

Abstract
Congenital aortic valve stenosis (AVS), coarctation of the aorta (COA) and hypoplastic left heart syndrome (HLHS) are congenital cardiovascular malformations that all involve the left ventricular outflow tract (LVOT). They are presumably caused by a similar developmental mechanism involving the developing endothelium. The exact etiology for most LVOT malformations is unknown, but a strong genetic component has been established. We demonstrate here that mutations in the gene NOTCH1, coding for a receptor in a developmentally important signaling pathway, are found across the spectrum of LVOT defects. We identify two specific mutations that reduce ligand (JAGGED1) induced NOTCH1 signaling. One of these mutations perturbs the S1 cleavage of the receptor in the Golgi. These findings suggest that the levels of NOTCH1 signaling are tightly regulated during cardiovascular development, and that relatively minor alterations may promote LVOT defects. These results also establish for the first time that AVS, COA and HLHS can share a common pathogenetic mechanism at the molecular level, explaining observations of these defects co-occurring within families.
INTRODUCTION
Aortic valve stenosis (AVS), coarctation of the aorta (COA), and hypoplastic left heart syndrome (HLHS) are congenital defects that constitute a clinically important group of cardiovascular malformations (CVMs) termed left ventricular outflow tract (LVOT) malformations. Together, LVOT malformations occur at a rate of 1/1000 live births (1–3). They account for a significant proportion of infant mortality and incur considerable financial burden (4,5). A presumed, although unproven, unifying pathogenesis for these malformations is a defect in the developing endothelium or endocardium that leads to subsequent blood flow obstruction and secondary effects.
Both environmental and genetic etiologies have been implicated in the development of LVOT defects. Prenatal exposure to solvents (3) or high phenylalanine (secondary to maternal phenylketonuria) (6) is associated with higher rates of these defects. Epidemiology studies have demonstrated rate differentials by sex, ethnicity and geography (2,3). These and other studies (7,8) have documented a high concordance for LVOT malformations among families with multiple CVMs, although not necessarily of the identical LVOT defect within the family. Our group and others (9,10) have also noted a much higher rate (∼5.5) of bicuspid aortic valve (BAV) among families with LVOT malformations compared to the general population, suggesting BAV may be a forme fruste of the more serious LVOT malformations.
We have previously demonstrated a strong genetic component for the LVOT malformations (11), with estimated sibling recurrence rates >30. Formal inheritance analyses are consistent with a complex genetic inheritance involving from one to six loci. These analyses also suggested an autosomal dominant inheritance with reduced penetrance may operate in some families.
Recently, two families were described with calcific aortic valve disease, usually in the context of a BAV, that had mutations in the NOTCH1 gene (12). One family had a member affected with mitral valve atresia, double outlet right ventricle and hypoplastic left ventricle, which could represent a variant of HLHS. Two subsequent studies have also found missense NOTCH1 mutations in sporadic cases of BAV (13,14).
Notch is a highly conserved local signaling pathway involved in multiple developmental processes (15). NOTCH1 (NCBI GeneID 4851) encodes a large, single-pass transmembrane receptor with 36 EGF-like repeats and three NOTCH/Lin repeats in the extracellular domain and an intracellular transactivating domain containing six ankyrin repeats. The receptor is cleaved in the Golgi (S1 cleavage) prior to being presented on the cell surface as a heterodimer of the extracellular domain with the transmembrane and intracellular domains (16). Multiple ligands (Jagged, Delta) interact with the Notch receptor, triggering two additional cleavage reactions, releasing the intracellular domain. This intracellular domain translocates to the nucleus, where it interacts with the CSL DNA binding protein (CBF1) to activate numerous transcription factor genes, particularly of the Hairy–Enhancer of Split family.
Given the above evidence for overlap between BAV and LVOT malformations, along with the identification of NOTCH1 mutations in familial and sporadic BAV patients, we hypothesized that NOTCH1 mutations may be implicated in AVS, COA, and HLHS as well. We identified novel missense NOTCH1 alleles that are associated with LVOT malformations. At least two of these alleles reduce the levels of JAGGED1-induced NOTCH1 activity in cell-culture assays, and one of these perturbs the S1 cleavage of the receptor. These results support a common genetic etiology for LVOT malformations across the phenotypic spectrum, and suggest that relatively minor alterations in NOTCH1 activity may predispose individuals to develop these cardiac defects.
RESULTS
NOTCH1 mutation analysis in patients with LVOT defects identifies novel missense variants
We collected a cohort of 91 unrelated European American subjects with LVOT malformations, diagnosed by echocardiography, cardiac catheterization and/or surgical observation. Subject genomic DNA was screened for mutations in NOTCH1. Eight different missense variants, six of them novel, were identified (Table 1 and Fig. 1A). Mutations were found across all phenotypes (AVS, 8/37; BAV, 1/5; COA, 2/35; HLHS, 3/14). One variant (G661S) was found in all three LVOT defects (AVS, COA and HLHS), as well as in a patient with BAV. There does not appear to be an obvious genotype–phenotype correlation, but all individuals with COA who have a NOTCH1 mutation also have a BAV. One additional change, G1476S, was observed among the controls but was not present in any of the cases. The proportion of missense mutations among the cases (14/91; 15.4%) versus the controls (8/208; 3.9%) was significantly increased (χ2 = 12.36, 1 d.f.; P < 0.001). The parents of those cases with missense variants were directly sequenced, and in all cases the same mutations were identified in one parent. Echocardiography data were available for 8/28 parents, none of whom had any abnormalities.
