

Abstract
Overexpression of amyloid precursor protein (APP), as well as mutations in the APP and presenilin genes, causes rare forms of Alzheimer’s disease (AD). These genetic changes have been proposed to cause AD by elevating levels of amyloid-β peptides (Aβ), which are thought to be neurotoxic. Since overexpression of APP also causes defects in axonal transport, we tested whether defects in axonal transport were the result of Aβ poisoning of the axonal transport machinery. Because directly varying APP levels also alters APP domains in addition to Aβ, we perturbed Aβ generation selectively by combining APP transgenes in Drosophila and mice with presenilin-1 (PS1) transgenes harboring mutations that cause familial AD (FAD). We found that combining FAD mutant PS1 with FAD mutant APP increased Aβ42/Aβ40 ratios and enhanced amyloid deposition as previously reported. Surprisingly, however, this combination suppressed rather than increased APP-induced axonal transport defects in both Drosophila and mice. In addition, neuronal apoptosis induced by expression of FAD mutant human APP in Drosophila was suppressed by co-expressing FAD mutant PS1. We also observed that directly elevating Aβ with fusions to the Familial British and Danish Dementia-related BRI protein did not enhance axonal transport phenotypes in APP transgenic mice. Finally, we observed that perturbing Aβ ratios in the mouse by combining FAD mutant PS1 with FAD mutant APP did not enhance APP-induced behavioral defects. A potential mechanism to explain these findings was suggested by direct analysis of axonal transport in the mouse, which revealed that axonal transport or entry of APP into axons is reduced by FAD mutant PS1. Thus, we suggest that APP-induced axonal defects are not caused by Aβ.
INTRODUCTION
Alzheimer’s disease (AD) is characterized by an insidious and progressive decline of cognitive functions eventually culminating in dementia (1). Post-mortem, AD brains are distinguished by amyloid deposits and neurofibrillary changes, enriched in amyloid-β peptides (Aβ) and microtubule-associated protein tau, respectively, in a parenchyma reflecting synaptic and neuronal loss in several brain regions, including the hippocampi, the entorhinal and the association cortices and the basal nuclei (BN), which all play a role in cognition (2–4). These pathological changes produce impairments in several neurotransmitter systems such as cholinergic depletion of the cortices owing to inadequate cholinergic input from the BN (5). Recent evidence suggests that cholinergic disconnection and the amyloid deposition observed in AD and AD models could be related to defects in axonal transport (6–8). In fact, the identification of axonal defects in early AD and in AD models is consistent with previously reported cytoskeletal and neuritic abnormalities in AD (9–14) and supports the hypothesis that impaired axonal transport and cytoskeletal alterations play a critical role in the pathogenesis of AD (15–17). It is also possible that defective axonal transport and axonal defects are related to Aβ exposure. Data reported so far support a role for Aβ in the axonal defect formation, in particular in the formation of dystrophic neurites associated with cored amyloid plaques (6,18) and provide evidence of amyloid deposits without axonal defects as well as of axonal defects either preceding amyloid deposition or in areas devoid of amyloid (7,19–23).
Familial AD (FAD) can be caused by mutations in genes encoding amyloid precursor protein (APP) and presenilins (PS1 and PS2). These mutations appear to act by altering the proteolytic processing of APP. Additional clues to the pathogenesis of AD are provided by the existence of AD-like pathology, possibly caused by APP overexpression, in Down’s syndrome in which chromosome 21, harboring the APP gene, is trisomic (24). Tenet that excess APP can cause AD is corroborated by the recent discovery of APP duplications in some forms of hereditary AD (25–27). Thus, some types of AD may be caused by overexpression of wild-type (WT) APP (28).
Intriguingly, recent work revealed that overexpression of WT APP or APP bearing FAD mutations in Drosophila and mice can cause impaired axonal transport and axonal defects (7,8,29,30). In this regard, it has also been reported that PS1 not only has a role in regulated intramembrane proteolysis (RIP) (31) of proteins such as APP (32) and Notch (33–35), but it may also play roles in the regulation of kinesin-I-mediated intracellular transport (36,37). These findings suggest that APP and PS1 functions may be intimately linked to the molecular motor kinesin-I during axonal transport (38–41). In fact, in both Drosophila and mice, APP-mediated axonal transport defects can be enhanced by genetic reductions in kinesin-I (7,29,30). These reductions also enhanced aberrant Aβ generation and amyloid deposition (7).
The finding that genetic manipulations of APP and presenilins can cause defects in axonal transport, coupled to reports that axonal transport may be defective both early and late in AD (7,13,42,43) led to the suggestion that defects in axonal transport could play a major role in the cause or progression of AD (16,17). Unresolved, however, is the issue of whether axonal transport can be directly poisoned by Aβ, or whether mutations that enhance aberrant Aβ generation can cause axonal transport defects independent of Aβ production. Although intentionally increasing or decreasing APP expression can alter Aβ levels, such manipulations also alter the amount of other critical APP domains that may play roles in axonal transport. To test the effects of perturbing Aβ on axonal transport in vivo, we altered Aβ42/Aβ40 ratios by combining well-characterized APP transgenes with transgenic FAD PS1 mutations or with fusions of Aβ40 or Aβ42 to the BRI protein linked to Familial British and Danish Dementia (44–47) and tested whether axonal transport and other phenotypes induced by APP were enhanced by changes in Aβ.
RESULTS
Increasing Aβ peptides does not enhance APP-induced axonal blockages in mice
Axonal blockages in Drosophila and mice are thought to correspond to aberrant accumulations of proteins, vesicles and organelles within axons that block or otherwise inhibit normal axonal transport (7,29,48–51). Similar axonal morphologies have been reported in AD and may be an early event in the pathogenic progression, possibly preceding amyloid deposition (7). Axonal blockages have been observed in WT and FAD mutant APP-overexpressing Drosophila larval neurons and in mouse models of AD (7,29). In the mouse models, axonal blockages were observed long before amyloid deposition. Formally, APP overexpression could cause axonal blockages and axonal transport defects by directly increasing the amount of Aβ peptides. Alternatively, axonal transport defects could be induced by perturbing the proteolytic generation of other regions of APP in the intact protein (38). Since intentionally increasing or decreasing APP expression alters levels of Aβ and other critical APP domains simultaneously, we sought to test the effects of aberrant Aβ generation on axonal transport by increasing Aβ42/Aβ40 ratios in the absence of changes in APP expression. To achieve this goal, we first increased Aβ42/Aβ40 ratios by combining well-characterized APP transgenes with transgenic FAD PS1 mutations, and tested whether axonal transport and other phenotypes induced by APP overexpression were enhanced. Thus, we first analyzed axonal blockage formation in 4-month-old WT, single transgenic FAD mutant APP overexpressing (Tg-swAPPPrp), single transgenic FAD mutant PS1 (Tg-A246EPS1Prp) and double transgenic FAD mutant APP and FAD mutant PS1 (Tg-swAPPPrp; Tg-A246EPS1Prp) mice in both the C57BL/6J/C3H/HeJ and C57BL/6J genetic backgrounds.
Since previous work on cholinergic fibers in the BN found that large diameter axonal varicosities correspond to axonal swellings, a bona fide equivalent of axonal blockages at the light microscope level (7), we measured diameters and lengths of varicosities and inter-varicosity shafts (Supplementary Material, Fig. S1). These measurements were made ‘blind’ to the genotypes of the sections, which were coded. Codes were broken only after completion of the data collection. As reported previously (7), single transgenic Tg-swAPPPrp mice had a significantly higher percentage of choline acetyltransferase-immunoreactive (ChAT-IR) fibers with varicosities having diameters between 2.7 and 3.6 µm, 3.6 and 4.5 µm and larger than 4.5 µm compared with WT littermates in both genetic backgrounds examined (Fig. 1A and D and Supplementary Material, Fig. S2). Single transgenic Tg-A246EPS1Prp mice displayed a slight elevation in the percentage of ChAT-IR fibers with large diameter varicosities in the 2.7–3.6 µm category compared with WT littermates in the C57BL/6J genetic background. To our surprise, however, there were no differences in the percentage of ChAT-IR fibers with varicosities in any of the diameter intervals when double transgenic Tg-swAPPPrp; Tg-A246EPS1Prp were compared to WT littermates in either of the genetic backgrounds. In agreement with these measurements are the average total sizes of the ChAT-IR varicosities and inter-varicosity shafts, which revealed a significant increase in the diameter and length of the varicosities in the Tg-swAPPPrp compared with the WT, Tg-A246EPS1Prp and Tg-swAPPPrp; Tg-A246EPS1Prp littermates (Supplementary Material, Table S1).
Figure 1.
