

Abstract
PTHR1-signaling pathway is critical for the regulation of endochondral ossification. Thus, abnormalities in genes belonging to this pathway could potentially participate in the pathogenesis of Ollier disease/Maffucci syndrome, two developmental disorders defined by the presence of multiple enchondromas. In agreement, a functionally deleterious mutation in PTHR1 (p.R150C) was identified in enchondromas from two of six unrelated patients with enchondromatosis. However, neither the p.R150C mutation (26 tumors) nor any other mutation in the PTHR1 gene (11 patients) could be identified in another study. To further define the role of PTHR1-signaling pathway in Ollier disease and Maffucci syndrome, we analyzed the coding sequences of four genes (PTHR1, IHH, PTHrP and GNAS1) in leucocyte and/or tumor DNA from 61 and 23 patients affected with Ollier disease or Maffucci syndrome, respectively. We identified three previously undescribed missense mutations in PTHR1 in patients with Ollier disease at the heterozygous state. Two mutations (p.G121E, p.A122T) were present only in enchondromas, and one (p.R255H) in both enchondroma and leukocyte DNA. Assessment of receptor function demonstrated that these three mutations impair PTHR1 function by reducing either the affinity of the receptor for PTH or the receptor expression at the cell surface. These mutations were not found in DNA from 222 controls. Including our data, PTHR1 functionally deleterious mutations have now been identified in five out 31 enchondromas from Ollier patients. These findings provide further support for the idea that heterozygous mutations in PTHR1 that impair receptor function participate in the pathogenesis of Ollier disease in some patients.
INTRODUCTION
Enchondromatosis (OMIM 166000) or Ollier disease (World Health Organization terminology) (1) is a developmental disorder defined by the presence of multiple enchondromas (2–5). Typically, these cartilaginous lesions have an asymmetric distribution, but important variability is seen in the age of onset of the disease and the size, number, location and evolution of the enchondromas. Most patients have bilateral enchondromatosis, but there is a tendency for one side of the body to be more severely affected. The condition in which multiple enchondromatosis is associated with vascular anomalies characterized by the presence of fusiform cells and high frequency of mesenchymal tumors is known as Maffucci syndrome (2,6). Enchondromas in Ollier disease present a significant risk of malignant transformation into chondrosarcoma, which usually occurs in young adults, and thus occurs at an earlier age than is observed in patients with isolated chondrosarcoma (2,7–9). The reported incidence of malignant transformation is even higher in Maffucci syndrome, the prognosis of which is more severe than that of Ollier disease (2,3). The association of Ollier disease with other tumors, particularly ovarian juvenile granulosa cell tumors, has been reported (2,10–12).
Enchondromas are almost exclusively localized in the metaphysis of long bones and in the small bones of the hands and feet (2,3,13). The tumors initially develop close to the growth plate cartilage where endochondral bone ossification occurs and then migrate progressively towards the diaphysis. Endochondral bone ossification is a highly regulated process that requires the differentiation of mesenchymal cells into hypertrophic chondrocytes and the subsequent replacement of a cartilaginous matrix by mineralized bone. It has been postulated that enchondromas result from abnormalities in signaling pathways controlling the proliferation and differentiation of chondrocytes, leading to the development of intraosseous cartilaginous foci. There are few reports of cytogenetic evaluation of benign enchondromas in Ollier disease, and no tumor-specific chromosomal abnormalities have been associated with enchondromas, or chondrosarcomas in these diseases (14–16). Studies evaluating isolated chondrosarcomas have usually revealed heterogeneous and complex changes (16), although loss of heterozygosity at chromosomal band 9p21 has been observed in several cases, and is associated with a loss of expression of the INK4A/p16 protein, a tumor-suppressor gene involved in control of the cell cycle (17). Expression of parathyroid-related peptide (PTHrP), its receptor PTHR1 and BCL2 may be correlated with the grade of malignancy in chondrosarcoma (18–21).
PTHrP and Indian Hedgehog (IHH) acting on their respective receptors PTHR1 and PTCH1 participate in a tightly coupled signaling relay that is critical for the regulation of chondrocyte differentiation and endochondral ossification (22). Thus, abnormalities in these genes could potentially participate in the pathogenesis of Ollier disease/Maffucci syndrome. In agreement with this hypothesis, a functionally deleterious mutation in PTHR1 (p.R150C) was identified in enchondromas from two of six unrelated patients with enchondromatosis (23). However, neither the p.R150C mutation (26 tumors) nor any other mutation in the PTHR1 gene (11 patients) could be identified in another study (24). These findings suggest that the molecular defects associated with enchondromatosis may be heterogeneous, but no other candidate genes, including those participating in the PTHrP/IHH pathway, have been evaluated.
To further define the role of PTHR1-signaling pathway in Ollier disease/Maffucci syndrome, we analyzed the coding sequences of PTHR1 and three other genes (PTHrP, IHH, Gsα) implicated in this pathway in two large cohorts of patients. In this study, we have (i) identified three new mutations in PTHR1 in patients with Ollier disease, (ii) characterized the abnormalities in receptor function resulting from these mutations and (iii) evaluated the impact of the PTHR1 mutations on the receptor function through modeling of the N-terminal ectodomain (N-ted).
RESULTS
Sequencing of candidate genes
PTHrP, IHH and Gsα were tested only in the French cohort. No variant alleles were identified in the coding sequence of the PTHrP gene in either leukocyte or tumor DNA in the 46 patients studied, whereas three previously described silent polymorphisms (rs3731878, p.T200T; rs3731881, p.P251P; rs394452, p.T376T) and one previously undescribed heterozygous variant (p.Y180Y/c.540T>C; HP 65) were identified in IHH. The allele frequency of the known polymorphisms did not differ from that reported in the NCBI SNP database (not shown). Three previously described silent polymorphisms (rs7121, p.I131I; rs3730171, p.I186I; rs8386, p.N371N) were identified in Gsα. Again, their allele frequency did not differ from that reported in the NCBI SNP database (not shown).
Seven heterozygous previously unreported PTHR1 SNPs, four silent (p.D30D, p.A72A, p.T163T, p.V455V) and three non-synonymous (p.G121E, p.A122T, p.R225H), were identified only in samples from the 84 patients with Ollier disease/Maffucci syndrome (Supplementary Material, Tables S1 and S2). The three previously undescribed non-synonymous mutations were all found in patients with Ollier disease. Two of these mutations (p.G121E, p.A122T) were observed in DNA from enchondromas, but not in leukocyte DNA; the R255H mutation was detected both in enchondroma and leukocyte DNA. In addition, two known non-synonymous polymorphisms (p.G100D, p.E546K) were identified in patients and controls (p.G100D, one patient and one control; p.E546K, three patients and six controls) (23,25). The allele frequency of the common synonymous PTHR1 polymorphism p.N463N (rs186987) was similar in the patient and control populations, and to that described in the NCBI SNP database. Two unreported heterozygous polymorphisms, one synonymous (p.S492S) and one non-synonymous (p.P581R), were each identified in one control subject.
Functional characterization of mutant and wild-type PTHR1
Functional studies evaluating the impact of all non-synonymous SNPs identified in patients with Ollier disease, including the three new alterations identified in this study (p.G121E, p.A122T, p.R225H), the mutation described by Hopyan et al. (p.R150C) and the two known polymorphisms (p.G100D, p.E546K), were performed (functional characterization of p.E546K, but not p.G100D, has been previously reported (23,25)). In addition, the p.H223R mutation identified in patients with Jansen’s metaphyseal chondrodysplasia was evaluated (26).
