

Abstract
Spinal muscular atrophy (SMA) is a common pediatric neuromuscular disorder caused by insufficient levels of the survival of motor neuron (SMN) protein. Studies involving SMA patients and animal models expressing the human SMN2 gene have yielded relatively little information about the earliest cellular consequences of reduced SMN protein. In this study, we have used severe- and mild-SMN2 expressing mouse models of SMA as well as material from human patients to understand the initial stages of neurodegeneration in the human disease. We show that the earliest structural defects appear distally and involve the neuromuscular synapse. Insufficient SMN protein arrests the post-natal development of the neuromuscular junction (NMJ), impairing the maturation of acetylcholine receptor (AChR) clusters into ‘pretzels’. Pre-synaptic defects include poor terminal arborization and intermediate filament aggregates which may serve as a useful biomarker of the disease. These defects are reflected in functional deficits at the NMJ characterized by intermittent neurotransmission failures. We suggest that SMA might best be described as a NMJ synaptopathy and that one promising means of treating it could involve maintaining function at the NMJ.
INTRODUCTION
Proximal spinal muscular atrophy (SMA) is a common autosomal recessive neurodegenerative disease in humans caused by mutations in the survival of motor neuron 1 (SMN1) gene (1). An almost identical copy gene, SMN2, unique to humans, is unable to compensate for the homozygous loss of SMN1 due to a C→T transition in exon 7 that affects splicing and replaces most of the full-length (FL) SMN transcript with an isoform, SMNΔ7 (2,3). SMNΔ7 protein is unstable and rapidly degraded. Low levels of FL SMN, expressed from SMN2 which is always retained in patients, are insufficient for the health and maintenance of the neuromuscular system resulting in neurodegeneration, muscular paralysis and, in severe cases, death. It is not yet clear why the neuromuscular system is selectively affected by insufficient SMN, a ubiquitously expressed protein. Due to the inherently unstable nature of the SMA locus, patients have been found to possess as many as eight copies of the SMN2 gene (4). Disease severity in humans is inversely correlated with SMN2 copy number (5,6). This finding has been directly confirmed in different lines of transgenic mice lacking the single murine Smn gene but carrying a varying number of copies of a genomic fragment that contains human SMN2 (7,8).
Based on the human phenotype, SMA has historically been described as a motor neuron disease characterized by a degeneration of the anterior horn cells of the spinal cord and skeletal muscle atrophy (reviewed in 9). The description of the tissue specific nature of SMA, particularly, the effect of reduced SMN on the lower motor neurons has relied on the analysis of end-stage disease patient material. While such studies have been informative, they provide but a snapshot of the cellular effects of reduced SMN on the neuromuscular system. More recently, investigators have utilized cell culture techniques to determine functions of the SMN protein that could be relevant to SMA pathology. These putative functions are based partly on localization studies and partly on the effects of depleting SMN in relevant cell types (10–12). In neurons and neuron-like cells, SMN was found to localize in neuritic processes (13,14). In muscle, the protein was found at neuromuscular junctions (NMJs) (13,15). Based on these findings it was suggested that SMN likely plays a hitherto un-described role in these sub-cellular compartments. SMN's putative role in muscle, particularly, at the NMJ has been bolstered by findings from experiments indicating that cultured muscle cells from SMA patients fail to cluster acetylcholine receptors (AChRs) at the junction (16). Recent reports in support of a role of SMN at the NMJ in vivo have utilized the fruit fly, Drosophila melanogaster and transgenic mice, respectively (17–19). However, in none of the studies were the model organisms engineered to express a constant low level of SMN as is characteristic of human SMA. This could, arguably complicate the interpretation of the observed neuromuscular phenotype. Thus, although the above-mentioned studies have provided tantalizing clues about the cellular processes that accompany neuromuscular dysfunction in SMA, a complete in vivo profile of the degenerative process under steady low levels of SMN protein has yet to be presented.
To gain insight into the molecular and cellular causes of neurodegeneration in SMA, mouse models of the disease were generated (7,8,20,21). The mice express uniformly low levels of the SMN protein from one or more genomic copies of human SMN2 with or without additional mutant SMN transgenes. In this study, we have used severe- and mild-SMA mice from two of the reports referred above (20,21) to determine the initial effects of reduced SMN on the neuromuscular system. Our analysis involved examining the effects of low SMN on the neuromuscular system of the mice over multiple time points as the disease progresses. We show for the first time that cellular defects appear prior to overt symptoms. We further show that such defects are initially restricted distally to the neuromuscular synapse. Finally, our studies indicate that reduced levels of the SMN protein impair the normal maturation of the NMJ and cause neurotransmission defects which likely account for the profound muscle weakness and motor neuron loss that characterize SMA.
RESULTS
Characterization of functional parameters of disease onset and progression in fully congenic FVB/N SMA mice
To understand the initial stages of neurodegeneration in SMA, we chose to focus on a mouse model of severe SMA (21). Although the mice were reported to have been backcrossed over six generations to the FVB/N strain, single nucleotide polymorphism (SNP) analysis using 175 markers covering the whole genome indicated that the mice still derived ∼11% of their genome from C57Bl/6J. Since mutant phenotypes and disease pathology often vary considerably in mixed strains of mice (22), transgenic carriers were backcrossed to the FVB/N strain over an additional five generations. SNP analysis on the resulting strain of mice indicates that they are >99% FVB/N. These fully congenic animals were subsequently used to generate SMA mice homozygous (SMN2+/+;SMNΔ7+/+;Smn−/− referred to henceforth as Δ7+/+ SMA mice) and hemizygous (SMN2+/+;SMNΔ7+/−;Smn−/− termed Δ7+/− SMA mice hereafter) for the SMNΔ7 transgene for our experiments. At P0, Δ7+/+ SMA mice are indistinguishable from normal littermates by weight (1.31 ± 0.11 versus 1.34 ± 0.21 g; n = 10, P > 0.5) whereas Δ7+/− SMA animals are significantly smaller (1.38 ± 0.07 versus 1.18 ± 0.07 g; n = 10; P < 0.001). At P2, even though Δ7+/+ SMA mice and normal littermates are of similar size, the affected animals are easily recognized by compromised righting ability, a sign of muscle weakness (Fig. 1A and B). Mutants become progressively weaker with age until they are unable to right themselves at P5. Body weight peaks at ∼P8 (Fig. 1C). Kaplan–Meier survival curves indicate that Δ7+/+ SMA mice survive for an average of 11.4 ± 0.4 days; n = 81. Affected Δ7+/− littermates survive, on an average, 6.4 ± 0.8 days; n = 15, consistent with lower amounts of SMN in them (Fig. 1D and E). Interestingly our data indicates that the N11 FVB/N Δ7+/+ SMA mice are more severely affected than those reported by Le et al. (2005). An independent analysis of the lifespan of the hybrid mice indicates an average survival of 17.7 ± 0.2 days; n = 90. The dramatic decrease in mean survival in our fully congenic mice of the same genotype indicates possible beneficial hybrid effects in the originally reported SMA animals. Southern blot analysis on the fully and partially congenic carrier mice indicates that there was no loss of transgene copy number in the former group while they were being backcrossed to FVB/N mice; western blots indicate that the two sets of mice express equivalent levels of SMN protein (Supplementary Material, Fig. S1A and B). Collectively these results suggest that factors that modify the phenotype and survival in the partially congenic strain of mice do so without affecting SMN levels. Assuming that these modifiers segregate, it would be prudent to use the fully congenic animals, particularly, in studies involving the evaluation of potential therapeutics. Since Δ7+/− SMA animals are extremely severely affected, precluding certain types of analyses, we focused our subsequent experiments on Δ7+/+ mice.
Figure 1.

Phenotypic characteristics of SMA mice. (A) Phenotypes of fully congenic FVB/N Δ7+/+ SMA mice and Control (Ctrl) littermates during the course of the disease. Scale bar: 2 cm. (B) Righting ability curves of SMA mice: SMN2+/+;SMNΔ7+/+;Smn−/− (black squares); SMN2+/+;SMNΔ7+/−;Smn−/− (red squares) and Ctrl: SMN2+/+;SMNΔ7+/+;Smn+/+ (black circles), SMN2+/+;SMNΔ7+/−;Smn+/+ (red circles). Scores were assigned as described in methods; n= 10 in each case. (C) Body weight graphs of SMA mice and Control littermates between birth and P12; n = 10 in each case. (D) Kaplan–Meier survival curves of SMA mice. The log-rank test indicated a significant difference (P < 0.0001) between mean survival of the fully congenic (N11 FVB/N) Δ7+/+ and partially congenic SMA mice of the same genotype. Δ7+/+ mice also live significantly longer than Δ7+/− SMA mice. Note: n = 15, 81 and 90 for the Δ7+/−, N11 FVB/N Δ7+/+ and N6 FVB/N Δ7+/+ SMA mice, respectively; survival expressed as mean ± SEM. (E) Western blots of SMN in the lumbar (L1–L5) spinal cords of P2 SMA mice and Ctrl.
SMN levels determine the extent of pre-synaptic defects at the NMJ; structural abnormalities precede the appearance of overt symptoms in SMA mice
Four days old Δ7+/+ SMA mice do not exhibit significant spinal motor neuron loss (21). Yet, muscle weakness is evident as early as post-natal day 2 (P2). To determine whether defects located more distally at the NMJ might explain the apparent muscle weakness in SMA mice, we examined junctions in the gastrocnemius muscle between P2 and P14. Antibodies against the neurofilament (NF) protein and synaptic vesicles (SV2) were used to visualize distal axons and nerve terminals, respectively. AChRs were stained with labeled bungarotoxin. At P2, NMJs in SMA and Control littermates were indistinguishable (Fig. 2A). However, by P5 ∼25% of all nerve terminals in SMA mice appeared thick and swollen with abnormal NF aggregates. With age, a progressive increase in the number of such defective terminal axons was noted in the SMA mice, reaching ∼90% by P14 (Fig. 2B). In addition to, or possibly because of, the abnormal accumulation of NF, SMA axons fail to form the fine terminal arbors characteristically found as early as P8 at normal NMJs. These results suggest that NF accumulation and poor terminal arborization at the NMJ are important and are previously unappreciated aspects of SMA pathology. A careful examination of the NMJs did not reveal greater than wild-type numbers of unoccupied AChR clusters. However, we did see an abnormal number (∼15%) of axons in end-stage disease animals terminating in retraction bulb-like structures. The vast majority (∼90%) of these axons were found to be apposed to relatively weakly staining AChR clusters, probably in the process of being disassembled (Fig. 2A; also see Fig. 6A). The absence of a significant number of unoccupied endplates in mutants is in contrast to recent findings (23). However, the absence of anatomically denervated endplates in our SMA mice does not necessarily suggest that the junctions are functional. Poly-neuronal to mono-neuronal innervation as assessed by the number of axons innervating individual endplates at P0, P6 and P14 did not differ between SMA mice and Control littermates suggesting that the process of synapse elimination is not affected in SMA (Fig. 2C).