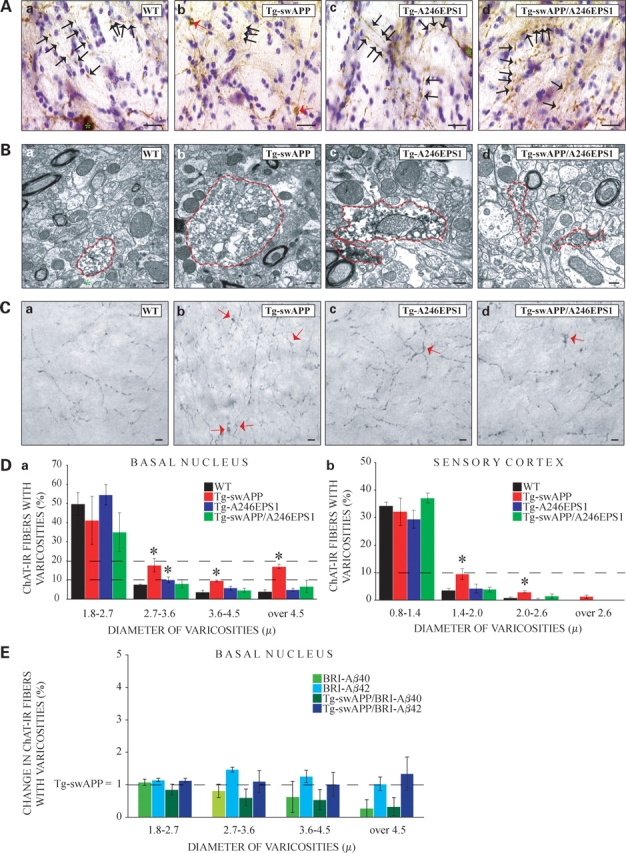
Mutant PS1 suppresses APP-induced axonal swellings or blockages in the baso-cortical cholinergic fibers of mice. (A) Representative light microscope images of cholinergic fibers in BN showing varicosities (black arrows) in all genotypes, while large axonal swellings (red arrows) were seen primarily in Tg-swAPPPrP (b), but not WT (a), Tg-A246EPS1PrP (c) or Tg-swAPPPrP; Tg-A246EPS1PrP (d) littermates (bar 15 µm). Perikarya (a and c) are marked with green asterisks. (B) Representative electron microscope images of cholinergic profiles in BN showing axonal blockages in Tg-swAPPPrP (b) and Tg-A246EPS1PrP (c), but not WT (a) or Tg-swAPPPrP; Tg-A246EPS1PrP (d) mice; axonal profiles are circled in red (bar 300 nm). (C) Representative light microscope images of cholinergic fibers in deep layers of the SC showing swellings (red arrows) in Tg-swAPPPrP (b), to a lesser extent in Tg-A246EPS1PrP (c) and Tg-swAPPPrP; Tg-A246EPS1PrP (d), but not in WT (a) mice (bar 15 µm). (D) (a) Increased percentage of cholinergic fibers in BN with varicosities of diameters between 2.7 and 3.6 µm, 3.6 and 4.5 µm and over 4.5 µm (swellings) in Tg-swAPPPrP (n = 3, * means P ≤ 0.05) and between 2.7 and 3.6 µm (swellings) in Tg-A246EPS1PrP (n = 3, * means P ≤ 0.05), but not WT (n = 3) or Tg-swAPPPrP; Tg-A246EPS1PrP mice (n = 3). (b) Increased percentage of cholinergic fibers in deep layers of the SC with varicosities of diameters between 1.4 and 2.0 µm and 2.0 and 2.6 (swellings) in Tg-swAPPPrP (n = 3, * means P ≤ 0.05), but not WT (n = 3), Tg-A246EPS1PrP (n = 3) or Tg-swAPPPrP; Tg-A246EPS1PrP mice (n = 3). (E) No significant difference in the percentage of cholinergic fibers in the BN with varicosities of diameters between 1.8 and 2.7 µm, 2.7–3.6 µm, 3.6–4.5 and over 4.5 µm between 10- and 11-month-old Tg-swAPPPrP (n = 6) and Tg- BRI-Aβ40Prp (n = 3), Tg-BRI-Aβ42Prp (n = 5), Tg-swAPPPrP; BRI-Aβ40Prp (n = 4) and Tg-swAPPPrP; BRI-Aβ42Prp (n = 4) littermates.
To confirm the nature of axonal swellings, and to test for additional fiber abnormalities, we examined cholinergic fibers in the BN by immuno-labeling for choline acetyltransferase (ChAT) followed by electron microscopy. ChAT-IR profiles of diameters less than 2.7 µm containing scarce vesicles and mitochondria were found in the BN of all genotypes. ChAT-IR profiles of large diameters containing numerous haphazardly distributed vesicles and mitochondria were identified almost exclusively in Tg-swAPPPrp, but not in WT and Tg-swAPPPrp; Tg-A246EPS1Prp, littermates (Fig. 1B, Supplementary Material, Fig. S3). As previously reported (7), Tg-swAPPPrp commonly exhibited axonal blockages that were not immunoreactive for ChAT and occasionally displayed electron dense axoplasm reminiscent of axonal degeneration (Supplementary Material, Figs S4 and S5).
We reasoned that if cholinergic axonal abnormalities were occurring close to the cell bodies in the BN, then we should also observe changes in cholinergic fibers at their termini. Thus, we investigated cholinergic terminals in the deep layers of the sensory cortex (SC). These data were also collected ‘blind’ to the genotypes of the sections by two independent observers (G.B.S. and E.M.R.) and produced comparable final results. At the level of the SC, fewer cholinergic fibers exhibited varicosities compared to the BN (Supplementary Material, Table S1). These ChAT-IR varicosities were substantially smaller compared to those sampled close to their origin in the BN. Again, however, only Tg-swAPPPrp mice contained significantly increased percentage of ChAT-IR fibers having varicosities of diameters between 1.4 and 2.0 microns, 2.0 and 2.6 microns and larger than 2.6 microns compared with WT, Tg-A246EPS1Prp and Tg-swAPPPrp; Tg-A246EPS1Prp littermates (Fig. 1C and D). Surprisingly, in contrast to fibers in the BN, we found that the length of cholinergic fibers per field at their termini was significantly increased in Tg-swAPPPrp and significantly decreased in Tg-swAPPPrp; Tg-A246EPS1Prp compared with WT and Tg-A246EPS1Prp littermates (Supplementary Material, Fig. S6). These data parallel those obtained for cholinergic synapses (52) and disclose possible aberrancies in the metabolism or transport of ChAT or in the structure of the termini. Taken together, however, and contrary to expectation, these data reveal that double transgenic Tg-swAPPPrp; Tg-A246EPS1Prp animals, which have perturbed Aβ generation, particularly increased Aβ42, have less severe axonal phenotypes than single transgenic Tg-swAPPPrp animals.
To corroborate these findings and to clarify whether suppression of axonal blockage phenotypes by transgenic FAD PS1 mutations was due to some other aspect of presenilin function, we turned to an alternative approach. Previous work revealed that fusions of BRI-Aβ42 chimeras lead to synthesis and secretion of Aβ42 and amyloid deposition (45). In particular, when Tg-BRI-Aβ42Prp mice were crossed with Tg-swAPPPrp (53), the double transgenic Tg- swAPPPrp; Tg-BRI-Aβ42Prp mice had significantly increased RIPA-insoluble formic acid extracted Aβ40 and Aβ42 as well as enhanced amyloid deposition in several brain areas including BN compared with single transgenic Tg-swAPPPrp, Tg-BRI-Aβ40Prp and BRI-Tg-Aβ42Prp or Tg-swAPPPrp; Tg-BRI-Aβ40Prp double transgenic littermates (45). Thus, to test whether Aβ40 or Aβ42 enhance axonal pathology observed in Tg-swAPPPrp, we crossed heterozygous Tg-swAPPPrp mice in the B6/SJL genetic background (53) with either heterozygous Tg-BRI-Aβ40Prp or heterozygous Tg-BRI-Aβ42Prp mice in the B6/C3 genetic background (45). Resulting mice were examined for Aβ production as well as amyloid deposition and scored for axonal defects. Aβ levels and amyloid deposition observed in WT, Tg-swAPPPrp, Tg-BRI-Aβ40Prp, Tg-BRI-Aβ42Prp, Tg-swAPPPrp; Tg-BRI-Aβ40Prp and Tg-swAPPPrp; Tg-BRI- Aβ42Prp littermates were similar to what previously reported (44,45). Specifically, 10–11-month-old Tg-swAPPPrp mice had 2935.4 ± 575.35 and 1561.2 ± 166.76 fmols/mg of wet brain of formic acid extractable Aβ40 and Aβ42, respectively. Tg-swAPPPrp; Tg-BRI-Aβ40Prp littermates had 603 ± 161.41 and 266.5 ± 129.93 fmols/mg of wet brain formic acid extractable Aβ40 and Aβ42, respectively, while Tg-swAPPPrp; Tg-BRI-Aβ42Prp littermates had 24499.67 ± 4841.7 and 9394.67 ± 2554.19 fmols/mg of wet brain formic acid extractable Aβ40 and Aβ42, respectively. All of these transgenic mice except for Tg-BRI-Aβ40Prp exhibited amyloid deposition with Tg-swAPPPrp; Tg-BRI-Aβ42Prp demonstrating the most extensive amyloid deposition. Axonal pathology was scored ‘blind’ to the genotypes by two independent observers (G.B.S. and E.M.R.). Data collected by either of the observers revealed comparable percentages of fibers with varicosities in all of the diameter intervals examined among Tg-swAPPPrp, Tg-BRI-Aβ40Prp and Tg-BRI-Aβ42Prp and no differences in the percentage of fibers with varicosities in any of the diameter intervals among Tg-swAPPPrp, Tg-swAPPPrp; Tg-BRI- Aβ40Prp and Tg-swAPPPrp; Tg-BRI-Aβ42Prp littermates despite dramatic changes in Aβ levels and amyloid deposition (Fig. 1E). Intriguingly, although BRI-Aβ40 and BRI-Aβ42 transgenes dramatically altered Aβ levels (6–9-fold increase in Aβ40 and Aβ42 in Tg-swAPPPrp; Tg-BRI-Ab42Prp versus Tg-swAPPPrp), they had no impact on the frequency of axonal pathology generated by the Tg-swAPPPrp transgene. Thus, enhancing Aβ accumulation per se does not lead to enhanced formation of axonal blockages, suggesting that the major route of generation of axonal phenotypes in APP transgenic mice is not a result of increasing levels of Aβ.