Table 1.
Missense NOTCH1 variants, by anatomic defect of subject
| Nucleotide change | Exon | Amino acid change | Domain | Defects | Case freq. | Control freq. |
|---|---|---|---|---|---|---|
| c.2058G→A | 12 | G661S | EGF-like repeat 17 | AVS, BAV (1); BAV (1); COA, BAVa (1); HLHS (1) | 4/91** | 1/212 |
| c.2123G→A | 13 | A683T | EGF-like repeat 18 | AVS (1); HLHS (1) | 2/91 | 0/216 |
| c.2156G→A | 13 | E694K | EGF-like repeat 18 | AVS | 1/91 | 0/212 |
| c.3912G→A | 23 | R1279H | EGF-like repeat 33 | AVS (3) | 3/91 | 4/207 |
| c.4104C→T | 25 | A1343V | EGF-like repeat 34 | HLHS | 1/91 | 1/207 |
| c.4125G→T | 25 | R1350L | EGF-like repeat 35 | AVS | 1/91 | 1/208 |
| c.4898G→A | 26 | R1608H | Extracellular | AVS | 1/91 | 0/208 |
| c.7682G→A | 34 | V2536I | Cytoplasmic | COA, BAV | 1/91 | 0/209 |
AVS, aortic valve stenosis; BAV, bicuspid aortic valve; COA, coarctation of the aorta; HLHS, hypoplastic left heart syndrome.
aThis individual also has the R1279H variant in cis.
**P = 0.028 (Fisher exact) comparing cases versus control frequency.
Figure 1.

NOTCH1 mutations in LVOT subjects. (A) Molecular characterization of NOTCH1 missense mutations identified in patients. DNA sequence analysis of the eight missense mutations is shown. (B) Schematic representation of the NOTCH1 protein indicating the localization of protein variants identified in this study. LNR=Lin12/Notch repeats. TM, transmembrane domain. (B) Cross species sequence comparison for the highly conserved non-synonymous changes G661S, A683T, R1279H and A1343V.
Four novel mutations (A683T, E694K, R1608H and V2536I) were not observed in more than 200 ethnically matched controls. The G661S mutation was found in one control, but it is significantly over-represented among the cases (Fisher’s exact P = 0.028). These mutations are likely to represent disease susceptibility alleles. Three additional alleles are likely to represent polymorphisms. The R1279H allele was observed in three patients and four controls. The A1343V mutation, observed here in one case and one control, has been previously described in 1/48 individuals with BAV and thoracic aortic aneurysm, but was not seen among 122 normal controls (13). Combining our study with that of McKellar et al. (13), the frequencies of this mutation in cases is 2/161 versus 1/329 controls (Fisher’s exact P = 0.25). The R1350L variant, seen here in one case and one control, has also been found previously in one case and one control.
Missense variants occurred across NOTCH1, a gene that in general is highly conserved. Some evidence of clustering appears with three variants in EGF repeats 17 and 18 and three variants among repeats 33 through 35 (Fig. 1B). Sequence comparison was made across nine species (chimp, rhesus macaque, mouse, dog, cow, chicken, zebrafish and Drosophila against human) to assess evolutionary conservation of the discovered variants (Fig. 1C). The variants G661S, A683T, R1279H and A1343V lie in EGF-like domains and exhibit high sequence conservation. While position 1608 also demonstrates high conservation, it is outside any specific protein domain in the extracellular region. Variants E694K, R1350L, A2331T and V2536I were not well conserved. The substituted amino acid noted here for positions 694, 2331 and 2536 was found at the corresponding location in at least one other species, indicating the change could be well tolerated.
LVOT-associated missense variants reduce JAGGED1-induced NOTCH1 signaling
Although missense mutations in NOTCH1 have previously been described in patients with BAV (13,14), no functional analysis of any NOTCH1 allele associated with cardiac disease has been described. We focused on likely disease susceptibility mutations that affect highly conserved amino acids and that were identified in more than one subject (G661S and A683T). Our analysis also included the A1343V mutation, which has previously been described as a disease-associated allele (although it was identified in a single control individual in this study, it likely represents a polymorphism). Finally, we assessed the effects of the R1279H variant. This variant affects a highly conserved amino acid in the extracellular EGF repeats of the receptor, and was found in multiple subjects, but as it was also identified in multiple control individuals it is likely to represent a polymorphism, and thus would not be predicted to affect protein activity. Equivalent mutations were introduced into a construct encoding rat NOTCH1 to assess whether the alleles associated with LVOT malformations affect NOTCH1 function. We first assessed Notch1 pathway activity in the absence of induction by ligand. NOTCH1 expression vectors and a CBF1 responsive luciferase reporter (17) were co-transfected into NIH3T3 cells. In the absence of activating ligand, control Notch1 drives low levels of luciferase activity (Fig. 2A). Similar levels were seen for constructs encoding NOTCH1G661S, NOTCHA683T, NOTCH1A1343V or NOTCH1R1279H, indicating that these alleles are unlikely to cause leaky activation in the absence of activating ligand (Fig. 2A).
Figure 2.