Figure 2.
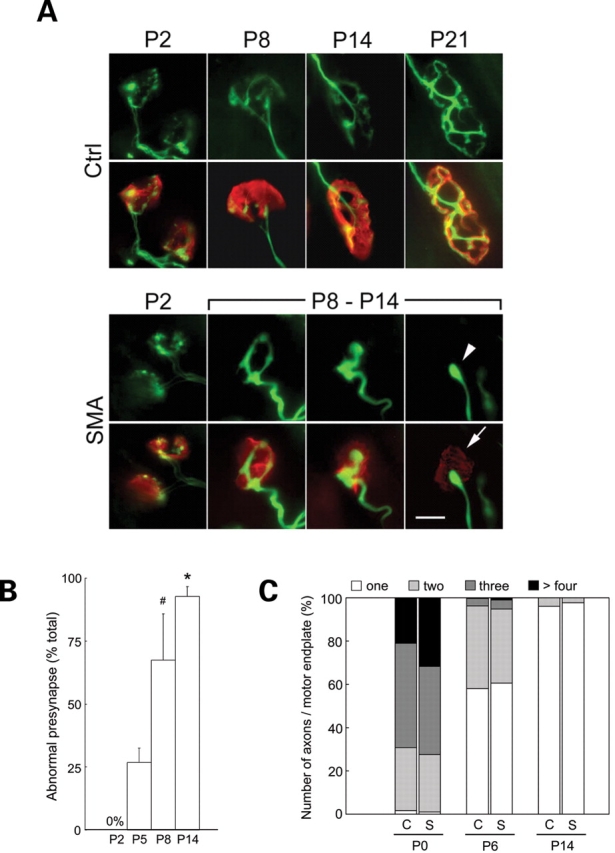
Pre-synaptic NMJ defects in severe-SMA mice. (A) Representative images of NMJs in the gastrocnemius muscle of Δ7+/+ SMA mice and a Control littermate highlighting defects in the mutant as the disease progresses. Green: pre-synapse (NF and SV2 staining). Red: motor endplates (rhodamine-α-BTX staining). Arrowhead: a severely shrunken (bulb-like) nerve terminal apposed to an endplate characterized by weakly staining AChR clusters (arrow). Scale bar: 10 µm. (B) Quantification of defective terminals in the gastrocnemius of Δ7+/+ SMA mice indicating increasing numbers of abnormal NMJs with age. Increases in defective terminals are statistically significant. ‘Asterisk' denotes P < 0.01 (versus P8, t-test); ‘Hash' indicates P < 0.01 (versus P5, t-test); n = 3. (C) The proportion of axons per motor endplate in the gastrocnemius muscles of Δ7+/+ SMA mice (S) and Ctrl (C) indicates normal timing of synapse elimination in affected mice.
Figure 6.

Post-synaptic NMJ defects in SMA mice. (A) Motor endplates in gastrocnemius muscles of a P14 Δ7+/+ SMA mouse and a Control (Ctrl) littermate indicate that the NMJs of affected animals appear immature (few perforations, weak staining, lack of synaptic folds). In some cases, disassembled AChRs were noted (3). NMJ perforations are indicated with asterisks. Scale bar: 10 µm. (B) Quantification of plaque-like (structurally immature motor endplates without perforations) AChR clusters in the gastrocnemius muscles of Δ7+/+ SMA mice (S) and Ctrl (C); n = 3. ‘Asterisk' denotes P < 0.01 (versus Control, Student's t-test). n.s.: not significant. (C) An increase in transcript levels of the embryonic (γ) subunit of the AChR in SMA mice. ‘Asterisk' denotes P < 0.01 (Student's t-test). (D) Motor endplates with less than three perforations in the gastrocnemius muscles of mild (A2G) SMA mice (S) and Ctrl (C) at different ages indicate early (P15) evidence of post-synaptic defects. (E) Representative images of motor endplates used to quantify post-synaptic defects in (D). Scale bar: 3.85 µm.
5q SMA is characterized by weakness in proximal muscles (24). To determine whether pre-synaptic defects at the NMJ preferentially affect these muscles in our SMA mice, we dissected and examined the following additional muscles: biceps brachii, oblique abdominal muscle and internal intercostal muscles—a group of proximal muscles relatively severely affected in human SMA and, the diaphragm, buccinator and the tongue—muscles that are relatively spared in patients. At P2, we saw no pre-synaptic defects in the gastrocnemius, biceps brachii, buccinator and tongue, while 12–25% of terminals in the intercostal muscles, diaphragm and abdominal muscles displayed an abnormal accumulation of NF protein (Fig. 3A). Although the diaphragm is thought to be relatively spared in human SMA, we found NMJ defects in this muscle in mice as well as in affected patients (see the following section). An examination of the above-mentioned muscles at P8 revealed pre-synaptic defects ranging from ∼25% of NMJs (buccinator) to ∼75% (intercostals, biceps, abdominal, tongue, diaphragm and gastrocnemius) suggesting that although proximal muscles are generally affected first in 5q SMA, many distal muscles are also affected as the disease progresses. This is consistent with findings in type I and severe type II SMA patients who survive into their second and third decades of life (25).
Figure 3.
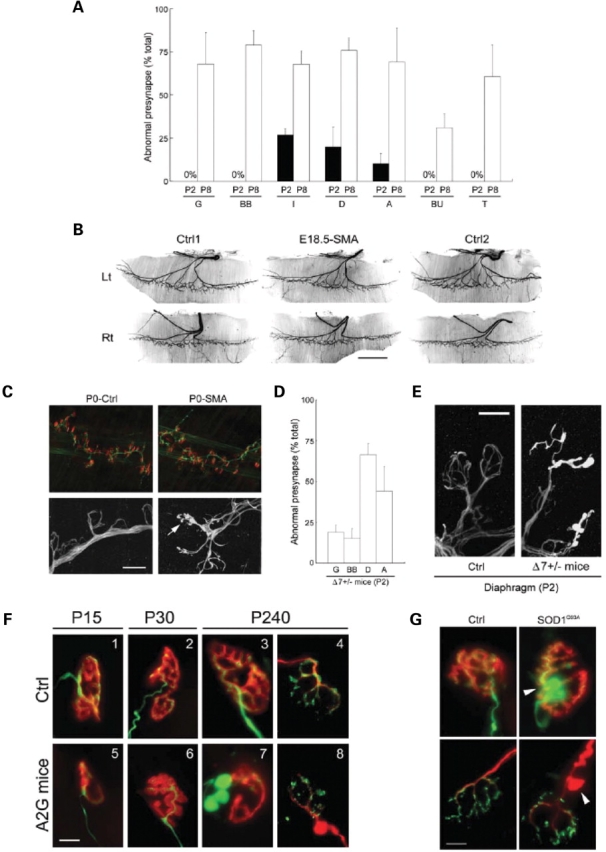
Onset of pre-synaptic NMJ defects in SMA mice. (A) Pre-synaptic defects in representative proximal and distal muscles of Δ7+/+ SMA mice depicting the relatively early (P2) appearance of defects in proximal muscle groups. G, gastrocnemius; BB, biceps brachii; I, internal intercostals; D, diaphragm; A, oblique abdominal; BU, buccinator, and T, tongue muscles. Data represented as mean ± SD (n = 3). (B) Hemi-diaphragms of E18.5 Δ7+/+ SMA and Control (Ctrl1,2) littermates stained with an antibody against NF protein depicting normal branching and innervation by branches of the phrenic nerve in mutants. Scale bar: 1 mm. (C) At birth (P0) NMJs in the diaphragms of Δ7+/+ SMA mice exhibit clear signs of pre-synaptic NF aggregates (arrow). Upper panels: green (NF staining), red (rhodamine-α-BTX staining). Lower panels: NF staining. Scale bar: 10 µm for lower panels, 56 µm for upper panels. (D) Onset of pre-synaptic defects is a correlate of SMN levels and appears in all muscles sampled in Δ7+/− SMA mice as early as P2. (E) Poor terminal arborization and aggregates of NF protein in the pre-synapse of NMJs in Δ7+/− SMA diaphragms. Scale bar: 10 µm. (F) NMJs in the gastrocnemius muscles of mild-SMA mice and Control (Ctrl) littermates depicting increasingly obvious defects as the disease progresses. (1–3, 5–7): green (NF and SV2 staining), red (rhodamine-α-BTX staining). (4, 8): green (SV2 staining), red (NF staining). Scale bar: 10 µm. (G) NF accumulation in pre-terminal axons in the gastrocnemius muscle of end-stage SOD1G93A mice (P166). Upper panels: green (NF and SV2 staining), red (rhodamine-α-BTX staining). Lower panels: green (SV2 staining), red (NF staining). Ctrl: age-matched non-transgenic mouse. Arrow heads: axonal swellings filled with NF. Scale bar: 10 µm.
