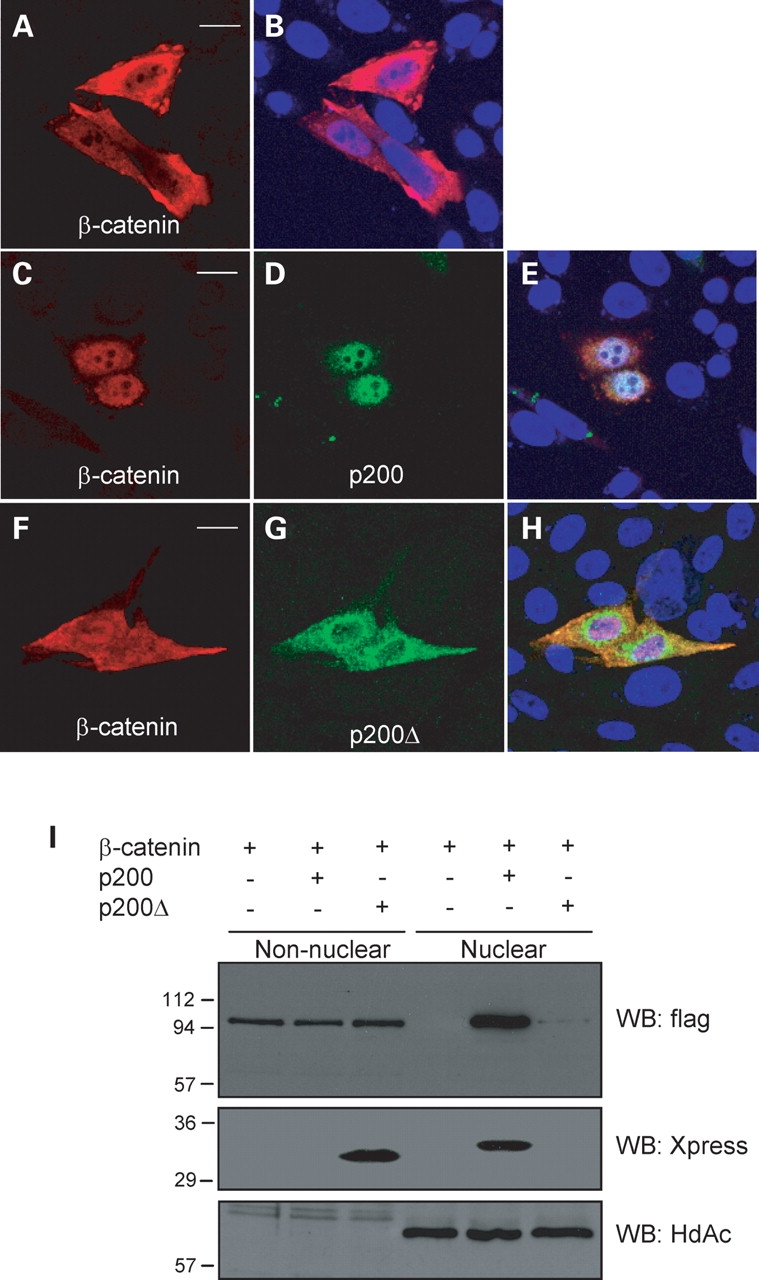
Expression of the PC1 CTT, p200, results in the nuclear accumulation of β-catenin. (A–H) CHO cells were co-transfected with β-catenin-flag and either control vector β-catenin (pcDNA3.1), p200 or p200Δ. p200 expression was largely restricted to the nucleus (D) and elicited a redistribution of β-catenin from the cytosol to the nucleus (C) when compared with control cells expressing β-catenin alone (A). p200Δ was confined to the cytoplasm (G) and did not significantly alter the pattern of β-catenin distribution (F). In merged images, nuclei were stained with DAPI (blue) (B, E and H). Bars 10 µm. (I) Representative western blot showing the subcellular fractionation of transfected CHO cells into nuclear and non-nuclear fractions. β-catenin was enriched in the nuclear compartment when co-expressed with p200. The purity and efficacy of the nuclear preparations was demonstrated using the nuclear protein histone deacetylase-1 (HdAc).
PC1 CTT associates with β-catenin
We next wished to determine whether the ability of p200 to elicit the nuclear accumulation of β-catenin might be conferred by a physical interaction between these two proteins. Using cells expressing β-catenin and p200 we found that β-catenin co-precipitates with p200 that has been affinity purified from cell lysates using the Xpress antibody (Fig. 3A). Co-immunoprecipitation experiments performed using lysates prepared from CHO cells transiently transfected with β-catenin alone confirmed that Xpress antibody does not pull down β-catenin in a non-specific immune complex. Furthermore, whereas p200Δ was effectively immunoprecipitated with Xpress antibody from cells co-transfected with β-catenin and p200Δ, the association of β-catenin with this immune complex was negligible compared to that observed in cells expressing β-catenin and p200. The total cell lysate input used for co-immunoprecipitation was 20-fold greater than the quantity of lysate loaded on the control lanes of the gels. Thus, we estimate that the efficacy of co-immunoprecipitation was ∼5–20% of total input. When co-expressed in a variety of other cell types (COS-7 and HEK 293T), p200 but not p200Δ was similarly able to co-immunoprecipitate β-catenin. When immunoprecipitations were performed using a mixture (50:50, by vol.) of lysates prepared from cells individually expressing β-catenin and p200, β-catenin did not co-immunoprecipitate with Xpress antibody (Fig. 3B). This result suggests that the interaction between β-catenin and p200 occurs in vivo and that formation of the complex may require an active cellular process. Notably, we also observed that p200 singly expressed in HEK 293T cells was able to immunoprecipitate β-catenin that is endogenously expressed in this cell type (Supplementary Material, Fig. S2).
Figure 3.

The PC1 CTT, p200, associates with β-catenin and inhibits its activation of the canonical Wnt signaling pathway. (A) Representative western blot of Xpress antibody immunoprecipitates from CHO cells transfected with the indicated constructs (Xpress recognizes p200 and p200Δ). The p200 protein, but not p200Δ was able to co-immunoprecipitate β-catenin. Arrowheads indicate IgG bands. (B) Reconstitution of cell lysates [1+2. β-catenin+p200 (mix)] obtained from CHO cells singly expressing either β-catenin (1) or p200 (2) followed by immunoprecipitation indicates that the p200/β-catenin complex does not form under these conditions. Cells co-transfected (co-tf) with β-catenin and p200 [3. β-catenin and p200 (co-tf)] served as control. Arrowhead: IgG. (C) Co-immunoprecipitation of β-catenin by p200, performed in the presence of ethidium bromide (EtBr), does not disrupt the ability of β-catenin to associate with p200. Arrowhead: IgG. (D) TCF-dependent gene activation is attenuated by p200 but not by p200Δ or membrane anchored PLAP-p200. TCF-dependent luciferase reporter activity was measured using HEK 293T cells transfected with TOPflash and β-galactosidase constructs and further complemented with DNA constructs as indicated. Data are presented as means ± SE of five independent experiments. P200 inhibits both baseline TCF activity (open bars) and TCF activity stimulated by β-catenin expression (solid bars). *P < 0.05 versus respective control condition. (E) Western blot of cell lysates used in a single, typical TOPflash reporter assay. β-catenin was detected with anti-flag, p200 and p200Δ with anti-Xpress and PLAP-p200 with anti-PLAP. (F) Cells null for PC1 (PC1−/−) present elevated levels of basal TCF-dependent reporter activity (TOP) when compared with heterozygous PC1+/− parental cells. This response was diminished at higher cell confluence. Data are presented as means ± SE of four independent experiments. *P < 0.05 versus respective confluence level in PC1+/− cells.
