

Abstract
Enthalpy arrays enable label-free, solution-based calorimetric detection of molecular interactions in a 96-detector array format. Compared with conventional calorimetry, enthalpy arrays achieve a significant reduction of sample volume and measurement time through the combination of the small size of the detectors and ability to perform measurements in parallel. The current capabilities of the technology for studying enzyme-catalyzed reactions are demonstrated by determining the kinetic parameters for reactions with three model enzymes. In addition, the technology has been used with two classes of enzymes to determine accurate inhibitor constants for competitive inhibitors from measurements at a single inhibitor concentration.
Keywords: nanocalorimetry, enzyme assay, label-free assay
Introduction
Enzyme assays can be performed with continuous spectrophotometric, discontinuous radiometric, antibody-dependent fluorescence/luminescence, chromatographic, or capillary electrophoresis assays. The photometric assays, while generally rapid and simple to perform, often require modification or substitution of the natural substrate with one that contains a fluorophore or chromophore or employ the coupling to a second enzymatic reaction to produce a spectroscopically detectable product. Chromatographic or electrophoretic techniques, while potentially label free, are far more complex to perform. A broadly applicable, rapid, label free, and simple assay should facilitate enzyme analysis.
Calorimetry enables one to perform a direct enzyme activity assay without modifying the substrate or using coupled reactions to produce the observed signal [1; 2]. In addition, a calorimetric measurement is unaffected by spectroscopically opaque solutions, which may occur with high concentration of substrate or product. While the use of isothermal titration calorimetry (ITC) to determine enzyme kinetics has been described [1; 2; 3], the measurements are performed one at a time and require quantities of reactants that are considered quite large in biochemical studies, making high throughput measurements or measurements with limited amounts of material unfeasible. Nanocalorimeters can in principle overcome these limitations of conventional ITC.
Enthalpy arrays are arrays of nanocalorimeters which allow scientists to measure thermodynamics and kinetics of molecular interactions using small sample volumes (250 nl) and short measurement times (typically 5–10 minutes) [4]. Enthalpy arrays achieve miniaturization and arrayed measurements by sacrificing some of the accuracy and capability of ITC instruments, which is a winning trade-off for many applications.
Previously, we demonstrated the use of enthalpy arrays to monitor enzymatic reactions and demonstrated basic assays for substrate specificity and enzyme inhibitor activity [5]. The data showed a linear relationship between initial substrate concentration and total integrated heat of the reaction, consistent with complete conversion of substrate to product. Although this information could be used to determine if a compound is a substrate or inhibitor for an enzyme, the total integrated heat does not provide any information on the enzyme turnover (kcat), KM of the substrate, or KI of the inhibitor.
Here we describe the use of enthalpy array technology to determine the kinetic parameters for enzymatic reactions and the mechanism of action of inhibitors, using enzymes from two EC classifications. This new ability to determine kinetic parameters was made possible by incorporating magnetic mixing into the enthalpy array measurements, thereby improving the mixing.
Material and methods
Rapid magnetic mixing of sample drops
The design and implementation of magnetic mixing on enthalpy arrays will be described in detail elsewhere. Briefly, magnetic stir bars were fabricated from a cobalt-based magnetic alloy (Metglas® 2714A; Metglas®, Inc., Conway, SC) and passivated by coating with SiON followed by PEGylation with mPEG silane from Creative PEGWorks (Winston Salem, NC). PEGylated magnetic stir bars (400 μm × 200 μm × 15 μm) were placed on the appropriate location of the detector region manually using an X-Y-Z micromanipulator (West Bond, Anaheim, CA). One bar was used for each pair of drops and was placed on the region where the enzyme or BSA drop would be placed during drop deposition.
Sample deposition and recording of data
Drops (250 nl) containing enzyme, substrate, buffer, or BSA were deposited on the appropriate location of the detector region using a Deerac spot-on liquid handling system (Deerac Fluidics, Dublin, Ireland) (Figure 1). The array was placed in a temperature-controlled measurement chamber on a block containing individually-addressable magnetic stirring mechanisms, and a polymer cap was applied over the array to minimize drop evaporation.
Figure 1.

Schematic of a single enthalpy array detector. Sample and reference regions are designated based on the material (substrate, enzyme or BSA) in the drops deposited on each region.
The drops were allowed to come to thermal equilibration (2–3 minutes) prior to activating the magnetic mixing device under the site to be measured. Upon activation, the magnetic stirring mechanism was rotated at approximately 1000 rpm and allowed to equilibrate for 30 seconds. After equilibration, the reaction was initiated by applying a voltage (180 V) across the merging electrodes to electrostatically merge the drops of interest. Specifically, two drops were merged in the sample region, and two similar but nonreacting drops were merged on the reference region. The nonreacting drops provided a reference for the differential measurement. The output voltage of the Wheatstone bridge was typically recorded for 30 s prior to the merge time and 3–5 min following initiation of the reaction. Magnetic mixing continued during the entire time.
Enzyme assays
A variety of enzyme reactions are reported here to demonstrate enthalpy array measurement of enzymatic reactions.