Reduced SMN causes motor axon defects at early embryonic stages in a fish model of SMA (11). To determine if similar defects also appear in SMA mice and to define the onset of structural abnormalities in the nerve terminals at NMJs, we examined the diaphragms of E18.5 embryos and newborn (P0) animals. The phrenic nerve that innervates the diaphragm does so in a stereotypical manner, branching medially to form the crural branch, dorsally to give rise to the costal branch and ventrally to the sternal branch. We examined 10 wild-type and three SMA embryos. Except for normal variations, we did not find any gross differences between the innervation patterns of the intramuscular nerves of the diaphragm (Fig. 3B). No abnormal NF accumulation was detected in pre-terminal axons or nerve terminals of SMA embryos. Nor did we see evidence of unoccupied AChR clusters (data not shown). This suggests that reduced levels of SMN typically seen in severe SMA do not cause motor defects during embryonic development, a result consistent with normal motor unit numbers in pre-symptomatic type I SMA patients (26). By P0, however, ∼10% of diaphragmatic NMJs displayed pre- but no apparent post-synaptic defects (Fig. 3C). This result indicates that the onset of cellular abnormalities in SMA precedes the appearance of overt symptoms characteristic of the disease. Furthermore, structural defects of the pre-synapse become apparent before those of the post-synapse in a severe form of the disease.
The onset of disease symptoms in SMA is a correlate of SMN levels. We surmised that SMN levels also determine when cellular defects first appear. An analysis of the NMJs in Δ7+/− SMA mice and those in mild-SMA mice co-expressing SMN2 and an SMN1 A2G missense mutation (20) indicates that this is indeed the case (Table 1). In the more severe mice, pre-synaptic defects as assessed by poor terminal arborization and NF accumulation at terminals appeared in distal (gastrocnemius – 19% of all NMJs) as well as proximal muscles (abdominal muscles – 44% of all NMJs; diaphragm – 70% of all NMJs) as early as P2 (Fig. 3D and E); in the mild-SMA mice, NF accumulation was not evident either at P15 or at P30 but was obvious at 8 months (Fig. 3F). Interestingly, the pattern of NF protein aggregates differs in severe- and mild-SMA junctions. While NF invades the pre-terminal axon as well as nerve terminals in severe mice, it is restricted to just the pre-terminal axons of mild-SMA mice. However, a majority (∼75%) of the NMJs in 8 month old mild-SMA mice exhibited these defects.
Table 1.
Percentage of NMJ defects as the disease progresses between P2 and P14 in SMA mice with increasingly severe phenotypes
| P2 | P8 (P5) | P14 | ||
|---|---|---|---|---|
| Δ7+/− | Proximal (abd.) | 44.2 ± 9.0% (+ −) | 95.5 ± 4.0% (+ +) at P5 | – |
| Distal (gastroc.) | 19.0 ± 4.0% (+ −) | 81.9 ± 5.0% (+ +) at P5 | – | |
| Δ7+/+ | Proximal (abd.) | 10.3 ± 5.1% (+ −) | 69.0 ± 11.5% (+ +) | 87.3 ± 9.5% (+ +) |
| Distal (gastroc.) | 0% (− −) | 67.5 ± 9.5% (+ +) | 92.7 ± 3.0% (+ +) | |
| A2G | Proximal (abd.) | 0% (− −) | 27.6 ± 6.9% (+ +)a | 68.0 ± 2.8% (+ +)a |
| Distal (gastroc.) | 0% (− −) | 9.5 ± 5.6% (+ +)a | 57.6 ± 3.0% (+ +)a |
Note: Oblique abdominal muscle (abd.) and the gastrocnemius (gastroc.) served as representative proximal and distal muscles, respectively, in this analysis. Δ7+/−, Δ7+/+ and A2G refer to the genotypes of the SMA mice as described in the main text. Symbols in parentheses represent pre- and post-synaptic defects respectively with ‘+’ indicating the presence of, and ‘−’, the absence of defects. Pre-synaptic defects include poor terminal arborization, NF accumulation in nerve terminals and/or pre-terminal axons; post-synaptic defects include AChR clusters of reduced complexity, smaller endplates and weakly staining AChRs indicating disassembly/dispersal of the receptors at the NMJ.
aIndicates that the pre-synaptic defects are restricted to poor terminal arborization, n ≥ 2.
Aggregation of NF protein in SMA is restricted to the distal ends of α-motor neurons
NF aggregates in the perikarya and axons of neurons are common to many neurodegenerative diseases (27) and may be a non-specific response in moribund cells. To determine if reduced SMN has a similar effect on spinal motor neurons, we examined the different sub-cellular compartments of these cells. Ultrastructural studies of the nerve terminals at the NMJs in diaphragmatic muscle from two P14 Δ7+/+ SMA animals corroborated our immunohistochemical findings. Ten of the 12 nerve terminals examined were found to be misshapen, swollen and contained large amounts of disorganized NFs (Fig. 4A). Synaptic vesicles in these terminals were not uniformly dispersed as in wild-type NMJs, but trapped at the periphery, not necessarily at active zones. This may be attributed to the presence of the large NF aggregates in the terminals. Vesicle number did not differ significantly between SMA and wild-type junctions, and western blot analysis for synaptic vesicle (SV2) protein in distal nerves indicated no detectable difference between affected animals and Control littermates (Supplementary Material, Fig. S2). Interestingly we found a decrease in the size (603 ± 225 µm2 Controls versus 416 ± 171 µm2 SMA; n = 44 in each case; P < 0.001) but not numbers of mitochondria in the nerve terminals of SMA mice. No such decrease in the size of post-synaptic mitochondria was observed (977 ± 300 µm2 Controls versus 859 ± 294 µm2 SMA; n = 38 in each case; P > 0.5). To determine whether NF accumulation in the pre-synapse is unique to SMA, we examined the NMJs in the gastrocnemius of symptomatic SOD1G93A mice. Numerous NMJs in this mouse model of amyotrophic lateral sclerosis (ALS) are denervated (28). Of those that remain innervated, ∼41% exhibit swollen pre-terminal axons that stain strongly with anti-NF antibodies (Fig. 3G). Nerve terminals, on the other hand, are devoid of excess NF protein, a pattern similar to NMJ pathology in mild-SMA mice. These results suggest that NF accumulation in the pre-synapse is unique only to severe forms of SMA and that aggregates in pre-terminal axons are a more general phenomenon of motor neuron disease.
Figure 4.
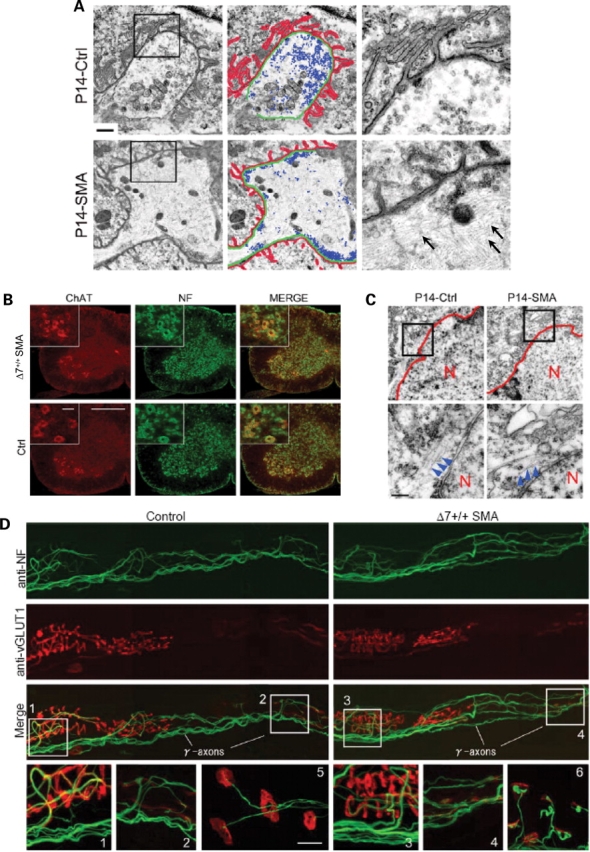
NF accumulation at the distal ends of SMA α-motor axons. (A) Electron micrographs of NMJs in the diaphragms of a P14 Δ7+/+ SMA mouse and Control (Ctrl) littermate depicting profound abnormalities of the pre- (swollen terminal filled with intermediate filaments, abnormal distribution of synaptic vesicles) as well as post-synapse (shallow junctional folds) in the mutant. Post-synaptic folds, terminal axons and synaptic vesicles are indicated in red, green and blue, respectively. High magnification images of insets are shown on the right with NF indicated by arrows. Scale bar: 1 µm. (B) Perikarya of C4 level spinal motor neurons from a P14 Δ7+/+ SMA mouse do not indicate abnormal accumulations of NF protein. Scale bar: 360 µm, inset: 10 µm. (C) Electron micrographs of the motor neurons confirmed the immunohistochemical results. Low levels of cytoplasmic NF found in Control and SMA motor neurons are indicated by the arrowheads; N, motor neuron nucleus. Scale bar: 300 nm. (D) Muscle spindles in the pronator teres muscle from a P14 Δ7+/+ SMA mouse indicating the apparent resistance of γ-motor axons to reduced SMN protein. Post-synapses were detected with rhodamine-α-BTX (red). Anulospiral endings of sensory axons around intrafusal fibers (1, 3) were detected using an anti-vesicular glutamate transporter 1 (red). NMJs of α-motor axons are indicated in 5 and 6. NMJs of γ-motor axons in the same muscle are indicated in 2 and 4. NF aggregates were found at α-motor nerve terminals but not in sensory axons or γ-motor terminals. Scale bar: 10 µm for the bottom panels (1–6).
To determine if reduced SMN also causes perikaryal accumulation of NF protein, we examined cervical spinal motor neurons of Δ7+/+ SMA mice immunohistochemically. In contrast to findings in ALS mice (29) and a neuronal SMA mutant (18), our analysis revealed no abnormal accumulation of NF in the perikarya (Fig. 4B). This was confirmed ultrastructurally (Fig. 4C). Nor did we find NF aggregates in proximal axons (data not shown). Western blot analysis of cervical spinal cord from SMA and Control mice corroborated this finding (Supplementary Material, Fig. S3). These results indicate that NF accumulation under constant low levels of SMN protein is restricted to the distal ends of motor neurons. Coupled with western blot data on NF levels in intramuscular nerves of 8 months old SMA mice, which indicates an increase in NF protein in older animals (Supplementary Material, Fig. S4), our results suggest that there is an up-regulation of intermediate filament expression as the disease progresses.