To assess whether the observed association of β-catenin and p200 could be a result of an indirect interaction mediated via binding to DNA, co-immunoprecipitation with Xpress antibody was performed in the presence of ethidium bromide, a DNA-intercalating compound that can dissociate proteins from DNA (23). As shown in Fig. 3C, β-catenin was readily detected in p200 immunoprecipitates performed when ethidium bromide was added 30 min prior to the antibody. This result indicates that the association of p200 and β-catenin is likely a result of a DNA-independent protein–protein interaction. Pre-treating cell extracts with DNaseI at 1 µg/ml prior to the immunoprecipitation, a strategy that induces DNA unwinding, was also without effect (data not shown).
PC1 inhibits TCF-dependent gene transcription
To determine whether there was a functional consequence of the observed interaction between β-catenin and p200, we performed TOPflash reporter assays to measure TCF-dependent gene transcription. This method has been used extensively to provide a specific and sensitive assessment of β-catenin-dependent Wnt signaling (7,9,12). Assays were carried out using HEK 293T cells transiently transfected with the TCF reporter gene. We found that, despite its propensity to induce the nuclear accumulation of β-catenin, p200 blunted the TCF-dependent transactivation detected under basal conditions as well as that induced by overexpression of β-catenin (Fig. 3D). Notably, the p200-mediated decrease of basal activity was typically 20–30%, whereas the p200-mediated inhibition of β-catenin-dependent TCF transactivation was 60–70%. In contrast, p200Δ did not inhibit TCF-dependent transcription. To further confirm that the capacity of p200 to accumulate in the nucleus plays a role in the inhibition of Wnt signaling, we took advantage of a p200 construct fused to a transmembrane anchor, composed of the placental alkaline phosphatase ectodomain and the vesicular stomatitis virus G protein transmembrane domain (PLAP-p200) (20). When co-expressed together with β-catenin, membrane anchored p200 was not able to attenuate TCF-dependent gene transcription. As depicted in Figure 3E, the observed effects of p200, p200Δ and PLAP-p200 were not due to disparate levels of expression of any of the constructs when singly- or co-expressed. As a control for the specificity of TCF-dependent transactivation and the effects of p200, we also made use of the FOPflash luciferase reporter plasmid containing mutated TCF-4 consensus binding sites. The FOPflash reporter did not respond to any of the experimental challenges. β-catenin did not activate the reporter and p200 itself did not cause any inhibition (data not shown). These data suggest that the inhibition of β-catenin-mediated TCF-dependent gene transcription by the last 200 amino acids of PC1 is pathway specific and that it requires that the CTT be soluble and have the capacity to enter the nucleus.
Taken together, the data presented here indicate that Wnt signaling is elevated in cystic tissue isolated from patients harboring pathogenic mutations in the gene encoding PC1 (Fig. 1) and that the expression of the PC1 CTT suppresses β-catenin-stimulated TCF-driven transcription (Fig. 3). If, as these data imply, the presence of the PC1 protein or its CTT serves to inhibit signaling in the Wnt pathway, then we would expect basal levels of TCF-driven transcription to be elevated in cells that express no PC1 when compared with comparable cells that express native levels of the PC1 protein. To test this possibility, we took advantage of a renal proximal tubule epithelial cell line derived from PKD1 (flox/−): TSLargeT mice (24). A true PKD1 null cell line was generated by transfecting the parental cell line, which is heterozygous for PC1 expression, with a cDNA encoding Cre recombinase (both cell lines were kindly provided by Dr S. Somlo, Yale University). TOPflash and FOPflash assays were performed on the parental heterozygous line and the null cells at various levels of confluence. As can be seen in Figure 3F, at low confluency, the PC1 null cells displayed a 2.5-fold elevation of the level of basal TCF-driven expression of the luciferase reporter construct when compared with the parental PC1 heterozygous cell line. This relative increase in unstimulated Wnt signaling detected in the null cells diminished at higher levels of confluence. Similar experiments performed using a cell line derived from thick ascending limb yielded similar results (data not shown). These results demonstrate that cell lines deficient in PC1 expression exhibit a higher basal level of Wnt signaling than do cells heterozygous for PC1, and this signal is contact-inhibited at high confluence. Thus, expression of native levels of PC1 acts to suppress Wnt signaling, and loss of PC1 expression results in aberrantly high levels of Wnt signaling, consistent with the microarray results presented in Figure 1.
The N-terminus of β-catenin is required for p200 binding
The primary structure of β-catenin includes a central core region composed of 12 imperfect armadillo repeats flanked on either side by N- and C-terminal segments (25). In most cases, it is the central armadillo repeat domain that facilitates the direct binding of β-catenin with interacting proteins. However, it has also been appreciated that the N- and C-termini of β-catenin can modulate the binding of the armadillo region to its partner proteins. To test the involvement of the N- and C-terminal domains in the binding of β-catenin to the CTT of PC1, we performed co-immunoprecipitation experiments using HEK 293T cells co-expressing N- or C-terminal deletion mutants of β-catenin along with p200 (Fig. 4A). Wild-type β-catenin, ΔN89β-catenin (missing the first 89 residues) and β-cateninΔC695 (missing the last 87 residues) were expressed in HEK 293T cells. Whereas immunoprecipitation of p200 brought down β-cateninΔC695, deletion of a portion of the N-terminus of β-catenin resulted in a marked reduction in its ability to associate with p200 (Fig. 4A). When examined by immunofluorescent imaging, we noted that p200 expression resulted in the redistribution and nuclear accumulation of β-cateninΔC695 (compare Fig. 5F with H). In contrast, and in concert with the immunoprecipitation results, expression of p200 did not appear to alter appreciably the subcellular localization of ΔN89β-catenin (compare Fig. 5C with A). When co-expressed with p200, ΔN89β-catenin was found localized throughout the cell body and nucleus and did not show intense nuclear staining. GST pull-down assays performed using the N-terminal 106 amino acids of β-catenin indicated that this N-terminal portion of the protein was not sufficient to precipitate p200 derived from cells expressing the protein (data not shown). These results suggest that while the N-terminus of β-catenin is required for association with the PC1 CTT, it is not in itself sufficient.