Ligand binding
Wild-type (WT) and mutant PTHR1 cell surface expression and affinity were assessed by the inhibition of [125I] hPTH1-34 binding by increasing concentrations of unlabeled hPTH1-34. For cells expressing WT PTHR1, half-maximal displacement of bound radiolabeled PTH was obtained with ∼3 × 10−10 m unlabeled hPTH1-34 (Fig. 1A and Table 1). Similar results were obtained for cells transfected with PTHR1 receptors carrying the non-synonymous mutations p.G100D, p.A122T, p.R150C and p.E546K (Fig. 1A and Table 1). In contrast, a marked reduction in [125I]hPTH(1–34) maximal radioligand binding and binding affinity was observed for cells expressing PTHR1 carrying the p.G121E and p.R255H mutations (Fig. 1A and Table 1). The IC50 for these mutants was at least 50-fold higher than that observed for WT PTHR1 (>15 nm), but a precise value could not be determined because binding affinities >15 nm cannot be determined using our approach. The binding properties of PTHR1 carrying the p.G121E, p.A122T and p.R150C mutations were also studied in COS-7 cells; results similar to those observed in CHO cells were obtained, except for an approximate one log right shift in the binding affinity observed for WT and mutant PTHR1. Thus, p.A122T and p.R150C PTHR1, maximal binding and binding affinity were similar to that of WT PTHR1; for p.G121E PTHR1, maximal binding and binding affinity were decreased. As described previously, maximal radioligand binding by the constitutively active p.H223R mutant was ∼50% lower than that of WT-PTHR1, with a similar half-maximal displacement of bound PTH (Fig. 1A and Table 1).
Figure 1.

Functional evaluation of WT and mutant PTHR1 expressed in CHO cells. (A) Binding of [125I] PTH 1–34 by cells incubated with radio ligand only (maximal binding) and in the presence of increasing concentrations of unlabeled hPTH 1–34. The maximal binding measured in cells transfected with the WT-PTHR1 was set as 100%. Results were corrected for non-specific binding measured in the presence of 8 × 10−7 m PTH 1–34. (B) Immunological assessment of cell-surface expression of WT and mutant PTHR1 expressed in CHO cells. Transfected cells were incubated sequentially with polyclonal rabbit anti-human PTHR1 antibody and [125I] -labeled anti-rabbit immunoglobin antibody. Results are expressed as % [125I]-labeled anti-rabbit immunoglobin antibody bound in cells transfected with the WT-PTHR1. p.G100D substitution disrupts the epitope recognition by the PTHR1 antibody (see Results). Results are the mean ± SEM of at least three experiments performed in duplicate with two plasmid preparations. *P < 0.01 compared with WT. Statistical analysis was performed using one-way ANOVA and comparisons between mutant and WT PTHR1 receptors using a Dunnett’s test.
Table 1.
Cell-surface expression, binding and signaling properties of WT and mutant PTHR1
| Cell-surface expression (% WT)a | Maximal PTH1–34 binding (% WT)b | IC50 (nm)c | Efficiency (% WT)d | EC50 (nm)c | |
|---|---|---|---|---|---|
| WT | 100 ± 6.8 | 100.0 ± 5.5 | 0.3 ± 0.01 | 100 ± 10 | 0.63 ± 0.02 |
| G100D | 3 ± 0.8e | 104.3 ± 13.7 | 0.6 ± 0.03 | 100 ± 9 | 0.43 ± 0.02 |
| G121E | 32 ± 3.3 | 17.2 ± 0.4 | ND | 77 ± 6 | 29.92 ± 3.3 |
| A122T | 106 ± 3.4 | 106.4 ± 8.2 | 0.14 ± 0.01 | 47 ± 3 | 0.67 ± 0.02 |
| R150C | 104 ± 17.8 | 94.8 ± 22.0 | 0.2 ± 0.02 | 48 ± 2.5 | 0.25 ± 0.02 |
| H223R | 77 ± 3.7 | 48.5 ± 6.1 | 0.6 ± 0.02 | 16 ± 1.4 | 2.9 ± 0.02 |
| R255H | 33 ± 1.5 | 14.0 ± 14.0 | ND | 95 ± 18 | 15 ± 0.8 |
| E546K | 88 ± 8.3 | 97.1 ± 20.4 | 0.1 ± 0.02) | 101 ± 10 | NA |
Results are the mean ± SEM of at least three experiments performed in duplicate with two plasmid preparations. 10−7 m PTH 1–34 led to maximal stimulation of cAMP production in WT-PTHR1 transfected cells (see Fig. 2A). ND, not detectable; NA, not available.
aTo determine cell-surface expression of WT and mutant PTHR1, transfected cells were incubated sequentially with polyclonal rabbit anti-human PTHR1 antibody and [125I]-labeled anti-rabbit immunoglobin antibody. Results are expressed as % [125I]-labeled anti-rabbit immunoglobin antibody bound in cells transfected with the WT-PTHR1.
bBinding assays utilized [125I]-Nle8,18, Tyr34-hPTH 1–34 as tracer radioligand and hPTH 1–34 as competitor ligand. Results were corrected for non-specific binding measured in the presence of 8 × 10−7 m PTH 1–34. Results are expressed as % maximal [125I]-PTH 1–34 bound in cells transfected with the WT-PTHR1.
cThe IC50 and EC50 values were calculated using GraphPad prism Software.
dThe % efficiency is the ratio of cAMP stimulation by 10−7 m PTH 1–34 in cells transfected with mutant PTHR1 over that obtained in cells transfected with WT-PTHR1.
eG100D substitution disrupts the epitope recognition by the PTHR1 antibody (see Fig. 1B and Results).
Immunological assessment of PTHR1 cell-surface expression
In order to quantify receptor expression, we assessed cell-surface expression of WT and mutant receptors using a polyclonal primary antibody directed against an extracellular epitope encoded by E2 of PTHR1, and a secondary iodinated antibody (27). Expression of p.A122T, p.R150C and p.E546K PTHR1 was similar to that of WT PTHR1 (Fig. 1B and Table 1), whereas expression of p.G121E and p.R255H PTHR1 were ∼70% lower than that of WT PTHR1 (Fig. 1B and Table 1). These results are consistent with the results obtained in the radioreceptor studies described earlier. Antibody binding for the p.G100D mutation was <5% of that observed for the WT receptor (Fig. 1B and Table 1). The p.G100D mutation is located within the epitope against which the antibody was raised. As these cells show normal binding of radiolabeled PTH, this result indicates that the p.G100D mutation disrupts the epitope, preventing recognition by this antibody. Expression of the p.H223R mutant was ∼25% lower than that of WT-PTHR1, results consistent with previous studies (26).
Analysis of PTHR1 expression using confocal microscopy
To evaluate the distribution of PTHR1 expression in CHO cells, cells were transfected with an expression vector producing GFP-tagged PTHR1, and 48 h after transfection, expression of PTHR1 on the surface of non-permeabilized cells was evaluated using the PTHR1 E2 antibody and a rhodamine-labeled secondary antibody. Cell-surface expression was clearly observed for WT PTHR1 and p.A122T, p.R150C and p.E546K PTHR1 (Fig. 3). In contrast, although some cell-surface expression was detected, a diffuse green labeling was observed in cells transfected with p.G121E and p.R255H PTHR1. This labeling pattern indicates that the receptors were expressed but not appropriately addressed to the plasma membrane. Similar results were observed in cells expressing p.H223R PTHR1, as previously reported. The thin green labeling observed at the cell periphery without cytoplasmic staining in cells transfected by p.G100D PTHR1 indicates that this mutant is correctly addressed to the cell surface.