In one set of examples, phosphorylation of Kemptide (LRRASLG) [6] by the catalytic subunit of human cAMP-dependent kinase (PKA) (EC 2.7.11.11) was measured at 21 °C in 100 mM Tris-HCl (pH 7.5), 20 mM MgCl2, 1 mM DTT, 0.1 mM EGTA, 0.005% Brij 35. PKA catalytic subunit was placed in the measurement buffer via buffer exchange in an Amicon Ultra centrifugal filter device (10 kDa MWCO). The sample region materials consisted of a drop of PKA catalytic subunit (4 μM) and a drop of substrate solution (2–3 mM Kemptide, 2.5–10 mM ATP). The reference region used a drop of BSA (0.02 mg/ml, 0.30 μM) and a drop of the same substrate solution (2–3 mM Kemptide, 2.5–10 mM ATP) used in the sample region (Figure 1). The purpose of the small amount of BSA is to give the reference drops wetting behavior similar to the sample drops, especially after merging. Immediately after merging, the combined drops in the sample region contained 2 μM PKA catalytic subunit and 1–1.5 mM Kemptide with 1.25–5 mM ATP. The combined drops in the reference region contained 0.15 μM BSA and 1–1.5 mM Kemptide with 1.25–5 mM ATP. Reactions with H-89 inhibitor were performed as above, except that the inhibitor was mixed with PKA and incubated at room temperature for 15 min prior to drop deposition. Reactions with staurosporine contained 2% DMSO in all drops. Reactions with inhibitor peptides were performed as above with the inhibitor included in the substrate (Kemptide and ATP) solution. The catalytic subunit of human cAMP dependent kinase (recombinant) was from Millipore/Upstate (Temecula, CA). Kemptide and inhibitor peptides (PKI-tide; IAAGRTGRRQAIHDILVAA and Ala-peptide; LRRAALG) were obtained from AnaSpec (San Jose, CA). All other reagents were obtained from Sigma–Aldrich and used without further purification.
In another set of examples, trypsin (EC 3.4.21.4) hydrolysis of benzoylarginine ethyl ester (BAEE) was measured at 21 °C in 200 mM Tris–HCl, pH 8.0, 50 mM CaCl2, 0.2% polyethylene glycol 8000. The sample region materials consisted of a drop of trypsin (10 μM, 0.73 BAEE units) and a drop of substrate solution (10 mM BAEE). The reference region used a drop of BSA (0.02 mg/ml, 0.30 μM) and a drop of the same substrate solution (10 mM BAEE) used in the sample region (Figure 1). Immediately after merging, the combined drops in the sample region contained 5 μM trypsin (0.73 BAEE units) and 5.0 mM BAEE, and the combined drops in the reference region contained 0.15 μM BSA and 5.0 mM BAEE. Reactions with trypsin inhibitors were performed as above, except that the inhibitor was mixed with trypsin and incubated at room temperature for 15 min prior to drop deposition. No binding of BAEE to BSA was detected in reactions containing 5 mM BAEE and 0.15 μM BSA in the sample region and 5 mM BAEE and buffer in the reference region. All reagents were obtained from Sigma–Aldrich and used without further purification.
In a third set of examples, hexokinase (EC 2.7.1.1) phosphorylation of glucose or fructose was measured at 21 °C in 100 mM Tris–HCl, pH 7.5, 20 mM MgCl2, 0.5 mM TCEP. The sample region materials consisted of a drop containing hexokinase (10–20 U/ml, ≈0.15–0.30 μM) and a drop containing both ATP (5–20 mM) and glucose (10–20 mM) or fructose (75 mM). The reference region used a drop of BSA (0.02 mg/ml, 0.30 μM) and a drop of the same substrate solution (5–20 mM ATP, 10–20 mM glucose or 75 mM fructose) used in the sample region (Figure 1). Immediately after merging, the combined drops in the sample region contained ≈0.08–0.15 μM hexokinase (0.0025–0.005 U), 2.5–10 mM ATP, and 5–10 mM glucose (or 37.5 mM fructose), and the combined drops in the reference region contained 0.15 μM BSA, 2.5–10 mM ATP, and 5–10 mM glucose (or 37.5 mM fructose). The molar concentration of hexokinase was calculated assuming that the enzyme has a monomer molecular weight of 54 kDa with a maximum specific activity of 600 U/mg. This specific activity is a literature value for purified hexokinase that has been recrystallized five times and contains a mixture of two forms of the enzyme (I and II), which are indistinguishable in specific activity and KM values [7; 8]. All reagents were obtained from Sigma–Aldrich and used without further purification.
Parameters for experiment
The temperature noise of the VOx detectors is 10–30 μK for the best-of-class devices that were used for the measurements presented here [5]. For any given enzyme-catalyzed reaction, the temperature signal is proportional to [E], so the enzyme concentration needs to be high enough to yield a sufficient signal (400 μK or 0.4 μW) at maximum reaction velocity Vmax to give a signal-to-noise ratio of at least 4:1. At the same time, the steady-state enzymatic reaction must last long in comparison to initial transients such as the substrate dilution heat, requiring that the ratio of enzyme to substrate not be too large. With magnetic mixing, these transients typically last less than 10 seconds, so a 60 second enzymatic reaction is usually sufficient. For the reactions presented here, we pushed the enzyme concentrations toward the lower end of the ranges that would result in a good signal-to-noise ratio at Vmax, and the substrate concentration was then adjusted to produce the length of reaction desired. Typically, four replicates of each measurement were performed and the average and standard error of the mean of the measurements is reported. Measurements with inhibitors had a corresponding set of control measurements without inhibitor performed at the same time.
With other assay techniques, rate versus substrate concentration curves often are produced by measuring the initial velocity at different initial concentrations of substrate. In contrast, the measurements presented here yield rate versus substrate concentration from a single continuous reaction, conferring advantages such as fewer solutions to prepare and fewer measurements to perform. As a practical matter, enthalpy arrays are also not sensitive enough for obtaining accurate rate versus substrate concentration curves by measuring the initial velocity at different initial concentrations of substrate. Consider, for example, the drop in rate for the curve in Figure 2A corresponding to no inhibitor. Information about KM is embedded in the steepness of the drop, which occur over less than 3–5 seconds. The drop is resolvable in Figure 2A because it occurs after any initial transients at the time of drop merging. If one attempted to acquire comparable data by measurements of initial velocity at different initial substrate concentrations, the initial velocity at the same enzyme concentration would not be resolvable at the same level of precision due to interference by initial transients. In principle one could overcome this problem by lowering the enzyme concentration, thereby increasing the time over which the velocity remains essentially unchanged, but the signal would be much lower and the enthalpy arrays are not sensitive enough to enable such measurements.