Figure 4.
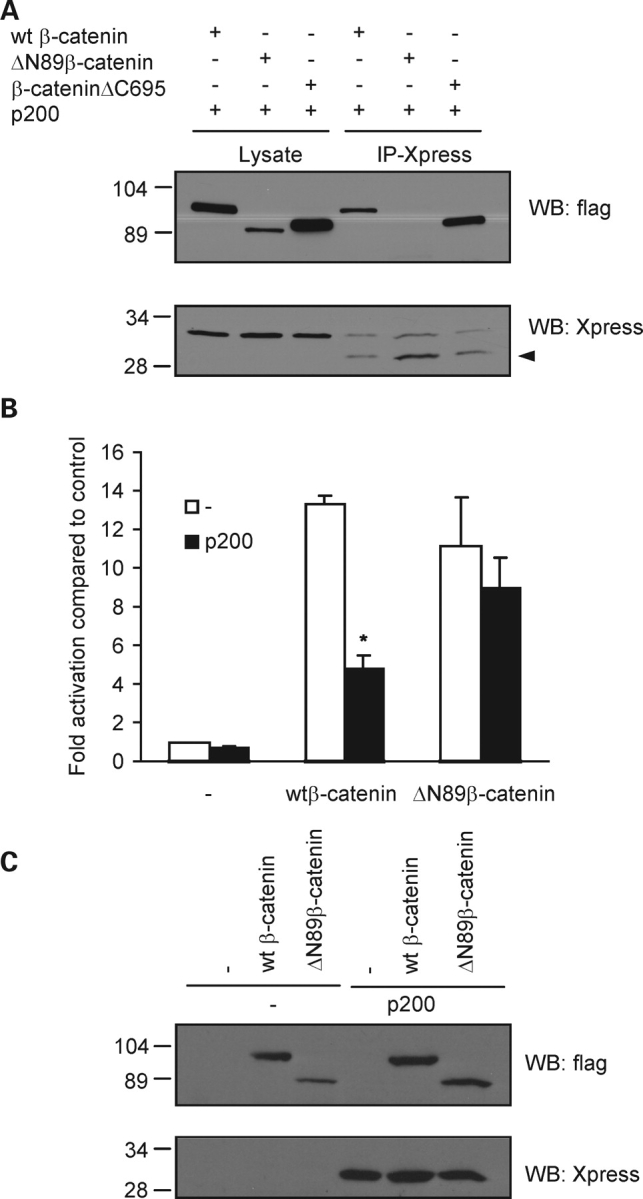
The N-terminus of β-catenin is required for p200 association as well as for the functional effects of p200 expression on Wnt signaling. (A) Deletion of the C-terminus of β-catenin (β-cateninΔC695) did not significantly disrupt the ability of p200 to bind β-catenin, whereas deletion of the N-terminus of β-catenin (ΔN89β-catenin) greatly reduced their interaction when compared with wild-type (wt) β-catenin. Arrowhead: IgG. (B) Removal of the N-terminal 89 amino acids of β-catenin prevents the inhibitory effects of p200 on TCF-dependent transcription. The TOPflash luciferase reporter assay was performed using HEK 293T cells transiently transfected with control DNA, wt β-catenin and ΔN89β-catenin in the presence (solid bars) or absence (open bars) of p200. Data are presented as means ± SE of four independent experiments. *P < 0.05 versus wtβ-catenin alone. (C) Western blot depicting the pattern of protein expression of each of the transfected gene products obtained from aliquots used in a representative TOPflash reporter assay. β-catenin was detected with anti-flag and p200 with anti-Xpress.
Figure 5.

p200 does not alter the subcellular distribution of β-catenin lacking its N-terminal domain. Representative immunofluorescent images of CHO cells expressing ΔN89β-catenin or β-cateninΔC695 either singly or in combination with p200 are shown. Expression of ΔN89β-catenin (C) in the presence of p200 (D) did not alter the distribution of ΔN89β-catenin from that observed when it was singly expressed (A). Coexpression of β-cateninΔC695 (H) together with p200 (I) elicited a redistribution of β-cateninΔC695 from the cytosol (F) to the nucleus. In merged images, nuclei were counterstained with DAPI (blue) (B, E, G and J). Bars 10 µm.
Since our earlier results suggested that interaction of p200 with β-catenin was responsible for its inhibitory effects on TCF-dependent transcription, we tested whether the inability of ΔN89β-catenin to bind to p200 altered its behavior in these functional assays (Fig. 4B). While both wtβ-catenin and ΔN89β-catenin robustly stimulated TCF-dependent gene transactivation, p200 was without marked inhibitory effect on activation mediated by the N-terminal truncation mutant of β-catenin. These results confirm that p200 association with β-catenin is necessary for its inhibition of TCF-dependent transcription. As shown in Fig. 4C, disparity in the expression levels of the transfected DNA constructs did not account for the observed functional effects.
PC1 CTT reduces β-catenin affinity for TCF
Recent studies suggest that β-catenin binding to its molecular partners may be regulated by its propensity to adopt specific conformational states formed under distinct biological conditions (13,26). Since p200 inhibits β-catenin-dependent signaling while it enhances β-catenin’s accumulation in the nucleus, we posited that p200 may alter the binding characteristics of β-catenin for TCF. To test this hypothesis, we performed an in vitro assay in which we assessed the binding of β-catenin, present in extracts prepared from HEK 293T cells, to a GST fusion protein that incorporates the β-catenin binding site of TCF (TCF–GST). In Figure 6, we show that p200 expression reduces the relative affinity of β-catenin for TCF–GST by a factor of about 10 when lysates prepared from cells transfected with β-catenin and p200 are compared to those prepared from cells expressing β-catenin alone. Co-expression of β-catenin with p200Δ did not recapitulate the effects of p200. Interestingly, we also noted that the relative β-catenin affinity for CAD–GST, a GST fusion protein comprising the β-catenin binding site present in E-cadherin, was also decreased in lysates prepared from β-catenin/p200 expressing cells. The magnitude of this reduction was smaller than that detected using TCF–GST (Fig. 6B). Pulldown experiments performed using GST alone produced no β-catenin binding, demonstrating the specificity of the observed interactions (data not shown).
Figure 6.

p200 expression reduces the apparent affinity of β-catenin for TCF–GST and CAD–GST. (A) Western blot of a pull-down assay depicting the relative binding affinity of β-catenin for TCF–GST and CAD–GST fusion proteins. Lysates used in the pull-down assay were obtained from cells expressing β-catenin alone or β-catenin and p200/ p200Δ. Lysate loading represents 20 µg of protein. Input for GST-binding assays was 100 µg. (B) Semi-quantitative densitometric analysis of β-catenin band density from four independent experiments. Values represent arbitrary units (a.u.) calculated as the ratio of β-catenin binding to TCF–GST or CAD–GST under each of the transfection schemes relative to its level in the corresponding input and are presented as means ± SE. *P < 0.05 versus other respective treatment conditions.