Mutant PS-1 modulates APP-induced phenotypes in Drosophila
Previous work showed that overexpression of WT and FAD mutant human APP and Drosophila APPL in the Drosophila larval nervous system causes axonal blockages reminiscent of those found in mutants for molecular motors (29,30,50). To test whether FAD mutant presenilin can suppress formation of these blockages as we observed in mice, we examined cysteine string protein stained segmental nerves of Drosophila larvae by immunofluorescence microscopy. Specifically, we used the Gal4-UAS system to express FAD mutant human APP (54) alone or in combination with a transgene encoding a FAD mutant Drosophila presenilin (PsnE280A), which is comparable to the FAD A246E PS1 transgene that we tested in mice (55). In previous work, we found that the expression of human APP carrying the Swedish FAD mutation caused axonal blockages within larval segmental nerves (29). Strikingly, expression of the Swedish mutant of APP in combination with PsnE280A (Fig. 2A and B) failed to induce axonal blockages. Note that the expression of PsnE280A alone had no obvious effect on axonal phenotypes. This observation suggested that, as in the mouse, FAD mutant presenilin suppressed, rather than enhanced the formation of axonal blockages caused by FAD mutant APP.
Figure 2.

FAD mutant Drosophila presenilin suppresses FAD mutant APP-induced axonal blockages in the segmental projection neurons of Drosophila larvae. (A) Cysteine string protein-immunoreactive (CSP) axonal blockages are observed (arrows) in larvae overexpressing FAD mutant human APP (SWE, n = 5), but not in larvae expressing FAD mutant Drosophila presenilin (PsnE280A, n = 5) or in larvae co-expressing FAD mutant Drosophila presenilin with FAD mutant APP (PsnE280A;SWE, n = 5, bar=10 µm). (B) Quantification of axonal blockages. (C) TUNEL analysis indicates apoptotic nuclei in larval brains expressing FAD mutant APP (SWE, n = 5, arrows), but not in larval brains expressing FAD mutant Drosophila presenilin (PsnE280A, n = 5) or in larval brains co-expressing FAD mutant Drosophila presenilin with FAD mutant APP (PsnE280A;SWE, n = 5, bar=10 µm). (D) Quantification of TUNEL positive nuclei.
In previous work (29), we found that the expression of the Swedish mutant of APP could induce apoptosis in Drosophila larval neurons. To test whether mutant presenilin-mediated suppression of FAD mutant APP-induced phenotypes included suppression of apoptosis in addition to suppression of axonal blockages, we compared TUNEL staining in larval brains expressing FAD mutant APP and FAD mutant presenilin singly or in combination. We observed TUNEL-positive neurons in animals expressing FAD mutant APP alone, but not in combination with FAD mutant presenilin (Fig. 2C and D). Thus, it appears that FAD mutant presenilin can suppress apoptosis induced by FAD mutant APP.
Mutant PS1 does not enhance APP-induced behavioral impairments in mice
It was surprising that modulating APP processing and/or perturbing the amounts of different Aβ species directly did not worsen of FAD mutant APP-induced axonal transport deficits in mice or Drosophila. We noted, however, that our failure to find any enhancement of APP-induced phenotypes by altering Aβ is consistent with a number of previous reports of small, if any, enhancements in phenotype when FAD APP overexpression alone was compared to FAD APP overexpression combined with an FAD mutant PS transgene (52,56–58). To extend our findings, we asked whether behavioral phenotypes were enhanced in our mice as would be expected if Aβ levels caused behavioral phenotypes on their own. We crossed heterozygous Tg-swAPPPrp and heterozygous Tg-A246EPS1Prp mice in the C57BL/6J/C3H/HeJ and C57BL/6J genetic backgrounds. WT, Tg-swAPPPrp, Tg-A246EPS1Prp and Tg- swAPPPrp; Tg-A246EPS1Prp littermates produced for behavioral analysis were coded for their genotypes at weaning. Upon reaching 4 months of age, these animals were tested with a battery of behavioral tasks over a 10 day period. In brief, day 1 consisted of an open field test and a set of three consecutive rotarod trials. Days 3, 5 and 8 were designated for similar rotarod trials. Days 9 and 10 were used for training and testing for fear conditioning, respectively. Codes were broken only upon completion of all tasks.
To assess open-field behavior, we measured the corner index (CI) defined as the number of times the mouse reaches any of the corners of the experimental box in 60 s. We found increases in the corner indices in Tg-A246EPS1Prp and Tg-swAPPPrp; Tg-A246EPS1Prp compared with WT and Tg-swAPPPrp mice in the C57BL/6J/C3H/HeJ genetic background and no change between genotypes in the C57BL/6J genetic background (Supplementary Material, Fig. S7).
When tested on the rotarod (Fig. 3A), Tg-swAPPPrP mice exhibited a substantial deficit compared to WT mice or mice in which FAD mutant PS1 was expressed on its own. Strikingly, not only did co-expression of FAD mutant PS1 with FAD mutant APP fail to enhance the deficit seen in Tg-swAPPPrP mice, but it actually produced partial suppression of the working performance deficit seen in Tg-swAPPPrp mice.
Figure 3.

Mutant PS1 suppresses APP-induced behavioral impairments in mice. (A) Significant decrease in the time spent on the rotating rod by Tg-swAPPPrP (n = 9 and 17, * means P ≤ 0.05) and Tg-swAPPPrP; Tg-A246EPS1PrP (n = 7 and 17, * means P ≤ 0.05) mice on trial days 1, 3, 5 and 8 and on trial day 8, respectively, in the C57BL/6J/C3H/HeJ (a) and on trial days 1, 3 and 8 and on trial days 1 and 3, respectively, in the C57BL/6J (b) genetic background compared with WT (n = 11 and 19) littermates. No difference in the time spent on the rotating rod between WT and Tg-A246EPS1PrP (n = 11 and 15) mice in either of the genetic backgrounds. (B) No difference in the percentage of freezing between WT (n = 11 and 19), Tg-swAPPPrP (n = 9 and 17), Tg-A246EPS1PrP (n = 11 and 15) and Tg-swAPPPrP; Tg-A246EPS1PrP (n = 7 and 17) littermates during fear conditioning training (a and b) in C57BL/6J/C3H/HeJ (a) and C57BL/6J (b) genetic backgrounds. Significantly reduced percentage of freezing in Tg-swAPPPrP (* means P ≤ 0.05), but not Tg-A246EPS1PrP or Tg-swAPPPrP; Tg-A246EPS1PrP, compared with WT littermates during evaluation of cued conditioning 24 h after training (c and d) in C57BL/6J/C3H/HeJ (c) and C57BL/6J (d) genetic backgrounds. In brackets, n corresponds to the number of mice tested with the first number referring to the C57BL/6J/C3H/HeJ and second to the C57BL/6J genetic background (EXP corresponds to 3 min exploration period, CS to 20 s conditioned stimulus and I to the 1 min interval between CS, for a description of the fear conditioning paradigm consult Behavioral analysis section of the Materials and Methods).
Since rotarod tests revealed changes in motor performance between genotypes, we chose not to use water maze testing and instead evaluated fear conditioning, a form of associative learning that does not rely on motor performance. We excluded possible alterations in pain and sound perception between genotypes by verifying that the minimal amount of current required to elicit a reaction to foot-shock and the sound required to elicit startle response were the same among genotypes and genetic backgrounds (data not shown). Following training (see Materials and Methods), we found that all four genotypes of mice could learn to associate a neutral conditioned stimulus to an aversive unconditioned stimulus (foot-shock; Fig. 3B).
The next day, we tested mice for contextual conditioning, and 2 h later, for cued conditioning. When compared to WT, Tg-A246EPS1Prp and Tg-swAPPPrp; Tg-A246EPS1Prp littermates in two genetic backgrounds (C57BL/6J/C3H/HeJ and C57BL/6J), Tg-swAPPPrp mice exhibited early changes in contextual conditioning that are not enhanced, and may even be mildly suppressed by expression of FAD mutant PS1 (Supplementary Material, Fig. S8). When tested for cued conditioning, all mice responded to the testing environment as a novel context, and all mice exhibited significant conditioning in response to the conditioned stimulus. Tg-swAPPPrp, but not Tg-A246EPS1Prp or Tg-swAPPPrp; Tg-A246EPS1Prp, mice exhibited deficits in conditioning compared to WT littermates, suggesting that these behavioral phenotypes were not enhanced by increasing Aβ generation.