Figure 2.

Trypsin hydrolysis of BAEE in the absence (black) and presence (red) of 1 mM benzamdine. Reactions contained 5 μM trypsin and 5 mM BAEE. (A) Rate of heat generation (Q) versus time.(B) Rate vs. remaining BAEE concentration, and fits to Eq. (5) (no inhibitor) and Eqs. (6)–(10) (1 mM benzamidine) shown as solid curves.
Data analysis
For each measurement we record the differential temperature as a function of time. We convert this information into an enthalpy by deconvolving Q(τ) from the equation
| (1) |
In this equation, T(t) is the temperature change (T) as a function of time (t) relative to the baseline temperature, Q(τ) is the rate of heat generation (e.g. due to a biochemical reaction) as a function of time, and 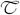 (t-τ), the “impulse response”, is the temperature rise that would occur at a time t given a Dirac delta function impulse of heat at time τ. To deconvolve Q(τ), we approximate Q(τ) as a piece-wise linear continuous function and use a least-squares regression to determine the fitting coefficients. Following calculation of Q(τ), we correct for thermal baseline shifts and integrate Q(τ) over time to obtain the total enthalpy qtotal [5].
(t-τ), the “impulse response”, is the temperature rise that would occur at a time t given a Dirac delta function impulse of heat at time τ. To deconvolve Q(τ), we approximate Q(τ) as a piece-wise linear continuous function and use a least-squares regression to determine the fitting coefficients. Following calculation of Q(τ), we correct for thermal baseline shifts and integrate Q(τ) over time to obtain the total enthalpy qtotal [5].
Next, Q(τ) is transformed into the reaction velocity as a function of substrate.
In the first step, we apply Savitzky-Golay filtering (order=4, frame size=31) to Q(τ) to smooth out higher frequency noise from the electronics [9].
In the second step, we fit the portion of the data corresponding to [S]»KM to
| (2) |
The [S]»KM range of the data is recognizable as a linear portion of Q(t) preceding the final drop-off (e.g. see Figure 2A). In Eq. (2), the variable kcat is the turnover number, [E] is the enzyme concentration, and ΔH is the enthalpy per mole of substrate reacted. For standard Michaelis-Menten kinetics and ideal solution enthalpies, β will be zero, but we show data (Figures 2A and 4A) for which there is a clear nonzero slope for Q versus [S] at [S] greater than the observed KM. The slope, quantified as a nonzero β, could arise either because the kinetics at high [S] do not follow Michaelis-Menten kinetics or because ΔH varies with concentration of substrate and/or product (i.e. non-ideal thermodynamics). In principle, either is possible. To distinguish one from the other, one can examine the total heat evolved over the entire course of the reaction,
Figure 4.

Effect of ATP concentration on KM, Kemptide. (A) Rate of heat generation (Q) versus time for phosphorylation of Kemptide by PKA. Reaction contained 2 μM PKA, 5 mM ATP, and 1 mM Kemptide. (B) Rate vs. remaining limiting substrate concentration, and fit to Eq. (5). (C) Reciprocal velocity versus 1/[Kemptide] at various [ATP]. The rate versus substrate data were fit using Eq. (5) and then converted to reciprocal velocity for display on the linear plot. 1.25 mM ATP (circles) gives KM, Kemptide of 50 μM, 2.5 mM ATP (squares) gives KM, Kemptide of 19 μM, and 5 mM ATP (triangles) gives KM, Kemptide of 16 μM.
| (3) |
for a range of initial substrate concentrations S0. If ΔH is constant, as it would be for ideal solutions, then the observed qtotal should vary linearly with S0. Thus, linear behavior of qtotal versus S0 can be regarded as evidence that a nonzero β in Eq. (2) corresponds to a deviation from simple Michaelis-Menten kinetics, rather than from non-ideal solution thermodynamics.
To fit β using Q(t) data and Eq. (2) requires [S] as a function of time t. Accordingly, we apply the fact that the rate of heat generation Q(t) equals the enthalpy of reaction per mole of substrate times the rate of substrate consumption to obtain the equation
| (4) |
We determine β and an initial estimate of kcat by performing a nonlinear least-squares regression at [S]»KM on Eq. (2) subject to Eq. (4). For this purpose we used the Matlab® function lsqcurvefit.
After we have determined β, we can fit the data for Q/([E]ΔH) versus [S] over the full range of [S], not just [S]»KM, to determine kinetic parameters. For example, one can determine kcat and KM for experiments with no inhibitor by fitting the data for Q/([E]ΔH) versus [S] to
| (5) |
In this equation, the (1+β[S]) term modifies the standard Michaelis-Menten kinetics. As a practical matter, the modification becomes significant at [S] »KM for the data we present below, or in other words, (1/β) »KM. Thus, it is clearly distinguishable from the effect of KM. As before, we use the Matlab® function lsqcurvefit to perform a regression on Eq. (5), using the value of β determined from the fit of Eq. (2) at [S]»KM. We also allow for the possibility that the values of [S] may be off by a small constant in the regression, which helps to accommodate for noise near the [S]=0 limit of the data.