Figure 3.
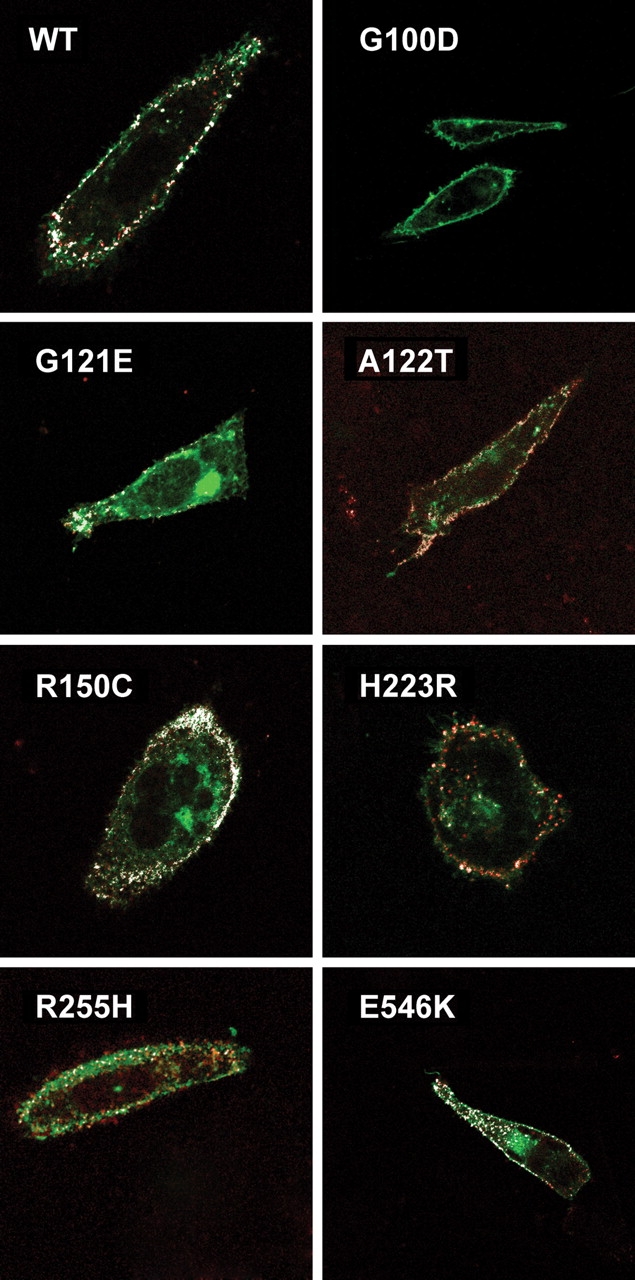
Confocal immunofluorescent microscopy analysis of WT and mutant PTHR1 tagged with GFP and using sequentially polyclonal rabbit anti-human PTHR1 antibody and rhodamine-labeled anti-rabbit immunoglobin antibody, without cellular permeabilization. Co-localization (originally yellow, and falsely colored in white for clarity) of green GFP-tagged PTHR1 and the rhodamine-labeled secondary antibody against the PTHR1 primary antibody (directed against the E2 extracellular epitope) indicates cell-surface expression. No co-localization was observed in cells transfected with the p.G100D PTHR1, indicating that the p.G100D mutation prevents PTHR1 recognition by the PTHR1 antibody (see Results). CHO cells were transfected with 1 µg plasmid DNA coding for the WT or mutant PTHR1, and confocal immunofluorescent microscopy analysis was performed as described in Material and Methods.
cAMP production
The functional consequences of the PTHR1 mutations were assessed by comparing basal and agonist-stimulated cAMP production in cells expressing WT and mutant receptors (Table 1). Basal cAMP expressed as picomoles/well was lower in cells expressing G121E, A122T and R255H PTHR1 than that in cells expressing WT receptors (Fig. 2C). However, except for the constitutionally active p.H223R PTHR1, basal cAMP production corrected for receptor expression in cells transfected with mutants PTHR1 was not significantly different from that measured for WT receptors (Fig. 2B).
Figure 2.
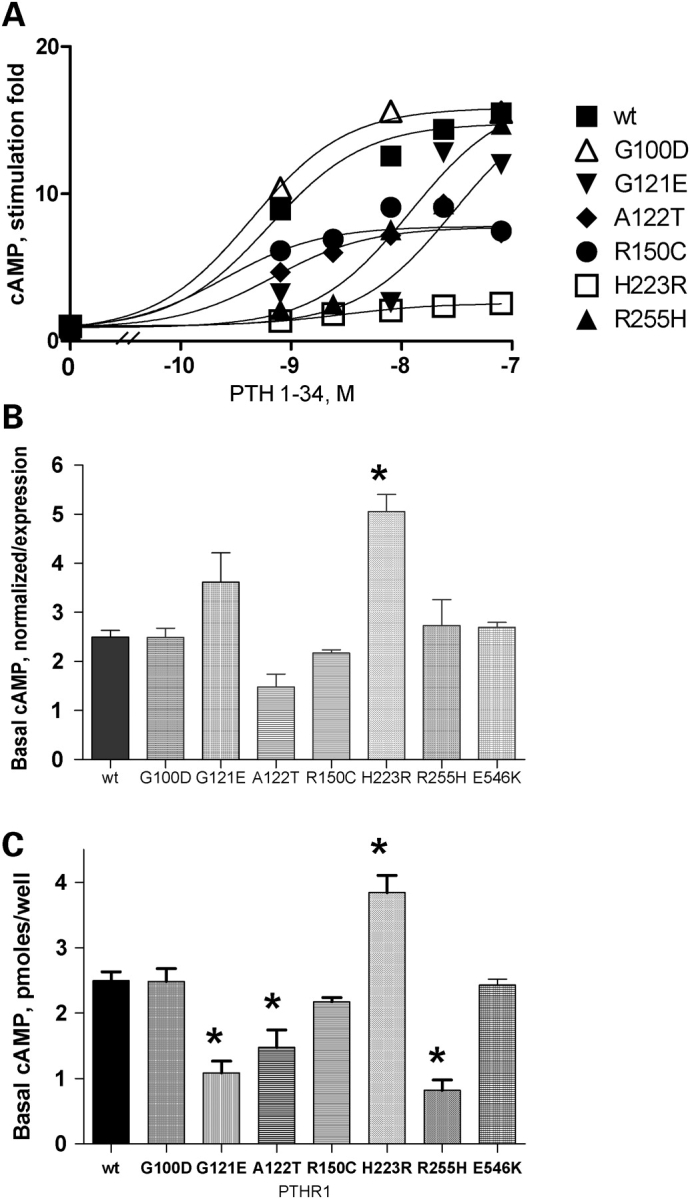
(A) cAMP accumulation in response to increasing concentrations of hPTH 1–34. Results are expressed as stimulation fold over basal. CHO cells were transfected with 1 µg plasmid DNA coding for the WT or mutant PTHR1, and functional studies were performed 48 h later as described in Materials and Methods. Results are the mean of at least three experiments performed in duplicate with two plasmid preparations. SB, specific binding. (B) Basal cAMP production in cells transfected with WT or mutant PTHR1. Basal cAMP accumulation is expressed as picomoles cAMP/well normalized to cell-surface expression as determined in Figure 1B. Expression of the mutant p.G100D PTHR1 was considered to be that of WT PTHR1 on the basis of results from radioligand binding and confocal immunofluorescence experiments (see Results). (C) Basal cAMP production in cells transfected with WT or mutant PTHR1 expressed as picomoles cAMP/well. CHO cells were transfected with 1 µg plasmid DNA coding for the WT or mutant PTHR1 and experiments performed 48 h later. Results are the mean ± SEM of at least three experiments performed in duplicate with two plasmid preparations. *P < 0.01 compared with WT. Statistical analysis was performed using one-way ANOVA and comparisons between mutant and WT PTHR1 receptors using a Dunnett’s test.