Mutant PS1 reduces axonal transport of APP in mice
Since recent evidence suggests that genetic manipulation of PS1 can lead to changes in the delivery of APP to cell surfaces and synapses (41,59), we asked whether FAD mutant PS1-mediated reduction of APP axonal transport could be responsible for reversing the observed APP-induced phenotypes. Axonal transport was evaluated in sciatic nerve ligation experiments in which gels and western blots were run and scanned by one investigator, then coded for genotypes and analyzed ‘blind’ by another investigator who broke the codes only upon completion of the experiments. In 4-month-old WT mice, we observed a significant increase in the levels of APP in the proximal and distal stumps in the ligated compared with unligated sciatic nerves (Fig. 4A and B). In 4-month-old Tg-swAPPPrp mice, although we observed an increase in APP accumulation at the proximal ligature site, the extent was less than in WT, suggesting a reduction in the anterograde axonal transport of APP upon its overexpression. This observation differs from the recent conclusion (40) that overexpressing APP does not lead to altered transport in the sciatic nerve. However, this paper did not compare Tg-APP to WT controls as we have done here. Our observation is consistent with the many published observations (7,29,30,60) that overexpressing APP and its homologues can poison axonal transport. Analysis of 4-month-old Tg-A246EPS1Prp and Tg-swAPPPrp; Tg-A246EPS1Prp ligations suggested that the expression of FAD mutant PS1 may reduce anterograde axonal transport of APP (Fig. 4C and D). Intriguingly, expression of FAD mutant APP with FAD mutant PS1 may operate additively in reducing anterograde axonal transport of APP. We tested the possibility that FAD mutant PS1 poisons anterograde APP axonal transport by causing release of kinesin-I from vesicles as recently proposed (40,41). We observed no change in the amount of kinesin-I associated with brain-derived membrane preparations either by flotation or by differential centrifugation between WT and Tg- A246EPS1Prp littermates (data not shown). Control experiments showed increased levels of the axonal anterograde molecular motor, kinesin-I, in the proximal stumps and increased levels of a bidirectionally transported scaffold protein, SYD/JIP3, in both proximal and distal stumps of the ligated compared with unligated sciatic nerves (Supplementary Material, Fig. S9) (30,38,39,61). Levels of molecules residing within Schwann cell membranes or those thought to be moving with the slow wave of axonal transport, such as myelin basic protein and tubulin, were used as loading controls. Collectively, these results suggest that, in addition to increasing Aβ42 generation (Supplementary Material, Fig. S10), expression of FAD mutant PS1 reduces the fast anterograde axonal transport of endogenous and transgenic APP.
Figure 4.

Mutant PS1 reduces axonal transport of APP in mice. (A) Representative western blots in the 6 h sciatic nerve ligation paradigm. In 4-month-old WT mice 6 h ligation of the sciatic nerve produced significant accumulation of APP in its proximal and distal stumps, but not in the dorsal root ganglion. Proteins undergoing slow axonal transport, such as tubulin, as well as proteins expressed by Schwann cells, such as myelin basic protein (MBP), were used as loading controls. (B) Average levels of APP were significantly increased after 6 h of ligation relative to unligated controls in the proximal stump of 4-month-old WT (n = 6, P ≤ 0.005), Tg-swAPPPrP (n = 7, P ≤ 0.005), Tg-A246EPS1PrP (n = 6, P ≤ 0.000005) and Tg-swAPPPrP; Tg-A246EPS1PrP (n = 7, P ≤ 0.005) and in the distal stump of 4-month-old WT (n = 6, P ≤ 0.05) and Tg-swAPPPrP (n = 7, P ≤ 0.05), but not Tg-A246EPS1PrP (n = 6) and Tg-swAPPPrP; Tg-A246EPS1PrP (n = 7). (C) Representative image (a) of significantly reduced average levels of APP (b) in the proximal stump of ligated sciatic nerves in 4-month-old Tg-A246EPS1PrP (blue; n = 6) compared with WT (black; n = 6) littermates (P ≤ 0.05). (D) Representative image (a) of significantly reduced average levels of APP (b) in the proximal stump of ligated sciatic nerves in 4-month-old Tg-swAPPPrP; Tg-A246EPS1PrP (green; n = 7) compared with Tg-swAPPPrP (red; n = 7) littermates (P ≤ 0.05). APP and tubulin bands from representative western blots (a in C and D) belong to the same membranes.
DISCUSSION
Overexpression of APP in Drosophila and mice gives rise to axonal defects akin to those found in AD (7,29,30). Formation of these defects was shown to be enhanced by genetic reductions in kinesin-1 in Drosophila (29,30) and in mice, where increased Aβ42/Aβ40 ratios and enhanced amyloid deposition were also observed (7,57). To test whether axonal defects are the result of aberrant Aβ generation, we combined FAD mutant APP with FAD mutant PS1 or BRI-Aβ chimeras in Drosophila or mice. We found that the generation of axonal defects was unchanged or suppressed in Drosophila and mice expressing FAD mutant PS1 combined with FAD mutant APP and that significant increases in Aβ produced by expression of BRI-Aβ chimeras in FAD APP mice failed to enhance formation of axonal defects. In addition, not only was the axonal blockage phenotype suppressed by combining FAD mutant PS1 with FAD APP overexpression, but also neuronal apoptosis was suppressed in Drosophila. In parallel with these findings, behavioral deficits were also not enhanced by perturbing Aβ42 generation. In fact, in several cases, behavioral defects may actually have been slightly suppressed. These observations suggest that there may be different mechanisms by which transgenic FAD mutations in APP and PS1 contribute to AD. Intriguingly, transgenic FAD PS1 mutations appear unable to produce axonal pathology as well as amyloid deposits on their own, while in combination with APP several phenotypes including axonal pathology and behavior are suppressed or unchanged despite enhanced amyloid plaque formation.
Although our findings are in apparent contradiction to the widely held view that phenotypic defects produced by APP overexpression are caused by elevated Aβ42, we note that some previous studies made similar observations on comparisons of single versus double transgenic animals, although these were not emphasized. For example, overexpression of mutant APP or mutant PS1, but not their combined overexpression, elevates cortical ChAT activity (62). Similarly, the density of cortical cholinergic synapses has been shown to increase in mice overexpressing FAD mutant APP and decreased in mice expressing FAD mutant APP combined with FAD mutant PS1 (52). Our findings are also consistent with research showing that increasing Aβ by expressing FAD mutant PS1 together with FAD mutant APP failed to produce substantial enhancements of the behavioral deficits observed in mice overexpressing FAD mutant APP alone (56–58). Indeed, these comparisons revealed an unexpectedly subtle, if any, increase in behavioral deficits despite large increases in Aβ42/Aβ40 ratios and amyloid deposition in double transgenic animals. Finally, our data are consistent with a recent report about transgenic mice overexpressing APP which lack a caspase cleavage site in the C-terminal cytoplasmic region of APP. These transgenic mice generate large amounts of Aβ peptides, but they do not exhibit the significant cellular, synaptic and behavioral changes seen in matched transgenic mice producing comparable levels of Aβ, but in which the caspase cleavage site is intact (63). Another recent report compared retrograde NGF transport in mice overexpressing a variety of WT versions of mouse and human APP as well as mice that were either single transgenic Tg-swAPPPrP or double transgenic Tg-swAPPPrp; Tg-A246EPS1Prp (8). The results suggest that NGF transport deficits can be caused by overexpression of APP, and that human Aβ40 or Aβ42 peptides are not needed to generate NGF transport deficits, although increasing human Aβ42 levels may minimally enhance NGF transport deficits caused by APP overexpression. Thus, in many experiments in addition to ours, levels of proteolytic processing of APP to Aβ peptides such as Aβ42 are not well correlated with the development of a number of cellular and behavioral deficits. We note that many phenotypes present in double transgenic, but not WT animals, have been reported and attributed solely to Aβ or APP processing, but most such experiments do not compare double APP and PS1 transgenic genotypes to the single transgenic APP genotype as we have reported here.
A recent in vivo dynamics study of cortical amyloid plaque formation found amyloid deposition prior to neuritic defects (6). Thus, Aβ is sufficient, but as we show here, and argue above, not necessary for the formation of neuritic defects. We conclude that Aβ is not the only APP- or PS1-related insult that can induce axonal transport deficits and produce neuronal changes. A mechanism by which FAD mutant PS1 might suppress APP-induced cellular defects is not yet clear, but could involve the proposed role of PS1 in gating axonal entry or axonal transport of APP (39,59,64–66). Such gating could lead to reduced amounts of APP entry into axons, and could be achieved by regulating proteolytic processing of APP as has been proposed for BACE overexpression (67–69) and might simultaneously lead to enhanced levels of APP processing to Aβ42. Alternatively, PS1 could control the association of APP with molecular motor proteins and the transport machinery via proteins such as glycogen synthase kinase 3β, which could lead to reduced axonal transport (41). PS1 could also regulate APP transport via its putative interaction with proteins such as CLIP170 (70), which is thought to play a role in the formation of the dynein complex and possibly in retrograde axonal transport.
In conclusion, it is surprising that although expressing FAD mutant PS1 with overexpression of FAD mutant APP enhances Aβ42 production and amyloid deposition, it suppresses axonal defects and gives mild suppression, as opposed to enhancement of other defects such as behavior. Our observation that increasing Aβ in animals expressing FAD APP does not enhance axonal defects, coupled to other reports, raise the possibility that the total amount or character of proteolytic processing of APP to Aβ is not the primary driver of detrimental phenotypic changes in axons. Instead, we suggest that either the cellular location of APP processing, or the axonal defects generated by APP overexpression are the major causes of neuronal and behavioral defects seen in mice overexpressing mutant APP. Further work is necessary to discriminate between these possibilities.