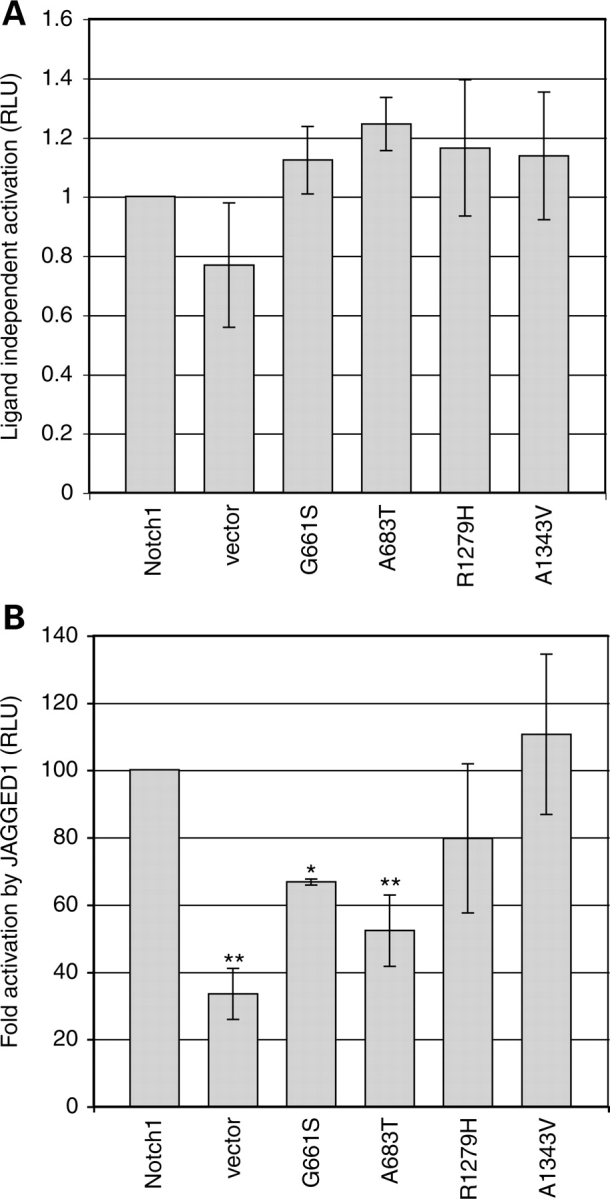
Mutations in the extracellular domain alter ligand-induced activation in variant NOTCH1 proteins. (A) Luciferase assays of Notch1 pathway activity in the absence of Jagged1 ligand. Low levels of NOTCH activity are seen in the wild-type and most variants, similar to that seen in the vector control. Results are presented as normalized luciferase values with wild-type Notch1 arbitrarily set to 1, and no significant differences are observed between the wild-type and any mutant (one way ANOVA, Dunnett’s post hoc, P > 0.05). (B) Luciferase assays of Notch1 pathway activity in the presence of Jagged1 ligand. Results are expressed as the fold Notch1 activation seen after culture with JAGGED1 cells compared to control L-cells, with wild-type NOTCH1 set to 100% (actual fold activation = 5.5 ± 1.4). Fold activation was significantly lower than wild-type NOTCH1 for NOTCH1G661S (66.72 ± 0.87) and NOTCH1A683T (52.2 ± 10.6). No significant change in activation was observed for NOTCH1R1279H (79.7 ± 22.2) or NOTCH1A1343V (110.6 ± 23.8). Error bars indicate the standard deviation from the mean. All experiments were performed at least three times in triplicate. Statistical significance was calculated by one-way ANOVA followed by Dunnett’s post hoc test. *P <.05, **P <.01.
To assess ligand-induced NOTCH1 activity, cells expressing NOTCH1 and the CBF luciferase reporter were co-cultured with control Ltk-mouse fibroblasts (hereafter referred to as L-cells) or with L-cells constitutively expressing JAGGED1 ligand. The fold induction of NOTCH1-dependent signaling induced by JAGGED1 was compared between wild-type NOTCH1 and mutant NOTCH1 (Fig. 2B). The closely spaced mutations in EGF repeats 17 and 18 (G661S and A683T), which we propose to be disease-associated alleles, each significantly reduce the induction of NOTCH1 by JAGGED1 compared to that seen with wild-type NOTCH1 (to 66.7% of control levels of induction for NOTCH1G661S (P < 0.05) and to 55.2% of control levels of induction for NOTCH1A683T (P < 0.01). In contrast, the NOTCH1R1279H and NOTCH1A1343V variants, which were identified at similar rates in controls and patients, do not significantly change the level of JAGGED1-induced activation induced, supporting the idea that they represent naturally occurring polymorphisms of NOTCH1.
The NOTCH1A683T variant perturbs S1 cleavage of the NOTCH1 receptor
One possible explanation for the reduction in Notch1 signaling observed in the NOTCH1G661S and NOTCH1A683T variants would be that the amino acid substitutions decrease either the expression level or the stability of the mutant proteins. If this is the case, we would expect to see a reduction in the total amount of protein expressed after transfection with vectors encoding NOTCH1G661S and NOTCH1A683T compared to vectors encoding wild-type NOTCH1, NOTCH1R1279H or NOTCH1A1343V. To address this question, we introduced these mutations into a vector encoding an N-terminally HA-tagged NOTCH1 receptor and assessed protein expression levels in transfected cells. As expected, we observe both a p300 and a p180 form of NOTCH1 using an antibody directed against a tag at the N-terminus. The p180 band represents NOTCH1 that has undergone S1 cleavage in the Golgi, while the p300 band is presumed to represent full-length NOTCH1. In cells expressing wild-type NOTCH1, we see robust levels of both the uncleaved p300 form of the receptor and the S1 cleaved p180 form of the receptor. We observe that total NOTCH1G661S and NOTCH1A683T proteins (p300 + p180) are observed at similar levels to wild-type, thus the reduction in JAGGED1-induced signaling through these mutant proteins is unlikely to be due to a simple reduction in receptor expression or stability (Fig. 3A and data not shown).