In cells expressing the WT PTHR1, cAMP production increased 15-fold over basal, and half-maximal stimulation of cAMP production was obtained with ∼6 × 10−10 m hPTH(1–34) (Fig. 2A and Table 1). When challenged with increasing amounts of hPTH(1–34), three patterns of cAMP accumulation were observed in cells expressing PTHR1 carrying non-synonymous polymorphisms: (i) For cells expressing p.A122T and p.R150C PTHR1, an ∼50% decrease in maximal ligand-induced cAMP production was observed, but the EC50 was similar to that of the WTPTHR1. As described previously, a similar decrease in the maximal stimulation fold of cAMP production without change in EC50 was also observed in cells expressing the H223R mutation. (ii) For cells expressing p.G121E and p.R255H PTHR1, a marked shift to the right in the dose–response curve was observed (EC50 29.9 × 10−9 and 15 × 10−9 m, respectively). cAMP stimulation fold over basal by 10−7 m hPTH(1–34) was 77 and 95%, respectively, for cells expressing p.G121E and p.R255H PTHR1 compared with that of cells expressing the WT PTHR1. (iii) For cells expressing the p.G100D and p.E546K polymorphisms, results were similar to those obtained for WT PTHR1, indicating that these non-synonymous mutations do not affect receptor function.
A summary of the impact of the non-synonymous mutations evaluated in this study on the expression and function of PTHR1 is shown in Table 2.
Table 2.
Summary of cell-surface expression, binding and signaling properties of mutant PTHR1 compared with WT PTHR1
| Cell-surface expression | PTH1–34 binding | Affinity | Maximal cAMP production | EC50 | |
|---|---|---|---|---|---|
| G100D | Normala | Normal | Normal | Normal | Normal |
| G121E | Reduced | Reduced | ND | Normal | Increased |
| A122T | Normal | Normal | Normal | Reduced | Normal |
| R150C | Normal | Normal | Normal | Reduced | Normal |
| R255H | Reduced | Reduced | ND | Normal | Increased |
| E546K | Normal | Normal | Normal | Normal | NA |
aNormal expression based on GFP-tagged G100D PTHR1 (Fig. 1). Expression could not be assessed using primary antibody against exon E2 epitope. ND, not detectable; NA, not available.
Location of mutations in the structure of the hPTHR1 N-ted complexed to PTH
In the structure described by Pioszak and Xu (28), the PTHR1 N-ted (PDB ID code 3C4M) contains one major α-helice (residues 33–57) and two antiparallel β-sheets (β-strands 1 and 2, residues 110–120; β-strands 3 and 4, residues 130–140) stabilized by three disulfide bonds linking Cys48–Cys117, Cys108–Cys148 and Cys131–Cys170 (Fig. 4). This structured core represents a typical sushi domain present in several structures of class B GPCR N-ted (28,29). The ligand-binding ridge is localized along the structured core of the N-ted (28) (Fig. 4). The exon E2 sequence present in the PTHR1 N-ted is not included within the structured core and lies in front of the first β-sheet (dotted line, Fig. 4).
Figure 4.

Representation of the recently resolved crystal structure of the PTH–PTHR1 N-ted complex (PDB ID code 3C4M) (28) and the position of the PTHR1 mutations identified in this and previous studies. The figures show ribbon representation of receptor N-ted. Light gray, PTHR1 N-ted main chain containing β-sheets (β1–β4); dashed line, exon E2 (residues 57–105); black, disulfide bonds between residues Cys48–Cys117, Cys108–Cys148 and Cys131–Cys170); dark gray, PTH. The mutated amino acids are indicated in medium gray. All mutated amino acids are located within the structured core.
The localization of amino acid changes present in the PTHR1 N-ted identified in this and previous studies, including the p.P132L mutation (Blomstrand chondrodysplasia) (30–32), and inducing changes in PTHR1 function is shown in Figure 4. These mutations are all included within the structured core of the N-ted of the PTH receptor, but outside the putative binding ridge. The p.G121E and p.A122T mutations, which, respectively, reduced ligand affinity and maximal cAMP production, are located in a loop between the β2 and β3 sheets. The p.R150C mutation, which reduced maximal cAMP production, is located in a loop between β3 sheet and the disulfide bridge Cys131–Cys170 (Fig. 4). The p.P132L mutation is localized close to a stretch of four residues (amino acid 135–138) involved in direct contacts between PTH and PTHR1 N-ted. In contrast, the p.G100D mutation, which does not modify PTHR1 function, is located in exon E2. Because the p.R255H mutation is not included in the PTHR1-N-ted, it could not be positioned in the structure.
DISCUSSION
To further define the role of PTHR1-signaling pathway in Ollier disease and Maffucci syndrome, we have analyzed the coding sequences and the intron–exon boundaries of four genes participating in this pathway in a large cohort of patients. In this study, we identified three previously undescribed missense mutations in PTHR1 in patients with Ollier disease. Two of these mutations were present only in enchondromas, and one in both enchondroma and leukocyte DNA. The assessment of receptor function demonstrated that all these mutations impair PTHR1 function by reducing either the affinity of the receptor for PTH or the receptor expression at the cell surface. Structural modeling of PTHR1 indicated that the deleterious mutations associated with Ollier disease and located within the N-ted all lie within the structured core of the N-ted. Functionally deleterious PTHR1 mutations have now been identified in five of 31 enchondromas from patients with Ollier disease (this study, French cohort: 1/11; Belgium cohort: 2/3; ref 23: 2/6; ref 24: 0/11), whereas such mutations have not be found in DNA from 222 control patients (this study) evaluated by the same techniques. These findings provide further support for the idea that heterozygous mutations in PTHR1 that impair receptor function can participate in the pathogenesis of Ollier disease in some patients.
PTHR1 mutations and the pathogenesis of Ollier disease
It has been suggested that enchondromas result from abnormalities in signaling pathways controlling proliferation and differentiation of chondrocytes, leading to the development of the intraosseous cartilaginous foci. In particular, PTHR1 regulates the switch from proliferating to hypertrophic chondrocytes, and thereby influences the number of cells expressing IHH. IHH is a member of a family of morphogen proteins that are important for embryonic patterning, and is highly expressed in the transition zone between proliferating and hypertrophic chondrocytes. This multifunctional protein stimulates chondrocyte proliferation, predominantly through a PTHrP-independent mechanism, and also delays their hypertrophy by increasing PTHrP synthesis by periarticular chrondrocytes. Thus, PTHR1- and IHH-signaling pathways are tightly coupled, although both exert functions independently of each other. Reduced PTHR1 signaling would be expected to impair this coupling process necessary for harmonious chondrocyte proliferation and differentiation, thereby contributing to the development of enchondromas.