We also examined inhibited reactions. We are able to observe a change in the apparent KM caused by a competitive inhibitor, allowing us to determine KI. To understand the meaning of an increase in apparent KM, it is useful to examine the Michaelis-Menten equation modified for competitive inhibition ([10], eqn. 3.32):
| (6) |
The apparent KM is seen to be the true KM multiplied by (1+[I]/KI). At inhibitor concentrations [I]>KI, the shift in apparent KM becomes significant. When [I]≫E0 holds, the concentration of free inhibitor [I] is close to the total concentration of inhibitor I0, making it reasonable to use I0 in the above equation in place of [I], the standard practice in enzymology. However, in our measurements [I]≫E0 is not always the case. Therefore, when [I]≫E0 does not hold, we perform the regression for KI using
| (7) |
where
| (8) |
| (9) |
| (10) |
At trace enzyme (E0≪I0), the right-hand side of Eq. (7) approaches I0, as expected, recovering the classic form of Eq. (6) for trace enzyme. Equation (7) derives from solving the following equations for [I] in terms of E0, I0, KM, and KI:
| (11) |
| (12) |
| (13) |
| (14) |
Thus, the regression analysis takes full account of compound depletion of inhibitor because it includes the mass-action consequences of the equilibria involving substrate and inhibitor.
Results
The design of the enthalpy array allows a set of sample and reference drops to be merged for each detector reaction (Figure 1). Subsequently, the temperature difference between the sample and reference region is recorded, beginning at the original substrate concentration (typically much greater than KM) and continuing beyond the point that substrate is completely consumed. As described in the methods, the temperature data was converted into the rate of heat generation as a function of time. Shown in Figure 2A is the rate of heat generation (Q) for trypsin hydrolysis of BAEE. Upon merging of the substrate and enzyme drops at t=0 sec, the signal rapidly increased because BAEE hydrolysis is exothermic and raised the temperature of the sample region relative to the reference region. As the enzyme converted substrate to product, the rate of heat generation showed a roughly linear behavior with a negative slope for approximately 70 seconds, after which time substrate depletion caused a decrease in rate and the signal returned to the baseline. The data in Figure 2A were integrated to give qtotal for the reaction. This value was used to determine the rate and the concentration of remaining substrate at any given time, as described in Material and Methods, yielding the black points in Figure 2B. Our measured value of ΔHapp (qtotal/moles of substrate) for hydrolysis of BAEE was −12.5 (±0.4) kcal/mol at 21 °C, compared to ΔHapp −11.5 kcal/mol reported at 25 °C [2]. Fitting the black points (no inhibitor) in Figure 2B yielded kcat=8.5 s−1 and KM=6.4 μM, in good agreement with the values reported in the literature for [BAEE]<1 mM(see Table 1) [11; 12] and with those obtained performing a standard spectrophotometric assay [13] using lower concentrations of the same trypsin and BAEE stock solutions used in the enthalpy array measurements (kcat=12 s−1 and KM=14 μM).
Table 1.
Kinetic parameters and inhibition constants obtained by enthalpy array versus published values.
| Enthalpy Array | Literature values | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Enzyme | Substrate | Inhibitor | KM (μM) | kcat (s−1) | KI (μM) | KM (μM) | kcat (s−1) | KI (μM) | |
| Trypsin | BAEE | None | 6.4 ±1.1 | 8.5 ±0.3 | 4a | 15a | [11; 12] | ||
| Trypsin | BAEE | benzamidine | 43 ±8b | 20 | [15] | ||||
| Trypsin | BAEE | leupeptin | 0.13 ±0.04c | 0.15 | [16] | ||||
| PKA | Kemptide | None | 16 ±4 | 8.5 ±0.5 | 5–16 | 11–14 | [6; 23] | ||
| PKA | Kemptide | Ala-peptide | 530 ±90b | 320–490 | [23; 29] | ||||
| PKA | Kemptide | PKI-tide | 0.66 ±0.04c | IC50= 0.2d | [32] | ||||
| Hexokinase | ATP, (glucose) | None | 180 ±30 | 250 ±10 | 95–200 | 270–450 | [8;20;21; 22] | ||
| Hexokinase | glucose | None | 94 ±8 | 250 ±10 | 84–167 | 270–450 | |||
| Hexokinase | ATP, (fructose) | None | 200 ±80 | 400 ±70 | 45–200 | 380–990e | [18;19; 21; 22] | ||
Values reported are the average of at least three measurements with the standard error of the mean indicated.
KM and kcat for [BAEE]<1mM [12].
KI values were derived using Equation (6), using the average value of KM in the absence of inhibitor.
KI values were derived using Equations (6)-(10), using the average value of KM in the absence of inhibitor.
IC50 value
Reported as Vmax relative to Vmax glucose (1.4−2.2X).
The nonzero slope of the linear region in Figure 2A is quantified by a nonzero β. As discussed in Materials and Methods, it is necessary to consider whether a nonzero β in this case corresponds to nonideal enthalpies or a deviation from Michaelis-Menten kinetics. From Figure 3B of Recht, et al [5], it can be seen that qtotal (described simply as “heat” in that reference) is linear in [BAEE] for hydrolysis of BAEE by trypsin. That result suggests that the nonzero β in our present results corresponds to a deviation from Michaelis-Menten kinetics. Indeed, upon recognizing this, we investigated the literature for hydrolysis of BAEE by trypsin more closely and found published evidence for substrate activation at [S]>KM [12; 14]. Under the conditions used here, we do not observe the plateau for the reaction rate because our highest substrate concentration is not high enough for that purpose, and therefore we could not fit the kinetic parameters for activation as described in the cited publications [12,14]. In theory, if much higher substrate concentration was used, one could obtain the kinetic constants for the activation, but this is beyond the scope of this study.