Figure 3.

The A683T mutation affects S1 cleavage of the NOTCH1 receptor. (A) Western blot analysis of N-terminally HA-tagged NOTCH1 protein. NIH3T3 cells transfected with Notch1 expression vectors were lysed and the expression of NOTCH1 protein was assessed with an anti-HA antibody. HA-NOTCH1 bands are observed at 300 and 180 kDa, representing full-length (p300) and S1 cleaved (p180) NOTCH1 receptor. All proteins are expressed robustly, and to similar levels as seen in wild-type. (B) The levels of p300 and p180 Notch1 proteins were quantitated in western blots, demonstrating that the NOTCH1A683T variant undergoes S1 cleavage at reduced levels. Results are expressed as the percent of total protein found in the p180 band. Error bars represent the standard deviation from the mean of at least four experiments. ** indicates P < 0.01 by ANOVA followed by Dunnett’s post hoc.
The relative amounts of p300 and p180 NOTCH1 in the cell reflect the efficiency of S1 cleavage of the protein in the Golgi before presentation on the cell surface. The S1 cleavage is generally considered as a prerequisite for ligand-induced, CBF1-mediated Notch signaling (16,18). Thus, one possible mechanism to reduce ligand-induced NOTCH1 signaling would to reduce the efficiency of S1 processing. In western blot analysis of the LVOT-associated mutations, we noted that some variants appear to exhibit a reduction in the ratio of p180 to p300, suggesting a possible reduction in S1 cleavage. To address this question, the amount of protein in the p300 and p180 bands was quantified in at least four independent experiments, and the percentage of total NOTCH1 protein that has undergone S1 cleavage was calculated. We find that the NOTCH1A683T variant significantly reduces the percentage of total NOTCH1 protein that undergoes S1 cleavage (from 44.5% in wild-type NOTCH1 to 33.4% in NOTCH1A683T P < 0.01). Thus, this mutation affects both protein processing and NOTCH1 signaling, and the reduction in cleavage efficiency may play a role in the decrease in JAGGED1-induced signaling.
DISCUSSION
We identified eight missense mutations, six of them novel, in patients with sporadic LVOT malformations. Of these, five were either completely absent or were under-represented in over 200 ethnically matched controls, suggesting they may be disease predisposition alleles. The frequency of mutations in our cohort (15.4%) exceeds that reported previously in subjects with BAV and thoracic aortic aneurysms (2/48, 4.2%) or BAV alone (2/48, 4.2%). If we consider only mutations found in cases at highly conserved sites that have functional alterations (A683T), our frequency is 2/91 (2.2%). G661S, which is present in one control but over-represented among the cases, also involves a conserved site and demonstrates functional change. The addition of that variant would increase the frequency to 6/91 (6.6%). The reason for the higher rate of missense changes in our group is unknown, and may be due to chance. Screening of a larger cohort will be required to better define the frequency of NOTCH1 mutations among individuals with LVOT defects.
A possible limitation of this study is the absence of echocardiography screening of the control group. While all controls are healthy without evidence of heart defects, there is a 1–2% possibility of asymptomatic BAV in this group (19–22). This could result in mutation ascertainment bias in the controls, causing a false negative result for specific mutations. We would estimate this rate to be very low, i.e. [rate of BAV in controls (0.02)] × [rate of mutation among individuals with BAV (0.04)]=rate of mutations in control group (0.0008).
We show in this study that mutations in NOTCH1 that alter function of the signaling pathway are found in individuals with AVS, COA and HLHS. The presumed common pathogenetic cause for LVOT defects is underscored by the finding that one variant (G661S) was found in all three LVOT defects (AVS, COA and HLHS), as well as in a patient with BAV. Interestingly, the previously reported R1108X mutation also caused a spectrum of LVOT phenotypes, including aortic valve disease and an individual with mitral valve atresia, double outlet right ventricle and hypoplastic left ventricle (a possible variation of HLHS) (12). These findings support the hypothesis that a wide spectrum of LVOT defects are developmentally related, and a single genetic defect can underlie diverse phenotypic outcomes. This is reminiscent of findings that mutations in NKX2–5 are observed in rare patients with non-syndromic LVOT defects, although functional studies of the identified mutation demonstrated minor effects (23–25). Taken together, these results support the suggestion that LVOT defects have a common developmental cause, and are governed by complex genetic inheritance.
We note that while G661S significantly decreases ligand induced signaling compared to wild-type, and R1279H does not have a statistically significant effect, the comparison between G661S and R1279 does not suggest a large difference in activity between these two mutants. The experiments reported here were specifically designed to allow comparison of the variant proteins to the wild-type control, thus future analyses will be required to determine whether G661S and R1279H actually have significantly different activities or whether there may be a subtle alteration in ligand-induced signaling through R1279H.