MATERIALS AND METHODS
Mice and Drosophila
Transgenic mice carrying the Swedish FAD double-mutation (Tg-swAPPPrP) were crossed with mice heterozygous for PS1 carrying the A246E FAD mutation (Tg-A246EPS1PrP) to produce WT mice, Tg-swAPPPrP mice, Tg-A246EPS1PrP mice and mice carrying both transgenes (Tg-swAPPPrP and Tg-A246EPS1PrP) (71,72). Mice were initially generated and tested in the original mixed C57BL/6J/C3H/HeJ genetic background. To confirm and extend preliminary findings obtained with mice in the C57BL/6J/C3H/HeJ genetic background, heterozygous Tg-swAPPPrP and Tg-A246EPS1PrP mice were crossed for five generations with C57BL/6J mice and then to each other to produce a cohort of transgenic mice in the C57BL/6J genetic background. Genetic crosses in both genetic backgrounds produced mice with the expected genotype ratios for the Mendelian mode of inheritance. Unless otherwise stated, the results shown refer to experiments performed with 4-month old mice in the C57BL/6J genetic background. Transgenic mice hemizygous for the Swedish FAD double-mutation of APP, Tg-2567 (Tg-swAPPPrP) (53), in the B6/SJL genetic background were crossed with hemizygous BRI-Aβ40Prp or BRI-Aβ42Prp mice in the B6/C3 genetic background (45). The resulting WT, Tg-swAPPPrP, Tg-BRI-Aβ40Prp, Tg-swAPPPrP/BRI-Aβ40Prp, and WT, Tg-swAPPPrP, Tg-BRI-Aβ42Prp and Tg-swAPPPrP/BRI-Aβ42Prp littermates were examined at 10–11 months.
For Drosophila transgene expression, Drosophila lines carrying an FAD mutant human APP transgene UAS-SWE (73) or a FAD mutant Drosophila presenilin transgene UAS-PsnE280A (74) were crossed to a strain carrying the APPL-GAL4 (29) driver at 29°C. Genetic interaction tests were performed as previously described (29). Males that were APPL-GAL4/Y;PsnE280A/B3 or APPL-GAL4/Y;SWE/B3 were crossed to female homozygous for UAS-SWE or UAS-PsnE280A; only females carrying APPL-GAL4 along with SWE;PsnE280A or PsnE280A;SWE were used for the analysis.
Histology and immunohistochemistry
WT, Tg-swAPPPrP, Tg-A246EPS1PrP and Tg-swAPPPrP/A246EPS1PrP littermates were transcardially perfused with 0.1m phosphate solution at pH 7.4 (Sörensen’s solution) followed by buffered 4% paraformaldehyde (PFA). Cerebra were separated from the brains, post-fixed in buffered 4% PFA, cryoprotected in 30% sucrose overnight and cut serially into 50 µm thick sections to obtain sets of random systematic coronal sections representative of the BN or of the SC. Floating section were quenched with 0.6% H2O2, blocked and permeabilized in 0.3% normal donkey serum (NDS), 1% bovine serum albumin (BSA), 0.25% Triton X-100 in Sörensen’s solution, incubated for 48 h with anti-choline acetyltransferase (ChAT) antibody (AB144P, Chemicon), then with the biotinylated secondary antibody and the Vectastain Elite ABC (Vector Labs) and visualized using diaminobenzidine (DAB) tetrachloride. Sets of sections adjacent to the ChAT-IR ones were stained with thionin.
Sections were imaged using an Axioplan microscope (Carl Zeiss) connected to an X–Y–Z stage (MSA 001–6, RSF Elektronik linked to Microcode II, Boeckler Instruments), a color video camera (Diagnostic Instruments) and analyzed by the Bioquant image analysis software (Bioquant Image Analysis Corporation). ChAT-IR neurons in the BN were counted using optical fractionator (75,76) and their volumes estimated with the isotropic rotator (77). Density of ChAT-IR fibers was approximated with a Weibel grid using an intersection approach (78,79). Length of ChAT-IR fibers was obtained using the strut analysis with the help of the Bioquant imaging analysis software. Diameters and lengths of shafts and varicosities of ChAT-IR fibers were measured by the Bioquant image analysis software.
WT, Tg-swAPPPrP, Tg- BRI-Aβ40Prp, Tg-BRI-Aβ42Prp and Tg-swAPPPrP/BRI-Aβ40Prp, Tg-swAPPPrP/BRI-Aβ42Prp brains were formalin fixed. Five micrometer thick paraffin-embedded random non-systematic coronal sections of cerebri and cerebelli were either stained with Thioflavine S or immunostained with anti-total Aβ antibody (33.1.1., courtesy of Dr T.E. Golde) on an autostainer (DAKO) or with anti-ChAT antibody and developed as previously described using streptavidin biotin peroxidase methods. WT samples could not be analyzed owing to poor staining quality of the samples.
Analysis of the data was conducted using the Kruskall–Wallis non-parametric analysis of variance (ANOVA) and the Mann–Whitney U-test for multiple comparisons at a significance of P ≤ 0.05. Part of the results of the comparison of the axonal blockages in WT and Tg-swAPPPrP, but not Tg-A246EPS1PrP and Tg-swAPPPrP; Tg-A246EPS1PrP, mice were reported previously (7).
Drosophila immunofluorescence and tunel assay
To examine axonal blockages, larvae were dissected, fixed and stained as previously described (29,49). The antibody recognizing the synaptic vesicle marker cysteine string protein (anti-CSP, courtesy of Dr K. Zinsmaier) was used at a dilution of 1:20. Anti-mouse FITC was used at a dilution of 1:200 and fixed preparations were mounted, imaged and analyzed as described (29,49). Larval brains from the different genotypes were dissected for TUNEL analysis and detection of apoptotic cells was preformed as previously described (29) using the fluorescein-based cell death kit (Roche). Quantification of dying neuronal cells was assayed as described (29).
Immuno-electron microscopy
Mice were transcardially perfused with Sörensen’s solution followed by phosphate-buffered 0.5% glutaraldehyde (GA), 4% PFA. Cerebra were removed, post-fixed in 0.5% GA, 4%FA and cut with a vibratome into 50 µm thick random systematic coronal sections representative of the entire BN. Floating sections were quenched with H2O2, permeabilized in 0.05% Triton X-100 for 30 min at 4°C, blocked in 0.1% NDS, 0.5% BSA, 1% gelatin in Sörensen’s solution, incubated for 48 h with anti-ChAT antibody followed by the biotinylated secondary antibody and the Vectastain Elite ABC (Vector Labs). Visualization of ChAT was achieved using DAB reaction and followed by 10 min incubation in buffered 2% GA prior to embedment into Epon 812 resin. Embedded sections were cut into semi-thin sections, stained with 0.25% toluidine blue in 0.1% sodium borate and 70–80 nm ultrathin sections that were mounted onto copper grids and stained with uranyl acetate and lead citrate. Images from semithin and ultrathin sections were collected using an Axioplan microscope (Carl Zeiss) and an EM208S electron microscope (Philips), respectively. Comparable areas of the BN were sampled with similar number of semithin and ultrathin sections from each mouse. ChAT-IR profiles were analyzed using Photoshop 5.5 (Adobe) and Bioquant image analysis software (Bioquant Image Analysis Corporation). Part of the results of the comparison of the axonal blockages in the WT and Tg-swAPPPrP, but not Tg-A246EPS1PrP and Tg-swAPPPrP; Tg-A246EPS1PrP, mice were reported previously (7).
Biochemical analysis
Sciatic nerves from 4-month-old WT, Tg-swAPPPrP, Tg-A246EPS1PrP and Tg-swAPPPrP; Tg-A246EPS1PrP littermate mice were ligated as previously described (38,39). Briefly, one sciatic nerve from each animal was ligated at the mid thigh and the other nerve left as an unligated control. Six hours after ligation, animals were sacrificed, and approximately 5 mm of the proximal and distal halves of the nerve flanking the ligature or from the unligated nerve were dissected and directly homogenized with 1.2x NUPAGE LDS Sample buffer. Additionally, L4 to L6 dorsal root ganglia from ligated and unligated sides were dissected and homogenized similar to the nerves. Protein concentration of each sample was calculated using RC DC protein assay (Bio-Rad). The same amount of protein per sample was electrophoresed on NuPAGE 4–12% Bis-Tris gels with MES buffer. Gels were transferred to nitrocellulose membranes for 2 h at 300 mA, blocked with 5% milk and probed with antibodies against APP (22C11; Sigma), tubulin (DM1A; Sigma), tubulin beta III (Sigma), myelin basic protein (Dako), KIF5B and Syd. Secondary antibodies were conjugated to HRP (Zymed) and developed by ECL (Pierce).
To generate a Kif5B antibody, we grew C-terminally truncated Kif5B-His fusion protein (pET-23B) (80) in pLysS BL21 (DE3) bacteria, induced with 0.5 mm IPTG and purified using Ni-NTA agarose. Gel slabs containing approximately 300 mg of Kif5B-HIS were injected into three rabbits to produce polyclonal sera against Kif5B. Characterization of the Kif5B antibodies showed that these antibodies recognize primarily Kif5B (based on the in vitro assays with Kif5A and Kif5C) and turned out to be exceptionally clean despite being crude sera (38,39). SYD antibody was generated previously (61,81). Quantification of APP, Kif5B and SYD was normalized for α-tubulin and myelin basic protein. Experimental samples with signal intensities within the linear range were quantified by two independent investigators, one blind to the genotypes, using Image J (NIH) or Scion Image software (Scion Corporation). Almost all samples were examined on multiple gels and each gel on average by three to five different exposures.