Figure 2A also shows the hydrolysis of BAEE in the presence of 1 mM benzamidine (red line), a known competitive inhibitor of trypsin [15]. Again, the rate of heat generation showed a roughly linear negative slope for approximately 60 seconds when [S]>KM. The signal returned to original levels as substrate was completely hydrolyzed, and in particular more slowly than the reaction in the absence of inhibitor. This difference reflects a change in apparent KM for BAEE in the presence of benzamidine. As expected, the areas described by the two curves (ΔHapp) were identical. Enzyme turnover vs. substrate concentration (Figure 2B) confirmed competitive inhibition insofar as the rates converged at high [BAEE], and a fit to Eqs. (6)–(10) yielded a KI of 43 μM (Table 1), using 6.4 μM as KM for the uninhibited reaction (the value determined from the earlier fit with no inhibitor).
The hydrolysis of BAEE in the presence of 10 μM leupeptin, a second known competitive inhibitor of trypsin, was also measured [16]. The data were fit to Eq. (6)–(10), again using 6.4 μM as KM for the uninhibited reaction, yielding a KI of 130 nM (Table 1). The KI values for benzamidine and leupeptin are in good agreement with the values reported in the literature [15; 16].
The hydrolysis of BAEE by trypsin is an example of an enzymatic reaction involving a single substrate. Many enzymes, such as kinases, involve two substrates. We now illustrate enthalpy array measurements on a two-substrate enzyme using hexokinase as the example. In the case of hexokinase, the two substrates are ATP and a hexose. The KM for one substrate can be determined by limiting its concentration and providing the second substrate at a high enough excess over its KM to assure that the concentration of the second substrate remains above ≈10x KM when the limiting substrate has reacted completely. Figure 3 shows rate vs. substrate data for hexokinase phosphorylation of glucose and fructose with limiting ATP substrate. Two types of experiments are shown in the figure: KM,ATP-glucose: limiting ATP (2.5 mM) with excess glucose (5 mM), and KM,ATP-fructose: limiting ATP (5 mM) with excess fructose (37.5 mM). The ΔHapp for phosphorylation of glucose was −13.9 (±0.9) kcal/mol at 21 °C, compared to ΔHapp ≈−14 kcal/mol reported at 25 °C (at pH 7.6) [17]. The ΔHapp for phosphorylation of fructose was −16.1 (±0.4) kcal/mol at 21 °C.
Figure 3.
Hexokinase phosphorylation of glucose or fructose. Rate vs. remaining limiting substrate concentration, and fits to Eq. (5) (shown as solid curves) for the phosphorylation of glucose (open circles) or fructose (squares). The glucose reaction contained 154 nM hexokinase, 10 mM ATP and 5 mM glucose. The fructose reaction contained 77 nM hexokinase, 5 mM ATP and 37.5 mM fructose.
As summarized in Table 1, for the glucose reaction we measured kcat = 250 s−1, KM,ATP-glucose = 180 μM, and KM,glucose = 94 μM. For the fructose reaction, we measured kcat = 400 s−1 and KM,ATP-fructose = 200 μM. These values are in reasonably good agreement with the range of values reported in the literature [8; 18; 19; 20; 21; 22].
Hexokinase has both a very high turnover number and substrate KM in the 100 μM range, and these characteristics allow us to choose both low enzyme and high substrate concentrations in enthalpy array measurements. Other cases can be more restrictive, as illustrated by our next example, enthalpy array measurements of phosphorylation by the cAMP dependent kinase PKA. PKA has kinetic parameters that better represent the majority of pharmaceutically relevant kinases, which typically have a lower turnover number (kcat < 10 s−1) and KM in the 10 μM range.
As shown in Figure 4A and 4B and summarized in Table 1, phosphorylation of Kemptide (1 mM) by PKA in the presence of excess ATP (5 mM) yielded KM, Kemptide and kcat values in good agreement with the range of values reported in the literature [6; 23]. The effect of varying the ATP concentration was measured and is summarized in Figure 4C. To produce a reaction of sufficient length to obtain kinetic data, the concentration of ATP was higher than the concentration of Kemptide (1 mM) and high above the reported KM of ATP (8 μM). For all ATP concentrations measured, the enzyme initially turns substrate at Vmax, but KM, Kemptide approaches the lowest value reported in the literature (5 μM) as the ATP concentration is increased.
Attempts to measure KM, ATP using limiting ATP (1–2.5 mM) and excess Kemptide (1.5–5 mM) did not produce data with the characteristic plateau in the rate achieved by an enzyme producing a steady-state turnover of substrate to product. It is possible that the high concentration of Kemptide used for these measurements (1.5–5 mM) resulted in substrate inhibition. It has been reported that Kemptide at high concentration (>1 mM) shows substrate inhibition of PKA, although those measurements were performed with a much lower ATP concentration than used here (1 μM versus 1–2.5 mM ATP) [23]. The kinetic mechanism of PKA reported in the literature suggests initiating the reaction by combining a drop containing Kemptide with a drop containing a mixture of PKA plus saturating ATP might alleviate the substrate inhibition by Kemptide [23; 24]. Reactions performed by combining a drop containing PKA and ATP with a drop containing Kemptide and ATP did not yield information on the KM of ATP (not shown). It is important to note that product inhibition by ADP (KI = 7.3 μM; [23]) might a factor because the enzyme concentration used here in the initial drop (4 μM) was high enough that the intrinsic ATPase activity of PKA catalytic subunit (0.74 min−1; [25]) produced significant ADP in the time between mixing the enzyme with ATP and the addition of the Kemptide.