Our results and those from the literature indicate that missense PTHR1 mutations may be involved in the development of enchondromas in a minority of patients. Three amino acid substitutions in PTHR1 were identified in the present study, which impaired the ability of the mutant receptors to stimulate cAMP production, due to either a decrease in receptor expression and ligand affinity (p.G121E and p.R255H) or suboptimal agonist-induced cAMP production despite normal PTHR1 expression (p.A122T). Two of these mutations were somatic and the third one likely germline. None of these receptors displayed constitutional activity, whereas, as expected, increased constitutional activity was observed for the PTHR1 expressing the p.H223R mutation identified in patients with Jansen’s metaphyseal chondrodysplasia. We also evaluated the functional properties of the p.R150C mutation, previously identified in two patients with Ollier disease by Hopyan et al. (23). Consistent with the results of Hopyan et al., we found that PTH-induced cAMP production was reduced in cells transiently expressing this receptor, but in contrast to the previous report, receptor expression was normal. Thus, the apparent increase in constitutional activity reported by these authors following correction for receptor expression (23) was not observed in our study. The reason as to why the p.R150C receptor expression was normal in our experiments and decreased in Hopyan et al.’s studies is not clear, but is probably due to differences in experimental conditions. Nevertheless, all mutant receptors identified exclusively in patients with Ollier disease are characterized by impaired ligand-induced cAMP production.
It is noteworthy that for three of the five patients with Ollier disease in whom PTHR1 mutations were identified in enchondromas, similar mutations were not detected in peripheral blood leukocytes (this study and reference) (25). Because multiple enchondromas were present in these patients, the findings are consistent with the hypothesis that the mutations occurred during development, resulting in genetic mosaicism in these individuals. Further studies evaluating multiple tumors and other tissues from such patients will be required to further support this idea. Nevertheless, sensitive screening for PTHR1 mutations in this disease appears to require the evaluation of tumoral tissue, because mutations have now been identified in five out of 31 enchondromas, but only two out of 58 samples of leukocytes DNA evaluated by the same approach (P < 0.05 by Fisher’s exact test).
In the course of these studies, we also evaluated samples obtained from patients with Maffucci syndrome (enchondromas·chondrosarcomas, n = 12, blood leukocytes, n = 13), but deleterious mutations in PTHR1 were not identified. Because of the small number of patients studied, however, the frequency of finding PTHR1 mutations was not significantly different comparing patients with Ollier disease and Maffucci syndrome (five out of 31 and none out of 12 positive tumors, respectively, P = 0.3 by Fisher’s exact test). Further work is required to assess the role, if any, of PTHR1 mutations in Maffucci syndrome.
Other findings are consistent with the conclusion that abnormalities in the PTHR1/IHH pathway can be linked to the development of endochondromas. Transgenic mice expressing the mutant p.R150C PTHR1 under the control of the collagen type II promoter develop tumors that are similar to those observed in human enchondromatosis (23). A contribution of a dysregulation of IHH-signaling pathway to the development of enchondromas is also supported by the development of enchondromas in mice overexpressing the Hedgehog (Hh) transcriptional regulator, Gli2, and the activation of a Hedgehog-responsive Gli2-luciferase reporter construct by the p.R150C PTHR1 mutant (23). Finally, downregulation of IHH/PTHrP signaling as a result of EXT mutation has been shown to play a role in osteochondroma formation (29).
Even when present, it is not certain that the PTHR1 mutations identified are sufficient enough to induce enchondromas. The p.R150C mutation has been identified both in a patient with Ollier disease and in a parent who had other skeletal abnormalities without enchondromas. In this regard, it will be interesting to evaluate family members of patients expressing the p.R255H PTHR1 mutation identified in this study, but such clinical samples are not currently available. Indeed, the PTHR1 loss of function mutations identified in Ollier disease are expressed in the heterozygous state. PTHR1 dimerization has not been documented for receptors of this family, rendering a dominant negative effect of the mutant receptor unlikely. These findings support the hypothesis that, even when a PTHR1 mutation is present, a combination of genetic events, germline and/or somatic, is required for the development of enchondromas.
The nature of such putative genetic abnormalities remains to be defined. Abnormalities involving other participants in the PTHR1 pathway are potential candidates. For three of these genes, PTHrP, IHH and GNAS1 (Gsalpha), no missense mutation was identified in these studies. Although these negative results do not exclude the possibility that genetic abnormalities at these loci can contribute to the development of multiple enchondromas, our findings suggest that such abnormalities are not a frequent cause.
Other PTHR1 polymorphisms
Currently, only two missense polymorphisms in PTHR1 have been described in population studies, p.G100D and p.E546K. In the course of these studies, both of these polymorphisms were identified with apparently similar frequency in controls and patients with Ollier disease/Maffucci syndrome. We evaluated the functional properties of receptors carrying both of these polymorphisms, but no abnormalities were identified. Amino acid p.G100 is located in exon E2 of the PTHR1; this exon is not present in other family II GPCR, and its deletion has been previously shown not to affect PTHR1 function (33). Therefore, the lack of deleterious effect of the p.G100D mutation was not unexpected. The p.E546K polymorphism has previously been extensively characterized by Schipani et al. (25), and no abnormalities were found.
Three-dimensional spatial location of mutations
In order to better analyze the mechanism through which mutations may affect receptor function, we represented the recently resolved crystal structure of the PTHR1 N-ted–PTH complex (PDB ID code 3C4M) (28) and positioned the PTHR1 mutations identified in this and previous studies.
The observation that the functionally deleterious PTHR1 mutations (p.G121E, p.A122T, p.R150C and p.P132L) are all located within the structured core but outside the putative ligand-binding ridge indicates that the mutations may not directly impact receptor–ligand interactions, but rather induce changes in the three-dimensional organization of the N-ted-structured core necessary for PTH recognition and receptor activation. In support of this, the p.P132L substitution (Blomstrand chondrodysplasia) (30–32) appears to disrupt an interaction required to hold the structure of the N-ted and PTH–PTH1R signaling (28). An analysis performed using a model of the VPAC1 receptor (also a class II GPCR) (34) supports this conclusion. In this model, proline-87 in the VPAC1 receptor, which corresponds to proline-132 in PTHR1, is also included within the structured core of the N-ted of the VPAC1 receptor, but outside the binding ridge. The substitution of P87 by alanine induced an important decrease in the affinity of VIP for its receptor (unpublished data). In contrast, substitution of proline-87 by glycine, which introduces a flexible point in the structured core, does not affect VPAC1 receptor function (35). The p.G100D mutation, which does not modify PTHR1 function, is not located within exon E2 because the p.R255H mutation is not included in the PTHTR1–N-ted; no structure-function correlates could be obtained for this mutation.
In conclusion, this study provides further evidence that functionally deleterious mutations in PTHR1 are present in a subset of patients with Ollier disease and therefore offers support to the idea that abnormalities in signaling pathways controlling chondrocyte proliferation and differentiation can contribute to the pathogenesis of this disorder. Our results emphasize that the genetic abnormalities responsible for this disease are likely to be both heterogeneous and multifactorial. The evaluation of additional candidate genes in large cohorts of patients may prove rewarding.