SMA pathology is defined by loss of the α-motor neurons in the spinal cord. To determine whether structural defects are restricted to these cells or whether neighboring γ-motor neurons are also vulnerable to reduced SMN protein, we examined the distal ends of both populations in the pronator teres muscle of P14 Δ7+/+ SMA mice. α-Motor nerve terminals terminate at NMJs and γ-motor nerve terminals terminate at muscle spindles. In contrast to α-motor terminals, >90% (n = 300) of which were found to exhibit distinct pre-synaptic defects including abnormal NF aggregates, none of the γ-motor terminals and Ia sensory fibers (n = 150) which innervate intrafusal muscle fibers displayed such defects (Fig. 4D). This result indicates that reduced SMN affects not just ventral spinal motor neurons, but a specific subset of these cells, the α-motor neurons.
Based on the striking defects found at the NMJs of SMA mice, we asked if similar abnormalities are also a characteristic of human SMA. Consistent with our findings in SMA mice, pre-terminal axons and nerve terminals in diaphragmatic muscle of type I SMA patients but not Controls are characterized by abnormal aggregates of NF proteins (Fig. 5). Approximately 70% (197/281) of the NMJs examined in tissue from affected patients had thick, poorly branched nerve terminals that stained intensely for NF protein. Motor endplates apposed to these terminals were small and structurally poorly developed. This result suggests that NMJ defects in SMA model mice are truly indicative of SMA pathology in the human disease. Furthermore, although the diaphragm may be functionally relatively spared in the human disease, it is characterized by profound structural defects of the NMJ, consistent with findings in the severely affected Δ7+/+ SMA mice.
Figure 5.

NMJ defects in SMA patients. NMJ defects similar to those found in SMA mice are apparent in human SMA patients. Approximately 70% of NMJs in the diaphragms of 6 months old type I SMA patients exhibit pre-synaptic defects characterized by NF accumulation and poor terminal arborization. Endplates are poorly structured and stain weakly for labeled-BTX. A few NMJs appear morphologically normal (arrow heads). Normal NMJs in the diaphragm of an age-matched Control (right-side panels). Scale bar: 20 µm.
Impaired maturation of the NMJ in SMA mice
As previously mentioned, NMJs in Δ7+/+ SMA mice at P2 or earlier are structurally indistinguishable from those in wild-type littermates. To determine whether reduced SMN affects the numbers of NMJs, endplates in representative areas (distal 600 µm2 of sternal nerve branch) of E18.5 SMA and Control left hemi-diaphragms were counted. No significant difference in numbers of endplates was found (221 ± 32 Control versus 241 ± 22 SMA; n = 3 in each case; P > 0.4). However, while wild-type NMJs increase in size and complexity, transitioning from a plaque-like structure to a pretzel shape, a preponderance of NMJs in SMA mice remains small and plaque-like with reduced numbers of AChRs (Fig. 6A). In P14 Δ7+/+ SMA mice, >50% of NMJs remain plaque-like, defined by the lack of perforations in the AChR clusters while only ∼10% of wild-type NMJs lacked perforations (Fig. 6B). SMA NMJs were, on an average, one-half the size of wild-type NMJs (117 ± 47 versus 245 ± 51 µm2; n > 100; P < 0.001), an observation that is likely reflective of the smaller size of the mutant mice. Thus, normal levels of SMN are dispensable for the formation of the NMJ, but required for its post-natal maturation. Impaired maturation of the NMJ is not unique to severe SMA. An examination of the AChR clusters in the gastrocnemius muscle of mild-SMA mice indicated similar albeit less severe defects. Mutant NMJs were found to be structurally less complex than wild-type junctions at all ages examined (Fig. 6B and D).
Changes in the geometric configuration of AChR clusters are but one aspect of NMJ maturation. A second important event that accompanies NMJ maturation involves the replacement of the fetal form of the AChR, containing a γ subunit (composition α2βγδ), with the ε subunit-containing adult form (α2βεδ) during the first 2 weeks of post-natal life in mice (30). To determine whether impaired NMJ maturation in SMA mice, as assessed by the persistence of plaque-like AChR clusters, is also reflected in perturbations of the γ→ε subunit switch, we isolated muscle from P11 Δ7+/+ mutants and Control littermates to quantify levels of the γ subunit transcript. In all the four muscle groups tested, the γ subunit was expressed at significantly higher levels in mutant tissue than in Control samples (Fig. 6C). Abdominal muscle exhibited particularly high levels of the γ subunit transcript, in keeping with early defects of the NMJs in it. This result further indicates the relatively immature status of SMA NMJs and the importance of the SMN protein in ensuring the proper post-natal maturation of the NMJ.
Neuromuscular transmission defects in SMA mice
Given the striking defects we observed at SMA junctions in patients as well as mice, it appeared likely that there would be associated alterations in neuromuscular transmission. We therefore performed intracellular electrophysiological recordings on the predominantly fast-twitch semitendinosus muscles of several 9 months old mild-SMA mice. This muscle is innervated by fast motor neurons that typically fire at 50–100 Hz. In response to repetitive stimulation for 1 s periods at 10, 20, 50 and 100 Hz, all wild-type junctions were able to maintain effective transmission despite a 50–70% depression by the end of the train (Fig. 7A, top trace). 63.3% of SMA junctions sampled responded similarly (Fig. 7A, bottom trace). However, the remaining 36.7% of NMJs exhibited intermittent transmission failures (Fig. 7B) characterized by a complete lack of responses or very small responses of relatively constant amplitude (Fig. 7A, second and third traces, respectively). These failures appeared at frequencies as low as 10 Hz and increased to ∼60% of all stimuli by 100 Hz (Fig. 7C).
Figure 7.
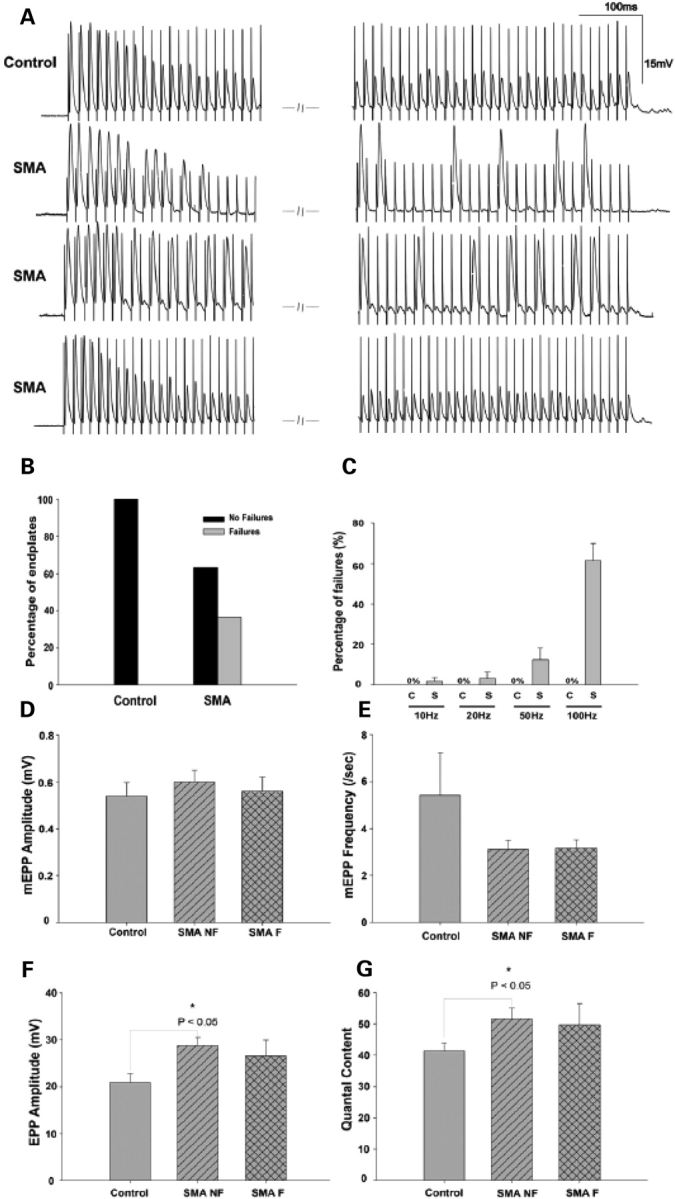
Functional characterization of NMJs in mild-SMA mice. (A) Trains of end-plate potentials (EPPs) at 100 Hz stimulation recorded from mutant and wild-type NMJs. Wild-type junctions displayed moderate depression but had no transmission failures (top trace). A proportion of mutant junctions responded similarly (bottom trace) while the rest (36.7%) exhibited intermittent transmission failures (second and third trace). (B) Graphical representation of mutant and wild-type junctions exhibiting failures (mutant NMJs, n = 30; wild-type NMJs, n = 25). (C) The failure rate (mean for all junctions) increased with stimulus frequency in the SMA mutant junctions; (10 Hz; 1.82 ± 1.82%, 20 Hz; 3.33 ± 2.88%, 50 Hz; 12.59 ± 5.70%, 100 Hz; 61.55±8.29%) while wild-type NMJs did not fail at any frequency tested. (C, wild-type; S, SMA). (D–G) Neurotransmission parameters in mutant (non-failing: SMA NF; failing: SMA F) and wild-type NMJs to single stimuli showing (D) miniature end-plate potentials (mEPP) size, (E) mEPP frequency, (F) EPP amplitude in response to a single suprathreshold stimulus to the nerve and (G) mean quantal content. No significant difference was detected in mEPP size and frequency between mutant and wild-type junctions (P > 0.05; one-way ANOVA); EPPs and quantal content in SMA NF junctions were significantly increased over wild-type values (P < 0.05; one-way ANOVA). Data represented as mean ± SEM.