PC1 CTT attenuates TCF-dependent gene transcription mediated by endogenous β-catenin
We have shown that an absence of PC1 correlates with elevated TCF-dependent gene transactivation (Fig. 3F) and that the CTT of PC1 is sufficient to blunt this response in cells expressing exogenous β-catenin (Fig. 3D). To establish whether the PC1 CTT was indeed able to act as an inhibitor of physiologically relevant TCF-dependent gene transcription, we applied two strategies to activate the canonical Wnt signaling pathway in HEK 293T. Lithium is an inhibitor of GSK-3β that results in the stabilization of cytosolic β-catenin. Consistent with our previous results showing that p200, and not p200Δ, was able to inhibit TCF-dependent gene transcription mediated by heterologously expressed β-catenin, we found that the PC1 CTT was also able to abrogate the stimulatory effects of lithium on the TOPflash assay (Fig. 7A). These results were specific for the TCF signaling pathway since the FOPflash reporter showed no response under these conditions (not shown). Lithium, as expected, increased the level of endogenous β-catenin (Fig. 7B), yet had no effect on the expression of the loading control, β-actin. Notably, p200 did not appear to exert its inhibitory effect on TCF-mediated transcription by down regulating endogenous β-catenin levels. We also transfected HEK 293T with Wnt-3a, one of the naturally occurring Wnt ligands that signal through the canonical Wnt pathway. TCF-dependent transcription of the luciferase reporter was increased greater than 15-fold by Wnt-3a expression (Fig. 7C). When Wnt-3a was co-expressed with p200, TCF-dependent gene transcription was attenuated. As shown in Figure 7D, Wnt-3a caused a moderate increase in β-catenin expression that was not markedly affected by co-expression of p200. These results indicate that the free CTT of PC1 can act as a negative regulator of endogenous β-catenin-dependent Wnt signaling.
Figure 7.

p200, but not p200Δ, blunts canonical Wnt signaling mediated by endogenous β-catenin. (A) Effect of LiAc on TCF-dependent transcription. HEK 293T cells were transfected with DNA constructs as indicated, and then treated with 20 µM LiAc for 3 h prior to TOPflash luciferase measurement. Results are presented as means ± SE of four experiments. *P < 0.05 versus respective control conditions.(B) Western blot of HEK293T cell lysates from aliquots of a representative TOPflash analysis described in (A), illustrating the increase of β-catenin expression induced by LiAc and the effect of p200 and p200Δ. (C) Activation of canonical Wnt signaling by expression of Wnt-3a ligand is partially blocked by p200 but not by p200Δ. HEK 293T cells were transfected with DNA constructs as indicated. TOPflash data are given as means ± SE of six experiments. *P < 0.05 versus respective control condition. (D)Western blot analysis of cell lysates from a single experiment as performed in (C). β-actin was used as a loading control.
DISCUSSION
Although numerous studies suggest that aberrant β-catenin-dependent signaling is an important contributor to the cyst formation that occurs in PKD, the molecular mechanisms accounting for this connection remain obscure (14–16,27,28). In the current study, we demonstrate through gene expression profiling that elements of the Wnt signaling pathway are consistently and substantially elevated in renal cystic tissue derived from patients with ADPKD attributable to germline mutations in the PKD1 gene. This activation of the Wnt pathway is not observed in minimally cystic or non-cystic tissue derived from the same patients, demonstrating that stimulation of this pathway occurs specifically in cells fated to undergo or involved in cyst formation. Evidence suggests that induction of each cyst in a patient carrying a heterozygous germline mutation in the PKD1 gene is the consequence of an independent loss of heterozygosity event, in which the acquisition of a second hit mutation in the wild-type allele of the PKD1 gene in a single renal epithelial cell eliminates polycystin-1 expression or function in that cell and its progeny (6). Thus, the activation of the Wnt pathway that we observed in cystic tissue suggests the possibility that the loss of both copies of the PKD1 gene, and of the functional expression of the polycystin-1 protein that it encodes, may remove a suppressive influence that maintains the Wnt pathway in a relatively inactive state under normal circumstances. Support for this interpretation is also found in our measurements of basal levels of Wnt signaling in cells that are heterozygous or null for PC1 expression. When these cells are subconfluent, TCF-driven transcription is substantially elevated in the PC1 null cells when compared with their heterozygous counterparts, indicating that the PC1 expression is clearly necessary and sufficient to inhibit basal Wnt signaling under these conditions. This difference decreases with confluency, suggesting that when these cells achieve sufficient density other, presumably cell–cell contact-dependent, mechanisms can stand in for the absent suppressive influence of PC1 to reduce basal Wnt signaling in the PC1 null cells. We are currently working towards assessing the expression of individual targets of the Wnt pathway in the PC1−/− cell lines. Our preliminary data (not shown) indicate that the answer will be complicated and interesting. While c-myc expression appears to be elevated in PC1−/− cells, it appears that cyclin D1 expression may actually be reduced in the PC1 null cells. In future studies, we will endeavor to understand the nature of this somewhat surprising ‘decoupling’ of Wnt targets in the context of a consistently elevated Topflash signal. We are intrigued by the possibility that PC1 may differentially regulate TCF-mediated transcription in a target-specific manner.
We find that the free CTT of PC1 physically associates with β-catenin and acts as an inhibitor of Wnt-dependent intracellular signaling in a manner consistent with its propensity to reduce the apparent affinity of β-catenin for the transcriptional co-factor, TCF. The inhibitory effect of the PC1 CTT requires a 21 amino acid sequence present in the PC1 CTT that embodies this protein’s nuclear localization signal, as well as the presence of the N-terminal portion of β-catenin. Based on our observations, we propose that in vivo, the release of the CTT of PC1 may act to attenuate β-catenin-dependent signaling, thereby providing a regulatory signal that is permissive for epithelial differentiation and maturation. Taken together, these data suggest that in human ADPKD, the loss of PC1 CTT-mediated inhibition of β-catenin activity causes epithelial cells to revert to a less differentiated phenotype that is typified by proliferation and cyst formation.
According to our model, the CTT should act as a stoichiometric inhibitor of β-catenin stimulated TCF-mediated transcription. At first glance, this model might appear to be inconsistent with the very low levels of cleaved CTT that are detected in normal kidney cells (20). It has been appreciated for some time, however, that the nuclear signaling form of β-catenin corresponds to that population of the protein that is N-terminally unphosphorylated at residues 37 and 41 (29), and is estimated to account for ∼1% of the total cytosolic pool of β-catenin in Wnt-activated cells (C.J. Gottardi, unpublished observations). This evidence that the signaling form of β-catenin constitutes a very small subset of the whole suggests that the low levels of cleaved CTT observed in wild-type cells may be sufficiently abundant to inhibit physiological levels of β-catenin-driven signaling.