MATERIALS AND METHODS
Patients
Informed consent was obtained for all patients. The study was performed in two series of patients. The first series comprised 46 patients (29 females, 17 males) from a French cohort. The clinical characteristics of the patients are presented in Supplementary Material, Table S1. Data were obtained by review of clinical charts, contact with physicians in charge of the patients and through a questionnaire sent to the patients in collaboration with the French association for Ollier disease/Maffucci syndrome. Patient age at diagnosis ranged from 1 to 36 years (median 6.4 years). All patients were affected by at least three enchondromas. Unilateral body involvement was present in 23 patients (52%), and bilateral involvement with a marked asymmetry was noted in nine patients. No patients had spinal involvement. Two patients (HP17 and HP55) were affected by chondrosarcomas. One patient (HP17) had Maffucci syndrome. For all patients, the diagnosis of exostosis was excluded. Sixteen females (59%) and four males (26%) had a height ≥50 percentile (P = 0.06 by Fisher’s exact test). In all patients, serum calcium and phosphorus levels were within the normal range for the age (data not shown). Leukocyte DNA was obtained from all patients; tumor DNA was available for 12 patients, including the patient with Maffucci syndrome (10 enchondromas and two chondrosarcomas).
The second series comprised 38 patients from a Belgium cohort, 16 patients affected by Ollier disease (nine females, seven males; age at the time of the study 6–38 years) and 22 patients affected by Maffucci syndrome (11 females, 11 males; age at the time of the study 15–52 years) (Supplementary Material, Table S2). Leukocyte DNA was obtained from all patients except one; tumor DNA was available for three patients affected with Ollier disease and 13 patients with Maffucci syndrome (10 enchondromas, two chondrosarcomas, one spindle cell hemangioendothelioma).
Genomic DNA from 222 Caucasians was used as a control group.
Sequence analysis
Genomic DNA was extracted from peripheral blood and/or tumors obtained at the time of surgery using a QIAamp DNA purification kit (Qiagen SA, Courtaboeuf, France). Intronic, and when required, exonic primers were used to amplify all coding exons and intron–exon junctions for the PTHR1, IHH, PTHrP and GNAS1 (Gsα exons 1–13) genes. PTHR1 gene was analyzed in all leukocyte and tumor DNA samples from the French and Belgium cohorts. IHH and PTHrP were analyzed in all leukocyte and tumor DNA from the French cohort. The GNAS1 gene (exons 1–13 of Gsa) was analyzed in tumor (nine samples) and leukocyte DNA from four patients from the French cohort. The sequences of primers are available upon request. PCR products were analyzed by direct sequencing (French patients and 112 controls) or by prescreening using SSCP/heteroduplex analysis combined with sequencing of abnormally migrating fragments (Belgium patients and 110 controls).
PCR products were sequenced in both directions. Sequencing reactions were performed using the BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Courtaboeuf, France) and analyzed using an ABI PRISM 3100 sequencer (Applied Biosystems) and Beckman CEQ2000 fluorescent capillary sequencer for the French and Belgium cohorts, respectively.
Nucleotide positions are numbered from the ATG start codon in the complementary DNA (cDNA) (sequence accession numbers: PTHR1, NM_000316; IHH, NM_002181; PTHrP transcript variant 1, NM_198965; PTHrP transcript variant 2, NM_002820; PTHrP transcript variant 3, NM_198964, GNAS1, NM_000516). All nucleotide changes (except for previously reported polymorphisms) were verified by resequencing of a different PCR product. Only silent and missense nucleotide changes in the coding region are reported. However, for all polymorphisms identified in introns, it was verified that they were not predicted to affect splicing (http://www.fruitfly.org/seq_tools/splice.html).
Site-directed mutagenesis
Full-length WT cDNA encoding the human PTHR1 (hPTHR1) was inserted into the pEGFP-N2 N-terminal fusion protein vector (Clontech Laboratories, Mountain View, CA, USA) upstream of the enhanced green fluorescent protein gene (30). Missense mutations were introduced in the WT PTHR1 cDNA construct using the Quick Change site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) and confirmed by complete sequencing of cDNA inserts (oligonucleotide sequences available on request).
Cell culture and transient transfection
The function of all mutated PTHR1 receptors was studied following transient expression in CHO cells. Mutant p.R150C, p.G121E, p.A122T PTHR1 were also studied following expression in COS-7. CHO and COS-7 cells were cultured in 24-well plates as described previously (36–38). Transfection of CHO cells were performed using 1.5 µl of FuGENE 6 transfection reagent (Roche Diagnostics, Indianapolis, IN, USA) with 1 µg pEGFPN2 plasmid encoding WT or mutant PTHR1. Transfection of COS-7 cells were performed using lipofectamine 2000 (Invitrogen SARL, Cergy Pontoise, France) and 500 ng DNA/well as described previously (36,38).
[125I] PTH 1–34 binding, immunological assessment of receptor expression and camp measurement
Techniques used to assess PTH 1–34 binding, PTHR1 cell membrane expression and cAMP production have been described (36–38). Briefly, the binding properties of WT and mutated hPTHR1 receptors were determined by competitive inhibition of [125I] (Nle8,18Tyr34) human PTH (1–34) amide ([125I]–PTH1–34) (Amersham Biosciences UK Ltd, Buckinghamshire, UK) binding to transfected cells by increasing concentrations of unlabeled (Nle8,18Tyr34) human PTH (1–34) (Bachem UK Ltd, Merseyside, UK) (PTH1–34) as described previously (30,38). The specific binding was calculated as the difference between [125I]-peptide bound and the non-specific binding (i.e. that measured in the presence of 8 × 10−7m unlabeled PTH 1–34). To determine the IC50, binding data were fitted to a sigmoid curve with variable slope (Graphpad Prism 4, GraphPad Software Inc., San Diego, CA, USA). Immunological assessment of cell-surface receptor expression was performed using a rabbit polyclonal antibody raised against a 17 amino acid epitope present in exon E2 of the human PTHR1 (90E-S-E-E-D-K-E-A-P-T-G-S-R-Y-R-G-106R) (PTHR1 E2 antibody) and a secondary [125I]-labeled donkey anti-rabbit immunoglobulin antibody (Amersham Biosciences UK Ltd) as described previously (27). For the determination of intracellular cAMP production, cells were incubated without or with hPTH1–34 under continuous agitation in 0.5 ml of culture medium containing 0.1% (w/v) bovine serum albumin and 1 mm 3-isobutyl-l-methylxanthine (Sigma-Aldrich, Saint-Quentin Fallavier, France). After a 20 min incubation at 37°C, the medium was removed and cells were lysed with 1 m perchloric acid. The cAMP present in the lysate was measured by radioimmunoassay as described (36). Results are expressed as pmol of cAMP/well. Experiments were performed at least three times in duplicate with two different plasmid preparations.
Confocal laser scanning microscopy
CHO cells were transiently transfected with the WT or mutant PTHR1 cDNA inserted into the pEGFP plasmid. Forty-eight hours after transfection, unpermeabilized cells were incubated for 30 min at 4°C with the PTHR1 antibody used for the assessment of PTHR1 expression (PTHR1 E2 antibody), washed extensively with phosphate-buffered saline and incubated for 30 min at 4°C with a secondary rhodamine-labeled donkey anti-rabbit immunoglobulin. Cells were fixed in 1% paraformaldehyde. Glass coverslides were mounted and examined by confocal laser scanning microscopy (CLSM-510-META, Zeiss, Germany) equipped with epifluorescent optics (x63 NA 1.3 oil-immersion objective). Simultaneous two-channel recording was performed with a pinhole size of 90 µm using excitation wavelengths of 488/588 nm, a 510/580 double-dichroic mirror and a 515–545 bandpass fluorescein isothiocyanate filter together with a 590 nm long-pass filter. Double-labeled cells were analyzed separately to avoid spillover between channels. In all experiments, omission of the primary antibody was confirmed to result in no detectable staining.