Intermittent transmission failures similar to the ones we observed were reported in NCAM (neural cell adhesion molecule) and CD24 mutant mice (31–34) and have been ascribed to a defect in the myosin alkali light chain kinase (MLCK) signaling pathway. Failures in these mutants were rescued by the activation of protein kinase C (PKC) with the phorbol ester, phorbol 12-myristate 13-acetate (PMA). Treatment of the SMA junctions with PMA failed to restore normal neurotransmission indicating that the underlying cause of the functional NMJ defects in this disease is not associated with the MLCK signaling pathway. Nevertheless, the results mentioned above indicate that reduced SMN is responsible not just for structural abnormalities of the NMJ but also for functional deficits at the synapse.
Measurements of additional parameters of transmission indicated that there were no significant alterations in miniature end-plate potentials (mEPP) size and frequency (Fig. 7D and E) between either mutant junctions that failed (0.56 ± 0.06 mV) or those that did not (0.60 ± 0.05 mV) compared with Controls (0.54 ± 0.06 mV). However, quantal content and end-plate potentials (EPPs) in response to single stimuli were increased at SMA junctions and reached statistical significance for NMJs that did not fail (Fig. 7F and G). These results may be indicative of a compensatory mechanism in response to an increased threshold for the generation of an action potential in SMA muscle. Finally, paired-pulse facilitation did not differ between Control and SMA junctions although at junctions that exhibited failures, the second stimulus failed to evoke a response at the shortest interval (5 ms) tested (data not shown).
A rostro-caudal gradient defines motor neuron loss in SMA mice
To determine the effect of reduced SMN on motor neuron pathology during the course of the disease in Δ7+/+ mice, we counted axon numbers in the dorsal and ventral roots and the phrenic nerve which is primarily a motor nerve. Motor neuron cell body counts were carried out in parallel. At P0, we saw no difference in motor neuron numbers at either lumbar or cervical levels between SMA and Control mice (data not shown). Neither was there any difference in the numbers of axons in the phrenic nerve (A fibers – SMA = 224 ± 10; Control = 199 ± 25, n = 2; P > 0.1) or of partially myelinated fibers (SMA = 3.8 ± 1.6%; Control = 4.6 ± 2.6%, n = 2; P > 0.1). However, by P14 we found an ∼30% reduction in the number of axons in the phrenic nerve and C4 ventral root of SMA animals (Fig. 8A–D). Interestingly, large caliber (diameter ≥ 3.5 µm) axons were preferentially lost in the ventral roots of SMA mice, a sign of axonal atrophy and suggestive of a selective vulnerability of larger motor neurons in the spinal cord (Fig. 8C). A similar loss of large caliber axons was observed in the phrenic nerve (Fig. 8D). Assuming that the large caliber axons arise from α-motor neurons, this observation is consistent with our findings at the terminals of α and γ motor neurons. No difference was noted in the number of axons in the dorsal root (Fig. 8A and B). The loss of axons in cervical ventral roots was paralleled by a decrease in numbers of choline acetyl transferase (ChAT) positive motor neurons in the cervical spinal cord of P14 SMA animals (Fig. 8E). Moreover, surviving motor neurons were significantly smaller in affected mice (Controls – 825 ± 20 µM2; SMA – 600 ± 20 µM2, n = 3; P < 0.01). Surprisingly, an assessment of lumbar motor neuron cell bodies and axons from ventral roots at the L4 level indicated no significant difference in the numbers between SMA and wild-type littermates (Fig. 8A and B). While we do not construe this as an indication of functional motor neurons at lumbar levels, our data is indicative of a caudal to cranial gradient in motor neuron loss in SMA and is consistent with early defects detected at NMJs in the diaphragm and intercostal muscles.
Figure 8.

Motor neuron and motor axon loss in SMA mice. (A) Quantification of myelinated axons in C4-dorsal roots (C4-d), C4-ventral roots (C4-v), PN (phrenic nerves), and L4-ventral roots (L4-v) in P14 Δ7+/+ SMA (S) mice and age-matched Controls (C). Data represented as mean ± SD (n = 3). ‘Asterisk' denotes P < 0.01 (Student's t-test). n.s.: not significant. (B) Transverse sections through the C4 and L4 roots and phrenic nerve from a P14 Δ7+/+ SMA mouse and a Control (Ctrl) indicating selective loss and atrophy of axons in the C4 root and phrenic nerve. Axons in the caudally located L4 root are spared. Scale bar: 100 µm. (C, D) Graphs indicating loss of large caliber axons in the C4 root and phrenic nerve in P14 Δ7+/+ SMA mice. (E) Motor neuron cell body counts indicate a selective and significant loss (∼30%) in the rostral but not caudal spinal cords of P14 Δ7+/+ SMA (S) mice; Controls (C). ‘Asterisk' denotes P < 0.01 (Student's t-test); n = 3. n.s.: not significant.
DISCUSSION
In this study we used animal models of SMA to investigate the earliest cellular consequences of reduced SMN and to chart the process of neuromuscular degeneration as the disease progresses. Our analysis has defined when and where reduced SMN causes disease pathology and, from a clinical perspective, provided us with important information to design effective treatments for the disease. Our main findings were: (1) Cellular defects precede overt phenotypic characteristics. (2) The first detectable structural consequences of reduced SMN are abnormalities at the distal end of the motor neuron – the NMJ suggesting that SMA may best be described as a NMJ synaptopathy. The defects first appear in the neonate and become progressively worse during the course of the disease. (3) Pre-synaptically, the defects are defined by abnormal NF accumulations in the nerve terminals and poor terminal arborization. In mild-SMA mice, NF aggregates are restricted to pre-terminal axons. (4) Post-synaptically, the defects are defined by the persistence of (a) immature plaque-like AChR clusters and (b) the embryonic (γ subunit-containing) form of the receptor. Together the pre- and post-synaptic abnormalities are suggestive of an impaired maturation of the NMJ and imply that SMN plays an important role in ensuring proper post-natal development of the neuromuscular synapse. Importantly, NMJ defects in SMA mice were confirmed in muscle tissue from human patients. (5) SMA is characterized by a selective vulnerability of the largest (α) motor neurons of the ventral spinal cord. γ-motor neurons as assessed by terminal NF aggregates are spared in the disease. Furthermore, abnormal NF aggregates in the α-motor neurons are restricted to the distal axon and never extend into the perikarya. (6) Structural defects at the NMJ are reflected in functional synaptic transmission defects. (7) SMA involves a selective degeneration of rostrally located motor neurons and their axons.
Structural abnormalities of the NMJ: a hallmark of SMA pathology
SMA has long been defined as a neuromuscular disease with an emphasis on the selective loss of spinal motor neurons and skeletal muscle atrophy (35). Surprisingly, the NMJ has not been examined in greater detail. Existing knowledge of disease pathology is based largely on post-mortem material and biopsies. While this does not explain why the NMJ has received so little attention, it did preclude an analysis of the earliest effects of reduced SMN and a detailed profile of the neurodegenerative process as the disease progresses. Here we sought to fill this gap using mouse models of SMA. In part the study was prompted by observations indicating a lag between the onset of disease symptoms and spinal motor neuron loss (21). Importantly, and unlike previous reports, our results are based on the analysis of transgenic mice that express a constant, low level of the SMN protein, akin to the human condition. This is an important distinction between the current study and previous reports that relied on models in which the SMN protein is eventually completely depleted, either generally or in selected cell types (18,19).
Our analysis of severe as well as mild-SMA mice indicates that NMJ defects appear well before the disease phenotype becomes apparent. Abnormalities include poor terminal arborization, accumulation of NF aggregates in nerve terminals and/or pre-terminal axons and poorly developed, immature looking AChR clusters that never attain the complex pretzel-shaped structures seen in wild-type animals. Although many of these defects are common to severe as well as mild forms of SMA, their extent and severity are not. NF accumulation is a late event in mild SMA and is restricted to the pre-terminal axon; in severe SMA, aggregates appear as early as P0 and invade not just the pre-terminal axon, but nerve terminals as well. It has been suggested that NF accumulation in the pre-synapse is specific to SMA (18). Our data suggests that while this may be true of severe SMA, it is not characteristic of milder forms of the disease. Indeed, the pattern of NF accumulation in mild SMA is common to at least two other neurodegenerative diseases – ALS (Fig. 3G) and Huntington's Disease (36), although in neither of these two mouse models do the NMJs exhibit poor terminal arborization and immature AChR clusters, features unique to severe as well as mild-SMA mice. We therefore suggest that while terminal NF accumulation is, (a) an important aspect of SMA pathology, (b) selectively affects α-motor neurons and (c) probably greatly detrimental to the normal functioning of the cell by disrupting processes such as axonal transport, it is nonetheless more likely a consequence rather than cause of motor neuron dysfunction. The absence of a significant increase in unoccupied AChR clusters we report in muscle from the Δ7+/+ SMA mice is not necessarily inconsistent with a decrease in fully occupied endplates (23), since the latter observation can be fully ascribed to an increase in partially occupied NMJs. Increased re-innervation in our Δ7+/+ SMA mice, an alternative explanation for why we fail to see more unoccupied endplates in the mutants is unlikely given that re-innervation of denervated neonatal/perinatal muscle is deficient and axonal sprouting absent (37).
Mechanisms that link reduced SMN to NF accumulation in distal axons and terminals have yet to be defined. Along with microtubules, these intermediate filaments play an important role in axon outgrowth and plasticity (38), but are cleared from terminals once the axon reaches its target, presumably by calcium-activated proteases (39). A recent report indicated that reduced SMN impairs the clustering of voltage-gated calcium channels (VGCCs) at the growth cone, resulting in decreased influx of Ca2+ ions into the distal axon (40). One intriguing possibility is that reduced Ca2+ in SMA terminals fails to activate calcium proteases at the synapse eventually causing a build-up of NF protein. Data to support this hypothesis will require investigating the distribution of VGCCs at the NMJs of SMA mice.