Our data suggest that the cleavage and release of the CTT from full-length PC1 in vivo (20) may be necessary components of a biological switch that acts to modulate Wnt-dependent signal transduction in renal epithelia. Whether changes in fluid flow, sensed by the primary cilium, serve as the physiologically relevant trigger for PC1 cleavage and nuclear translocation remains to be conclusively determined. It has recently been suggested, however, that inversin, a protein component of the primary cilium, can act as an important signal that terminates β-catenin-dependent canonical Wnt signaling in response to fluid flow during late stage renal development and thus permits terminal differentiation of the tubular epithelia (28). Inversin also appears to stimulate non-canonical Wnt signaling, which has led to the hypothesis that inversin-mediated ciliary signaling can act as a switch between the canonical and non-canonical Wnt signaling pathways. In this context, it is interesting to note that the free CTT of PC1 is a potent activator of JNK, which is a central participant in a non-canonical Wnt signaling pathway (20). It is tempting to suggest, therefore, that the PC1 CTT shares inversin’s ability to act as a molecular switch between the canonical and non-canonical Wnt signaling pathways. Despite this intriguing similarity, however, the activities of inversin and the PC1 CTT are clearly not identical. Inversin acts upstream of β-catenin (at the level of the disheveled protein) to inhibit canonical Wnt signaling (28), whereas our data demonstrate that the effects of the PC1 CTT on this pathway involve a direct interaction between the CTT and β-catenin.
Heterologous expression of the CTT of PC1 fused to a membrane anchor has previously been described to either activate the canonical Wnt signaling pathway or to have no effect on it (7,9,21). Under the experimental conditions employed here, we observed only a marginal stimulatory effect of PLAP-p200 on β-catenin-stimulated TCF-dependent gene transcription. In contrast, our results obtained using free PC1 CTT clearly indicate that the final 200 amino acids of PC1 act to attenuate Wnt signaling. We did not observe significant expression of the CTT of PC1 in the nucleus of cells overexpressing PLAP-p200 (unpublished observations). These data indicate that additional, upstream amino acids are necessary for the cleavage of the PC1 CTT and/or that the cellular machinery involved in the cleavage process does not recognize the PLAP-p200 protein as a substrate under the experimental conditions employed. Thus, the distinct effects of membrane-tagged and free PC1 CTT on β-catenin-dependent signaling may be accounted for by the differing extents to which these constructs permit the CTT to enter the nucleus.
It has recently been shown that the expression of a construct composed of the extreme 112 amino acids of the CTT of PC1 localizes to the nucleus and that it has no inhibitory effect on TCF-dependent gene transcription (21). While the individual amino acids responsible for the nuclear localization of this C-terminal fragment remain unknown, this construct does not contain the 21 amino acid putative nuclear localization motif that we have identified in the full length PC1 CTT fragment. We find that this 21 residue sequence is required for the association between the PC1 CTT and β-catenin. Thus, the nuclear targeting domain may be uniquely responsible for determining the inhibitory action of p200, and not p112, on β-catenin-dependent signaling. Studies aimed at identifying the cellular components that participate in the processing and cleavage of PC1 to release various sized fragments of the CTT, and the nature of their substrate specificities and functional parameters, will undoubtedly help to define the biological relevance of PC1 CTT signal transduction.
The 21 amino acid nuclear targeting motif present in p200 permits not only the nuclear localization of the PC1 CTT, but also its propensity to form a molecular complex with β-catenin. While these residues are clearly necessary for the association of β-catenin with p200, it remains to be established whether this short sequence is in itself sufficient to mediate this interaction. It has previously been suggested that full length PC1 is present at the adherens junction in a mulitprotein complex containing both β-catenin and E-cadherin (17,18). In lysates prepared from ADPKD epithelia, however, β-catenin also appears able to associate with PC1 in the absence of E-cadherin (18). Our findings support the notion that PC1 CTT association with β-catenin does not require the participation of cadherin and, more significantly, that the assembly of such a molecular complex can occur in the nucleus. Under physiological conditions, it seems likely that the interaction of the PC1 CTT with β-catenin in the nucleus is determined by the relative sizes of the pools of both free PC1 CTT and of soluble, signaling-competent β-catenin.
Our results indicate that the N-terminus of β-catenin is required for this protein’s association with PC1 CTT. Most proteins known to bind directly to β-catenin typically do so through molecular interactions mediated via the armadillo domain of the β-catenin molecule. The N-terminal domain is the primary site at which β-catenin undergoes post-translational phosphorylation and it is this phosphorylation that contributes to the regulation of the cellular stability of the full-length protein. Although our results indicate that the N-terminus of β-catenin is required for its association with PC1 CTT, we were unable to detect an in vitro association between p200 and a GST fusion protein encoding the β-catenin N-terminus. It is interesting to speculate, therefore, that the N-terminus of β-catenin may be involved in mediating an association with PC1 CTT by modulating the binding characteristics of PC1 CTT or other proteins to the armadillo region of the β-catenin molecule. Such a scenario is supported by studies indicating that the two termini of β-catenin can regulate the binding properties of proteins to the armadillo region (26,30,31).
We and others have previously found in vivo evidence indicating that the CTT of PC1 is weakly expressed in the nuclei of renal tubular epithelial cells of normal tissue and that nuclear expression of this PC1 fragment is elevated in some of the renal epithelial cells lining cysts derived from both transgenic mice overexpressing PC1 and human ADPKD subjects (20,21). It should also be noted, however, that only a subset of cyst-lining epithelial cells exhibit nuclear accumulation of the PC1 CTT and not all human ADPKD patients express detectable levels of PC1. Furthermore, most germline PKD1 mutations reported to date are predicted to truncate the CTT of PC1 (32). In the current study, we suggest that the loss of nuclear PC1 CTT may remove an inhibitory constraint to Wnt/β-catenin-dependent signaling. Indeed, experimental data obtained from transgenic mice indicates that both the absence of full-length PC1 as well as it overexpression is sufficient to elicit cyst formation (33,34). The complete extent to which the soluble CTT of PC1 is involved in both normal renal epithelial cell biology as well as renal cystic pathophysiology will require the generation of novel mutant PC1 mouse models.
Based on our results, it appears that one of the roles of the PC1 CTT is to negatively regulate β-catenin-dependent induction of TCF-mediated transcription. This is relevant since it has previously been shown that the β-catenin/TCF-regulated oncogene, c-myc, is overexpressed in human ADPKD (35,36) as well as in other varieties of renal cystic diseases (37,38) and that enhanced expression of c-myc in transgenic mice is by itself sufficient to induce a renal cystic phenotype (39). The data presented here expand the list of Wnt and proliferation-related genes whose expression is inappropriately elevated in human ADPKD cysts. It has been proposed that PC1 acts as a tumor suppressor and that cysts are the benign tumors that result when normal PC1 functional expression is inactivated (2,40). According to our observations, it seems likely that this tumor suppressive function may be at least partially dependent upon the ability of the free PC1 CTT to inhibit β-catenin-dependent Wnt signaling. Further analysis of this inhibition may reveal new strategies not only for PKD therapy, but also for the treatment of other pathophysiological conditions, such as various cancers, that are characterized by overactive β-catenin signaling.