There are a number of ATP-competitive inhibitors of PKA. The isoquinolinesulfonamide H-89 has been shown to act as an ATP-competitive inhibitor of the phosphorylation of histone IIb by PKA with a KI of 48 nM [26]. Although enthalpy array measurements could not be made with limiting ATP, we did observe inhibition of the phosphorylation of Kemptide by PKA in the presence of H-89. Figure 5A shows enzyme turnover vs. substrate for a series of measurements with increasing concentration of H-89 in the presence of 2.5 mM ATP. The reactions showed a clear decrease in kcat, but KM, Kemptide did not change significantly with increasing H-89 concentration (Table 2). When the reactions were performed at 5 mM ATP instead of 2.5 mM ATP, there was a smaller reduction in kcat and no significant change in KM, Kemptide as the concentration of H-89 was increased (Table 2). The pattern of inhibition was consistent with non-competitive inhibition to Kemptide (Figure 5B), but, as described above, the conditions necessary to measure inhibition in competition to ATP did not yield interpretable data in the absence of inhibitor.
Figure 5.

Effect of inhibitors on the phosphorylation of Kemptide by PKA. (A) Rate vs. remaining limiting substrate concentration in the presence of increasing concentration of H-89, and fits to Eq. (5) Reactions contained 2 μM PKA, 2.5 mM ATP, and 1 mM Kemptide. Inhibitor was added as indicated. No inhibitor (circles), 5 μM H-89 (squares) and 25 μM H-89 (diamonds). (B) Reciprocal velocity versus 1/[Kemptide] at 2.5 mM ATP with varying H-89. Lines are the non-linear fit to the data shown in (A). (C). Rate of heat generation (Q) versus time for phosphorylation of Kemptide by PKA in the absence (black) and presence of 4 μM staurosporine. Reactions contained 2 μM PKA, 5 mM ATP, and 1.5 mM Kemptide, 2% DMSO.
Table 2.
Inhibition of PKA by H-89. PKA present at 2 μM, Kemptide at 1 mM, ATP at the concentration indicated.
| 2.5 mM ATP | 5 mM ATP | |||
|---|---|---|---|---|
| [H-89] (μM) | kcat (s−1) | KM (μM) | kcat (s−1) | KM (μM) |
| 0 | 7 ±0.3 | 19 ±5 | 8.5 ±0.4 | 16 ±4 |
| 2.5 | 5.3 ±1.2 | 18 ±5 | 6.4 ±1.2 | 14 ±0.2 |
| 5 | 4.4 ±1.2 | 17 ±10 | 7.4 ±1.2 | 20 ±5 |
| 25 | 3.1 ±0.2 | 21 ±8 | 7.3 ±0.5 | 15 ±0.5 |
A second inhibitor, staurosporine, has been shown to be a potent ATP competitive inhibitor of PKA, with a reported KI of 5 nM [27]. As shown in Figure 5C, addition of staurosporine at the lowest concentration tested (4 μM) resulted in complete inhibition of the reaction, indicating the limit for which a quantitative assessment of KI can be made under these conditions.
For comparison with the ATP competitive inhibitors described above, reactions were performed with limiting Kemptide and excess ATP (5 mM) in the presence of 0.5 mM ADP. The reaction in the presence of ADP showed a >20% decrease in kcat, but no change in KM, Kemptide was observed, consistent with non-competitive inhibition to Kemptide (not shown).
In addition to the ATP-competitive inhibitors, a number of peptide-competitive inhibitors of PKA have been identified [28; 29; 30; 31; 32]. Figure 6A shows enzyme turnover versus Kemptide substrate in the presence of a 19 residue peptide (IAAGRTGRRQAIHDILVAA; PKI-tide) derived from the heat-stable inhibitor of PKA reported by Smith et al 1990 [32]. Fitting of the data to Eq. (6)–(10) confirms competitive inhibition with a KI for PKI-tide of 660 nM, consistent with the IC50 of 200 nM reported in the literature (Table 1) [32].
Figure 6.

Effect of peptide inhibitors on the phosphorylation of Kemptide by PKA. (A) Rate of enzyme turnover vs. remaining limiting substrate in the absence and presence of PKI-tide. Reactions contained 2 μM PKA, 5 mM ATP, and 1.5 mM Kemptide. Inhibitor was added as indicated. No inhibitor (circles), 4 μM PKI-tide (triangles). Data shown were fit to Eq. (5) or Eq. (6)-(10). (B) Rate of enzyme turnover vs. remaining limiting substrate in the absence and presence of increasing concentration of Ala-peptide, and fits to Eq (6). Reactions contained 2 μM PKA, 5 mM ATP, and 1 mM Kemptide. Inhibitor was added as indicated. (C) Reciprocal velocity versus 1/[Kemptide] at 5 mM ATP with varying Ala-peptide. Lines are the non-linear fit to the data shown in (B).
The alanine-substituted nonphosphorylatable analog of Kemptide (Ala-peptide) inhibits the phosphorylation of Kemptide [29]. At saturating ATP, Ala-peptide was competitive with Kemptide (Figure 6B and 6C) with a KI of 530 μM (Table 1). This is consistent with the KI values for Ala-peptide reported in the literature, which are between 320 and 490 μM depending on assay conditions [23; 29].
Discussion
We have presented examples of enzyme kinetics studies using enthalpy array technology with two different classes of enzymes. Using a continuous assay, a single experiment taking a total of 8 minutes (drop deposition, equilibration and enzymatic reaction) can be used to accurately determine KM and kcat for an enzymatic reaction that does not exhibit substrate or product inhibition. This assay has been used here to measure kcat from 10 s−1 (PKA with Kemptide) to 400 s−1 (hexokinase with fructose), as well as determine KM from 6 μM (trypsin with BAEE) to 200 μM (hexokinase with ATP), in good agreement with the published values for each of these enzymes measured by other techniques.