Our observations do not allow us to conclusively determine whether pre-synaptic defects precede post-synaptic abnormalities or not. In severe SMA, pre-synaptic defects as assessed by poor terminal arborization and NF accumulation in motor nerve terminals are the first structurally detectable abnormalities at the NMJ, arguing that impaired assembly of the post-synaptic specialization follows nerve defects. However, post-synaptic defects in neonates may simply be too subtle to detect by light microscopy and a recent report (23) suggests that post-synaptic changes occur independently of those in the nerve. Furthermore, in mildly affected mice, poor terminal arborization was always detected in conjunction with defects at the post-synapse defined by AChR clusters lacking in structural complexity. This suggests that reduced SMN in muscle may independently contribute to the SMA phenotype by perturbing post-synaptic maturation. One possible explanation for the differences in defects at the NMJs of the severe- and mild-SMA mice follows from the assumption that motor neurons express much higher levels of the SMN protein than muscle and that small fluctuations in protein levels have a more profound effect on the former cell type. By extension, one may argue that an increase from acutely low levels of SMN as seen in severe SMA to a mild decrease in protein as seen in type III/IV SMA is disproportionately beneficial to the motor neurons versus the muscle. Thus in mild forms of the disease, dysfunction in nerve and muscle occur in parallel, while in severe-SMA, defects in nerve (pre-synapse) precede those in muscle (post-synapse). One might postulate that these observations reflect different functions of the SMN protein, which are disrupted in a hierarchical manner based on relative levels in a cell type. If true, the influence of the pre-synapse on the post-synaptic specialization and vice versa that may be playing out in SMA is consistent with a complex signaling pathway between the two halves of the NMJ that ensure maturation and maintenance of the structure (41).
Functional deficits at the NMJ in SMA
The most obvious functional defect was the occurrence, in approximately a third of the SMA mutant junctions studied, of cyclical periods of transmission failures during brief trains of repetitive stimulation at frequencies similar to the in vivo firing rates of fast limb innervating motor neurons. Such failures would cause many muscle fibers to be ineffectively activated for the production of maximal tension during locomotor movements, and thus contribute to muscle weakness. We showed that the failures are not linked to abnormal activation of MLCK/myosyin II pathway that is required for effective synaptic vesicle cycling. An alternative possibility, compatible with the complete absence of post-synaptic responses or with very small responses, would be the blockade of action potentials proximal to the junction. Although mouse action potentials do not actively propagate into the nerve terminal, which is normally electronically depolarized by currents from the last hemi-node (42), more proximal blockade of action potentials or alterations in the passive electrical properties of the pre-terminal axon could produce the observed responses and might be caused by the large, NF-filled swellings of pre-terminal axons we observed in the mild-SMA mutant junctions. Epineural recordings (33) will be needed to confirm this mechanism.
In contrast to trains, responses to single stimuli were generally normal at SMA junctions, even in those that exhibited transmission failures. Normal mEPP amplitude suggests a normal density of post-synaptic AChRs while normal paired-pulse facilitation suggests that neither the probability of release to the first stimulus, nor Ca2+ dynamics in the terminal is greatly altered. These conclusions are further supported by the fact that quantal content was not reduced below wild-type levels, and that the synchrony of vesicle release during EPPs was unaltered. The reason for the slight increase in quantal content is currently unclear.
In the growth cones of severe-SMA mice, alterations in the clustering of N-type VGCCs and reductions in Ca2+ transients in response to synaptic (β-2 laminin) were reported (40). These could cause defects in early synaptic transmission and contribute to the regression of synapses that we reported here for these mice. However, the relatively normal transmission parameters in the mild-SMA mice suggest that synapse formation has occurred normally and that the density and distribution of the P/Q type VGCCs responsible for adult transmission are not greatly altered. Taken together the results suggest that different levels of functional SMN may be required for different aspects of synapse formation and maintenance. Nevertheless, despite relatively normal synapse formation in the mild-SMA mice, functional defects in synaptic transmission occur later in life and are likely to contribute to weakness and eventual death. It will be interesting to compare any alterations in functional transmission in the severe-SMA mice with those we have described for the weaker phenotype.
Selective vulnerability of rostrally located spinal motor neurons in SMA mice
Given the results (21) demonstrating motor neuron loss in the lumbar spinal cord, we were surprised to find no difference in cell body and ventral root axon counts between diseased (P14 Δ7+/+ SMA) and age-matched Controls at L4 levels. Interestingly, motor neuron and motor axon loss at cervical levels is significant suggesting a selective vulnerability of rostrally located spinal motor neurons to neurodegeneration. Based on NMJ defects, it has also been suggested that certain sub-populations of motor neurons but not others innervating the same muscle are preferentially affected in SMA mice (23). The vulnerable motor neurons conform to those defined as FaSyn (fast-synapsing) (43) while a neighboring pool from the same motor nucleus, shown to be DeSyn (delayed-synapsing), was relatively spared. Although it is tempting to extrapolate these findings and conclude that all FaSyn motor neurons are vulnerable and those that are DeSyn resistant, we suggest caution. As discussed earlier, the diaphragm, which is DeSyn and therefore expected to be unaffected, instead exhibits profound defects of the NMJs in SMA mice and human patients. Phrenic nerve axons innervating the diaphragm also degenerate. Functionally, however, this muscle is thought to be relatively spared, a paradox that could be explained if the diaphragm is capable of tolerating a far greater loss of healthy motor units than other proximal muscle groups. Our findings only highlight the need for additional studies to identify exactly which motor neurons are resistant and which are susceptible to degeneration in SMA. Nonetheless, having broadly classified relatively refractory (lumbar) versus vulnerable (cervical) motor neurons along the rostro-caudal axis and within pools (γ-motor neurons versus α-motor neurons), one may at least begin to ask what distinguishes them from one another molecularly as a way to further elucidate the function of the SMN protein in SMA.
An explanation for the selective vulnerability of motor neurons remains elusive, but likely has to do with the different SMN complexes known to exist (44–46). Whether motor neuronal defects are solely responsible for the SMA phenotype or whether perturbations in other cell types contribute to disease pathology continues to be debated. Numerous reports have implied a role for SMN in muscle that is relevant to the SMA phenotype (16,47–50) and it has recently been suggested that the protein may be important in myofiber maturation (51). On the other hand over-expressing SMN in muscle alone fails to alleviate the SMA phenotype in SMA mice (52). Selectively depleting SMN in muscle and nerve of transgenic mice while ensuring the expression of low levels of the protein in these cell types will determine whether SMA is truly a cell-autonomous disease or not.
In conclusion, we have used transgenic mice expressing constant low levels of the SMN protein to demonstrate that NMJ defects are a key component of SMA pathology. Importantly, the findings were confirmed in human SMA patients. Our analysis has revealed that reduced SMN acting either through the pre-synaptic apparatus and/or directly at the post-synapse impairs the maturation of the NMJ. These early and profound defects at the neuromuscular synapse very likely explain muscle weakness in SMA and warrant the search for strategies that would maintain function at the NMJs as a means of treating the disease.
MATERIALS AND METHODS
Generation of transgenic mice and breeding strategy
SMA carrier mice, SMN2+/+;SMNΔ7+/+;Smn+/− and SMN2+/+;Smn+/− were used to generate severe Δ7+/+ and Δ7+/− SMA mice. Mild-SMA mice (SMN2+/+;SMN1 A2G+/−;Smn−/−) were produced by interbreeding SMN2+/+;SMN1 A2G+/−;Smn+/− with SMN2+/+;Smn+/− animals. The partially congenic SMN2+/+;SMNΔ7+/+;Smn+/− carriers (21) and the SOD1G93A mutant mice were obtained from Jackson Laboratory (Bar Harbor, ME, USA). Genotypes were determined as described previously (7,20,21). All animal procedures were performed in accordance with institutional guidelines.
Motor behavioral analysis
Righting reflex was used to estimate muscle strength in SMA and Control mice as previously described (53). Each mouse was placed on its back and the time taken to place all four limbs on the ground was measured. The following arbitrary scores were used to quantify impaired righting ability: 5 for 0–2 s righting ability, 4 for 3–5 s, 3 for 6–10 s, 2 for 11–30 s, 1 for 31–60 s and 0 if >61 s. The procedure was repeated six times for each animal, and the sum of the scores was designated as righting ability score. Body weight was measured at the same time.
Southern and western blotting analysis
SMNΔ7 transgene copy number in fully (N11 FVB/N) and partially (N6 FVB/N) congenic Δ7+/+ mice used in the study was determined by Southern blotting as described previously (7). The single murine Smn knockout allele served as a loading control. Band intensities were quantified using Image J software (NIH, Bethesda, MD, USA). Western blotting was performed according to standard procedures and as previously described (7). Primary antibodies used were anti-SMN (MANSMA19; 1:2500); rabbit polyclonal anti-human FL-SMN (1:500), anti-NF M (1:1000; Chemicon International, Temecula, CA, USA), anti-synaptic vesicle protein 2 (1:500; Developmental Studies Hybridoma Bank, Iowa City, IA, USA) and anti-α-tubulin (1:1000; Sigma-Aldrich, Saint Louis, MO, USA).
Human diaphragm muscle
Diaphragm muscle was removed post-mortem from Type I SMA patients at John Hopkins University, frozen immediately and stored at −80°C without fixation. Non-SMA Control diaphragm muscle samples were obtained at Columbia University from patients with diaphragmatic hernia and immediately processed. All samples were obtained from patients <6 months of age (SMA, n = 3; Control, n = 3) in accordance with institutional guidelines and IRB approved protocols.
Immunohistochemical and ultrastructural analysis of NMJs
NMJ analysis was carried out on whole muscle, fixed and permeabilized for 5 min in 100% methanol, and incubated with 3% BSA to prevent any non-specific binding. Tissue was then incubated with the primary antibody for 48 h at 4°C, washed with PBS-Tween and incubated for 24 h at 4°C with appropriate fluorescently conjugated secondary antibody and/or rhodamine-α-bungarotoxin (BTX , 1:1000, Molecular Probes, Eugene, OR, USA). After further washing, larger muscles were teased to separate muscle fibers before mounting in Vectashield (Vector Laboratories, Burlingame, CA, USA). Primary antibodies used were: anti-NF 160 kDa (1:600; Chemicon International), anti-synaptic vesicle protein 2 (1:150; Developmental Studies Hybridoma Bank, Iowa City, IA, USA), anti-NF-M (1:1200; Chemicon International), anti-vesicular glutamate transporter 1 (1:2000; Chemicon International). Abnormal pre-synapses were defined as having thick, poorly branched terminals staining strongly for NF protein. To objectively distinguish between normal and abnormal pre-synapses, mean pixel density of the nerve terminals following NF staining was assessed. Samples were imaged under identical gain and exposure settings. All terminals with a mean pixel density ≥150 arbitrary units were deemed abnormal.