MATERIALS AND METHODS
Renal cyst and control tissues
Renal cysts of different sizes were obtained from five polycystic kidneys that were removed according to strict medical indications. The kidneys were kept on ice immediately after nephrectomy and throughout the tissue retrieval procedure in the surgical pathology suite, where the renal capsule was stripped and individual cysts identified. The volume of each intact cyst was determined by withdrawing all the cyst fluid into an appropriately sized syringe via a 21-gauge butterfly needle. Small cysts (SC) were defined as less than 1 ml, medium cysts (MC) between 10 and 25 ml and large cysts (LC) greater than 50 ml. MCT, which might have contained a few microscopic cysts from the renal cortex, was obtained as PKD control tissue from the above kidneys. Additionally, non-cancerous renal cortical tissue from three nephrectomized kidneys with isolated renal cell carcinoma was used as normal control tissue. All tissues were dissected within 30 min of nephrectomy, washed in cold PBS, snap-frozen in liquid N2 and stored at −80°C. For the microarray study, 13 cyst (SC: each pooled from 4 different SC, n = 5; MC, n = 5; LC, n = 3), 5 MCT and 3 normal renal cortical tissue samples were used. For the validation of specific genes of interest by real-time RT–PCR (qPCR), an expanded sample set (SC: n = 16; MC, n = 19; LC, n = 3; MCT, n = 5) from the above PKD kidneys was used. All the study patients were shown to have PKD1 by DNA linkage or documentation of a pathogenic mutation identified through DNA sequencing by Athena Diagnostics™. Informed consent was obtained from all five patients and the Institutional Review Board of the hospital where the nephrectomy was performed approved the research protocol used for this study.
RNA extraction
Total RNA was extracted from each sample using Absolutely RNA RT–PCR Miniprep Kit (Stratagene) with an on-column DNA digestion step to minimize genomic DNA contamination. The integrity of the RNA was assessed using the RNA 6000 Nano Assay on 2100 Bioanalyzer (Agilent Technologies) to ensure that the ratio of 28S to 18S rRNA was at least 2. As a negative control for potential DNA contamination, we routinely performed 45 cycles of PCR for β-actin pseudogenes and did not detect any amplification products.
Microarray analysis of cyst and control tissues
Detailed methods for the microarray analysis, including data preprocessing and normalization, hierarchical cluster analysis, GSEA, SAM and the validation of the microarray results by qPCR analysis, are presented in the Supplemental Information.
Cell culture and plasmids
CHO, HEK 293T cells (20) and PKD1 (flox/−) TSLargeT renal proximal tubule cells (24) were maintained as described. DNA plasmids for p200, p200Δ and PLAP-p200 (20), β-catenin (wild type, ΔN89β-catenin, β-cateninΔC695) (26), and the cadherin cytoplasmic domain (CAD–GST) and TCF β-catenin binding region (TCF–GST) fusion proteins (26) are described as noted. A cDNA encoding a GST fusion protein of the N-terminal amino acids 1–106 of β-catenin was kindly provided by M. Dunach, Universitat Autonoma de Barcelona. TOPflash and FOPflash plasmids were purchased from Upstate Biotechnology. Lipofectamine 2000 (Invitrogen) was used for all transfections according to the manufacturer’s instructions.
Co-immunoprecipitation studies
Transiently transfected CHO or HEK 293T cells grown in six well plates were rinsed 2X with PBS and lysed on ice for 30 min with shaking in 500 µl cell lysis buffer [50 mm Tris–HCl (pH 7.4) 150 mm NaCl, 1% Triton-X100, 1 mm EDTA and Complete protease inhibitor tablet (Roche)]. Lysates harvested by scraping were sonicated with three pulses of 5 s each using a Branson sonifier set at 40%. An aliquot was kept for determination of total protein expression and western blotting, and the remainder was incubated overnight with 1 µl Xpress antibody (Invitrogen) followed by incubation with washed protein G-sepharose (Amersham) beads for 2 h. Pelleted beads were washed extensively with lysis buffer and resuspended in 2X Laemmli sample buffer. Supernatants were boiled and separated by SDS-gel electrophoresis.
Cell fractionation and western blot analysis
Preparation of nuclear (1% triton-insoluble) and non-nuclear (1% triton-soluble) cell fractions and western blotting were performed as described (20). Nitrocellulose membranes were cut in half horizontally (at approximately the 50 kDa marker); the upper half was incubated with anti-β-catenin antibody and the lower half with anti-Xpress antibody. All primary antibodies and their respective dilutions are listed in the Supplemental information.
TOPflash luciferase reporter assay
HEK 293T cells grown in 12-well culture plates were transiently transfected with TOPflash TCF reporter plasmid (0.2 µg), β-galactosidase expression vector (0.2 µg) and various expression plasmids as indicated for individual experiments. The FOPflash reporter plasmid containing a mutated TCF/LEF binding site was also used as a negative control. The total quantity of DNA (1.6 µg) added to each well was held constant by adding ‘mock’ DNA (pcDNA3.1) where necessary. Cells were maintained in serum replete medium and then harvested 24–30 h post-transfection in 300 µl cell lysis buffer (Promega). Luciferase activity was determined using a luciferase assay system (Promega) and luminometer according to the manufacturer’s specifications. β-galactosidase activity was measured spectrophotometrically according to standard techniques (Promega) and was used to normalize for transfection efficiency. Aliquots of the cell lysates from each well were used to assess protein expression by western blotting. β-actin expression was used as a loading control. Experiments performed using PC1+/− and PC1−/− cells (kind gift of Dr S. Somlo, Yale University) were carried out in a similar manner except that a Renilla luciferase reporter was employed as an internal control for transfection efficiency.
Confocal immunofluorescence microscopy
Cells were grown on 12 mm glass cover slips and transiently transfected according to the various treatment conditions. Cells were fixed, permeabilized and incubated with primary antibodies as described (20). β-catenin was recognized using rabbit anti-flag (1:200) and p200/p200Δ were detected with mouse anti-Xpress antibody (1:200). Alexa 546-conjugated anti-rabbit IgG (1:200) and Alexa 488-conjugated anti-mouse IgG (1:200) (Molecular Probes) were used as secondary antibodies. Nuclear counterstaining was performed using DAPI (Molecular Probes). Cover slips were mounted in DakoCytomation fluorescent mounting media (Dako). Images were recorded using a Zeiss LSM510 inverted confocal microscope. Images are the product of 4-fold line averaging and contrast and brightness settings were chosen to ensure that all pixels were in the linear range.