WT, Tg-swAPPPrP, Tg-A246EPS1PrP and Tg-swAPPPrP/A246EPS1PrP hemibrains were Dounce homogenized using 5M guanidine hydrochloride, diluted 1 to 10 in 1X Dulbecco’s phosphate-buffered saline containing 5% BSA and protease inhibitors and centrifuged at 10,000 g for 20 min. Supernatants were loaded into well-established commercially available human-specific anti-Aβ40 and anti-Aβ42 sandwich ELISA plates (Biosource International) and processed according to the manufacturer’s instructions (7). WT, Tg-swAPPPrP, Tg-BRI-Aβ40Prp, Tg-BRI-Aβ42Prp and Tg-swAPPPrP; BRI-Aβ40Prp, Tg-swAPPPrP; BRI-Aβ42Prp hemibrains were homogenized in RIPA; RIPA-insoluble fractions were further solubilized in formic acid, neutralized with Tris base buffer and appropriately diluted. Aβ levels were determined by end-specific sandwich-ELISA as previously described (45,82).
Behavioral analysis
The open field test was performed in a 50 × 35 cm box with the floor consisting of alternative white and red 5 × 5 cm quadrants. Upon adjusting to the new room environment for 15 min, the mice were placed in the center of the box and the CI (83), defined as the number of times the mice entered corner quadrants with any two extremities in 1 min, established.
Rotarod apparatus (SD Instruments) consisted in ribbed plastic rotating rod separated into four sections with ‘beam-break’ sensors close to the bottom of the enclave. There were four testing days and during each testing day there were three consecutive trials. In each trial, the mice were placed on the rod facing away from the experimenter and towards the direction of the rod rotations. The rotation of the rod was accelerated from 0 to 45 rpm in 2 min and then kept rotating at 45 rpm for additional 6 min. Latencies before mice fell from the rod were recorded by the control unit of the apparatus. With rare exceptions, we tested one mouse per genotype per trial simultaneously and the position of each mouse changed between the trial days.
For fear conditioning, we used a custom-made apparatus (SD Instruments) consisting in two open top quadrangular Plexiglas enclosures with a lid each placed in a chamber with front doors and lateral observation window. First enclosure contained a wire grid floor and ‘beam-break’ sensors and was positioned in a white chamber harboring a light bulb and a speaker. This enclosure was used for training and to test contextual conditioning. The second enclosure had a different odor compared to the first one, contained ‘beam-break’ sensors, bedding and a light bulb and was positioned in a black chamber harboring a speaker, but in a location opposite to the one in the first chamber. This enclosure was used to test for cued conditioning. Visual presentation of the room where fear conditioning took place changed between the training and both testing session with curtains. Training started with a 3 min novel context exploration period followed by three consecutive conditioned stimulus (CS)/unconditioned stimulus (US) pairings separated by 1 min intervals (CS/US interval) where one CS/US pairing consisted in a 20 s long tone (CS: 77 dB, 2.8 Hz) coupled to a foot shock during its last 2 s (US: 0.75 mA) (84). Testing took place 24 hours after training. To test for contextual conditioning, we measured freezing in response to a 3 min exposure to the training environment. Testing cued conditioning took place 2 h later. To test for cued conditioning, we measured freezing in response to a 3 min novel context exploration (control) and to three consecutive CS separated by 1 min intervals (CS interval). Freezing responses were measured by visual scoring and by computer-aided analysis of the video frame capture (‘beam-breaks’). These two freezing response measurement methods are complimentary and since results from both data sets were consistent with each other only data obtained by visual scoring are shown.
Analysis of the data was conducted using the analysis of variance (ANOVA) and the Scheffe’s post hoc test for multiple comparisons at a significance of P ≤ 0.05.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
FUNDING
Supported by an Ellison Medical Foundation Senior Scholar Award in Aging Research and NIH grant GM35252 (L.S.B.G.), NIH grants EY13408 and EY07042 (D.S.W.), NIA grant AGO22595 (E.M.), a Boehringer-Ingelheim Fonds fellowship (G.B.S.), an Ellison Medical Foundation Senior postdoctoral fellowship (S.G), and a new investigator grant from the Alzheimer Association (S.G).
ACKNOWLEDGEMENTS
We thank Drs Fred H. Gage, Todd E. Golde, Edward H. Koo, Klara Limbäck-Stokin and Mark Mayford for helpful discussions. L.S.B.G. is an investigator of the Howard Hughes Medical Institute.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Katzman R. Alzheimer’s disease. N. Engl. J. Med. 1986;314:964–973. doi: 10.1056/NEJM198604103141506. [DOI] [PubMed] [Google Scholar]
- 2.Davies P., Maloney A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. 1976;2:1403. doi: 10.1016/s0140-6736(76)91936-x. [DOI] [PubMed] [Google Scholar]
- 3.Hyman B.T., Van Horsen G.W., Damasio A.R., Barnes C.L. Alzheimer’s disease: cell-specific pathology isolates the hippocampal formation. Science. 1984;225:1168–1170. doi: 10.1126/science.6474172. [DOI] [PubMed] [Google Scholar]
- 4.Perry E.K., Perry R.H., Blessed G., Tomlinson B.E. Necropsy evidence of central cholinergic deficits in senile dementia. Lancet. 1977;1:189. doi: 10.1016/s0140-6736(77)91780-9. [DOI] [PubMed] [Google Scholar]
- 5.Samuel W., Terry R.D., DeTeresa R., Butters N., Masliah E. Clinical correlates of cortical and nucleus basalis pathology in Alzheimer dementia. Arch. Neurol. 1994;51:772–778. doi: 10.1001/archneur.1994.00540200048015. [DOI] [PubMed] [Google Scholar]
- 6.Meyer-Luehmann M., Spires-Jones T.L., Prada C., Garcia-Alloza M., de Calignon A., Rozkalne A., Koenigsknecht-Talboo J., Holtzman D.M., Bacskai B.J., Hyman B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stokin G.B., Lillo C., Falzone T.L., Brusch R.G., Rockenstein E., Mount S.L., Raman R., Davies P., Masliah E., Williams D.S., et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 8.Salehi A., Delcroix J.D., Belichenko P.V., Zhan K., Wu C., Valletta J.S., Takimoto-Kimura R., Kleschevnikov A.M., Sambamurti K., Chung P.P., et al. Increased App expression in a mouse model of Down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006;51:29–42. doi: 10.1016/j.neuron.2006.05.022. [DOI] [PubMed] [Google Scholar]
- 9.Lampert P. Fine structural changes of neurites in Alzheimer’s disease. Acta. Neuropathol. (Berl.) 1971;5(Suppl. 5):49–53. doi: 10.1007/978-3-642-47449-1_6. [DOI] [PubMed] [Google Scholar]
- 10.Price D.L., Altschuler R.J., Struble R.G., Casanova M.F., Cork L.C., Murphy D.B. Sequestration of tubulin in neurons in Alzheimer’s disease. Brain. Res. 1986;385:305–310. doi: 10.1016/0006-8993(86)91077-2. [DOI] [PubMed] [Google Scholar]
- 11.Rasool C.G., Svendsen C.N., Selkoe D.J. Neurofibrillary degeneration of cholinergic and noncholinergic neurons of the basal forebrain in Alzheimer’s disease. Ann. Neurol. 1986;20:482–488. doi: 10.1002/ana.410200407. [DOI] [PubMed] [Google Scholar]
- 12.Terry R.D. The fine structure of neurofibrillary tangles in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 1963;22:629–641. doi: 10.1097/00005072-196310000-00005. [DOI] [PubMed] [Google Scholar]
- 13.Smith K.D., Kallhoff V., Zheng H., Pautler R.G. In vivo axonal transport rates decrease in a mouse model of Alzheimer’s disease. Neuroimage. 2007;35:1401–1408. doi: 10.1016/j.neuroimage.2007.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Minoshima S., Cross D. In vivo imaging of axonal transport using MRI: aging and Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging. 2008;35(Suppl. 1):S89–S92. doi: 10.1007/s00259-007-0707-8. [DOI] [PubMed] [Google Scholar]
- 15.Stokin G.B., Goldstein L.S. Linking molecular motors to Alzheimer’s disease. J. Physiol. Paris. 2006;99:193–200. doi: 10.1016/j.jphysparis.2005.12.085. [DOI] [PubMed] [Google Scholar]
- 16.Stokin G.B., Goldstein L.S. Axonal transport and Alzheimer’s disease. Annu. Rev. Biochem. 2006;75:607–627. doi: 10.1146/annurev.biochem.75.103004.142637. [DOI] [PubMed] [Google Scholar]
- 17.Terry R.D. The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J. Neuropathol. Exp. Neurol. 1996;55:1023–1025. [PubMed] [Google Scholar]