Motor endplate morphology was assessed in BTX (1:1200) stained 50 µm longitudinal sections of the gastrocnemius. Approximately 100 endplates per section were randomly selected to determine structure and size. Post-synaptic structural complexity in the severe-SMA mice was defined by the number of endplates lacking perforations (plaque-like motor endplates), and in the mild-SMA mice the number of endplates with <3 perforations. Endplate size was measured using SPOT advanced image analysis software (Diagnostic Instruments, Starling Heights, MI, USA). Images were taken using a Nikon Eclipse 80i fluorescence microscope (Nikon, Tokyo, Japan) equipped with a Spot Flex digital camera (Diagnostic Instruments, Starling Heights, MI, USA). Confocal images were obtained on a laser scanning confocal (BioRad Laboratory, Hercules, CA, USA).
NMJs in the diaphragm were examined ultrastructurally. Mice were perfused with 4% PFA and the diaphragm dissected out and post-fixed in 2.5% glutaraldehyde (24 h, 4°C). Tissue was then immersed in 0.1% osmium tetroxide, embedded in epoxy resin and sectioned at 60–90 nm. Sections were stained with uranyl acetate and lead citrate and NMJs imaged on a JEOL JEM-1200 EXII electron microscope equipped with a Hamamatsu OCRA XR-60 camera. Electron micrographs were taken at 80 kV.
Quantitative PCR
To quantify levels of the γ subunit of the AChR, RNA was isolated with Trizol (Invitrogen, CA, USA) according to the manufacturer's instructions. Following cDNA synthesis, quantitative PCR was carried out in triplicate on the Mx3000P system with accompanying software (Stratagene, CA, USA). β-actin was used as a normalizer to account for differences in cDNA input. Relative quantification of AChRγ mRNA, corrected for the quantity of β-actin mRNA was divided by a calibrator value (wild-type value) to calculate transcript levels. Primers used were AChRγF: 5′-GACCAACCTCATCTCCCTGA-3′ and AChRγR: 5′-GAGAGCCACCTCGAAGACAC-3′.
Morphological analysis of spinal motor neurons and motor axons
Cervical spinal cords dissected from mice perfused transcardially with 4% PFA were post-fixed in the same solution (2 h at 4°C) and then cryoprotected in 30% sucrose. Transverse sections, 20 µm, were incubated with anti-ChAT (1:100, Chemicon International) and/or anti-NF-M (1:1200) for 48 h at 4°C. After washing, sections were incubated with the appropriate fluorescently conjugated secondary antibody. To calculate the average number of ChAT-positive motor neurons within the anterior horn, 12 sections from each mouse were randomly selected and counted. This figure was then multiplied by the length of the spinal cord segment/section thickness. The perikaryal size of ChAT-positive motor neurons was measured using SPOT advanced image analysis software. Transmission electron microscopy (EM) was used to characterize the ultrastructure of the spinal motor neurons. Samples were prepared and analyzed as described earlier.
To analyze axons, the C4 and L4 ventral and dorsal roots as well as phrenic nerves were harvested from animals perfused as described earlier. Tissue was post-fixed in 2.5% glutaraldehyde for 24 h at 4°C, dehydrated in graded alcohols and embedded in epoxy resin. 0.6 µm sections were cut and stained with toluidine blue to assess axon morphology and number. To assess the number of A fibers in P0 phrenic nerves, transverse sections were stained and viewed using transmission EM as described earlier.
Intracellular recordings
Standard electrophysiological techniques were used to record from acutely isolated semitendinosus muscles as described previously (34). The muscle preparation was perfused with well-oxygenated Tyrode's solution (125 mm NaCl, 5 mm KCl, 24 mm NaHCO3, 1 mm MgCl2, 10 mm Glucose) with physiological (2 mm) Ca2+ levels to evoke in vivo levels of transmitter release. The solution also contained 1–2 µM μ-conotoxin GIIIB (Alomone Labs, Jerusalem, Israel), a specific blocker of muscle voltage-gated sodium channels, to prevent muscle contraction. Sharp glass electrodes were pulled (10–20 MΩ), filled with 3 M KCl, and single muscle fibers were impaled near the motor nerve endings. Potentials were recorded via an intracellular amplifier (World Precision Instruments, Sarasoto, FL, USA) using Axoscope software (40 KHz sampling rate; Molecular Devices, Sunnyvale, CA, USA). The nerves were stimulated via a suction electrode pulled from polyethylene tubing (PE-190; Becton Dickinson, sparks, MD, USA) and an SQ38 stimulator and PSIU6B stimulus isolation unit (Grass Technologies, West Warwick, RI, USA). Quantal content was calculated as mean EPP amplitude/mean mEPP amplitude while paired-pulse facilitation was assessed at intervals ranging from 5 to 200 ms as the ratio of the amplitude of the second EPP to the first EPP.
Statistics
The log-rank test was used to determine if mean survival values between N11 FVB/N Δ7+/+, Δ7+/− and N6 FVB/N Δ7+/+ mice was significantly different. Data in the manuscript is represented as mean ± SD unless indicated. The two-tailed Student's t-test or one-way ANOVA where indicated were used to compare means for statistical significance.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
FUNDING
SMA Foundation; Families of SMA; Muscular Dystrophy Association of America; American Academy of Neurology/SMA Foundation Young Investigator Award to U.M. National Institutes of Health (NS23678 and NS19640) to L.T.L.
ACKNOWLEDGEMENTS
We thank Drs A. Burghes, D.C. De Vivo and M. Winberg for comments and useful discussions, Dr T.M. Jessell for advice, reagents and access to essential equipment, Dr G.E. Morris for SMN antibodies and Dr T. Crawford for patient tissue. We are deeply grateful to Dr C.E. Henderson for advice, suggestions and a critical reading of this manuscript. R. Mauricio, C. Neeb and K. Brown provided technical help.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 2.Lorson C.L., Hahnen E., Androphy E.J., Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl Acad. Sci. USA. 1999;96:6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Monani U.R., Lorson C.L., Parsons D.W., Prior T.W., Androphy E.J., Burghes A.H., McPherson J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 4.Vitali T., Sossi V., Tiziano F., Zappata S., Giuli A., Paravatou-Petsotas M., Neri G., Brahe C. Detection of the survival motor neuron (SMN) genes by FISH: further evidence for a role for SMN2 in the modulation of disease severity in SMA patients. Hum. Mol. Genet. 1999;8:2525–2532. doi: 10.1093/hmg/8.13.2525. [DOI] [PubMed] [Google Scholar]
- 5.McAndrew P.E., Parsons D.W., Simard L.R., Rochette C., Ray P.N., Mendell J.R., Prior T.W., Burghes A.H. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet. 1997;60:1411–1422. doi: 10.1086/515465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feldkotter M., Schwarzer V., Wirth R., Wienker T.F., Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time light Cycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002;70:358–368. doi: 10.1086/338627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Monani U.R., Sendtner M., Coovert D.D., Parsons D.W., Andreassi C., Le T.T., Jablonka S., Schrank B., Rossol W., Prior T.W., et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 8.Hsieh-Li H.M., Chang J.G., Jong Y.J., Wu M.H., Wang N.M., Tsai C.H., Li H. A mouse model for spinal muscular atrophy. Nat. Genet. 2000;24:66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 9.Monani U.R. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48:885–896. doi: 10.1016/j.neuron.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Pagliardini S., Giavazzi A., Setola V., Lizier C., Di Luca M., DeBiasi S., Battaglia G. Subcellular localization and axonal transport of the survival motor neuron (SMN) protein in the developing rat spinal cord. Hum. Mol. Genet. 2000;9:47–56. doi: 10.1093/hmg/9.1.47. [DOI] [PubMed] [Google Scholar]
- 11.McWhorter M.L., Monani U.R., Burghes A.H., Beattie C.E. Knockdown of the survival motor neuron (smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J. Cell Biol. 2003;162:919–931. doi: 10.1083/jcb.200303168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shafey D., Cote P.D., Kothary R. Hypomorphic smn knockdown C2C12 myoblasts reveal intrinsic defects in myoblast fusion and myotube morphology. Exp. Cell Res. 2005;311:49–61. doi: 10.1016/j.yexcr.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 13.Fan L., Simard L.R. Survival motor neuron (SMN) protein: role in neurite outgrowth and neuromuscular maturation during neuronal differentiation and development. Hum. Mol. Genet. 2002;11:1605–1614. doi: 10.1093/hmg/11.14.1605. [DOI] [PubMed] [Google Scholar]
- 14.Zhang H.L., Pan F., Hong D., Shenoy S.M., Singer R.H., Bassell G.J. Active transport of the survival motor neuron protein and the role of exon-7 in cytoplasmic localization. J. Neurosci. 2003;23:6627–6637. doi: 10.1523/JNEUROSCI.23-16-06627.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Broccolini A., Engel W.K., Askanas V. Localization of survival motor neuron protein in human apoptotic-like and regenerating muscle fibers, and neuromuscular junctions. Neuroreport. 1999;10:1637–1641. doi: 10.1097/00001756-199906030-00003. [DOI] [PubMed] [Google Scholar]
- 16.Arnold A.S., Gueye M., Guettier-Sigrist S., Courdier-Fruh I., Coupin G., Poindron P., Gies J.P. Reduced expression of nicotinic AChRs in myotubes from spinal muscular atrophy I patients. Lab. Invest. 2004;84:1271–1278. doi: 10.1038/labinvest.3700163. [DOI] [PubMed] [Google Scholar]