Location of mutations in the structure of the hPTHR1 N-ted complexed to PTH
In order to locate the spatial position of the PTHR1 mutations, we plotted each residue in the recently published structure of the N-ted of PTHR1 (PDB ID code 3C4M) bound to PTH (15–34) obtained by crystallization and X-ray diffraction (28).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
FUNDING
These studies were supported by grants from INSERM (RBM 0430), the réseaux DHOS, the Fédération des Maladies Orphelines, et l’Association Ollier Maffucci (to C.S. and B.G.), and by the Interuniversity Attraction Poles initiated by the Belgian Federal Science Policy, network 5/25 and 6/05; Concerted Research Actions (ARC)—Convention Nos 02/07/276 and 07/12-005 of the Belgian French Community Ministry; the National Institute of Health, Program Project P01 AR48564; EU FW6 integrated project LYMPHANGIOGENOMICS, LSHG-CT-2004-503573 and the FNRS (Fonds national de la recherche scientifique) (to M.V., a ‘Maître de recherches du F.N.R.S.’). V.W. was supported by a fellowship from FRIA (Fonds pour la formation à la recherche dans l’industrie et dans l’agriculture) and Patrimoine UCL.
ACKNOWLEDGEMENTS
We are indebted to all the family members, the Association Ollier Maffucci and referring physicians for their invaluable contributions. We thank Cécile Pouzet for performing the confocal microscopy at the Institut fédératif de recherche 02 (Inserm, Université Paris VII, CHU Xavier Bichat). We are grateful to Cécile Jullier for sharing PTHR1 sequences from controls, and Augen Pioszak and Eric Xu for the availability of the atomic coordinates and structure factors of the extracellular domain of human PTH1R bound to PTH before publication in the protein data bank.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Fletcher C.D.M., Unni K., Mertens F., editors. World Health Organization Classification of Tumors. Pathology and Genetics. Tumors of Soft Tissue and Bone. Lyon: IARCPress; 2002. [Google Scholar]
- 2.Maroteaux P., Le Merrer M. Les maladies osseuses de l’enfant. 4ème ed. Paris: Médecine-Sciences, Flammarion; 2002. [Google Scholar]
- 3.Unni K.K. Cartilaginous lesions of bone. J. Orthop. Sci. 2001;6:457–472. doi: 10.1007/s007760170015. [DOI] [PubMed] [Google Scholar]
- 4.Whyte M. Acquired Disorders of Cartilage and Bone. 5th edn. Washington, DC: American Society for Bone and Mineral Research; 2003. [Google Scholar]
- 5.Silve C., Juppner H. Ollier disease. Orphanet J. Rare Dis. 2006;1:37. doi: 10.1186/1750-1172-1-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Casanova D., Boon L.M., Vikkula M. Venous malformations: clinical characteristics and differential diagnosis. Ann. Chir. Plast. Esthet. 2006;51:373–387. doi: 10.1016/j.anplas.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 7.Rozeman L.B., Hogendoorn P.C., Bovee J.V. Diagnosis and prognosis of chondrosarcoma of bone. Expert Rev. Mol. Diagn. 2002;2:461–472. doi: 10.1586/14737159.2.5.461. [DOI] [PubMed] [Google Scholar]
- 8.Schaison F., Anract P., Coste F., De Pinieux G., Forest M., Tomeno B. Chondrosarcoma secondary to multiple cartilage diseases. Study of 29 clinical cases and review of the literature. Rev. Chir. Orthop. Reparatrice Appar. Mot. 1999;85:834–845. [PubMed] [Google Scholar]
- 9.Schwartz H.S., Zimmerman N.B., Simon M.A., Wroble R.R., Millar E.A., Bonfiglio M. The malignant potential of enchondromatosis. J. Bone Joint Surg. Am. 1987;69:269–274. [PubMed] [Google Scholar]
- 10.Vaz R.M., Turner C. Ollier disease (enchondromatosis) associated with ovarian juvenile granulosa cell tumor and precocious pseudopuberty. J. Pediatr. 1986;108:945–947. doi: 10.1016/s0022-3476(86)80936-2. [DOI] [PubMed] [Google Scholar]
- 11.Tamimi H.K., Bolen J.W. Enchondromatosis (Ollier’s disease) and ovarian juvenile granulosa cell tumor. Cancer. 1984;53:1605–1608. doi: 10.1002/1097-0142(19840401)53:7<1605::aid-cncr2820530731>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 12.Mahafza W.S. Multiple enchondromatosis Ollier’s disease with two primary brain tumors. Saudi Med. J. 2004;25:1261–1263. [PubMed] [Google Scholar]
- 13.Tiet T.D., Alman B.A. Developmental pathways in musculoskeletal neoplasia: involvement of the Indian Hedgehog-parathyroid hormone-related protein pathway. Pediatr. Res. 2003;53:539–543. doi: 10.1203/01.PDR.0000054688.93486.18. [DOI] [PubMed] [Google Scholar]
- 14.Bovee J.V., Cleton-Jansen A.M., Rosenberg C., Taminiau A.H., Cornelisse C.J., Hogendoorn P.C. Molecular genetic characterization of both components of a dedifferentiated chondrosarcoma, with implications for its histogenesis. J. Pathol. 1999;189:454–462. doi: 10.1002/(SICI)1096-9896(199912)189:4<454::AID-PATH467>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 15.Bovee J.V., van Roggen J.F., Cleton-Jansen A.M., Taminiau A.H., van der Woude H.J., Hogendoorn P.C. Malignant progression in multiple enchondromatosis (Ollier’s disease): an autopsy-based molecular genetic study. Hum. Pathol. 2000;31:1299–1303. doi: 10.1053/hupa.2000.19308. [DOI] [PubMed] [Google Scholar]
- 16.Sandberg A.A. Genetics of chondrosarcoma and related tumors. Curr. Opin. Oncol. 2004;16:342–354. doi: 10.1097/01.cco.0000129678.72521.e5. [DOI] [PubMed] [Google Scholar]
- 17.van Beerendonk H.M., Rozeman L.B., Taminiau A.H., Sciot R., Bovee J.V., Cleton-Jansen A.M., Hogendoorn P.C. Molecular analysis of the INK4A/INK4A-ARF gene locus in conventional (central) chondrosarcomas and enchondromas: indication of an important gene for tumour progression. J. Pathol. 2004;202:359–366. doi: 10.1002/path.1517. [DOI] [PubMed] [Google Scholar]
- 18.Amling M., Posl M., Hentz M.W., Priemel M., Delling G. PTHrP and Bcl-2: essential regulatory molecules in chondrocyte differentiation and chondrogenic tumors. Verh. Dtsch. Ges. Pathol. 1998;82:160–169. [PubMed] [Google Scholar]