In vitro GST binding analysis
For TCF–GST and CAD–GST in vitro binding affinity experiments, 0.1% Triton X-100 lysates (100 µg) were prepared from HEK 293T cells transiently transfected with β-catenin in the presence or absence of p200/p200Δ. β-catenin was released from the insoluble fraction by sonication and then incubated with TCF–GST and CAD–GST fusion proteins prebound to glutathione-sepharose beads (Amersham). TCF–GST and CAD–GST fusion proteins were harvested from BL21 competent bacteria according to standard procedures (Stratagene) and equal loading on beads was determined by SDS–PAGE and coomassie staining. Following incubation for 2–4 h, the bound complexes were thoroughly rinsed, extracted with 2X Laemmli buffer, electrophoresed and western blotted, as described above.
Statistical analysis
Data are expressed as means ± SE. Differences between means were evaluated by Student’s t-test and analysis of variance. Statistical significance was accepted at P < 0.05.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
FUNDING
This work was supported by NIH grant DK57328 to M.J.C. and CIHR grant MOP77806 to Y.P., fellowships from the Wenner-Gren Foundation and Svenska Sällskapet för Medicinsk Forskning to M.L. and a PKD Foundation Fellowship grant (05b2f) to V.C.
ACKNOWLEDGEMENTS
We are grateful to Drs O. Ibraghimov-Beskrovnaya, S. Somlo and M. Dunach for the gift of reagents and to the members of the Caplan and Pei laboratories for helpful discussions and support.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Igarashi P., Somlo S. Genetics and pathogenesis of polycystic kidney disease. J. Am. Soc. Nephrol. 2002;13:2384–2398. doi: 10.1097/01.asn.0000028643.17901.42. [DOI] [PubMed] [Google Scholar]
- 2.Wilson P.D. Polycystic kidney disease. N. Engl. J. Med. 2004;350:151–164. doi: 10.1056/NEJMra022161. [DOI] [PubMed] [Google Scholar]
- 3.Grantham J.J., Geiser J.L., Evan A.P. Cyst formation and growth in autosomal dominant polycystic kidney disease. Kidney Int. 1987;31:1145–1152. doi: 10.1038/ki.1987.121. [DOI] [PubMed] [Google Scholar]
- 4.Calvet J.P., Grantham J.J. The genetics and physiology of polycystic kidney disease. Semin. Nephrol. 2001;21:107–123. doi: 10.1053/snep.2001.20929. [DOI] [PubMed] [Google Scholar]
- 5.Ong A.C., Harris P.C. Molecular pathogenesis of ADPKD: the polycystin complex gets complex. Kidney Int. 2005;67:1234–1247. doi: 10.1111/j.1523-1755.2005.00201.x. [DOI] [PubMed] [Google Scholar]
- 6.Pei Y. A ‘two-hit’ model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol. Med. 2001;7:151–156. doi: 10.1016/s1471-4914(01)01953-0. [DOI] [PubMed] [Google Scholar]
- 7.Kim E., Arnould T., Sellin L.K., Benzing T., Fan M.J., Gruning W., Sokol S.Y., Drummond I., Walz G. The polycystic kidney disease 1 gene product modulates Wnt signaling. J. Biol. Chem. 1999;274:4947–4953. doi: 10.1074/jbc.274.8.4947. [DOI] [PubMed] [Google Scholar]
- 8.Arnould T., Kim E., Tsiokas L., Jochimsen F., Gruning W., Chang J.D., Walz G. The polycystic kidney disease 1 gene product mediates protein kinase C alpha-dependent and c-Jun N-terminal kinase-dependent activation of the transcription factor AP-1. J. Biol. Chem. 1998;273:6013–6018. doi: 10.1074/jbc.273.11.6013. [DOI] [PubMed] [Google Scholar]
- 9.Le N.H., van der Bent P., Huls G., van de Wetering M., Loghman-Adham M., Ong A.C., Calvet J.P., Clevers H., Breuning M.H., van Dam H., et al. Aberrant polycystin-1 expression results in modification of activator protein-1 activity, whereas Wnt signaling remains unaffected. J. Biol. Chem. 2004;279:27472–27481. doi: 10.1074/jbc.M312183200. [DOI] [PubMed] [Google Scholar]
- 10.Parnell S.C., Magenheimer B.S., Maser R.L., Zien C.A., Frischauf A.M., Calvet J.P. Polycystin-1 activation of c-Jun N-terminal kinase and AP-1 is mediated by heterotrimeric G proteins. J. Biol. Chem. 2002;277:19566–19572. doi: 10.1074/jbc.M201875200. [DOI] [PubMed] [Google Scholar]
- 11.Puri S., Magenheimer B.S., Maser R.L., Ryan E.M., Zien C.A., Walker D.D., Wallace D.P., Hempson S.J., Calvet J.P. Polycystin-1 activates the calcineurin/NFAT (nuclear factor of activated T-cells) signaling pathway. J. Biol. Chem. 2004;279:55455–55464. doi: 10.1074/jbc.M402905200. [DOI] [PubMed] [Google Scholar]
- 12.Moon R.T., Kohn A.D., De Ferrari G.V., Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat. Rev. Genet. 2004;5:691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- 13.Gottardi C.J., Gumbiner B.M. Adhesion signaling: how beta-catenin interacts with its partners. Curr. Biol. 2001;11:R792–R794. doi: 10.1016/s0960-9822(01)00473-0. [DOI] [PubMed] [Google Scholar]
- 14.Qian C.N., Knol J., Igarashi P., Lin F., Zylstra U., Teh B.T., Williams B.O. Cystic renal neoplasia following conditional inactivation of apc in mouse renal tubular epithelium. J. Biol. Chem. 2005;280:3938–3945. doi: 10.1074/jbc.M410697200. [DOI] [PubMed] [Google Scholar]
- 15.Saadi-Kheddouci S., Berrebi D., Romagnolo B., Cluzeaud F., Peuchmaur M., Kahn A., Vandewalle A., Perret C. Early development of polycystic kidney disease in transgenic mice expressing an activated mutant of the beta-catenin gene. Oncogene. 2001;20:5972–5981. doi: 10.1038/sj.onc.1204825. [DOI] [PubMed] [Google Scholar]
- 16.Sorenson C.M. Nuclear localization of beta-catenin and loss of apical brush border actin in cystic tubules of bcl-2−/− mice. Am. J. Physiol. 1999;276:F210–F217. doi: 10.1152/ajprenal.1999.276.2.F210. [DOI] [PubMed] [Google Scholar]