Our results indicate that LVOT-associated mutations reduce ligand-activated NOTCH1 signaling. This is consistent with two NOTCH1 mutations found previously to cause familial calcific aortic valve disease (R1108X and H1505del) which are proposed to be null mutations, as they truncate the protein in the extracellular domain (12). These findings suggest that familial aortic valve disease can be caused by haploinsufficiency for the NOTCH1 receptor. The reduction of ligand-induced signaling observed through the NOTCH1G661S and NOTCH1A683T variants would support a similar model for the spectrum of non-syndromic LVOT defects, namely that Notch signaling levels must be tightly regulated and that relatively minor alterations in Notch1 activity levels may promote LVOT defects.
The molecular mechanisms by which these mutations affect NOTCH1 activation are, as yet, unclear. NOTCH1 mutations observed in leukemia have been shown to increase ligand-independent NOTCH1 activation by promoting S2 and S3 cleavage (26). In contrast, in this system where ligand-dependent activation is reduced, we observe a reduction in S1 cleavage of the A683T variant. These findings are typical of developmentally important genes, where gain-of-function mutations are associated with cancers and loss-of-function mutations cause congenital defects. This alteration in processing may change the cellular localization of the receptor, or may simply reduce the amount of S1 cleaved heterodimeric NOTCH1 available for ligand-induced signaling. This may be similar in some respects to mutations identified in Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL), in which mutations in the NOTCH3 receptor have been variously reported to affect NOTCH3 signaling, protein processing and cellular localization (27–29). Further experimentation will be required to assess the functional implications of the LVOT-associated NOTCH1 mutants identified here.
The mechanism(s) by which NOTCH1 mutations and the subsequent presumed reduction of NOTCH1 signaling promote LVOT defects remains unclear. Notch signaling has been proposed to play multiple functions during cardiac development, including cardiomyocyte differentiation, valve formation and outflow tract remodeling (30). In mice, Notch1 activation promotes epithelial to mesenchymal transisition (EMT) during valve development through the activation of Snail, and this may represent a developmental stage where NOTCH1 dosage is critically important (31). Interestingly, some NOTCH1 mutations may play additional roles in cardiac disease. Garg et al. (12) showed that RUNX2 levels are repressed by Notch1, suggesting that the de-repression of RUNX2 may contribute to valve calcification in some BAV patients. None of our subjects showed evidence of aortic valve calcification, but all are young and may not have had time to develop calcification. It is not yet known whether the variants identified here could additionally contribute to calcification phenotypes.
The findings of an increased proportion of NOTCH1 variants among LVOT malformation cases, compared to controls, suggest that these mutations are indeed LVOT disease susceptibility alleles. This is further supported by the findings that two variants perturb ligand-induced signaling. Clearly, as LVOT-associated variants are found in unaffected parents of our patients, the NOTCH1 mutations with their relatively subtle effects on signaling are not sufficient in and of themselves to perturb cardiac development. The variants presumably act in concert with genetic background, other specific gene mutations and/or developmental insults in order to cause phenotypes. The presence of a mutation in a normal parent, indicating reduced penetrance, is typical of many complex genetic diseases. This requirement for additional genetic or environmental causes may also partially explain why no cardiac defects have been reported in Notch1+/− mice, while NOTCH1 haploinsufficiency has been suggested as a cause of familial BAV.
In summary, we note for the first time a common molecular mechanism for AVS, BAV, COA and HLHS. This substantiates the observations from familial studies demonstrating a higher concordance for these LVOT defects in families with multiple individuals affected with a CVM. Notch signaling is involved in endothelial–mesenchymal transformation in the ventricular chamber (32), atrioventricular valves (31) and vasculature (33). We hypothesize that altered Notch1 signaling affecting this transformation is the unifying event in the pathogenesis of the LVOT disorders. Future studies will focus effort on candidate genes important in endothelial–mesenchymal transformation and on further defining the functional mechanisms of these NOTCH1 mutations.
MATERIALS AND METHODS
Study subjects
We collected a cohort of 91 unrelated European American subjects with LVOT malformations, diagnosed by echocardiography, cardiac catheterization and/or surgical observation. All subjects were enrolled after informed consent, per an IRB-approved protocol. Both parents of the subjects were also enrolled. The case anatomic lesions included AVS (n = 24), AVS with BAV (n = 13), BAV alone (n = 5), COA (n = 24), COA with BAV (n = 9), COA with AVS (n = 2) and HLHS (n = 14). All subjects had isolated non-syndromic heart defects. None of the subjects’ parents had a congenital heart defect. Echocardiography data were available on a limited number of parents (n = 50). All parental echocardiography examinations were normal. Blood samples were collected, and peripheral lymphocytes were transformed by EBV into lymphoblastoid cell lines. Genomic DNA was extracted using the PureGene kit (Gentra Systems). Controls consisted of 216 race and ethnically matched samples.
Mutation screening
Cases and controls were screened for mutations in the entire NOTCH1 gene by denaturing high performance liquid chromatography (dHPLC). Exons and flanking introns were amplified by PCR using primers designed with Primer Express software (Applied Biosystems, Inc). Melt curves were generated with the Navigator software and dHPLC was performed on a WAVE MD system (Transgenomic, Inc). PCR primers and melt temperatures are available on request.
Amplicons with chromatograms that differed from controls were sequenced bidirectionally by the dideoxy method using Big Dye Terminator v3.1 chemistry (Applied Biosystems) and run on an ABI 3730 sequencer to ascertain the presence and location of any mutation.