Once the KM for the reaction in the absence of inhibitor is known, the KI for competitive inhibitors can be assayed, as demonstrated here for benzamidine and leupeptin with trypsin and the Ala-peptide and PKI-tide with PKA. The limit on the KI that can be accurately measured is determined by the concentration of enzyme that is necessary to obtain a good signal-to-noise ratio for the uninhibited reaction. For weak inhibitors, such as benzamidine and the Ala-peptide, the inhibitor is present in high excess over enzyme (100 to 1000-fold) and the standard analysis using the increase in the observed KM for the substrate in the presence of inhibitor may be employed to calculate KI. For strong inhibitors, the inhibitor should be present at a slight excess (1.5 to 2-fold) over enzyme to ensure that all enzyme has inhibitor bound, and the data is analyzed using Eqs. (6) through (10). We estimate that the lower limit for which KI can be determined for a potent inhibitor is ≈1/50 the enzyme concentration, consistent with the data presented here for leupeptin (KI ≈ 1/40 enzyme concentration) and PKI-tide (KI ≈ 1/4 enzyme concentration).
One of the advantages calorimetry has over photometric assays is the ability to measure spectroscopically opaque solutions. Due to the relatively high initial substrate concentrations used for the measurements described here, it is important to note that deviations from simple Michaelis-Menten kinetics may be observed. These deviations may take the form of substrate activation, as previously reported for the trypsin/BAEE reaction [12], substrate inhibition, as previously reported for the PKA/Kemptide reaction [23], or product inhibition, also observed for PKA. The effects of substrate activation and substrate inhibition have been described in the results. If product inhibition occurs during the continuous assay, it could affect the observed KM, as the concentration of product can approach the millimolar range at the end of the reaction. If one is assaying a well-characterized enzyme and the product inhibition constant is known, this term could be included in a reaction mechanism-based model to fit the data rather than relying on a simple Michaelis-Menten model. This may provide an explanation why the apparent KM of Kemptide is lower with a higher concentration of ATP even though all the ATP concentrations used were in vast excess of KM(ATP). Product inhibition by ADP (KI = 7.3 μM; [23]) may alter the apparent KM of Kemptide derived from the fitting of the data to a simple Michaelis-Menten model because there is an insufficient concentration of ATP compared to the ADP concentration to keep the enzyme saturated with ATP during the complete continuous assay. Toward the latter part of the assay, the rate of the reaction will be reduced due to limiting ATP in addition to the rate decrease due to depletion of Kemptide.
Further improvements in the sensitivity of the technology will allow use of lower enzyme and substrate concentrations, reduction or elimination of the effects of product inhibition, and expansion of the number of enzymes and inhibitors that can be assayed with the technology. Accordingly, enthalpy arrays should prove very useful in drug discovery by providing label-free secondary assays for hit validation and lead characterization.
Acknowledgments
The authors thank Lorenz M. Mayr for support in establishing this collaboration as well as helpful discussion, Peter Kuhn for assistance in initiating this collaboration, Lai Wong, Vicki Geluz-Aguilar and Mary Anne Rosenthal for their assistance in device fabrication and Sasha Tuganov for assistance with the implementation of magnetic mixing.
Footnotes
This work was supported by NIH grant R01GM077435.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bianconi ML. Calorimetry of enzyme-catalyzed reactions. Biophys Chem. 2007;126:59–64. doi: 10.1016/j.bpc.2006.05.017. [DOI] [PubMed] [Google Scholar]
- 2.Todd MJ, Gomez J. Enzyme kinetics determined using calorimetry: a general assay for enzyme activity? Anal Biochem. 2001;296:179–87. doi: 10.1006/abio.2001.5218. [DOI] [PubMed] [Google Scholar]
- 3.Olsen SN. Applications of isothermal titration calorimetry to measure enzyme kinetics and activity in complex solutions. Thermochimica Acta. 2006;448:12–18. [Google Scholar]
- 4.Torres FE, Kuhn P, De Bruyker D, Bell AG, Wolkin MV, Peeters E, Williamson JR, Anderson GB, Schmitz GP, Recht MI, Schweizer S, Scott LG, Ho JH, Elrod SA, Schultz PG, Lerner RA, Bruce RH. Enthalpy arrays. Proc Natl Acad Sci U S A. 2004;101:9517–22. doi: 10.1073/pnas.0403573101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Recht MI, De Bruyker D, Bell AG, Wolkin MV, Peeters E, Anderson GB, Kolatkar AR, Bern MW, Kuhn P, Bruce RH, Torres FE. Enthalpy array analysis of enzymatic and binding reactions. Anal Biochem. 2008;377:33–9. doi: 10.1016/j.ab.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kemp BE, Graves DJ, Benjamini E, Krebs EG. Role of multiple basic residues in determining the substrate specificity of cyclic AMP-dependent protein kinase. J Biol Chem. 1977;252:4888–94. [PubMed] [Google Scholar]