- 19.Bovee J.V., van den Broek L.J., Cleton-Jansen A.M., Hogendoorn P.C. Up-regulation of PTHrP and Bcl-2 expression characterizes the progression of osteochondroma towards peripheral chondrosarcoma and is a late event in central chondrosarcoma. Lab. Invest. 2000;80:1925–1934. doi: 10.1038/labinvest.3780202. [DOI] [PubMed] [Google Scholar]
- 20.Kunisada T., Moseley J.M., Slavin J.L., Martin T.J., Choong P.F. Co-expression of parathyroid hormone-related protein (PTHrP) and PTH/PTHrP receptor in cartilaginous tumours: a marker for malignancy? Pathology. 2002;34:133–137. doi: 10.1080/003130201201117936. [DOI] [PubMed] [Google Scholar]
- 21.Pateder D.B., Gish M.W., O’Keefe R.J., Hicks D.G., Teot L.A., Rosier R.N. Parathyroid hormone-related peptide expression in cartilaginous tumors. Clin. Orthop. 2002:198–204. doi: 10.1097/00003086-200210000-00029. [DOI] [PubMed] [Google Scholar]
- 22.Kronenberg H.M. Developmental regulation of the growth plate. Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 23.Hopyan S., Gokgoz N., Poon R., Gensure R.C., Yu C., Cole W.G., Bell R.S., Juppner H., Andrulis I.L., Wunder J.S., et al. A mutant PTH/PTHrP type I receptor in enchondromatosis. Nat. Genet. 2002;30:306–310. doi: 10.1038/ng844. [DOI] [PubMed] [Google Scholar]
- 24.Rozeman L.B., Sangiorgi L., Bruijn I.H., Mainil-Varlet P., Bertoni F., Cleton-Jansen A.M., Hogendoorn P.C., Bovee J.V. Enchondromatosis (Ollier disease, Maffucci syndrome) is not caused by the PTHR1 mutation p.R150C. Hum. Mutat. 2004;24:466–473. doi: 10.1002/humu.20095. [DOI] [PubMed] [Google Scholar]
- 25.Schipani E., Weinstein L.S., Bergwitz C., Iida-Klein A., Kong X.F., Stuhrmann M., Kruse K., Whyte M.P., Murray T., Schmidtke J., et al. Pseudohypoparathyroidism type Ib is not caused by mutations in the coding exons of the human parathyroid hormone (PTH)/PTH-related peptide receptor gene. J. Clin. Endocrinol. Metab. 1995;80:1611–1621. doi: 10.1210/jcem.80.5.7745008. [DOI] [PubMed] [Google Scholar]
- 26.Schipani E., Kruse K., Jüppner H. A constitutively active mutant PTH-PTHrP receptor in Jansen-type metaphyseal chondrodysplasia. Science. 1995;268:98–100. doi: 10.1126/science.7701349. [DOI] [PubMed] [Google Scholar]
- 27.Jobert A.S., Fernandes I., Turner G., Coureau C., Prie D., Nissenson R.A., Friedlander G., Silve C. Expression of alternatively spliced isoforms of the parathyroid hormone (PTH)/PTH-related peptide receptor messenger RNA in human kidney and bone cells. Mol. Endocrinol. 1996;10:1066–1076. doi: 10.1210/mend.10.9.8885241. [DOI] [PubMed] [Google Scholar]
- 28.Pioszak A.A., Xu H.E. Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proc. Natl Acad. Sci. USA. 2008;105:5034–5039. doi: 10.1073/pnas.0801027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rozeman L.B., Hameetman L., Cleton-Jansen A.M., Taminiau A.H., Hogendoorn P.C., Bovee J.V. Absence of IHH and retention of PTHrP signalling in enchondromas and central chondrosarcomas. J. Pathol. 2005;205:476–482. doi: 10.1002/path.1723. [DOI] [PubMed] [Google Scholar]
- 30.Zhang P., Jobert A.S., Couvineau A., Silve C. A homozygous inactivating mutation in the parathyroid hormone/parathyroid hormone-related peptide receptor causing Blomstrand chondrodysplasia. J. Clin. Endocrinol. Metab. 1998;83:3365–3368. doi: 10.1210/jcem.83.9.5245. [DOI] [PubMed] [Google Scholar]
- 31.Hoogendam J., Farih-Sips H., Wynaendts L.C., Lowik C.W., Wit J.M., Karperien M. Novel mutations in the parathyroid hormone (PTH)/PTH-related peptide receptor type 1 causing Blomstrand osteochondrodysplasia types I and II. J. Clin. Endocrinol. Metab. 2007;92:1088–1095. doi: 10.1210/jc.2006-0300. [DOI] [PubMed] [Google Scholar]
- 32.Karaplis A.C., Bin He M.T., Nguyen A., Young I.D., Semeraro D., Ozawa H., Amizuka N. Inactivating mutation in the human parathyroid hormone receptor type 1 gene in Blomstrand chondrodysplasia. Endocrinology. 1998;139:5255–5258. doi: 10.1210/endo.139.12.6522. [DOI] [PubMed] [Google Scholar]
- 33.Lee C., Gardella T.J., Abou-Samra A.B., Nussbaum S.R., Segre G.V., Potts J.T., Jr, Kronenberg H.M., Juppner H. Role of the extracellular regions of the parathyroid hormone (PTH)/PTH-related peptide receptor in hormone binding. Endocrinology. 1994;135:1488–1495. doi: 10.1210/endo.135.4.7523099. [DOI] [PubMed] [Google Scholar]
- 34.Ceraudo E., Murail S., Tan Y.V., Lacapere J.J., Neumann J.M., Couvineau A., Laburthe M. The vasoactive intestinal peptide alpha helix up to C-terminus interacts with the N-terminal ectodomain of the human VPAC1 receptor: photoaffinity, molecular modeling and dynamics. Mol. Endocrinol. 2008;22:147–155. doi: 10.1210/me.2007-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Couvineau A., Gaudin P., Maoret J.J., Rouyer-Fessard C., Nicole P., Laburthe M. Highly conserved aspartate 68, tryptophane 73 and glycine 109 in the N-terminal extracellular domain of the human VIP receptor are essential for its ability to bind VIP. Biochem. Biophys. Res. Commun. 1995;206:246–252. doi: 10.1006/bbrc.1995.1034. [DOI] [PubMed] [Google Scholar]
- 36.Couvineau A., Lacapere J.J., Tan Y.V., Rouyer-Fessard C., Nicole P., Laburthe M. Identification of cytoplasmic domains of hVPAC1 receptor required for activation of adenylyl cyclase. Crucial role of two charged amino acids strictly conserved in class II G protein-coupled receptors. J. Biol. Chem. 2003;278:24759–24766. doi: 10.1074/jbc.M301916200. [DOI] [PubMed] [Google Scholar]
- 37.Jobert A.S., Leroy C., Butlen D., Silve C. Parathyroid hormone-induced calcium release from intracellular stores in a human kidney cell line in the absence of stimulation of cyclic adenosine 3′,5′-monophosphate production. Endocrinology. 1997;138:5282–5292. doi: 10.1210/endo.138.12.5556. [DOI] [PubMed] [Google Scholar]
- 38.Jobert A.S., Zhang P., Couvineau A., Bonaventure J., Roume J., Le Merrer M., Silve C. Absence of functional receptors for parathyroid hormone and parathyroid hormone-related peptide in Blomstrand chondrodysplasia. J. Clin. Invest. 1998;102:34–340. doi: 10.1172/JCI2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.