- 17.Frugier T., Tiziano F.D., Cifuentes-Diaz C., Miniou P., Roblot N., Dierich A., Le Meur M., Melki J. Nuclear targeting defect of SMN lacking the C-terminus in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2000;9:849–858. doi: 10.1093/hmg/9.5.849. [DOI] [PubMed] [Google Scholar]
- 18.Cifuentes-Diaz C., Nicole S., Velasco M.E., Borra-Cebrian C., Panozzo C., Frugier T., Millet G., Roblot N., Joshi V., Melki J. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum. Mol. Genet. 2002;11:1439–1447. doi: 10.1093/hmg/11.12.1439. [DOI] [PubMed] [Google Scholar]
- 19.Chan Y.B., Miguel-Aliaga I., Franks C., Thomas N., Trulzsch B., Sattelle D.B., Davies K.E., van den Heuvel M. Neuromuscular defects in a drosophila survival motor neuron gene mutant. Hum. Mol. Genet. 2003;12:1367–1376. doi: 10.1093/hmg/ddg157. [DOI] [PubMed] [Google Scholar]
- 20.Monani U.R., Pastore M.T., Gavrilina T.O., Jablonka S., Le T.T., Andreassi C., DiCocco J.M., Lorson C., Androphy E.J., Sendtner M., et al. A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J. Cell Biol. 2003;160:41–52. doi: 10.1083/jcb.200208079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Le T.T., Pham L.T., Butchbach M.E., Zhang H.L., Monani U.R., Coovert D.D., Gavrilina T.O., Xing L., Bassell G.J., Burghes A.H. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 22.Nadeau J.H. Modifier genes and protective alleles in humans and mice. Curr. Opin. Genet. Dev. 2003;13:290–295. doi: 10.1016/s0959-437x(03)00061-3. [DOI] [PubMed] [Google Scholar]
- 23.Murray L.M., Comley L.H., Thomson D., Parkinson N., Talbot K., Gillingwater T.H. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2007 doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- 24.Munsat T.L., Davies K.E. International SMA consortium meeting (26–28 June 1992, Bonn, Germany) Neuromuscul. Disord. 1992;2:423–428. doi: 10.1016/s0960-8966(06)80015-5. [DOI] [PubMed] [Google Scholar]
- 25.Bach J.R., Saltstein K., Sinquee D., Weaver B., Komaroff E. Long-term survival in werdnig-hoffmann disease. Am. J. Phys. Med. Rehabil. 2007;86:339–345. doi: 10.1097/PHM.0b013e31804a8505. Quiz 346–348, 379. [DOI] [PubMed] [Google Scholar]
- 26.Swoboda K.J., Prior T.W., Scott C.B., McNaught T.P., Wride M.C., Reyna S.P., Bromberg M.B. Natural history of denervation in SMA: relation to age, SMN2 copy number, and function. Ann. Neurol. 2005;57:704–712. doi: 10.1002/ana.20473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Norgren N., Rosengren L., Stigbrand T. Elevated neurofilament levels in neurological diseases. Brain Res. 2003;987:25–31. doi: 10.1016/s0006-8993(03)03219-0. [DOI] [PubMed] [Google Scholar]
- 28.Fischer L.R., Culver D.G., Tennant P., Davis A.A., Wang M., Castellano-Sanchez A., Khan J., Polak M.A., Glass J.D. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 29.Tu P.H., Gurney M.E., Julien J.P., Lee V.M., Trojanowski J.Q. Oxidative stress, mutant SOD1, and neurofilament pathology in transgenic mouse models of human motor neuron disease. Lab. Invest. 1997;76:441–456. [PubMed] [Google Scholar]
- 30.Missias A.C., Chu G.C., Klocke B.J., Sanes J.R., Merlie J.P. Maturation of the acetylcholine receptor in skeletal muscle: regulation of the AChR gamma-to-epsilon switch. Dev. Biol. 1996;179:223–238. doi: 10.1006/dbio.1996.0253. [DOI] [PubMed] [Google Scholar]
- 31.Jevsek M., Jaworski A., Polo-Parada L., Kim N., Fan J., Landmesser L.T., Burden S.J. CD24 is expressed by myofiber synaptic nuclei and regulates synaptic transmission. Proc. Natl Acad. Sci. USA. 2006;103:6374–6379. doi: 10.1073/pnas.0601468103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Polo-Parada L., Bose C.M., Landmesser L.T. Alterations in transmission, vesicle dynamics, and transmitter release machinery at NCAM-deficient neuromuscular junctions. Neuron. 2001;32:815–828. doi: 10.1016/s0896-6273(01)00521-9. [DOI] [PubMed] [Google Scholar]
- 33.Polo-Parada L., Bose C.M., Plattner F., Landmesser L.T. Distinct roles of different neural cell adhesion molecule (NCAM) isoforms in synaptic maturation revealed by analysis of NCAM 180 kDa isoform-deficient mice. J. Neurosci. 2004;24:1852–1864. doi: 10.1523/JNEUROSCI.4406-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polo-Parada L., Plattner F., Bose C., Landmesser L.T. NCAM 180 acting via a conserved C-terminal domain and MLCK is essential for effective transmission with repetitive stimulation. Neuron. 2005;46:917–931. doi: 10.1016/j.neuron.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 35.Crawford T.O., Pardo C.A. The neurobiology of childhood spinal muscular atrophy. Neurobiol. Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- 36.Ribchester R.R., Thomson D., Wood N.I., Hinks T., Gillingwater T.H., Wishart T.M., Court F.A., Morton A.J. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the huntington's disease mutation. Eur. J. Neurosci. 2004;20:3092–3114. doi: 10.1111/j.1460-9568.2004.03783.x. [DOI] [PubMed] [Google Scholar]
- 37.Trachtenberg J.T., Thompson W.J. Schwann cell apoptosis at developing neuromuscular junctions is regulated by glial growth factor. Nature. 1996;379:174–177. doi: 10.1038/379174a0. [DOI] [PubMed] [Google Scholar]
- 38.Lasek R.J., Hoffman P.N. In: Cell Motility. Goldman R., Pollard T., Rosesnbaum J., editors. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1976. p. 1021. [Google Scholar]
- 39.Roots B.I. Neurofilament accumulation induced in synapses by leupeptin. Science. 1983;221:971–972. doi: 10.1126/science.6192501. [DOI] [PubMed] [Google Scholar]
- 40.Jablonka S., Beck M., Lechner B.D., Mayer C., Sendtner M. Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J. Cell Biol. 2007;179:139–149. doi: 10.1083/jcb.200703187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanes J.R., Lichtman J.W. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat. Rev. Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- 42.Brigant J.L., Mallart A. Presynaptic currents in mouse motor endings. J. Physiol. 1982;333:619–636. doi: 10.1113/jphysiol.1982.sp014472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pun S., Sigrist M., Santos A.F., Ruegg M.A., Sanes J.R., Jessell T.M., Arber S., Caroni P. An intrinsic distinction in neuromuscular junction assembly and maintenance in different skeletal muscles. Neuron. 2002;34:357–370. doi: 10.1016/s0896-6273(02)00670-0. [DOI] [PubMed] [Google Scholar]
- 44.Sharma A., Lambrechts A., Hao Le T., Le T.T., Sewry C.A., Ampe C., Burghes A.H., Morris G.E. A role for complexes of survival of motor neurons (SMN) protein with gemins and profilin in neurite-like cytoplasmic extensions of cultured nerve cells. Exp. Cell Res. 2005;309:185–197. doi: 10.1016/j.yexcr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 45.Zhang H., Xing L., Rossoll W., Wichterle H., Singer R.H., Bassell G.J. Multiprotein complexes of the survival of motor neuron protein SMN with gemins traffic to neuronal processes and growth cones of motor neurons. J. Neurosci. 2006;26:8622–8632. doi: 10.1523/JNEUROSCI.3967-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hao Le T., Fuller H.R., Lam Le T., Le T.T., Burghes A.H., Morris G.E. Absence of gemin5 from SMN complexes in nuclear cajal bodies. BMC Cell Biol. 2007;8:28. doi: 10.1186/1471-2121-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henderson C.E., Hauser S.L., Huchet M., Dessi F., Hentati F., Taguchi T., Changeux J.P., Fardeau M. Extracts of muscle biopsies from patients with spinal muscular atrophies inhibit neurite outgrowth from spinal neurons. Neurology. 1987;37:1361–1364. doi: 10.1212/wnl.37.8.1361. [DOI] [PubMed] [Google Scholar]
- 48.Braun S., Croizat B., Lagrange M.C., Warter J.M., Poindron P. Constitutive muscular abnormalities in culture in spinal muscular atrophy. Lancet. 1995;345:694–695. doi: 10.1016/s0140-6736(95)90869-2. [DOI] [PubMed] [Google Scholar]
- 49.Cifuentes-Diaz C., Frugier T., Tiziano F.D., Lacene E., Roblot N., Joshi V., Moreau M.H., Melki J. Deletion of murine SMN exon 7 directed to skeletal muscle leads to severe muscular dystrophy. J. Cell Biol. 2001;152:1107–1114. doi: 10.1083/jcb.152.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rajendra T.K., Gonsalvez G.B., Walker M.P., Shpargel K.B., Salz H.K., Matera A.G. A drosophila melanogaster model of spinal muscular atrophy reveals a function for SMN in striated muscle. J. Cell Biol. 2007;176:831–841. doi: 10.1083/jcb.200610053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nadeau A., D'Anjou G., Debray G., Robitaille Y., Simard L.R., Vanasse M. A newborn with spinal muscular atrophy type 0 presenting with a clinicopathological picture suggestive of myotubular myopathy. J. Child Neurol. 2007;22:1301–1304. doi: 10.1177/0883073807307105. [DOI] [PubMed] [Google Scholar]
- 52.Gavrilina T.O., McGovern V.L., Workman E., Crawford T.O., Gogliotti R.G., DiDonato C.J., Monani U.R., Morris G.E., Burghes A.H. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum. Mol. Genet. 2008;17:1063–1075. doi: 10.1093/hmg/ddm379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Butchbach M.E., Edwards J.D., Burghes A.H. Abnormal motor phenotype in the SMN Delta7 mouse model of spinal muscular atrophy. Neurobiol. Dis. 2007;27:207–219. doi: 10.1016/j.nbd.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.