- 17.Huan Y., van Adelsberg J. Polycystin-1, the PKD1 gene product, is in a complex containing E-cadherin and the catenins. J. Clin. Invest. 1999;104:1459–1468. doi: 10.1172/JCI5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roitbak T., Ward C.J., Harris P.C., Bacallao R., Ness S.A., Wandinger-Ness A. A polycystin-1 multiprotein complex is disrupted in polycystic kidney disease cells. Mol. Biol. Cell. 2004;15:1334–1346. doi: 10.1091/mbc.E03-05-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boca M., D’Amato L., Distefano G., Polishchuk R.S., Germino G.G., Boletta A. Polycystin-1 induces cell migration by regulating phosphatidylinositol 3-kinase-dependent cytoskeletal rearrangements and GSK3beta-dependent cell cell mechanical adhesion. Mol. Biol. Cell. 2007;18:4050–4061. doi: 10.1091/mbc.E07-02-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chauvet V., Tian X., Husson H., Grimm D.H., Wang T., Hiesberger T., Igarashi P., Bennett A.M., Ibraghimov-Beskrovnaya O., Somlo S., et al. Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J. Clin. Invest. 2004;114:1433–1443. doi: 10.1172/JCI21753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Low S.H., Vasanth S., Larson C.H., Mukherjee S., Sharma N., Kinter M.T., Kane M.E., Obara T., Weimbs T. Polycystin-1, STAT6 and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev. Cell. 2006;10:57–69. doi: 10.1016/j.devcel.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 22.Bafico A., Gazit A., Pramila T., Finch P.W., Yaniv A., Aaronson S.A. Interaction of frizzled related protein (FRP) with Wnt ligands and the frizzled receptor suggests alternative mechanisms for FRP inhibition of Wnt signaling. J. Biol. Chem. 1999;274:16180–16187. doi: 10.1074/jbc.274.23.16180. [DOI] [PubMed] [Google Scholar]
- 23.Lai J.S., Herr W. Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc. Natl Acad. Sci. USA. 1992;89:6958–6962. doi: 10.1073/pnas.89.15.6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joly D., Ishibe S., Nickel C., Yu Z., Somlo S., Cantley L.G. The polycystin 1-C-terminal fragment stimulates ERK-dependent spreading of renal epithelial cells. J. Biol. Chem. 2006;281:26329–26339. doi: 10.1074/jbc.M601373200. [DOI] [PubMed] [Google Scholar]
- 25.Huber A.H., Nelson W.J., Weis W.I. Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell. 1997;90:871–882. doi: 10.1016/s0092-8674(00)80352-9. [DOI] [PubMed] [Google Scholar]
- 26.Gottardi C.J., Gumbiner B.M. Distinct molecular forms of beta-catenin are targeted to adhesive or transcriptional complexes. J. Cell. Biol. 2004;167:339–349. doi: 10.1083/jcb.200402153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muto S., Aiba A., Saito Y., Nakao K., Nakamura K., Tomita K., Kitamura T., Kurabayashi M., Nagai R., Higashihara E., et al. Pioglitazone improves the phenotype and molecular defects of a targeted Pkd1 mutant. Hum. Mol. Genet. 2002;11:1731–1742. doi: 10.1093/hmg/11.15.1731. [DOI] [PubMed] [Google Scholar]
- 28.Simons M., Gloy J., Ganner A., Bullerkotte A., Bashkurov M., Kronig C., Schermer B., Benzing T., Cabello O.A., Jenny A., et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat. Genet. 2005;37:537–543. doi: 10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Staal F.J., Noort Mv M., Strous G.J., Clevers H.C. Wnt signals are transmitted through N-terminally dephosphorylated beta-catenin. EMBO Rep. 2002;3:63–68. doi: 10.1093/embo-reports/kvf002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castano J., Raurell I., Piedra J.A., Miravet S., Dunach M., Garcia de Herreros A. Beta-catenin N- and C-terminal tails modulate the coordinated binding of adherens junction proteins to beta-catenin. J. Biol. Chem. 2002;277:31541–31550. doi: 10.1074/jbc.M204376200. [DOI] [PubMed] [Google Scholar]
- 31.Solanas G., Miravet S., Casagolda D., Castano J., Raurell I., Corrionero A., de Herreros A.G., Dunach M. Beta-Catenin and plakoglobin N- and C-tails determine ligand specificity. J. Biol. Chem. 2004;279:49849–49856. doi: 10.1074/jbc.M408685200. [DOI] [PubMed] [Google Scholar]
- 32.Rossetti S., Consugar M.B., Chapman A.B., Torres V.E., Guay-Woodford L.M., Grantham J.J., Bennett W.M., Meyers C.M., Walker D.L., Bae K., et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2007;18:2143–2160. doi: 10.1681/ASN.2006121387. [DOI] [PubMed] [Google Scholar]
- 33.Thivierge C., Kurbegovic A., Couillard M., Guillaume R., Cote O., Trudel M. Overexpression of PKD1 causes polycystic kidney disease. Mol. Cell. Biol. 2006;26:1538–1548. doi: 10.1128/MCB.26.4.1538-1548.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu W., Shen X., Pavlova A., Lakkis M., Ward C.J., Pritchard L., Harris P.C., Genest D.R., Perez-Atayde A.R., Zhou J. Comparison of Pkd1-targeted mutants reveals that loss of polycystin-1 causes cystogenesis and bone defects. Hum. Mol. Genet. 2001;10:2385–2396. doi: 10.1093/hmg/10.21.2385. [DOI] [PubMed] [Google Scholar]
- 35.Husson H., Manavalan P., Akmaev V.R., Russo R.J., Cook B., Richards B., Barberio D., Liu D., Cao X., Landes G.M., et al. New insights into ADPKD molecular pathways using combination of SAGE and microarray technologies. Genomics. 2004;84:497–510. doi: 10.1016/j.ygeno.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 36.Lanoix J., D’Agati V., Szabolcs M., Trudel M. Dysregulation of cellular proliferation and apoptosis mediates human autosomal dominant polycystic kidney disease (ADPKD) Oncogene. 1996;13:1153–1160. [PubMed] [Google Scholar]
- 37.Cowley B.D., Jr, Smardo F.L., Jr, Grantham J.J., Calvet J.P. Elevated c-myc protooncogene expression in autosomal recessive polycystic kidney disease. Proc. Natl Acad. Sci. USA. 1987;84:8394–8398. doi: 10.1073/pnas.84.23.8394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harding M.A., Gattone V.H., 2nd, Grantham J.J., Calvet J.P. Localization of overexpressed c-myc mRNA in polycystic kidneys of the cpk mouse. Kidney Int. 1992;41:317–325. doi: 10.1038/ki.1992.44. [DOI] [PubMed] [Google Scholar]
- 39.Trudel M., D’Agati V., Costantini F. C-myc as an inducer of polycystic kidney disease in transgenic mice. Kidney Int. 1991;39:665–671. doi: 10.1038/ki.1991.80. [DOI] [PubMed] [Google Scholar]
- 40.Smyth B.J., Snyder R.W., Balkovetz D.F., Lipschutz J.H. Recent advances in the cell biology of polycystic kidney disease. Int. Rev. Cytol. 2003;231:51–89. doi: 10.1016/s0074-7696(03)31002-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.