- 7.Darrow RA, Colowick SP. Hexokinase from Baker’s yeast: ATP+Hexose-->ADP+Hexose-6-phosphate+H+ Methods Enzymol. 1962;5:226. [Google Scholar]
- 8.Kaji A, Trayser KA, Colowick SP. Multiple forms of yeast hexokinase. Ann N Y Acad Sci. 1961;94:798–811. doi: 10.1111/j.1749-6632.1961.tb35574.x. [DOI] [PubMed] [Google Scholar]
- 9.Savitzky A, Golay MJE. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Analytical Chemistry. 1964;36:1627. [Google Scholar]
- 10.Fersht A. Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. W.H. Freeman; New York: 1999. [Google Scholar]
- 11.Inagami T, Sturtevant JM. Nonspecific catalyses by alpha-chymotrypsin and trypsin. J Biol Chem. 1960;235:1019–23. [PubMed] [Google Scholar]
- 12.Trowbridge CG, Krehbiel A, Laskowski M., Jr Substrate Activation Of Trypsin. Biochemistry. 1963;2:843–50. doi: 10.1021/bi00904a037. [DOI] [PubMed] [Google Scholar]
- 13.Schwert GW, Takenaka Y. A spectrophotometric determination of trypsin and chymotrypsin. Biochim Biophys Acta. 1955;16:570–5. doi: 10.1016/0006-3002(55)90280-8. [DOI] [PubMed] [Google Scholar]
- 14.Trenholm HL, Spomer WE, Wootton JF. Substrate Activation of Trypsin. The Effect of Enzyme Acetylation1. Journal of the American Chemical Society. 1966;88:4281. [Google Scholar]
- 15.East EJ, Trowbridge CG. Binding of benzoamidine and protons to trypsin as measured by difference spectra. Arch Biochem Biophys. 1968;125:334–43. doi: 10.1016/0003-9861(68)90669-3. [DOI] [PubMed] [Google Scholar]
- 16.Umezawa H. Low-molecular-weight enzyme inhibitors of microbial origin. Annu Rev Microbiol. 1982;36:75–99. doi: 10.1146/annurev.mi.36.100182.000451. [DOI] [PubMed] [Google Scholar]
- 17.Bianconi ML. Calorimetric determination of thermodynamic parameters of reaction reveals different enthalpic compensations of the yeast hexokinase isozymes. J Biol Chem. 2003;278:18709–13. doi: 10.1074/jbc.M211103200. [DOI] [PubMed] [Google Scholar]
- 18.Berger L, Slein MW, Colowick SP, Cori CF. Isolation of hexokinase from baker’s yeast. J Gen Physiol. 1946;29:379. [PMC free article] [PubMed] [Google Scholar]
- 19.DelaFuente G, Sols A. The kinetics of yeast hexokinase in the light of the induced fit involved in the binding of its sugar substrate. Eur J Biochem. 1970;16:234–9. doi: 10.1111/j.1432-1033.1970.tb01076.x. [DOI] [PubMed] [Google Scholar]
- 20.Fromm HJ, Zewe V. Kinetic studies of yeast hexokinase. J Biol Chem. 1962;237:3027–32. [PubMed] [Google Scholar]
- 21.Slein MW, Cori GT, Cori CF. A comparative study of hexokinase from yeast and animal tissues. J Biol Chem. 1950;186:763–80. [PubMed] [Google Scholar]
- 22.Zewe V, Fromm HJ, Fabiano R. The Effect Of Manganous Ion On The Kinetics And Mechanism Of The Yeast Hexokinase Reaction. J Biol Chem. 1964;239:1625–34. [PubMed] [Google Scholar]
- 23.Whitehouse S, Feramisco JR, Casnellie JE, Krebs EG, Walsh DA. Studies on the kinetic mechanism of the catalytic subunit of the cAMP-dependent protein kinase. J Biol Chem. 1983;258:3693–701. [PubMed] [Google Scholar]
- 24.Adams JA, Taylor SS. Divalent metal ions influence catalysis and active-site accessibility in the cAMP-dependent protein kinase. Protein Sci. 1993;2:2177–86. doi: 10.1002/pro.5560021217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Armstrong RN, Kondo H, Kaiser ET. Cyclic AMP-dependent ATPase activity of bovine heart protein kinase. Proc Natl Acad Sci U S A. 1979;76:722–5. doi: 10.1073/pnas.76.2.722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–72. [PubMed] [Google Scholar]
- 27.Meggio F, Donella Deana A, Ruzzene M, Brunati AM, Cesaro L, Guerra B, Meyer T, Mett H, Fabbro D, Furet P, et al. Different susceptibility of protein kinases to staurosporine inhibition. Kinetic studies and molecular bases for the resistance of protein kinase CK2. Eur J Biochem. 1995;234:317–22. doi: 10.1111/j.1432-1033.1995.317_c.x. [DOI] [PubMed] [Google Scholar]
- 28.Cheng HC, Kemp BE, Pearson RB, Smith AJ, Misconi L, Van Patten SM, Walsh DA. A potent synthetic peptide inhibitor of the cAMP-dependent protein kinase. J Biol Chem. 1986;261:989–92. [PubMed] [Google Scholar]
- 29.Feramisco JR, Krebs EG. Inhibition of cyclic AMP-dependent protein kinase by analogues of a synthetic peptide substrate. J Biol Chem. 1978;253:8968–71. [PubMed] [Google Scholar]
- 30.Glass DB, Cheng HC, Kemp BE, Walsh DA. Differential and common recognition of the catalytic sites of the cGMP-dependent and cAMP-dependent protein kinases by inhibitory peptides derived from the heat-stable inhibitor protein. J Biol Chem. 1986;261:12166–71. [PubMed] [Google Scholar]
- 31.Glass DB, Cheng HC, Mende-Mueller L, Reed J, Walsh DA. Primary structural determinants essential for potent inhibition of cAMP-dependent protein kinase by inhibitory peptides corresponding to the active portion of the heat-stable inhibitor protein. J Biol Chem. 1989;264:8802–10. [PubMed] [Google Scholar]
- 32.Smith MK, Colbran RJ, Soderling TR. Specificities of autoinhibitory domain peptides for four protein kinases. Implications for intact cell studies of protein kinase function. J Biol Chem. 1990;265:1837–40. [PubMed] [Google Scholar]