Numbering of basepairs and amino acids was based on the reference sequence NM_017617.3 [gi:148833507] from the NCBI, with the first basepair of the transcript being the ‘A’ nucleotide of the ATG start codon.
Mutagenesis of NOTCH1
Four mutations were examined (G661S, A683T, R1279H and A1343V). Equivalent mutations were introduced into a rat Notch1 expression vector (34), by overlap extension PCR with nucleotide changes incorporated into PCR primers. Each mutagenic primer was used in a PCR reaction with a primer flanking the region of interest. The resulting products were used as templates in a second round of PCR using only the flanking primers, creating a PCR product with the desired mutation (primer sequences are found in Table 2). Mutated PCR products were TA cloned into pCRII-Topo (Invitrogen) and sequenced before conventional restriction digest and ligation were used to incorporate the mutated fragment into the rat Notch1 expression vector. Final constructs were confirmed by sequencing. For western blot analysis, mutations were similarly introduced into an N-terminally HA-tagged Notch1 expression allele (35).
Table 2.
Primer sequences for mutagenesis of rat NOTCH1
| Missense mutation | Flanking primer | Mutagenic primer |
|---|---|---|
| G661S (5′ fragment) | 5′ atgactgcccaggaaacaac | 5′ caagatcgatAgctacgagtgtg |
| G661S (3′ fragment) | 5′agggcagttgcatttgtacc | 5′ cacactcgtagcTatcgatcttg |
| A683T (5′ fragment) | 5′ atgactgcccaggaaacaac | 5′ gacgaatgtAcgggcagcccctg |
| A683T (3′ fragment) | 5′agggcagttgcatttgtacc | 5′ caggggctgcccgTacattcgtc |
| R1279H (5′ fragment) | 5′ tgagtccaacccttgtgtca | 5′ ctgtgacccacAtggcacccag |
| R1279H (3′ fragment) | 5′ tcattgaagttgagggagca | 5′ ctgggtgccaTgtgggtcacag |
| A1343V (5′ fragment) | 5′ tgagtccaacccttgtgtca | 5′ cttcgagggtgTcacttgtg |
| A1343V (3′ fragment) | 5′ tcattgaagttgagggagca | 5′ cacaagtgAcaccctcgaag |
Mutated nucleotides indicated in upper case.
NOTCH1 signaling assay
The activity of mutant Notch1 receptors was assessed utilizing previously described signaling assays (34,36). As in reference (34), NIH3T3 cells were employed as the signal receiving cells. NIH3T3 cells were maintained in DMEM+10% FCS. Control and Jagged1 Ltk-mouse fibroblasts (L-cells) (36) were obtained from Gerry Weinmaster and maintained in DMEM+10% FBS. 4 × 104 NIH 3T3 cells were plated in 24 well plates and transfected using Lipofectamine 2000 (Invitrogen) with 100 ng NOTCH1 expression vector (encoding wild-type or mutant NOTCH1) or control pEFBOS vector, 200 ng CBF1 responsive luciferase reporter (17) and 10 ng pSV Renilla as a transfection control. Total DNA concentration was brought to 1 µg with pcDNA3. Sixteen hours after, transfection cells were overlain with 1.24 × 106 control or JAGGED1-expressing L-cells, or fresh media as a control. After 24 h, cells were lysed and luciferase and Renilla levels were assayed using the dual luciferase kit (Promega). Luciferase values were normalized to Renilla expression to control for transfection variability. Notch-responsive activation of the luciferase reporter is expressed as a ratio of normalized luciferase values from coculture with JAGGED1-expressing cells to normalized luciferase values from coculture with control L-cells, with wild-type activation set to 100%. Notch1 pathway signaling activity between wild-type and each mutant was compared by ANOVA followed by Dunnett’s post hoc tests.
Western blot analysis
2.5 × 106 cells were transfected with NOTCH1 expression vectors in six well plates. After 24 h, cells were lysed in 500 µl RIPA buffer containing protease inhibitors. Samples were run on a 6% SDS–PAGE gel, transferred to nitrocellulose, and NOTCH1 protein was detected with mouse anti-HA antibody (HA-7, 1:1000, Sigma-Aldrich). Alexafluor anti-mouse 680 secondary antibody was used for detection (1:20 000, Invitrogen) and band intensity was quantified using Li-Cor Odyssey2.1 software. Blots were re-probed using a monoclonal anti-tubulin antibody (1:1000, Sigma) as a loading control. p300 and p180 bands were quantified, and the percent total protein cleaved was calculated as p180/(p180 + p300). Each experiment was performed at least four times, and statistical analysis was performed (one way ANOVA, Dunnett’s post hoc).
FUNDING
This study was supported by funding from the National Institutes of Health (HL70823 and HD43372) and the Research Institute at Nationwide Children’s Hospital to K.L.M., and the National Institutes of Health (HD39056) to J.W.B.