- 18.Masliah E., Mallory M., Deerinck T., DeTeresa R., Lamont S., Miller A., Terry R.D., Carragher B., Ellisman M. Re-evaluation of the structural organization of neuritic plaques in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 1993;52:619–632. doi: 10.1097/00005072-199311000-00009. [DOI] [PubMed] [Google Scholar]
- 19.Dickson T.C., King C.E., McCormack G.H., Vickers J.C. Neurochemical diversity of dystrophic neurites in the early and late stages of Alzheimer’s disease. Exp. Neurol. 1999;156:100–110. doi: 10.1006/exnr.1998.7010. [DOI] [PubMed] [Google Scholar]
- 20.Cork L.C., Masters C., Beyreuther K., Price D.L. Development of senile plaques. Relationships of neuronal abnormalities and amyloid deposits. Am. J. Pathol. 1990;137:1383–1392. [PMC free article] [PubMed] [Google Scholar]
- 21.Dickson D.W., Farlo J., Davies P., Crystal H., Fuld P., Yen S.H. Alzheimer’s disease. A double-labeling immunohistochemical study of senile plaques. Am. J. Pathol. 1988;132:86–101. [PMC free article] [PubMed] [Google Scholar]
- 22.Masliah E., Mallory M., Hansen L., Alford M., DeTeresa R., Terry R. An antibody against phosphorylated neurofilaments identifies a subset of damaged association axons in Alzheimer’s disease. Am. J. Pathol. 1993;142:871–882. [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X.H., Johnson V.E., Uryu K., Trojanowski J.Q., Smith D.H. A lack of amyloid beta plaques despite persistent accumulation of amyloid beta in axons of long-term survivors of traumatic brain injury. Brain Pathol. 2008 doi: 10.1111/j.1750-3639.2008.00176.x. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burger P.C., Vogel F.S. The development of the pathologic changes of Alzheimer’s disease and senile dementia in patients with Down’s syndrome. Am. J. Pathol. 1973;73:457–476. [PMC free article] [PubMed] [Google Scholar]
- 25.Cabrejo L., Guyant-Marechal L., Laquerriere A., Vercelletto M., De la Fourniere F., Thomas-Anterion C., Verny C., Letournel F., Pasquier F., Vital A., et al. Phenotype associated with APP duplication in five families. Brain. 2006;129:2966–2976. doi: 10.1093/brain/awl237. [DOI] [PubMed] [Google Scholar]
- 26.Rovelet-Lecrux A., Hannequin D., Raux G., Le Meur N., Laquerriere A., Vital A., Dumanchin C., Feuillette S., Brice A., Vercelletto M., et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 27.Sleegers K., Brouwers N., Gijselinck I., Theuns J., Goossens D., Wauters J., Del-Favero J., Cruts M., van Duijn C.M., Van Broeckhoven C. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain. 2006;129:2977–2983. doi: 10.1093/brain/awl203. [DOI] [PubMed] [Google Scholar]
- 28.Oyama F., Cairns N.J., Shimada H., Oyama R., Titani K., Ihara Y. Down’s syndrome: up-regulation of beta-amyloid protein precursor and tau mRNAs and their defective coordination. J. Neurochem. 1994;62:1062–1066. doi: 10.1046/j.1471-4159.1994.62031062.x. [DOI] [PubMed] [Google Scholar]
- 29.Gunawardena S., Goldstein L.S. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron. 2001;32:389–401. doi: 10.1016/s0896-6273(01)00496-2. [DOI] [PubMed] [Google Scholar]
- 30.Torroja L., Chu H., Kotovsky I., White K. Neuronal overexpression of APPL, the Drosophila homologue of the amyloid precursor protein (APP), disrupts axonal transport. Curr. Biol. 1999;9:489–492. doi: 10.1016/s0960-9822(99)80215-2. [DOI] [PubMed] [Google Scholar]
- 31.Brown M.S., Ye J., Rawson R.B., Goldstein J.L. Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell. 2000;100:391–398. doi: 10.1016/s0092-8674(00)80675-3. [DOI] [PubMed] [Google Scholar]
- 32.De Strooper B., Saftig P., Craessaerts K., Vanderstichele H., Guhde G., Annaert W., Von Figura K., Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 33.De Strooper B., Annaert W., Cupers P., Saftig P., Craessaerts K., Mumm J.S., Schroeter E.H., Schrijvers V., Wolfe M.S., Ray W.J., et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 34.Struhl G., Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 1999;398:522–525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- 35.Ye Y., Lukinova N., Fortini M.E. Neurogenic phenotypes and altered Notch processing in Drosophila Presenilin mutants. Nature. 1999;398:525–529. doi: 10.1038/19096. [DOI] [PubMed] [Google Scholar]
- 36.Naruse S., Thinakaran G., Luo J.J., Kusiak J.W., Tomita T., Iwatsubo T., Qian X., Ginty D.D., Price D.L., Borchelt D.R., et al. Effects of PS1 deficiency on membrane protein trafficking in neurons. Neuron. 1998;21:1213–1221. doi: 10.1016/s0896-6273(00)80637-6. [DOI] [PubMed] [Google Scholar]
- 37.Nishimura M., Yu G., Levesque G., Zhang D.M., Ruel L., Chen F., Milman P., Holmes E., Liang Y., Kawarai T., et al. Presenilin mutations associated with Alzheimer disease cause defective intracellular trafficking of beta-catenin, a component of the presenilin protein complex. Nat. Med. 1999;5:164–169. doi: 10.1038/5526. [DOI] [PubMed] [Google Scholar]
- 38.Kamal A., Stokin G.B., Yang Z., Xia C.H., Goldstein L.S. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 2000;28:449–459. doi: 10.1016/s0896-6273(00)00124-0. [DOI] [PubMed] [Google Scholar]
- 39.Kamal A., Almenar-Queralt A., LeBlanc J.F., Roberts E.A., Goldstein L.S. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- 40.Lazarov O., Morfini G.A., Pigino G., Gadadhar A., Chen X., Robinson J., Ho H., Brady S.T., Sisodia S.S. Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer’s disease-linked mutant presenilin 1. J. Neurosci. 2007;27:7011–7020. doi: 10.1523/JNEUROSCI.4272-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pigino G., Morfini G., Pelsman A., Mattson M.P., Brady S.T., Busciglio J. Alzheimer’s presenilin 1 mutations impair kinesin-based axonal transport. J. Neurosci. 2003;23:4499–4508. doi: 10.1523/JNEUROSCI.23-11-04499.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cash A.D., Aliev G., Siedlak S.L., Nunomura A., Fujioka H., Zhu X., Raina A.K., Vinters H.V., Tabaton M., Johnson A.B., et al. Microtubule reduction in Alzheimer’s disease and aging is independent of tau filament formation. Am. J. Pathol. 2003;162:1623–1627. doi: 10.1016/s0002-9440(10)64296-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Terry R.D., Gonatas N.K., Weiss M. Ultrastructural studies in Alzheimer’s presenile dementia. Am. J. Pathol. 1964;44:269–287. [PMC free article] [PubMed] [Google Scholar]
- 44.Kim J., Onstead L., Randle S., Price R., Smithson L., Zwizinski C., Dickson D.W., Golde T., McGowan E. Abeta40 inhibits amyloid deposition in vivo. J. Neurosci. 2007;27:627–633. doi: 10.1523/JNEUROSCI.4849-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGowan E., Pickford F., Kim J., Onstead L., Eriksen J., Yu C., Skipper L., Murphy M.P., Beard J., Das P., et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vidal R., Frangione B., Rostagno A., Mead S., Revesz T., Plant G., Ghiso J. A stop-codon mutation in the BRI gene associated with familial British dementia. Nature. 1999;399:776–781. doi: 10.1038/21637. [DOI] [PubMed] [Google Scholar]
- 47.Vidal R., Revesz T., Rostagno A., Kim E., Holton J.L., Bek T., Bojsen-Moller M., Braendgaard H., Plant G., Ghiso J., et al. A decamer duplication in the 3′ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred. Proc. Natl Acad. Sci. USA. 2000;97:4920–4925. doi: 10.1073/pnas.080076097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernstein M., Lichtman J.W. Axonal atrophy: the retraction reaction. Curr. Opin. Neurobiol. 1999;9:364–370. doi: 10.1016/s0959-4388(99)80053-1. [DOI] [PubMed] [Google Scholar]
- 49.Gunawardena S., Her L.S., Brusch R.G., Laymon R.A., Niesman I.R., Gordesky-Gold B., Sintasath L., Bonini N.M., Goldstein L.S. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- 50.Hurd D.D., Saxton W.M. Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila. Genetics. 1996;144:1075–1085. doi: 10.1093/genetics/144.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lampert P.W. A comparative electron microscopic study of reactive, degenerating, regenerating and dystrophic axons. J. Neuropathol. Exp. Neurol. 1967;26:345–368. doi: 10.1097/00005072-196707000-00001. [DOI] [PubMed] [Google Scholar]