ACKNOWLEDGEMENTS
We thank Gerry Weinmaster for reagents, and members of the Cole and McBride labs for helpful comments.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Pradat P., Francannet C., Harris J.A., Robert E. The epidemiology of cardiovascular defects, part I: a study based on data from three large registries of congenital malformations. Pediatr. Cardiol. 2003;24:195–221. doi: 10.1007/s00246-002-9401-6. [DOI] [PubMed] [Google Scholar]
- 2.McBride K.L., Marengo L., Canfield M., Langlois P., Fixler D., Belmont J.W. Epidemiology of noncomplex left ventricular outflow tract obstruction malformations (aortic valve stenosis, coarctation of the aorta, hypoplastic left heart syndrome) in Texas, 1999–2001. Birth Defects Res. A Clin. Mol. Teratol. 2005;73:555–561. doi: 10.1002/bdra.20169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferencz C., Loffredo C.A., Corea-Vilasenor A., Wilson P.D. In: Genetic and Environmental Risk Factors of Major Cardiovascular Malformations. Anderson R., editor. Vol. 5. Armonk, NY: Futura Publishing Co. Inc; 1997. pp. 165–225. [Google Scholar]
- 4.Centers for Disease Control and Prevention (CDC) Hospital stays, hospital charges, and in-hospital deaths among infants with selected birth defects—United States 2003. MMWR Morb. Mortal. Wkly Rep. 2007;56:25–29. [PubMed] [Google Scholar]
- 5.Minino A.M., Heron M.P., Smith B.L. Deaths: preliminary data for 2004. Natl. Vital Stat. Rep. 2006;54:1–49. [PubMed] [Google Scholar]
- 6.Levy H.L., Guldberg P., Guttler F., Hanley W.B., Matalon R., Rouse B.M., Trefz F., Azen C., Allred E.N., de la Cruz F., et al. Congenital heart disease in maternal phenylketonuria: report from the Maternal PKU Collaborative Study. Pediatr. Res. 2001;49:636–642. doi: 10.1203/00006450-200105000-00005. [DOI] [PubMed] [Google Scholar]
- 7.Wessels M.W., Berger R.M., Frohn-Mulder I.M., Roos-Hesselink J.W., Hoogeboom J.J., Mancini G.S., Bartelings M.M., Krijger R., Wladimiroff J.W., Niermeijer M.F., et al. Autosomal dominant inheritance of left ventricular outflow tract obstruction. Am. J. Med. Genet. A. 2005;134:171–179. doi: 10.1002/ajmg.a.30601. [DOI] [PubMed] [Google Scholar]
- 8.Menahem S. Familial aggregation of defects of the left-sided structures of the heart. Int. J. Cardiol. 1990;29:239–240. doi: 10.1016/0167-5273(90)90227-v. [DOI] [PubMed] [Google Scholar]
- 9.Lewin M.B., McBride K.L., Pignatelli R., Fernbach S., Combes A., Menesses A., Lam W., Bezold L.I., Kaplan N., Towbin J.A., et al. Echocardiographic evaluation of asymptomatic parental and sibling cardiovascular anomalies associated with congenital left ventricular outflow tract lesions. Pediatrics. 2004;114:691–696. doi: 10.1542/peds.2003-0782-L. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loffredo C.A., Chokkalingam A., Sill A.M., Boughman J.A., Clark E.B., Scheel J., Brenner J.I. Prevalence of congenital cardiovascular malformations among relatives of infants with hypoplastic left heart, coarctation of the aorta, and d-transposition of the great arteries. Am. J. Med. Genet. 2004;124A:225–230. doi: 10.1002/ajmg.a.20366. [DOI] [PubMed] [Google Scholar]
- 11.McBride K.L., Pignatelli R., Lewin M., Ho T., Fernbach S., Menesses A., Lam W., Leal S.M., Kaplan N., Schliekelman P., et al. Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: segregation, multiplex relative risk, and heritability. Am. J. Med. Genet. A. 2005;134:180–186. doi: 10.1002/ajmg.a.30602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garg V., Muth A.N., Ransom J.F., Schluterman M.K., Barnes R., King I.N., Grossfeld P.D., Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- 13.McKellar S.H., Tester D.J., Yagubyan M., Majumdar R., Ackerman M.J., Sundt T.M., 3rd Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2007;134:290–296. doi: 10.1016/j.jtcvs.2007.02.041. [DOI] [PubMed] [Google Scholar]
- 14.Mohamed S.A., Aherrahrou Z., Liptau H., Erasmi A.W., Hagemann C., Wrobel S., Borzym K., Schunkert H., Sievers H.H., Erdmann J. Novel missense mutations (p.T596M and p.P1797H) in NOTCH1 in patients with bicuspid aortic valve. Biochem. Biophys. Res. Commun. 2006;345:1460–1465. doi: 10.1016/j.bbrc.2006.05.046. [DOI] [PubMed] [Google Scholar]
- 15.Artavanis-Tsakonas S., Rand M.D., Lake R.J. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 16.Logeat F., Bessia C., Brou C., LeBail O., Jarriault S., Seidah N.G., Israel A. The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc. Natl Acad. Sci. USA. 1998;95:8108–8112. doi: 10.1073/pnas.95.14.8108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsieh J.J., Henkel T., Salmon P., Robey E., Peterson M.G., Hayward S.D. Truncated mammalian Notch1 activates CBF1/RBPJk-repressed genes by a mechanism resembling that of Epstein-Barr virus EBNA2. Mol. Cell. Biol. 1996;16:952–959. doi: 10.1128/mcb.16.3.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bush G., diSibio G., Miyamoto A., Denault J.B., Leduc R., Weinmaster G. Ligand-induced signaling in the absence of furin processing of Notch1. Dev. Biol. 2001;229:494–502. doi: 10.1006/dbio.2000.9992. [DOI] [PubMed] [Google Scholar]