- 52.Wong T.P., Debeir T., Duff K., Cuello A.C. Reorganization of cholinergic terminals in the cerebral cortex and hippocampus in transgenic mice carrying mutated presenilin-1 and amyloid precursor protein transgenes. J. Neurosci. 1999;19:2706–2716. doi: 10.1523/JNEUROSCI.19-07-02706.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hsiao K., Chapman P., Nilsen S., Eckman C., Harigaya Y., Younkin S., Yang F., Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 54.Fossella J., Samant S.A., Silver L.M., King S.M., Vaughan K.T., Olds-Clarke P., Johnson K.A., Mikami A., Vallee R.B., Pilder S.H. An axonemal dynein at the Hybrid Sterility 6 locus: implications for t haplotype-specific male sterility and the evolution of species barriers. Mamm. Genome. 2000;11:8–15. doi: 10.1007/s003350010003. [DOI] [PubMed] [Google Scholar]
- 55.Ye Y., Fortini M.E. Apoptotic activities of wild-type and Alzheimer’s disease-related mutant presenilins in Drosophila melanogaster. J. Cell Biol. 1999;146:1351–1364. doi: 10.1083/jcb.146.6.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dineley K.T., Xia X., Bui D., Sweatt J.D., Zheng H. Accelerated plaque accumulation, associative learning deficits, and up-regulation of alpha 7 nicotinic receptor protein in transgenic mice co-expressing mutant human presenilin 1 and amyloid precursor proteins. J. Biol. Chem. 2002;277:22768–22780. doi: 10.1074/jbc.M200164200. [DOI] [PubMed] [Google Scholar]
- 57.Holcomb L., Gordon M.N., McGowan E., Yu X., Benkovic S., Jantzen P., Wright K., Saad I., Mueller R., Morgan D., et al. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 58.Holcomb L.A., Gordon M.N., Jantzen P., Hsiao K., Duff K., Morgan D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav. Genet. 1999;29:177–185. doi: 10.1023/a:1021691918517. [DOI] [PubMed] [Google Scholar]
- 59.Cai D., Leem J.Y., Greenfield J.P., Wang P., Kim B.S., Wang R., Lopes K.O., Kim S.H., Zheng H., Greengard P., et al. Presenilin-1 regulates intracellular trafficking and cell surface delivery of beta-amyloid precursor protein. J. Biol. Chem. 2003;278:3446–3454. doi: 10.1074/jbc.M209065200. [DOI] [PubMed] [Google Scholar]
- 60.Rusu P., Jansen A., Soba P., Kirsch J., Lower A., Merdes G., Kuan Y.H., Jung A., Beyreuther K., Kjaerulff O., et al. Axonal accumulation of synaptic markers in APP transgenic Drosophila depends on the NPTY motif and is paralleled by defects in synaptic plasticity. Eur. J. Neurosci. 2007;25:1079–1086. doi: 10.1111/j.1460-9568.2007.05341.x. [DOI] [PubMed] [Google Scholar]
- 61.Cavalli V., Kujala P., Klumperman J., Goldstein L.S. Sunday Driver links axonal transport to damage signaling. J. Cell Biol. 2005;168:775–787. doi: 10.1083/jcb.200410136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hernandez D., Sugaya K., Qu T., McGowan E., Duff K., McKinney M. Survival and plasticity of basal forebrain cholinergic systems in mice transgenic for presenilin-1 and amyloid precursor protein mutant genes. Neuroreport. 2001;12:1377–1384. doi: 10.1097/00001756-200105250-00018. [DOI] [PubMed] [Google Scholar]
- 63.Galvan V., Gorostiza O.F., Banwait S., Ataie M., Logvinova A.V., Sitaraman S., Carlson E., Sagi S.A., Chevallier N., Jin K., et al. Reversal of Alzheimer’s-like pathology and behavior in human APP transgenic mice by mutation of Asp664. Proc. Natl Acad. Sci. USA. 2006;103:7130–7135. doi: 10.1073/pnas.0509695103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaether C., Lammich S., Edbauer D., Ertl M., Rietdorf J., Capell A., Steiner H., Haass C. Presenilin-1 affects trafficking and processing of betaAPP and is targeted in a complex with nicastrin to the plasma membrane. J. Cell Biol. 2002;158:551–561. doi: 10.1083/jcb.200201123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim S.H., Leem J.Y., Lah J.J., Slunt H.H., Levey A.I., Thinakaran G., Sisodia S.S. Multiple effects of aspartate mutant presenilin 1 on the processing and trafficking of amyloid precursor protein. J. Biol. Chem. 2001;276:43343–43350. doi: 10.1074/jbc.M108245200. [DOI] [PubMed] [Google Scholar]
- 66.Leem J.Y., Saura C.A., Pietrzik C., Christianson J., Wanamaker C., King L.T., Veselits M.L., Tomita T., Gasparini L., Iwatsubo T., et al. A role for presenilin 1 in regulating the delivery of amyloid precursor protein to the cell surface. Neurobiol. Dis. 2002;11:64–82. doi: 10.1006/nbdi.2002.0546. [DOI] [PubMed] [Google Scholar]
- 67.Cai D., Zhong M., Wang R., Netzer W.J., Shields D., Zheng H., Sisodia S.S., Foster D.A., Gorelick F.S., Xu H., et al. Phospholipase D1 corrects impaired betaAPP trafficking and neurite outgrowth in familial Alzheimer’s disease-linked presenilin-1 mutant neurons. Proc. Natl Acad. Sci. USA. 2006;103:1936–1940. doi: 10.1073/pnas.0510710103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee E.B., Zhang B., Liu K., Greenbaum E.A., Doms R.W., Trojanowski J.Q., Lee V.M. BACE overexpression alters the subcellular processing of APP and inhibits Abeta deposition in vivo. J. Cell Biol. 2005;168:291–302. doi: 10.1083/jcb.200407070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu H., Saura C.A., Choi S.Y., Sun L.D., Yang X., Handler M., Kawarabayashi T., Younkin L., Fedeles B., Wilson M.A., et al. APP processing and synaptic plasticity in presenilin-1 conditional knockout mice. Neuron. 2001;31:713–726. doi: 10.1016/s0896-6273(01)00417-2. [DOI] [PubMed] [Google Scholar]
- 70.Tezapsidis N., Merz P.A., Merz G., Hong H. Microtubular interactions of presenilin direct kinesis of Abeta peptide and its precursors. FASEB J. 2003;17:1322–1324. doi: 10.1096/fj.02-0980fje. [DOI] [PubMed] [Google Scholar]
- 71.Borchelt D.R., Thinakaran G., Eckman C.B., Lee M.K., Davenport F., Ratovitsky T., Prada C.M., Kim G., Seekins S., Yager D., et al. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 72.Borchelt D.R., Ratovitski T., van Lare J., Lee M.K., Gonzales V., Jenkins N.A., Copeland N.G., Price D.L., Sisodia S.S. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 73.Fossgreen A., Bruckner B., Czech C., Masters C.L., Beyreuther K., Paro R. Transgenic Drosophila expressing human amyloid precursor protein show gamma-secretase activity and a blistered-wing phenotype. Proc. Natl Acad. Sci. USA. 1998;95:13703–13708. doi: 10.1073/pnas.95.23.13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ye Y., Fortini M.E. Proteolysis and developmental signal transduction. Semin. Cell Dev. Biol. 2000;11:211–221. doi: 10.1006/scdb.2000.0167. [DOI] [PubMed] [Google Scholar]
- 75.Gundersen H.J. Stereology of arbitrary particles. A review of unbiased number and size estimators and the presentation of some new ones, in memory of William R. Thompson. J. Microsc. 1986;143:3–45. [PubMed] [Google Scholar]
- 76.West M.J., Slomianka L., Gundersen H.J. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat. Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- 77.Jensen E.B., Gundersen H.J. The rotator. J. Microsc. 1993;170:35–44. [Google Scholar]
- 78.Geula C., Mesulam M.M. Systematic regional variations in the loss of cortical cholinergic fibers in Alzheimer’s disease. Cereb. Cortex. 1996;6:165–177. doi: 10.1093/cercor/6.2.165. [DOI] [PubMed] [Google Scholar]
- 79.Geula C., Mesulam M.M. Cortical cholinergic fibers in aging and Alzheimer’s disease: a morphometric study. Neuroscience. 1989;33:469–481. doi: 10.1016/0306-4522(89)90399-0. [DOI] [PubMed] [Google Scholar]
- 80.Xia C., Rahman A., Yang Z., Goldstein L.S. Chromosomal localization reveals three kinesin heavy chain genes in mouse. Genomics. 1998;52:209–213. doi: 10.1006/geno.1998.5427. [DOI] [PubMed] [Google Scholar]
- 81.Bowman A.B., Kamal A., Ritchings B.W., Philp A.V., McGrail M., Gindhart J.G., Goldstein L.S. Kinesin-dependent axonal transport is mediated by the Sunday driver (SYD) protein. Cell. 2000;103:583–594. doi: 10.1016/s0092-8674(00)00162-8. [DOI] [PubMed] [Google Scholar]
- 82.Kawarabayashi T., Younkin L.H., Saido T.C., Shoji M., Ashe K.H., Younkin S.G. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hsiao K.K., Borchelt D.R., Olson K., Johannsdottir R., Kitt C., Yunis W., Xu S., Eckman C., Younkin S., Price D., et al. Age-related CNS disorder and early death in transgenic FVB/N mice overexpressing Alzheimer amyloid precursor proteins. Neuron. 1995;15:1203–1218. doi: 10.1016/0896-6273(95)90107-8. [DOI] [PubMed] [Google Scholar]
- 84.Limback-Stokin K., Korzus E., Nagaoka-Yasuda R., Mayford M. Nuclear calcium/calmodulin regulates memory consolidation. J. Neurosci. 2004;24:10858–10867. doi: 10.1523/JNEUROSCI.1022-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.