- 19.Gray G.W., Salisbury D.A., Gulino A.M. Echocardiographic and color flow Doppler findings in military pilot applicants. Aviat. Space Environ. Med. 1995;66:32–34. [PubMed] [Google Scholar]
- 20.Pauperio H.M., Azevedo A.C., Ferreira C.S. The aortic valve with two leaflets—a study in 2,000 autopsies. Cardiol. Young. 1999;9:488–498. doi: 10.1017/s1047951100005400. [DOI] [PubMed] [Google Scholar]
- 21.Roberts W.C. The congenitally bicuspid aortic valve. A study of 85 autopsy cases. Am. J. Cardiol. 1970;26:72–83. doi: 10.1016/0002-9149(70)90761-7. [DOI] [PubMed] [Google Scholar]
- 22.Steinberger J., Moller J.H., Berry J.M., Sinaiko A.R. Echocardiographic diagnosis of heart disease in apparently healthy adolescents. Pediatrics. 2000;105:815–818. doi: 10.1542/peds.105.4.815. [DOI] [PubMed] [Google Scholar]
- 23.McElhinney D.B., Geiger E., Blinder J., Benson D.W., Goldmuntz E. NKX2.5 mutations in patients with congenital heart disease. J. Am. Coll. Cardiol. 2003;42:1650–1655. doi: 10.1016/j.jacc.2003.05.004. [DOI] [PubMed] [Google Scholar]
- 24.Goldmuntz E., Geiger E., Benson D.W. NKX2.5 mutations in patients with tetralogy of fallot. Circulation. 2001;104:2565–2568. doi: 10.1161/hc4601.098427. [DOI] [PubMed] [Google Scholar]
- 25.Kasahara H., Lee B., Schott J.J., Benson D.W., Seidman J.G., Seidman C.E., Izumo S. Loss of function and inhibitory effects of human CSX/NKX2.5 homeoprotein mutations associated with congenital heart disease. J. Clin. Invest. 2000;106:299–308. doi: 10.1172/JCI9860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malecki M.J., Sanchez-Irizarry C., Mitchell J.L., Histen G., Xu M.L., Aster J.C., Blacklow S.C. Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol. Cell Biol. 2006;26:4642–4651. doi: 10.1128/MCB.01655-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haritunians T., Chow T., De Lange R.P., Nichols J.T., Ghavimi D., Dorrani N., St Clair D.M., Weinmaster G., Schanen C. Functional analysis of a recurrent missense mutation in Notch3 in CADASIL. J. Neurol. Neurosurg. Psychiatry. 2005;76:1242–1248. doi: 10.1136/jnnp.2004.051854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Low W.C., Santa Y., Takahashi K., Tabira T., Kalaria R.N. CADASIL-causing mutations do not alter Notch3 receptor processing and activation. Neuroreport. 2006;17:945–949. doi: 10.1097/01.wnr.0000223394.66951.48. [DOI] [PubMed] [Google Scholar]
- 29.Peters N., Opherk C., Zacherle S., Capell A., Gempel P., Dichgans M. CADASIL-associated Notch3 mutations have differential effects both on ligand binding and ligand-induced Notch3 receptor signaling through RBP-Jk. Exp. Cell Res. 2004;299:454–464. doi: 10.1016/j.yexcr.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 30.High F.A., Epstein J.A. The multifaceted role of Notch in cardiac development and disease. Nat. Rev. Genet. 2008;9:49–61. doi: 10.1038/nrg2279. [DOI] [PubMed] [Google Scholar]
- 31.Timmerman L.A., Grego-Bessa J., Raya A., Bertran E., Perez-Pomares J.M., Diez J., Aranda S., Palomo S., McCormick F., Izpisua-Belmonte J.C., et al. Notch promotes epithelial–mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004;18:99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grego-Bessa J., Luna-Zurita L., del Monte G., Bolos V., Melgar P., Arandilla A., Garratt A.N., Zang H., Mukouyama Y.S., Chen H., et al. Notch signaling is essential for ventricular chamber development. Dev. Cell. 2007;12:415–429. doi: 10.1016/j.devcel.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krebs L.T., Xue Y., Norton C.R., Shutter J.R., Maguire M., Sundberg J.P., Gallahan D., Closson V., Kitajewski J., Callahan R., et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14:1343–1352. [PMC free article] [PubMed] [Google Scholar]
- 34.Hicks C., Johnston S.H., diSibio G., Collazo A., Vogt T.F., Weinmaster G. Fringe differentially modulates Jagged1 and Delta1 signalling through Notch1 and Notch2. Nat. Cell Biol. 2000;2:515–520. doi: 10.1038/35019553. [DOI] [PubMed] [Google Scholar]
- 35.Ladi E., Nichols J.T., Ge W., Miyamoto A., Yao C., Yang L.T., Boulter J., Sun Y.E., Kintner C., Weinmaster G. The divergent DSL ligand Dll3 does not activate Notch signaling but cell autonomously attenuates signaling induced by other DSL ligands. J. Cell Biol. 2005;170:983–992. doi: 10.1083/jcb.200503113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindsell C.E., Shawber C.J., Boulter J., Weinmaster G. Jagged: a mammalian ligand that activates Notch1. Cell. 1995;80:909–917. doi: 10.1016/0092-8674(95)90294-5. [DOI] [PubMed] [Google Scholar]